
Role of m6A RNA Methylation in Thyroid Cancer Cell Lines

 , and
, and 
Abstract
:1. Introduction
2. Results
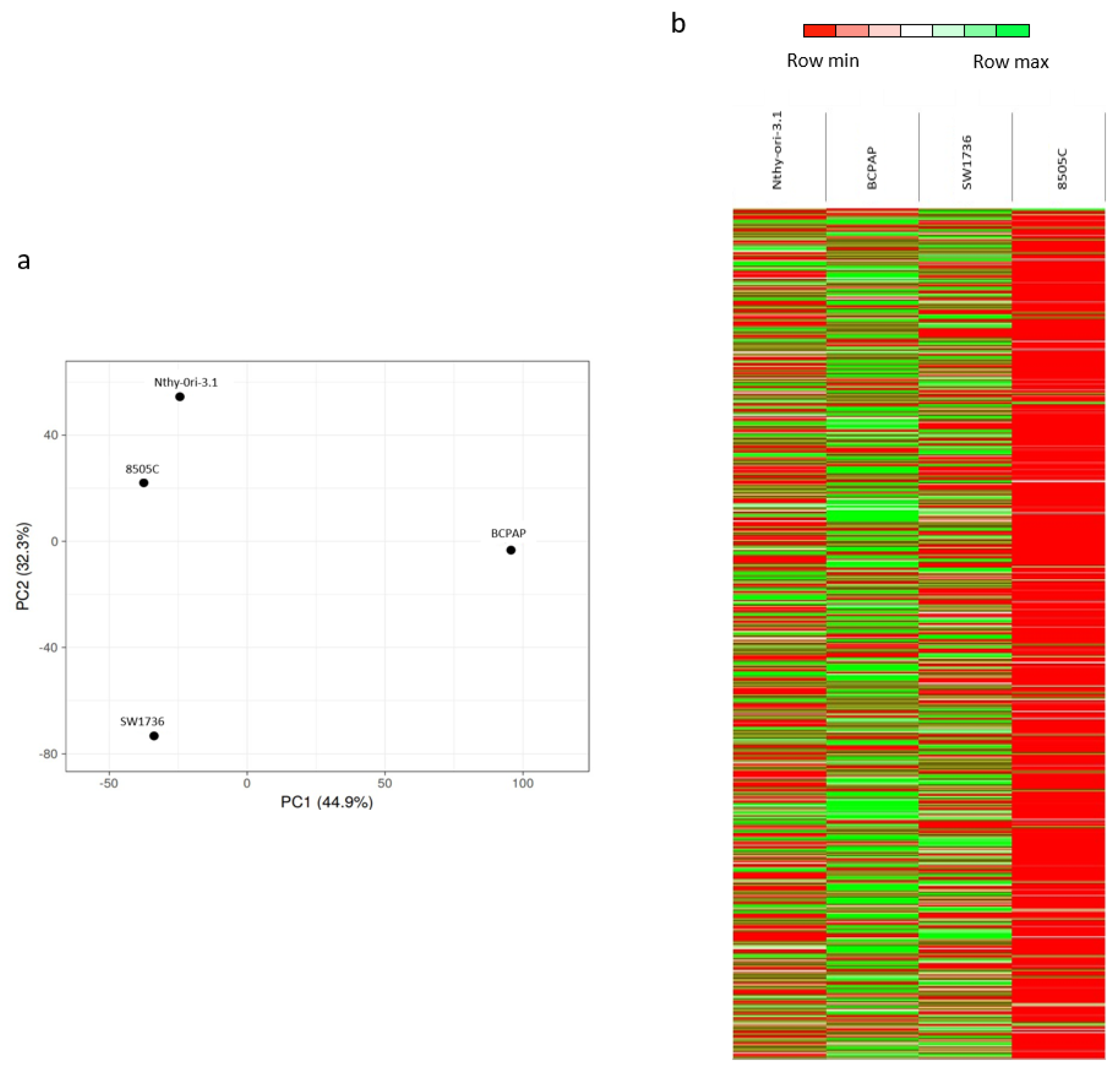
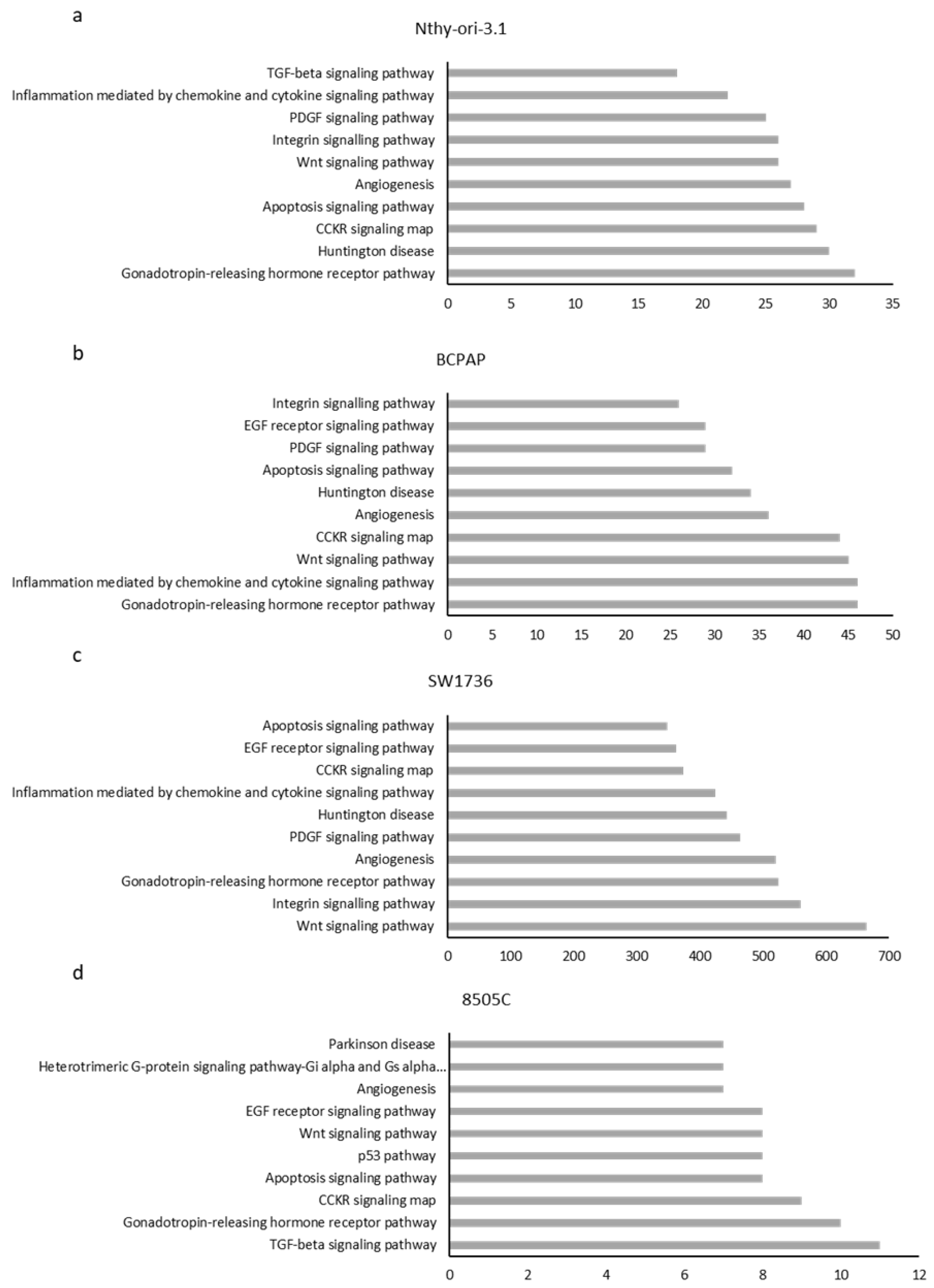
2.1. m6A Patterns in Thyroid Cells
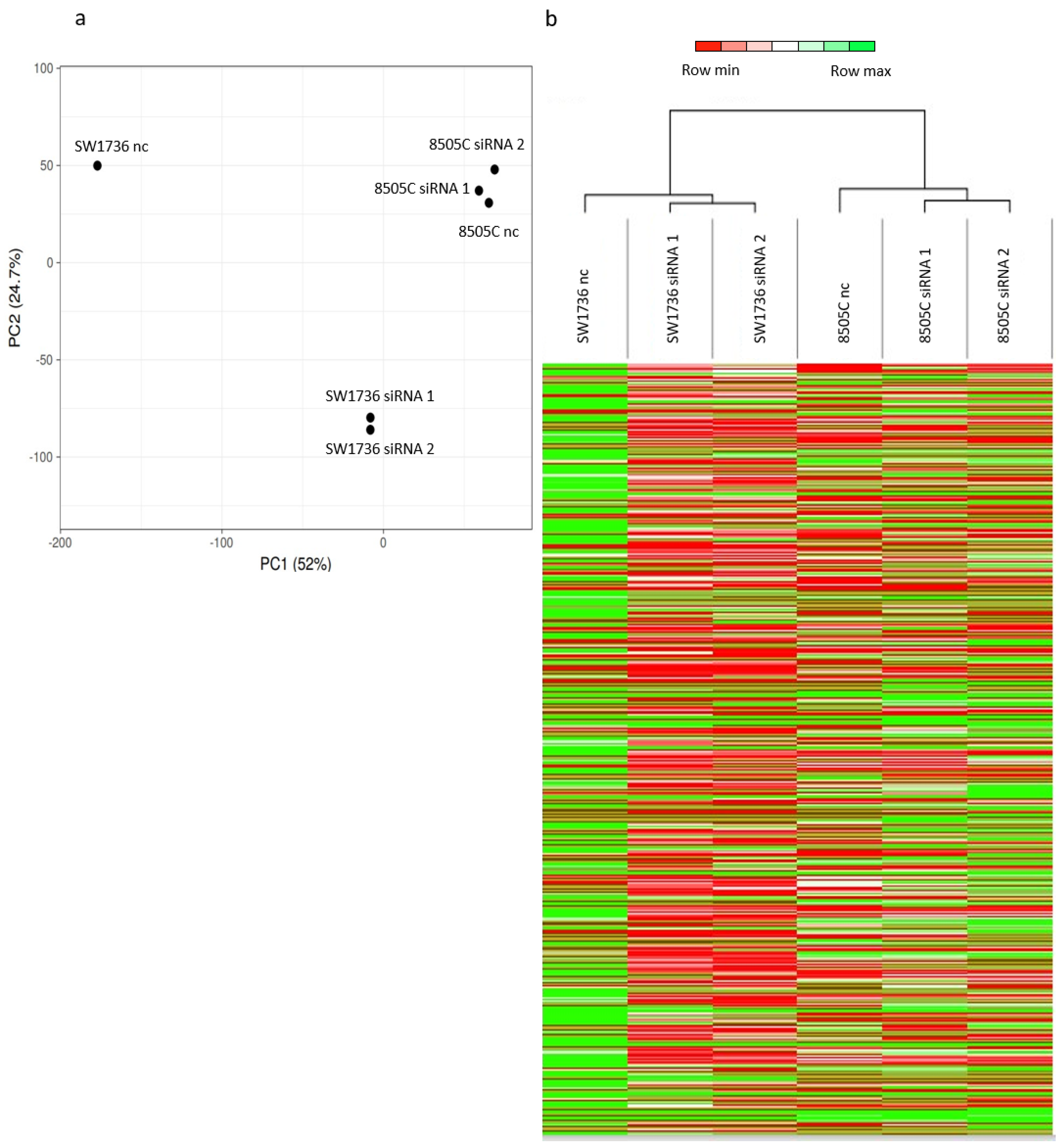
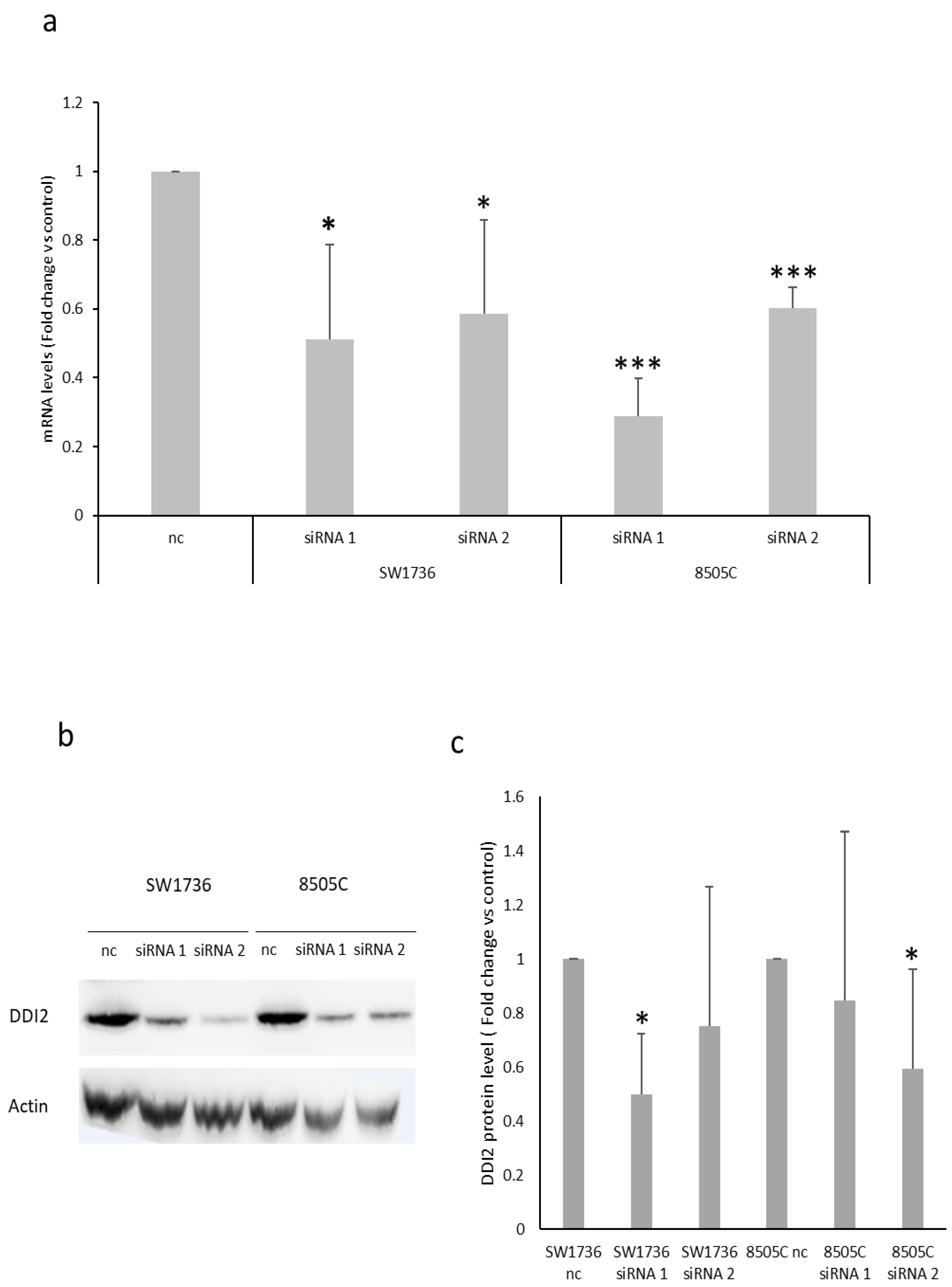
2.2. Effects of METTL3 Silencing in ATC Cell Lines
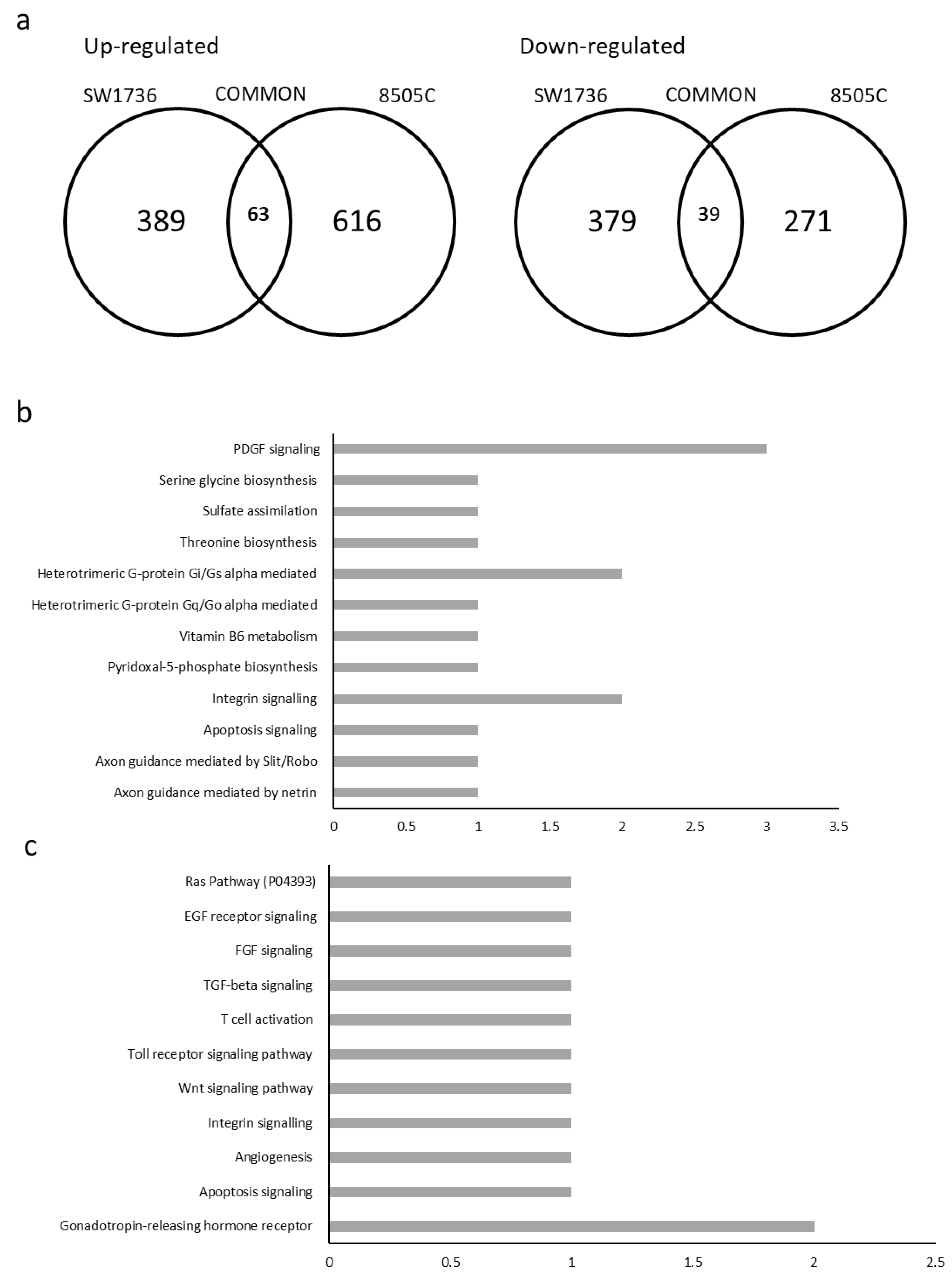
2.3. METTL3 Silencing Effects on Gene Expression
2.4. Relations between meRIP-seq and Effects of METTL3 Silencing on Gene Transcription
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Methylated RNA Immunoprecipitation
4.3. meRIP-seq Library Preparation and Sequencing
4.4. meRIP-Seq Bioinformatics Analysis
4.5. RNA Extraction and High-throughput Sequencing
4.6. METTL3 Silencing
4.7. Total m6A Abundance Assay
4.8. Cell Viability
4.9. Target Gene Expression Assay
4.10. Protein Extraction and Western Blot
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Abe, I.; Lam, A.K.-Y. Anaplastic thyroid carcinoma: Updates on WHO classification, clinicopathological features and staging. Histol. Histopathol. 2021, 36, 239–248. [Google Scholar] [PubMed]
- Lim, H.; Devesa, S.S.; Sosa, J.A.; Check, D.; Kitahara, C.M. Trends in Thyroid Cancer Incidence and Mortality in the United States, 1974–2013. JAMA 2017, 317, 1338–1348. [Google Scholar] [CrossRef] [PubMed]
- Cabanillas, M.E.; McFadden, D.G.; Durante, C. Thyroid cancer. Lancet 2016, 388, 2783–2795. [Google Scholar] [CrossRef]
- O’Neill, J.P.; Shaha, A.R. Anaplastic thyroid cancer. Oral Oncol. 2013, 49, 702–706. [Google Scholar] [CrossRef] [PubMed]
- Landa, I.; Ibrahimpasic, T.; Boucai, L.; Sinha, R.; Knauf, J.A.; Shah, R.H.; Dogan, S.; Ricarte-Filho, J.C.; Krishnamoorthy, G.P.; Xu, B.; et al. Genomic and transcriptomic hallmarks of poorly differentiated and anaplastic thyroid cancers. J. Clin. Investig. 2016, 126, 1052–1066. [Google Scholar] [CrossRef]
- Porter, A.; Wong, D.J. Perspectives on the Treatment of Advanced Thyroid Cancer: Approved Therapies, Resistance Mechanisms, and Future Directions. Front. Oncol. 2021, 10, 592202. [Google Scholar] [CrossRef]
- Subbiah, V.; Kreitman, R.J.; Wainberg, Z.A.; Cho, J.Y.; Schellens, J.H.M.; Soria, J.C.; Wen, P.Y.; Zielinski, C.; Cabanillas, M.E.; Urbanowitz, G.; et al. Dabrafenib and Trametinib Treatment in Patients With Locally Advanced or Metastatic BRAF V600–Mutant Anaplastic Thyroid Cancer. J. Clin. Oncol. 2018, 36, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Bible, K.C.; Kebebew, E.; Brierley, J.; Brito, J.P.; Cabanillas, M.E.; Clark, T.J., Jr.; Di Cristofano, A.; Foote, R.; Giordano, T.; Kasperbauer, J.; et al. 2021 American Thyroid Association Guidelines for Management of Patients with Anaplastic Thyroid Cancer. Thyroid 2021, 31, 337–386. [Google Scholar] [CrossRef]
- Alobuia, W.; Gillis, A.; Kebebew, E. Contemporary Management of Anaplastic Thyroid Cancer. Curr. Treat. Options Oncol. 2020, 21, 78. [Google Scholar] [CrossRef]
- Krug, R.M.; Morgan, M.A.; Shatkin, A.J. Influenza viral mRNA contains internal N6-methyladenosine and 5’-terminal 7-methylguanosine in cap structures. J. Virol. 1976, 20, 45–53. [Google Scholar] [CrossRef] [Green Version]
- Beemon, K.; Keith, J. Localization of N6-methyladenosine in the Rous sarcoma virus genome. J. Mol. Biol. 1977, 113, 165–179. [Google Scholar] [CrossRef]
- Dai, D.; Wang, H.; Zhu, L.; Jin, H.; Wang, X. N6-methyladenosine links RNA metabolism to cancer progression. Cell Death Dis. 2018, 9, 124. [Google Scholar] [CrossRef] [PubMed]
- Batista, P.J. The RNA Modification N 6 -methyladenosine and Its Implications in Human Disease. Genom. Proteom. Bioinform. 2017, 15, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; I Gregory, R. RNAmod: An integrated system for the annotation of mRNA modifications. Nucleic Acids Res. 2019, 47, W548–W555. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Zhou, K.I.; Parisien, M.; Dai, Q.; Diatchenko, L.; Pan, T. N6-methyladenosine alters RNA structure to regulate binding of a low-complexity protein. Nucleic Acids Res. 2017, 45, 6051–6063. [Google Scholar] [CrossRef]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012, 485, 201–206. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Y.; Toth, J.I.; Petroski, M.D.; Zhang, Z.; Zhao, J.C. N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat. Cell Biol. 2014, 16, 191–198. [Google Scholar] [CrossRef]
- Jia, G.; Fu, Y.; Zhao, X.; Dai, Q.; Zheng, G.; Yang, Y.; Yi, C.; Lindahl, T.; Pan, T.; Yang, Y.-G.; et al. N6-Methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 2011, 7, 885–887. [Google Scholar] [CrossRef]
- Liu, J.; Yue, Y.; Han, D.; Wang, X.; Fu, Y.; Zhang, L.; Jia, G.; Yu, M.; Lu, Z.; Deng, X.; et al. A METTL3–METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 2014, 10, 93–95. [Google Scholar] [CrossRef]
- Chen, X.; Xu, M.; Xu, X.; Zeng, K.; Liu, X.; Sun, L.; Pan, B.; He, B.; Pan, Y.; Sun, H.; et al. RETRACTED: METTL14 Suppresses CRC Progression via Regulating N6-Methyladenosine-Dependent Primary miR-375 Processing. Mol. Ther. J. Am. Soc. Gene Ther. 2019, 28, 599–612. [Google Scholar] [CrossRef] [Green Version]
- Bokar, J.A.; Shambaugh, M.E.; Polayes, D.; Matera, A.G.; Rottman, F.M. Purification and cDNA cloning of the AdoMet-binding subunit of the human mRNA (N6-adenosine)-methyltransferase. RNA 1997, 3, 1233–1247. [Google Scholar] [PubMed]
- Wang, X.; Huang, J.; Zou, T.; Yin, P. Human m6A writers: Two subunits, 2 roles. RNA Biol. 2017, 14, 300–304. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Feng, J.; Xue, Y.; Guan, Z.; Zhang, D.; Liu, Z.; Gong, Z.; Huang, J.; Tang, C.; Zou, T.; et al. Structural basis of N(6)-adenosine methylation by the METTL3–METTL14 complex. Nature 2016, 534, 575–578. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Kong, S.; Tao, M.; Ju, S. The potential role of RNA N6-methyladenosine in Cancer progression. Mol. Cancer 2020, 19, 88. [Google Scholar] [CrossRef]
- Bokar, J.A.; Rath-Shambaugh, M.E.; Ludwiczak, R.; Narayan, P.; Rottman, F. Characterization and partial purification of mRNA N6-adenosine methyltransferase from HeLa cell nuclei. Internal mRNA methylation requires a multisubunit complex. J. Biol. Chem. 1994, 269, 17697–17704. [Google Scholar] [CrossRef]
- Luo, J.; Liu, H.; Luan, S.; He, C.; Li, Z. Aberrant Regulation of mRNA m6A Modification in Cancer Development. Int. J. Mol. Sci. 2018, 19, 2515. [Google Scholar] [CrossRef]
- Chang, G.; Leu, J.-S.; Ma, L.; Xie, K.; Huang, S. Methylation of RNA N6-methyladenosine in modulation of cytokine responses and tumorigenesis. Cytokine 2019, 118, 35–41. [Google Scholar] [CrossRef]
- He, L.; Li, H.; Wu, A.; Peng, Y.; Shu, G.; Yin, G. Functions of N6-methyladenosine and its role in cancer. Mol. Cancer 2019, 18, 176. [Google Scholar] [CrossRef]
- Haugen, B.R.; Alexander, E.K.; Bible, K.C.; Doherty, G.M.; Mandel, S.J.; Nikiforov, Y.E.; Pacini, F.; Randolph, G.W.; Sawka, A.M.; Schlumberger, M.; et al. 2015 American Thyroid Association Management Guidelines for Adult Patients with Thyroid Nodules and Differentiated Thyroid Cancer: The American Thyroid Association Guidelines Task Force on Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid Off. J. Am. Thyroid Assoc. 2016, 26, 1–133. [Google Scholar] [CrossRef]
- Saini, S.; Tulla, K.; Maker, A.V.; Burman, K.D.; Prabhakar, B.S. Therapeutic advances in anaplastic thyroid cancer: A current perspective. Mol. Cancer 2018, 17, 154. [Google Scholar] [CrossRef] [Green Version]
- Chintakuntlawar, A.V.; Foote, R.L.; Kasperbauer, J.L.; Bible, K.C. Diagnosis and Management of Anaplastic Thyroid Cancer. Endocrinol. Metab. Clin. N. Am. 2019, 48, 269–284. [Google Scholar] [CrossRef] [PubMed]
- Allegri, L.; Domenis, R.; Navarra, M.; Celano, M.; Russo, D.; Capriglione, F.; Damante, G.; Baldan, F. Dihydrotanshinone exerts antitumor effects and improves the effects of cisplatin in anaplastic thyroid cancer cells. Oncol. Rep. 2021, 46, 204. [Google Scholar] [CrossRef] [PubMed]
- Allegri, L.; Baldan, F.; Roy, S.; Aubé, J.; Russo, D.; Filetti, S.; Damante, G. The HuR CMLD-2 inhibitor exhibits antitumor effects via MAD2 downregulation in thyroid cancer cells. Sci. Rep. 2019, 9, 7374. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Zhou, M.; Yin, J.; Wan, J.; Chu, J.; Jia, J.; Sheng, J.; Wang, C.; Yin, H.; He, F. METTL3 restrains papillary thyroid cancer progression via m6A/c-Rel/IL-8-mediated neutrophil infiltration. Mol. Ther. J. Am. Soc. Gene Ther. 2021, 29, 1821–1837. [Google Scholar] [CrossRef]
- Zhu, Y.; Peng, X.; Zhou, Q.; Tan, L.; Zhang, C.; Lin, S.; Long, M. METTL3-mediated m6A modification of STEAP2 mRNA inhibits papillary thyroid cancer progress by blocking the Hedgehog signaling pathway and epithelial-to-mesenchymal transition. Cell Death Dis. 2022, 13, 358. [Google Scholar] [CrossRef]
- Allegri, L.; Mio, C.; Russo, D.; Filetti, S.; Baldan, F. Effects of HuR downregulation on anaplastic thyroid cancer cells. Oncol. Lett. 2018, 15, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Tomei, S.; Marchetti, I.; Zavaglia, K.; Lessi, F.; Apollo, A.; Aretini, P.; Di Coscio, G.; Bevilacqua, G.; Mazzanti, C. A molecular computational model improves the preoperative diagnosis of thyroid nodules. BMC Cancer 2012, 12, 396. [Google Scholar] [CrossRef]
- Zhang, Z.; Chen, L.Q.; Zhao, Y.L.; Yang, C.G.; Roundtree, I.A.; Zhang, Z.; Ren, J.; Xie, W.; He, C.; Luo, G.Z. Single-base mapping of m6A by an antibody-independent method. Sci. Adv. 2019, 5, eaax0250. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [Green Version]
- Metsalu, T.; Vilo, J. ClustVis: A web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 2015, 43, W566–W570. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Total m6A-Enriched mRNAs | Specific m6A-Enriched mRNAs |
|---|---|---|
| Nthy-ori-3.1 | 4672 | 710 |
| BCPAP | 6296 | 1758 |
| SW1736 | 5134 | 1115 |
| 8505C | 1626 | 97 |
| ATC | 1286 | 74 |
| Nthy-ori-3.1 | BCPAP | SW1736 | 8505C | ||||
|---|---|---|---|---|---|---|---|
| mRNA | log 2 (m6A/IgG) | mRNA | log 2 (m6A/IgG) | mRNA | log 2 (m6A/IgG) | mRNA | log 2 (m6A/IgG) |
| MRPS24 | 23.5 | VKORC1 | 23.4 | NDUFB10 | 20.8 | CBS | 17.9 |
| SAT2 | 22.9 | NDUFA7 | 23.4 | COX5B | 20.5 | EMC6 | 17.8 |
| APRT | 22.8 | SPANXB1 | 23.2 | MRPL40 | 20.3 | UTP14C | 17.3 |
| RNF181 | 22.2 | NCBP2 | 22.9 | MRPS24 | 20.0 | PILRB | 17.2 |
| KCTD14 | 22.2 | SWI5 | 22.9 | MRPL14 | 19.8 | RDH14 | 17.1 |
| MRPL52 | 22.1 | C12orf45 | 22.6 | CD3EAP | 19.5 | NPIPA5 | 16.7 |
| U2AF1 | 21.9 | HNRNPH2 | 22.6 | SDF2L1 | 19.4 | SAPCD1 | 16.2 |
| ZNF593 | 21.9 | IFI6 | 22.6 | NR2F6 | 19.4 | SMIM11 | 16.0 |
| MED11 | 21.8 | AAMDC | 22.4 | RPP21 | 19.4 | PPAN | 15.9 |
| SURF2 | 21.7 | MYL6B | 22.3 | MRPS17 | 19.3 | C21orf33 | 15.7 |
| 21.6 | SURF2 | 22.3 | PALM2 | 19.2 | ICAM2 | 15.6 | |
| PAM16 | 21.6 | DUSP23 | 22.3 | APRT | 19.0 | NPIPA2 | 15.6 |
| DUS1L | 21.5 | TRPT1 | 22.2 | MND1 | 19.0 | GPR89B | 15.5 |
| MRPL55 | 21.5 | NKAP | 22.2 | MITD1 | 18.9 | C9orf116 | 15.5 |
| SSNA1 | 21.4 | C17orf49 | 22.2 | ISOC2 | 18.9 | TRIM34 | 15.5 |
| POLR2M | 21.4 | MED11 | 22.1 | GPKOW | 18.9 | C1orf53 | 15.4 |
| NUPR1 | 21.4 | PSMB8 | 22.1 | U2AF1 | 18.9 | DNLZ | 15.3 |
| CSNK1E | 21.3 | RP9 | 22.0 | FKBP11 | 18.8 | GSTT2B | 15.3 |
| SDF2 | 21.3 | U2AF1 | 21.9 | FDX1L | 18.7 | LIN7B | 15.2 |
| FDX1L | 21.3 | EAPP | 21.9 | MRPS12 | 18.7 | FAM156A | 15.2 |
| Up-Regulated | Down-Regulated | |
|---|---|---|
| SW1736 | 122 | 94 |
| 8505C | 81 | 33 |
| Common | 6 | 1 |
| Gene | Regulation |
|---|---|
| ELF2 | Up-regulated |
| MUC1 | Up-regulated |
| ROM1 | Up-regulated |
| TCEANC | Up-regulated |
| THNSL1 | Up-regulated |
| ZNF263 | Up-regulated |
| DDI2 | Down-regulated |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Allegri, L.; Baldan, F.; Molteni, E.; Mio, C.; Damante, G. Role of m6A RNA Methylation in Thyroid Cancer Cell Lines. Int. J. Mol. Sci. 2022, 23, 11516. https://doi.org/10.3390/ijms231911516
Allegri L, Baldan F, Molteni E, Mio C, Damante G. Role of m6A RNA Methylation in Thyroid Cancer Cell Lines. International Journal of Molecular Sciences. 2022; 23(19):11516. https://doi.org/10.3390/ijms231911516
Chicago/Turabian StyleAllegri, Lorenzo, Federica Baldan, Elisabetta Molteni, Catia Mio, and Giuseppe Damante. 2022. "Role of m6A RNA Methylation in Thyroid Cancer Cell Lines" International Journal of Molecular Sciences 23, no. 19: 11516. https://doi.org/10.3390/ijms231911516
APA StyleAllegri, L., Baldan, F., Molteni, E., Mio, C., & Damante, G. (2022). Role of m6A RNA Methylation in Thyroid Cancer Cell Lines. International Journal of Molecular Sciences, 23(19), 11516. https://doi.org/10.3390/ijms231911516