


Crosstalk between CXCR4/ACKR3 and EGFR Signaling in Breast Cancer Cells



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
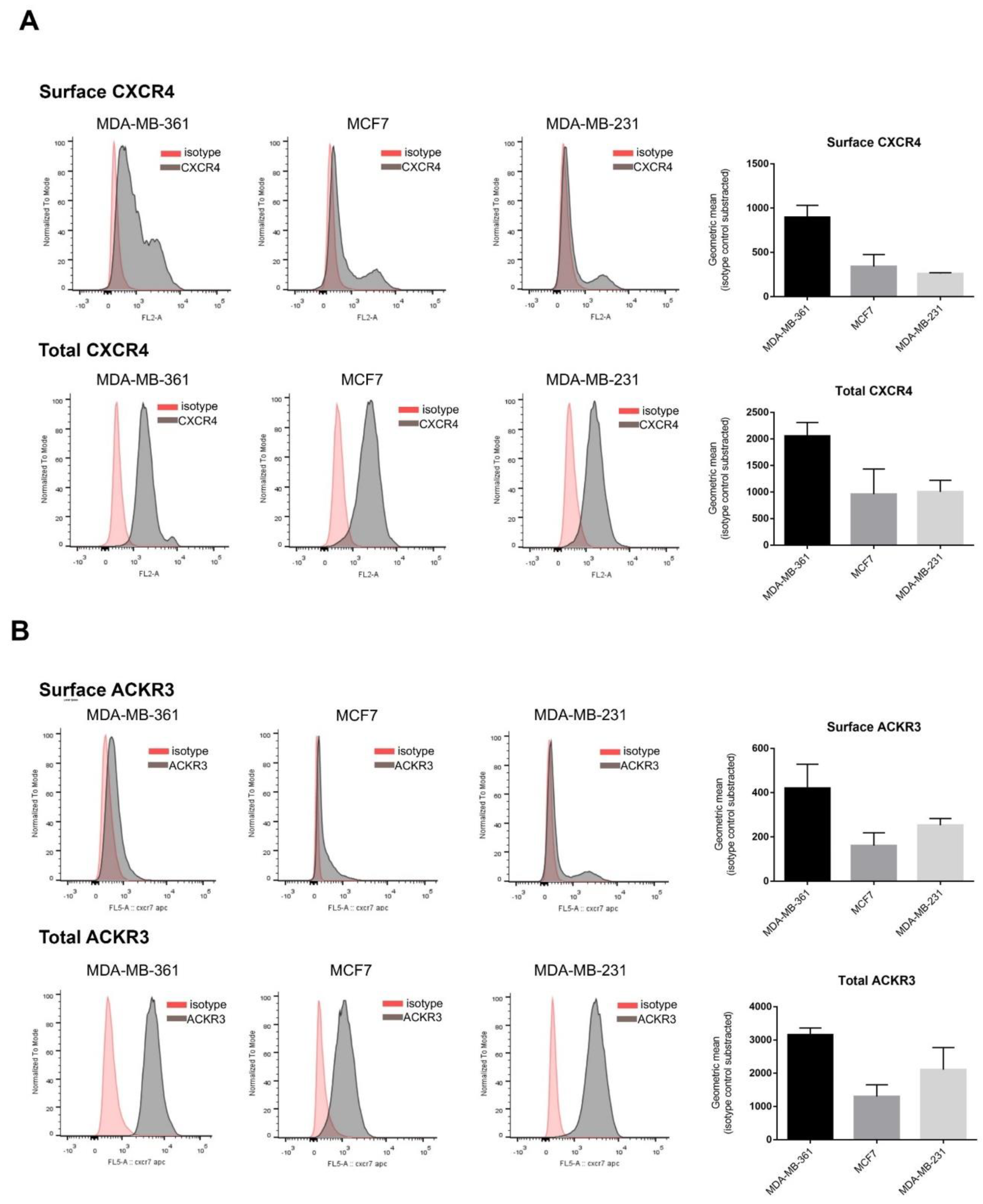
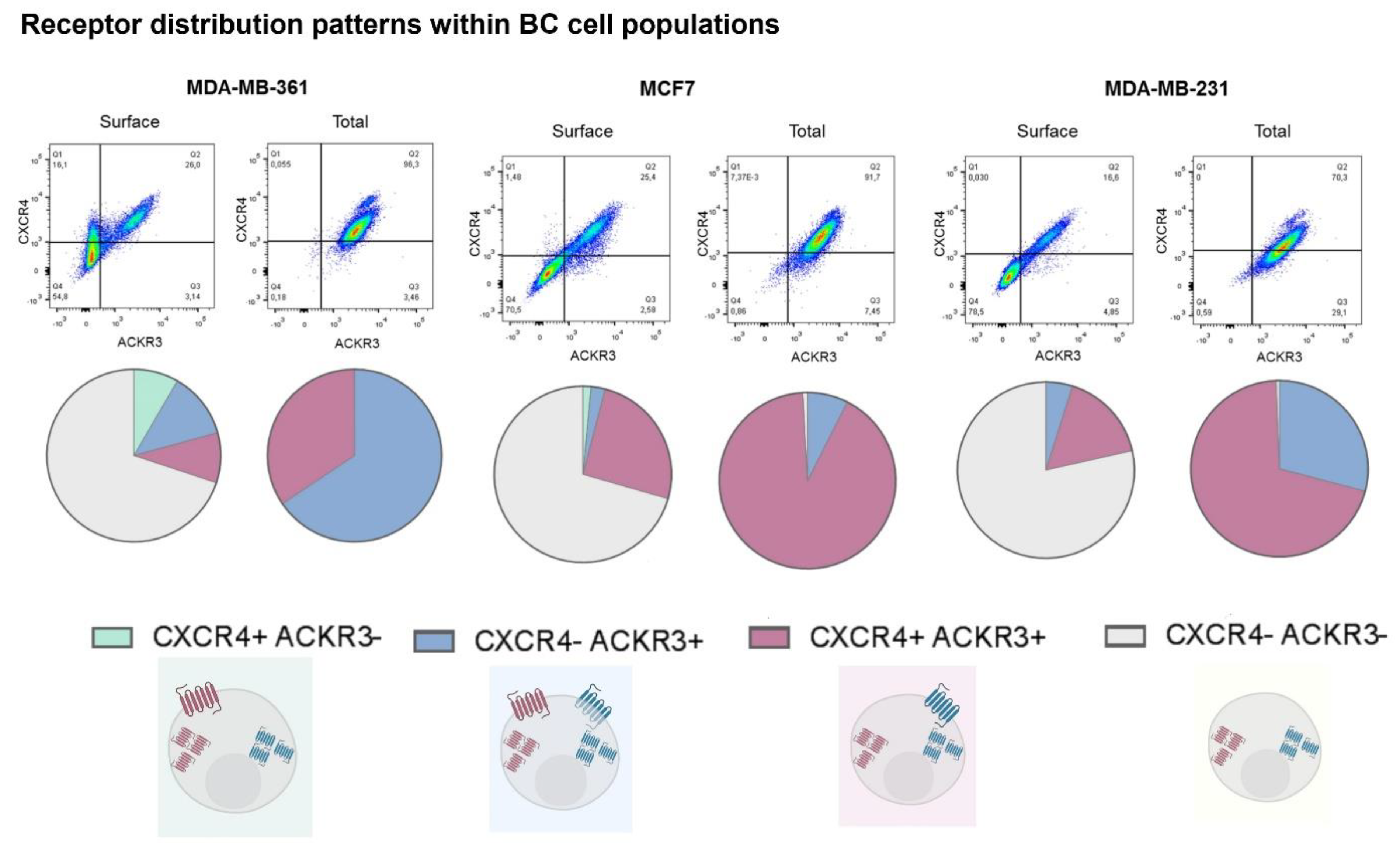
2.1. Breast Cancer Cell Lines Display Heterogeneous Patterns of CXCR4/ACKR3 Distribution
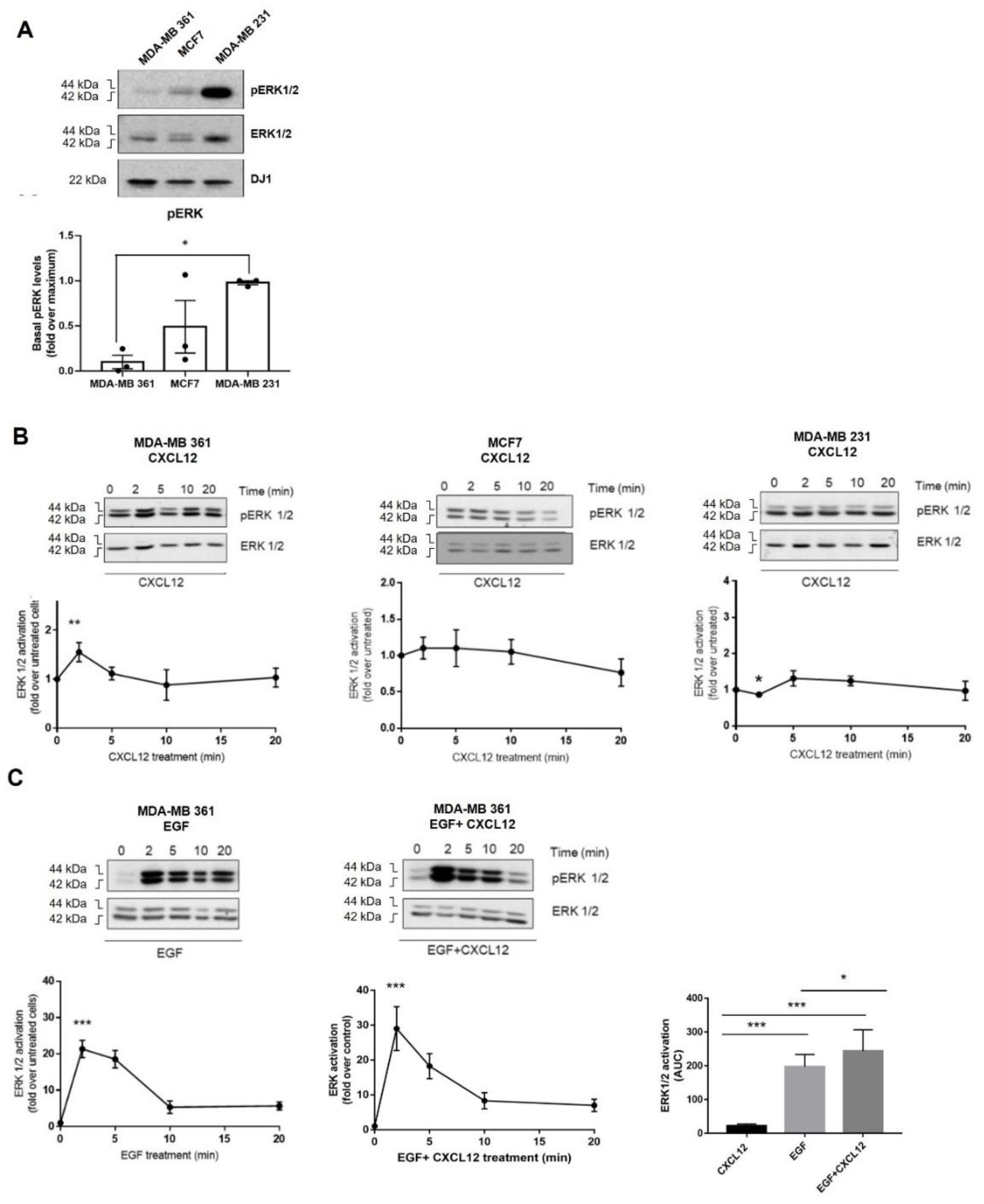
2.2. Breast Cancer Cells Differentially Activate ERK in Response to CXCL12 and ErbB Receptor Ligands
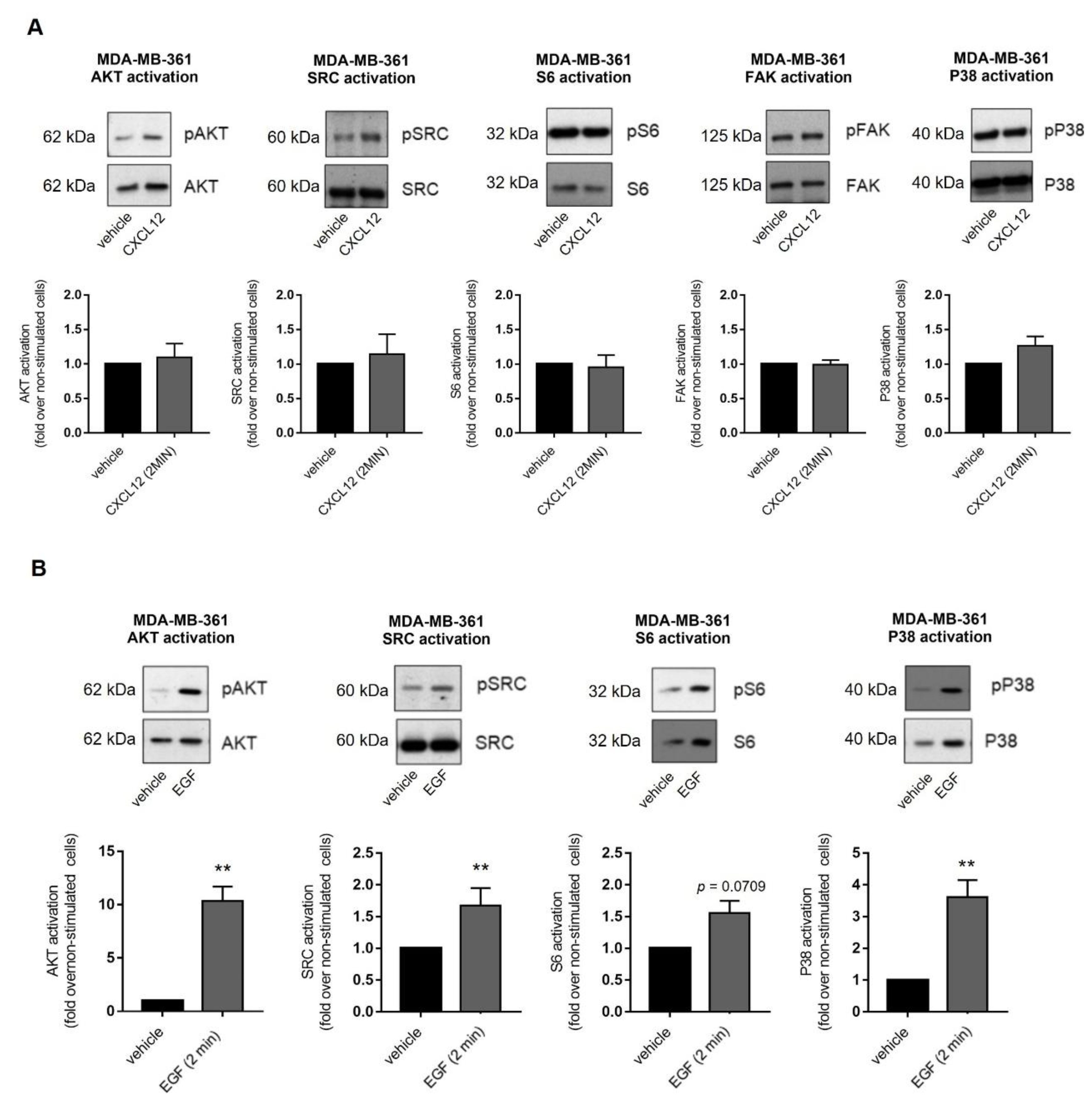
2.3. Exogenous CXCL12 Fails to Activate Other Key Transducing Molecules Related to Oncogenic Pathways in MDA-MB-361 Cells
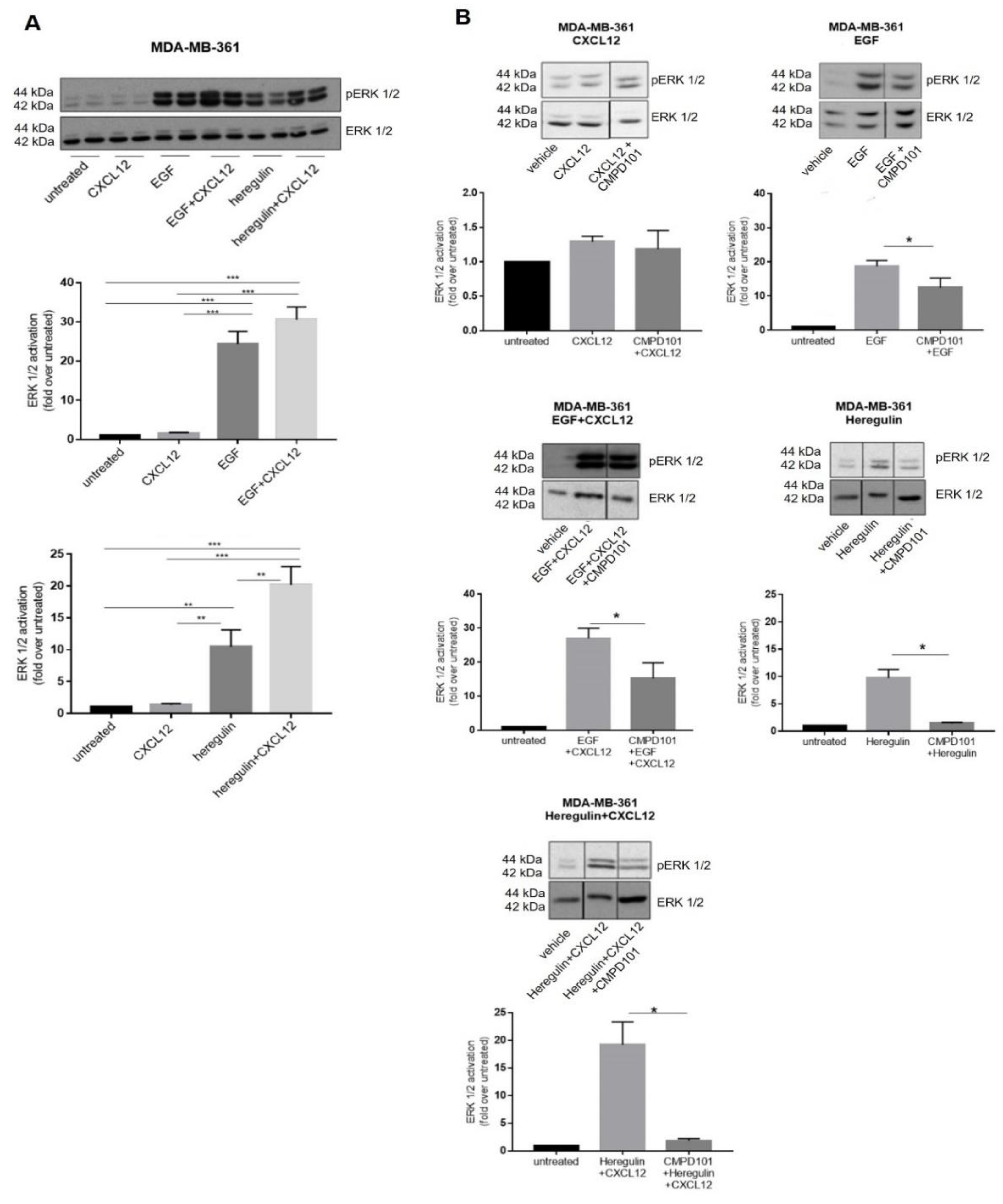
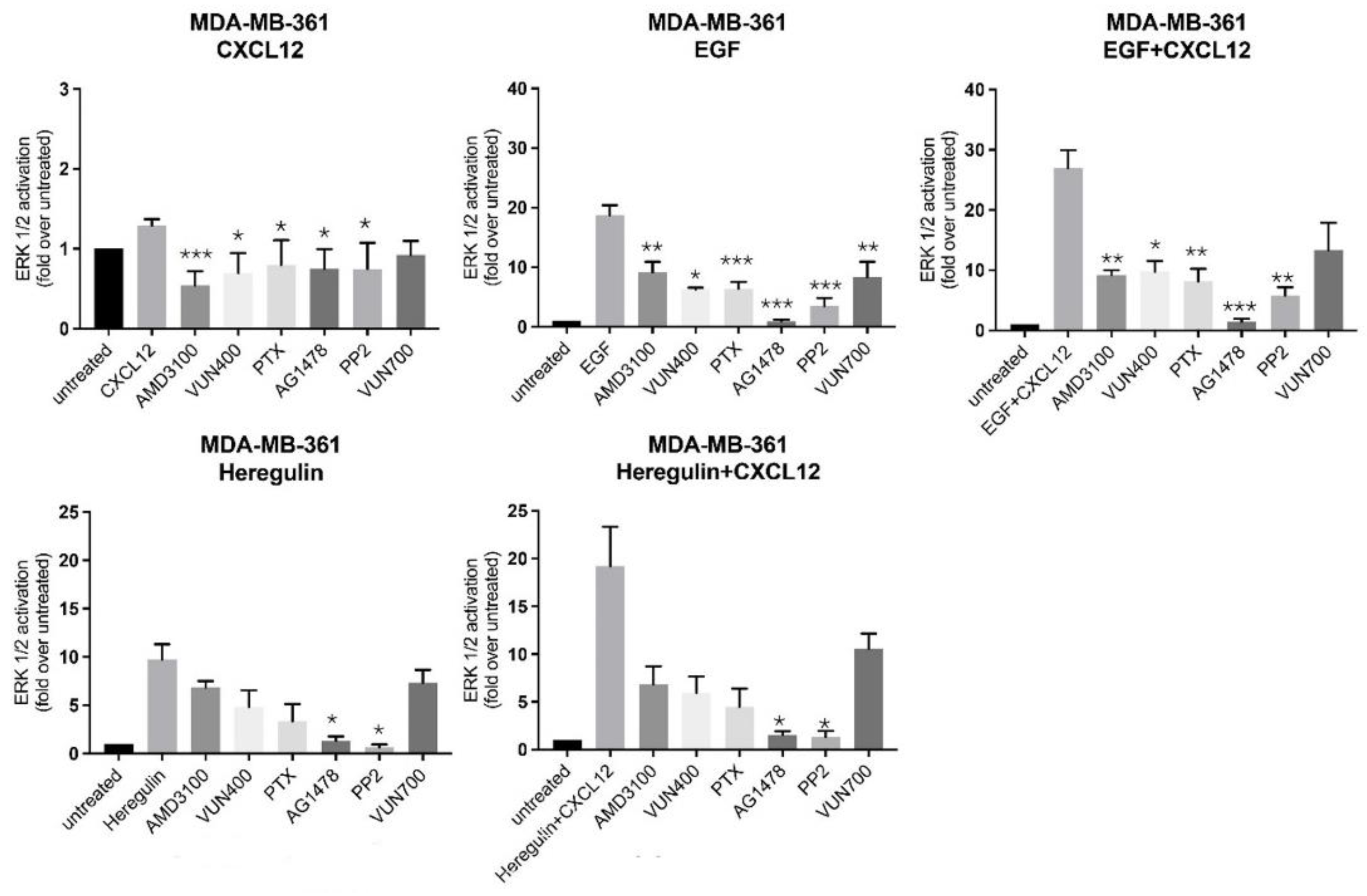
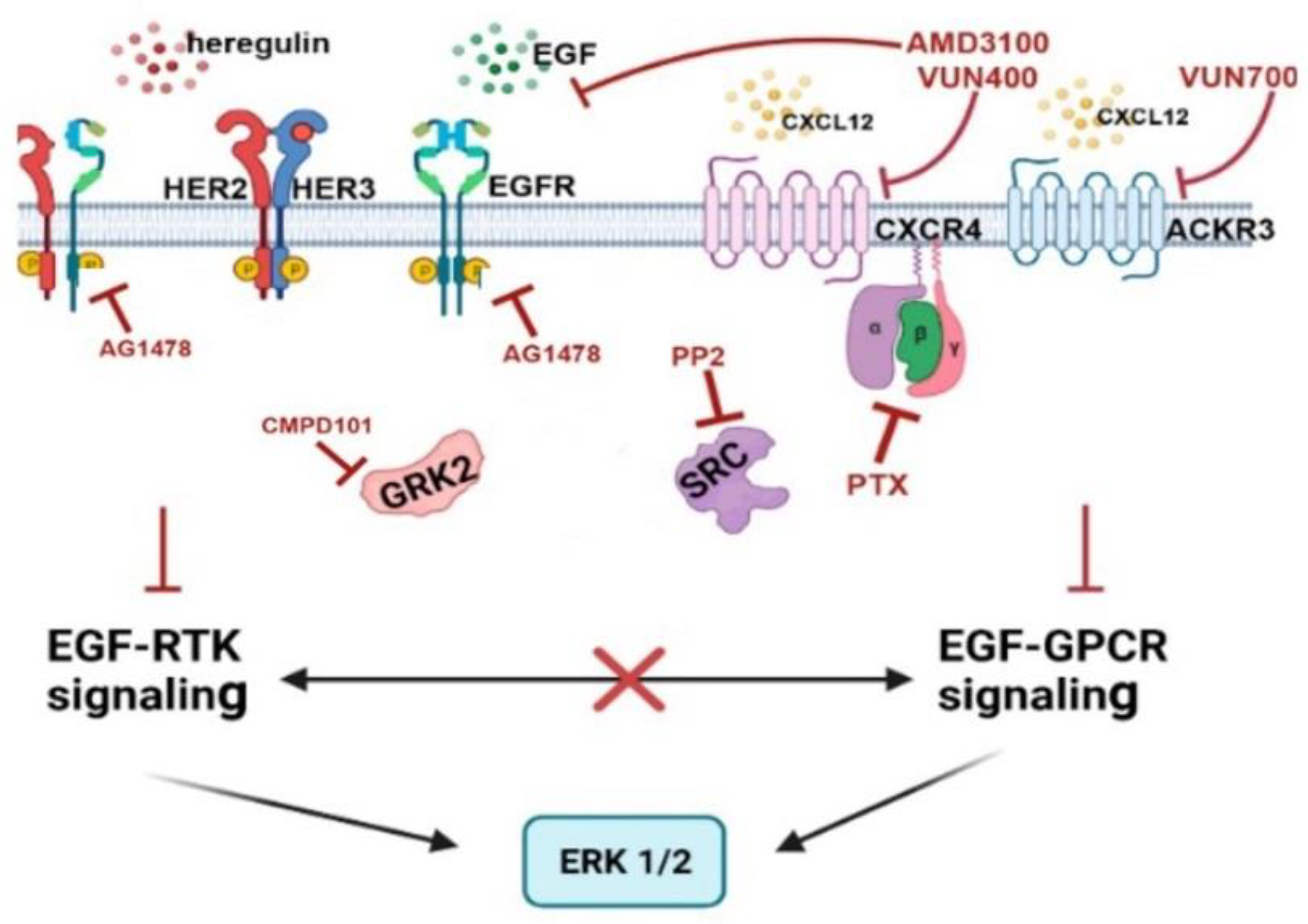
2.4. Mechanisms Involved in the Crosstalk between the CXCL12/CXCR4/ACKR3 and EGFR Family Signaling in Breast Cancer Cells Lines
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Cellular Treatments
4.2. Immunoblotting
4.3. Flow Cytometry
4.4. mRNA Analysis and Quantification
4.5. Quantification of CXCL12 Levels by ELISA
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.R.L.; Miller, D.K.; Jemal, A.; Miller, K.D.; Jemal, A. Cancer Statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellsworth, R.E.; Seebach, J.; Field, L.A.; Heckman, C.; Kane, J.; Hooke, J.A.; Love, B.; Shriver, C.D. A Gene Expression Signature That Defines Breast Cancer Metastases. Clin. Exp. Metastasis 2009, 26, 205–213. [Google Scholar] [CrossRef]
- Holliday, D.L.; Speirs, V. Choosing Correct Breast Cancer Cell Line. Breast Cancer Res. 2011, 13, 215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perou, C.M.; Sùrlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H. Molecular Portraits of Human Breast Tumours. Letters to Nature 748. Nature 2000, 533, 747–752. [Google Scholar]
- Sørlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; Van De Rijn, M.; Jeffrey, S.S.; et al. Gene Expression Patterns of Breast Carcinomas Distinguish Tumor Subclasses with Clinical Implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [Green Version]
- Fougner, C.; Bergholtz, H.; Norum, J.H.; Sørlie, T. Re-Definition of Claudin-Low as a Breast Cancer Phenotype. Nat. Commun. 2020, 11, 1787. [Google Scholar] [CrossRef] [Green Version]
- Haque, M.M.; Desai, K.V. Pathways to Endocrine Therapy Resistance in Breast Cancer. Front. Endocrinol. 2019, 10, 573. [Google Scholar] [CrossRef]
- Szostakowska, M.; Trębińska-Stryjewska, A.; Grzybowska, E.A.; Fabisiewicz, A. Resistance to Endocrine Therapy in Breast Cancer: Molecular Mechanisms and Future Goals. Breast Cancer Res. Treat. 2019, 173, 489–497. [Google Scholar] [CrossRef] [Green Version]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The Tumor Microenvironment at a Glance. J. Cell Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef] [Green Version]
- Nagarsheth, N.; Wicha, M.S.; Zou, W. Chemokines in the Cancer Microenvironment and Their Relevance in Cancer Immunotherapy. Nat. Rev. Immunol. 2017, 17, 559–572. [Google Scholar] [CrossRef] [Green Version]
- Smit, M.J.; Schlecht-Louf, G.; Neves, M.; van den Bor, J.; Penela, P.; Siderius, M.; Bachelerie, F.; Mayor, F., Jr. The CXCL12/CXCR4/ACKR3 Axis in the Tumor Microenvironment: Signaling, Crosstalk, and Therapeutic Targeting. Annu. Rev. Pharmacol. Toxicol. 2021, 61, 541–563. [Google Scholar] [CrossRef]
- Hattermann, K.; Mentlein, R. An Infernal Trio: The Chemokine CXCL12 and Its Receptors CXCR4 and CXCR7 in Tumor Biology. Ann. Anat. 2013, 195, 103–110. [Google Scholar] [CrossRef]
- Scala, S. Molecular Pathways: Targeting the CXCR4-CXCL12 Axis-Untapped Potential in the Tumor Microenvironment. Clin. Cancer Res. 2015, 21, 4278–4285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neves, M.; Fumagalli, A.; van den Bor, J.; Marin, P.; Smit, M.J.; Mayor, F. The Role of ACKR3 in Breast, Lung, and Brain Cancer. Mol. Pharmacol. 2019, 96, 819–825. [Google Scholar] [CrossRef] [PubMed]
- Heuninck, J.; Viciano, C.P.; Işbilir, A.; Caspar, B.; Capoferri, D.; Briddon, S.J.; Durroux, T.; Hill, S.J.; Lohse, M.J.; Milligan, G.; et al. Context-Dependent Signaling of CXC Chemokine Receptor 4 and Atypical Chemokine Receptor 3. Mol. Pharmacol. 2019, 96, 778–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fumagalli, A.; Zarca, A.; Neves, M.; Caspar, B.; Hill, S.J.; Mayor, F.; Smit, M.J.; Marin, P. CXCR4/AckR3 Phosphorylation and Recruitment of Interacting Proteins: Key Mechanisms Regulating Their Functional Status. Mol. Pharmacol. 2019, 96, 794–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamaruzman, N.I.; Aziz, N.A.; Poh, C.L.; Chowdhury, E.H. Oncogenic Signaling in Tumorigenesis and Applications of SiRNA Nanotherapeutics in Breast Cancer. Cancers 2019, 11, 632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Citri, A.; Skaria, K.B.; Yarden, Y. The Deaf and the Dumb: The Biology of ErbB-2 and ErbB-3. Exp. Cell Res. 2003, 284, 54–65. [Google Scholar] [CrossRef]
- Hynes, N.E.; Lane, H.A. ERBB Receptors and Cancer: The Complexity of Targeted Inhibitors. Nat. Rev. Cancer 2005, 5, 341–354. [Google Scholar] [CrossRef]
- Banerji, S.; Cibulskis, K.; Rangel-Escareno, C.; Brown, K.K.; Carter, S.L.; Frederick, A.M.; Lawrence, M.S.; Sivachenko, A.Y.; Sougnez, C.; Zou, L.; et al. Sequence Analysis of Mutations and Translocations across Breast Cancer Subtypes. Nature 2012, 486, 405–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nogués, L.; Palacios-García, J.; Reglero, C.; Rivas, V.; Neves, M.; Ribas, C.; Penela, P.; Mayor, F. G Protein-Coupled Re-ceptor Kinases (GRKs) in Tumorigenesis and Cancer Progression: GPCR Regulators and Signaling Hubs; Elsevier Ltd.: Amsterdam, The Netherlands, 2018; Volume 48, ISBN 3491196442. [Google Scholar]
- Nogués, L.; Reglero, C.; Rivas, V.; Neves, M.; Penela, P.; Mayor, F. G-Protein-Coupled Receptor Kinase 2 as a Potential Modulator of the Hallmarks of Cancer. Mol. Pharmacol. 2017, 91, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Penela, P.; Ribas, C.; Sánchez-Madrid, F.; Mayor, F. G Protein-Coupled Receptor Kinase 2 (GRK2) as a Multifunctional Signaling Hub. Cell. Mol. Life Sci. 2019, 76, 4423–4446. [Google Scholar] [CrossRef] [Green Version]
- Salcedo, A.; Mayor, F.; Penela, P. Mdm2 Is Involved in the Ubiquitination and Degradation of G-Protein-Coupled Receptor Kinase 2. EMBO J. 2006, 25, 4752–4762. [Google Scholar] [CrossRef] [Green Version]
- Nogués, L.; Reglero, C.; Rivas, V.; Salcedo, A.; Lafarga, V.; Neves, M.; Ramos, P.; Mendiola, M.; Berjón, A.; Stamatakis, K.; et al. G Protein-Coupled Receptor Kinase 2 (GRK2) Promotes Breast Tumorigenesis Through a HDAC6-Pin1 Axis. eBioMedicine 2016, 13, 132–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busillo, J.M.; Armando, S.; Sengupta, R.; Meucci, O.; Bouvier, M.; Benovic, J.L. Site-Specific Phosphorylation of CXCR4 Is Dynamically Regulated by Multiple Kinases and Results in Differential Modulation of CXCR4 Signaling. J. Biol. Chem. 2010, 285, 7805–7817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, J.; Busillo, J.M.; Stumm, R.; Benovic, J.L. G Protein-Coupled Receptor Kinase 3 and Protein Kinase C Phosphorylate the Distal C-Terminal Tail of the Chemokine Receptor CXCR4 and Mediate Recruitment of β-Arrestin. Mol. Pharmacol. 2017, 91, 554–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sosa, M.S.; Lopez-Haber, C.; Yang, C.; Wang, H.B.; Lemmon, M.A.; Busillo, J.M.; Luo, J.; Benovic, J.L.; Klein-Szanto, A.; Yagi, H.; et al. Identification of the Rac-GEF P-Rex1 as an Essential Mediator of ErbB Signaling in Breast Cancer. Mol. Cell 2010, 40, 877–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, K.; Feng, G.; Yan, Q.; Sun, L.; Zhang, K.; Shen, F.; Shen, M.; Ruan, S. CXCR4 and CXCR3 Are Two Distinct Prognostic Biomarkers in Breast Cancer: Database Mining for CXCR Family Members. Mol. Med. Rep. 2019, 20, 4791–4802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Cheng, G.; Hao, M.; Zheng, J.; Zhou, X.; Zhang, J.; Taichman, R.S.; Pienta, K.J.; Wang, J. CXCL12 / CXCR4 / CXCR7 Chemokine Axis and Cancer Progression. Cancer Metastasis Rev. 2010, 29, 709–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Riese, D.J.; Shen, J. The Role of the CXCL12/CXCR4/CXCR7 Chemokine Axis in Cancer. Front. Pharmacol. 2020, 11, 574667. [Google Scholar] [CrossRef]
- Huynh, C.; Dingemanse, J.; Meyer zu Schwabedissen, H.E.; Sidharta, P.N. Relevance of the CXCR4/CXCR7-CXCL12 Axis and Its Effect in Pathophysiological Conditions. Pharmacol. Res. 2020, 161, 105092. [Google Scholar] [CrossRef]
- Spinosa, P.C.; Humphries, B.A.; Mejia, D.L.; Buschhaus, J.M.; Linderman, J.J.; Luker, G.D.; Luker, K.E. Short-Term Cel-lular Memory Tunes the Signaling Responses of the Chemokine Receptor CXCR4. Sci. Signal. 2019, 12, eaaw4204. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Zhao, H.; Chen, H.; Yao, Q. CXCR4 in Breast Cancer: Oncogenic Role and Therapeutic Targeting. Drug Des. Dev. Ther. 2015, 9, 4953–4964. [Google Scholar] [CrossRef] [Green Version]
- Del Molino del Barrio, I.; Wilkins, G.C.; Meeson, A.; Ali, S.; Kirby, J.A. Breast Cancer: An Examination of the Potential of ACKR3 to Modify the Response of CXCR4 to CXCL12. Int. J. Mol. Sci. 2018, 19, 3592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, M.; Huang, K.; Zhou, J.; Yan, D.; Tang, Y.L.; Zhao, T.C.; Miller, R.J.; Kishore, R.; Losordo, D.W.; Qin, G. A Critical Role of Src Family Kinase in SDF-1/CXCR4-Mediated Bone-Marrow Progenitor Cell Recruitment to the Ischemic Heart. J. Mol. Cell. Cardiol. 2015, 81, 49–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, S.; Tang, J.; Wang, C.; Liu, J.; Fu, Y.; Luo, Y. CXCR7 Promotes Melanoma Tumorigenesis via Src Kinase Signaling. Cell Death Dis. 2019, 10, 191. [Google Scholar] [CrossRef] [Green Version]
- Ieranò, C.; Santagata, S.; Napolitano, M.; Guardia, F.; Grimaldi, A.; Antignani, E.; Botti, G.; Consales, C.; Riccio, A.; Na-nayakkara, M.; et al. CXCR4 and CXCR7 Transduce through MTOR in Human Renal Cancer Cells. Cell Death Dis. 2014, 5, e1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esencay, M.; Sarfraz, Y.; Zagzag, D. CXCR7 Is Induced by Hypoxia and Mediates Glioma Cell Migration towards SDF-1α. BMC Cancer 2013, 13, 347. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, D.; Zhao, J. The Role of Chemokine Receptor CXCR4 in Breast Cancer Metastasis Debarati. Am. J. Cancer Res. 2013, 3, 46–57. [Google Scholar]
- Chatterjee, M.; Seizer, P.; Borst, O.; Schönberger, T.; MacK, A.; Geisler, T.; Langer, H.F.; May, A.E.; Vogel, S.; Lang, F.; et al. SDF-1o: Induces Differential Trafficking of CXCR4-CXCR7 Involving Cyclophilin A, CXCR7 Ubiquitination and Promotes Platelet Survival. FASEB J. 2014, 28, 2864–2878. [Google Scholar] [CrossRef]
- Wang, C.; Chen, W.; Shen, J. CXCR7 Targeting and Its Major Disease Relevance. Front. Pharmacol. 2018, 9, 641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neves, M.; Perpiñá-Viciano, C.; Penela, P.; Hoffmann, C.; Mayor, F. Modulation of CXCR4-Mediated Gi1 Activation by EGF Receptor and GRK2. ACS Pharmacol. Transl. Sci. 2020, 3, 627–634. [Google Scholar] [CrossRef] [PubMed]
- Bobkov, V.; Arimont, M.; Zarca, A.; de Groof, T.W.M.; van der Woning, B.; de Haard, H.; Smit, M.J. Antibodies Targeting Chemokine Receptors CXCR4 and ACKR3. Mol. Pharmacol. 2019, 96, 753–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheeler, D.L.; Iida, M.; Dunn, E.F. The Role of Src in Solid Tumors. Oncol. 2009, 14, 667–678. [Google Scholar] [CrossRef]
- Zielińska, K.A.; Katanaev, V.L. The Signaling Duo CXCL12 and CXCR4: Chemokine Fuel for Breast Cancer Tumorigenesis. Cancers 2020, 12, 3071. [Google Scholar] [CrossRef]
- Koch, C.; Engele, J. Functions of the CXCL12 Receptor ACKR3/CXCR7-What Has Been Perceived and What Has Been Overlooked. Mol. Pharmacol. 2020, 98, 577–585. [Google Scholar]
- Inaguma, S.; Riku, M.; Ito, H.; Tsunoda, T.; Ikeda, H.; Kasai, K. GLI1 Orchestrates CXCR4/CXCR7 Signaling to Enhance Migration and Metastasis of Breast Cancer Cells. Oncotarget 2015, 6, 33648–33657. [Google Scholar] [CrossRef] [Green Version]
- Luker, K.E.; Lewin, S.A.; Mihalko, L.A.; Schmidt, B.T.; Winkler, J.S.; Coggins, N.L.; Thomas, D.G.; Luker, G.D. Scavenging of CXCL12 by CXCR7 Promotes Tumor Growth and Metastasis of CXCR4-Positive Breast Cancer Cells. Oncogene 2012, 31, 4750–4758. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Mao, X.; Fan, C.; Liu, C.; Guo, A.; Guan, S.; Jin, Q.; Li, B.; Yao, F.; Jin, F. CXCL12-CXCR4 Axis Promotes the Natural Selection of Breast Cancer Cell Metastasis. Tumor Biol. 2014, 35, 7765–7773. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Xiao, Q.; Bai, X.; Yu, Z.; Sun, M.; Zhao, H.; Mi, X.; Wang, E.; Yao, W.; Jin, F.; et al. Activation of STAT3 Is Involved in Malignancy Mediated by CXCL12-CXCR4 Signaling in Human Breast Cancer. Oncol. Rep. 2014, 32, 2760–2768. [Google Scholar] [CrossRef] [Green Version]
- Shiah, Y.J.; Tharmapalan, P.; Casey, A.E.; Joshi, P.A.; McKee, T.D.; Jackson, H.W.; Beristain, A.G.; Chan-Seng-Yue, M.A.; Bader, G.D.; Lydon, J.P.; et al. A Progesterone-CXCR4 Axis Controls Mammary Progenitor Cell Fate in the Adult Gland. Stem Cell Rep. 2015, 4, 313–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, P.; Mueller, W.; Schütz, D.; MacKay, F.; Thelen, M.; Zhang, P.; Stumm, R. CXCR7 Prevents Excessive CXCL12-Mediated Downregulation of CXCR4 in Migrating Cortical Interneurons. Development 2014, 141, 1857–1863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, H.T.; Reyes-Alcaraz, A.; Yong, H.J.; Nguyen, L.P.; Park, H.K.; Inoue, A.; Lee, C.S.; Seong, J.Y.; Hwang, J.I. CXCR7: A β-Arrestin-Biased Receptor That Potentiates Cell Migration and Recruits β-Arrestin2 Exclusively through Gβγ Subunits and GRK2. Cell Biosci. 2020, 10, 134. [Google Scholar] [CrossRef]
- Delcourt, N.; Bockaert, J.; Marin, P. GPCR-Jacking: From a New Route in RTK Signalling to a New Concept in GPCR Activation. Trends Pharmacol. Sci. 2007, 28, 602–607. [Google Scholar] [CrossRef] [PubMed]
- Crudden, C.; Ilic, M.; Suleymanova, N.; Worrall, C.; Girnita, A.; Girnita, L. The Dichotomy of the Insulin-like Growth Factor 1 Receptor: RTK and GPCR: Friend or Foe for Cancer Treatment? Growth Horm. IGF Res. 2015, 25, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Hynes, N.E.; Gattelli, A. P-Rex1, a Guanine Exchange Factor That Is Overexpressed in Breast Cancer, Is a Convergence Node for ErbB and CXCR4 Signaling. Mol. Cell 2011, 41, 5–7. [Google Scholar] [CrossRef]
- Mueller, W.; Schütz, D.; Nagel, F.; Schulz, S.; Stumm, R. Hierarchical Organization of Multi-Site Phosphorylation at the CXCR4 C Terminus. PLoS ONE 2013, 8, e64975. [Google Scholar] [CrossRef] [Green Version]
- Woerner, B.M.; Warrington, N.M.; Kung, A.L.; Perry, A.; Rubin, J.B. Widespread CXCR4 Activation in Astrocytomas Revealed by Phospho-CXCR4-Specific Antibodies. Cancer Res. 2005, 65, 11392–11399. [Google Scholar] [CrossRef] [Green Version]
- Mustafi, R.; Dougherty, U.; Mustafi, D.; Ayaloglu-Butun, F.; Fletcher, M.; Adhikari, S.; Sadiq, F.; Meckel, K.; Haider, H.I.; Khalil, A.; et al. ADAM17 Is a Tumor Promoter and Therapeutic Target in Western Diet-Associated Colon Cancer. Clin. Cancer Res. 2017, 23, 549–561. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wang, B.; Chu, H.; Yao, Y. Intrinsic Resistance to EGFR Tyrosine Kinase Inhibitors in Advanced Non-Small-Cell Lung Cancer with Activating EGFR Mutations. OncoTargets Ther. 2016, 9, 3711–3726. [Google Scholar] [CrossRef] [Green Version]
- Cabioglu, N.; Summy, J.; Miller, C.; Parikh, N.U.; Sahin, A.A.; Tuzlali, S.; Pumiglia, K.; Gallick, G.E.; Price, J.E. CXCL-12/Stromal Cell-Derived Factor-1α Transactivates HER2-Neu in Breast Cancer Cells by a Novel Pathway Involving Src Kinase Activation. Cancer Res. 2005, 65, 6493–6497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pattarozzi, A.; Gatti, M.; Barbieri, F.; Wu, R.; Porcile, C.; Lunardi, G.; Ratto, A.; Favoni, R.; Bajetto, A.; Ferrari, A.; et al. 17β-Estradiol Promotes Breast Cancer Cell Proliferation- Inducing Stromal Cell-Derived Factor-1-Mediated Epidermal Growth Factor Receptor Transactivation: Reversal by Gefitinib Pretreatment. Proteins 2008, 73, 191–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarnago, S.; Elorza, A.; Mayor, F. Agonist-Dependent Phosphorylation of the G Protein-Coupled Receptor Kinase 2 (GRK2) by Src Tyrosine Kinase. J. Biol. Chem. 1999, 274, 34411–34416. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neves, M.; Marolda, V.; Mayor, F., Jr.; Penela, P. Crosstalk between CXCR4/ACKR3 and EGFR Signaling in Breast Cancer Cells. Int. J. Mol. Sci. 2022, 23, 11887. https://doi.org/10.3390/ijms231911887
Neves M, Marolda V, Mayor F Jr., Penela P. Crosstalk between CXCR4/ACKR3 and EGFR Signaling in Breast Cancer Cells. International Journal of Molecular Sciences. 2022; 23(19):11887. https://doi.org/10.3390/ijms231911887
Chicago/Turabian StyleNeves, Maria, Viviana Marolda, Federico Mayor, Jr., and Petronila Penela. 2022. "Crosstalk between CXCR4/ACKR3 and EGFR Signaling in Breast Cancer Cells" International Journal of Molecular Sciences 23, no. 19: 11887. https://doi.org/10.3390/ijms231911887
APA StyleNeves, M., Marolda, V., Mayor, F., Jr., & Penela, P. (2022). Crosstalk between CXCR4/ACKR3 and EGFR Signaling in Breast Cancer Cells. International Journal of Molecular Sciences, 23(19), 11887. https://doi.org/10.3390/ijms231911887