



Deciphering the Role and Signaling Pathways of PKCα in Luminal A Breast Cancer Cells

 , , ,
, , ,  , and
, and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
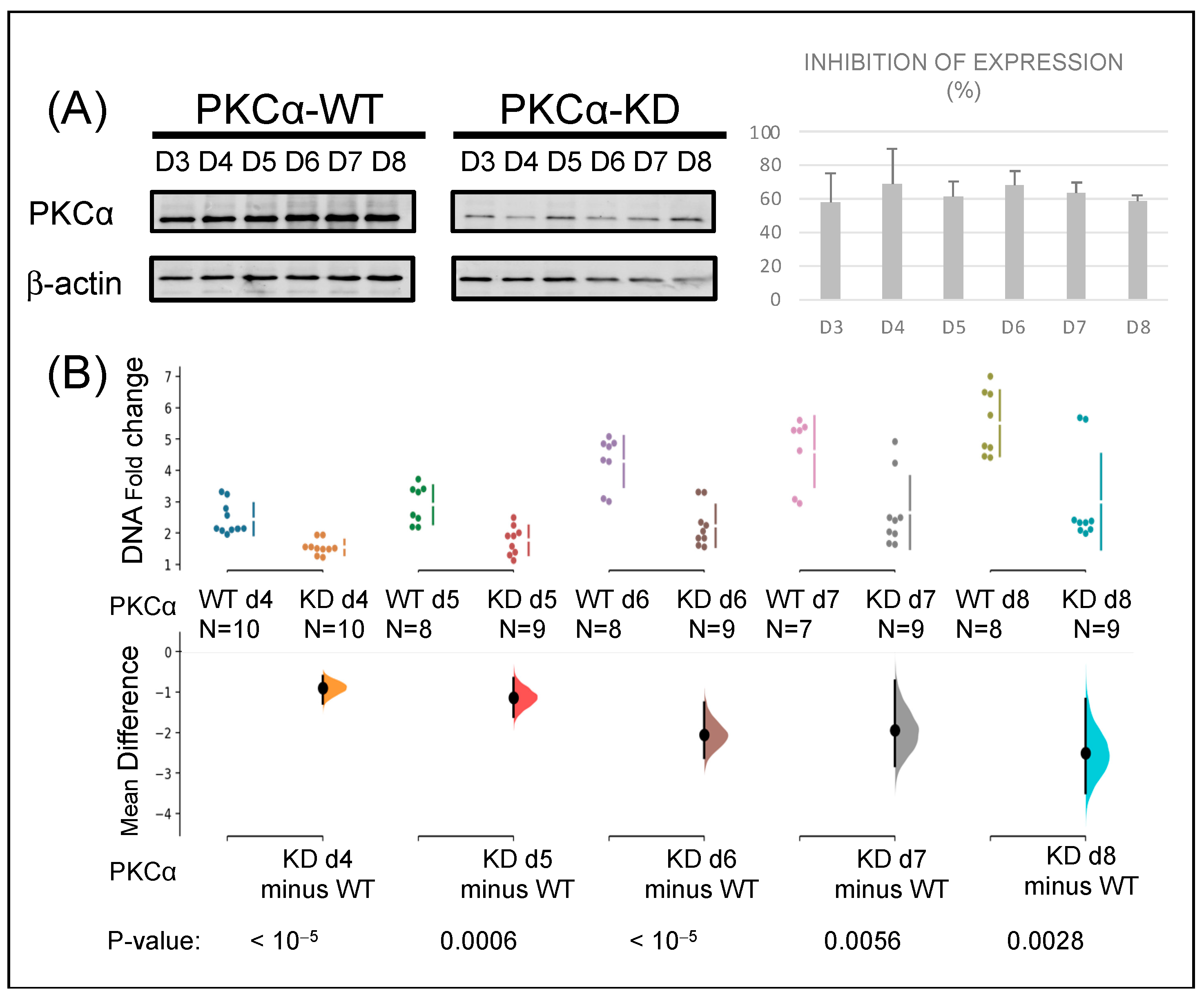
2.1. Knock-Down of PKCα in MCF-7 Cells Affects Their Tumorigenic Capacity
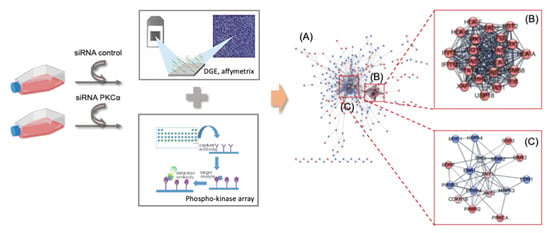
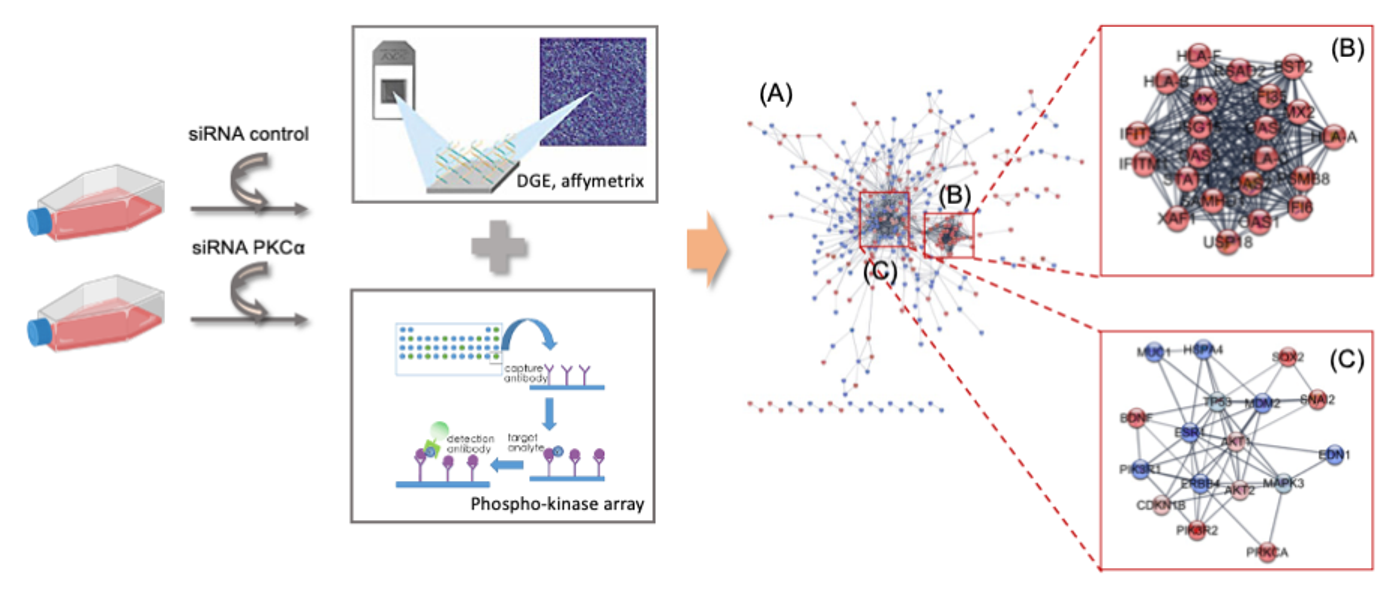
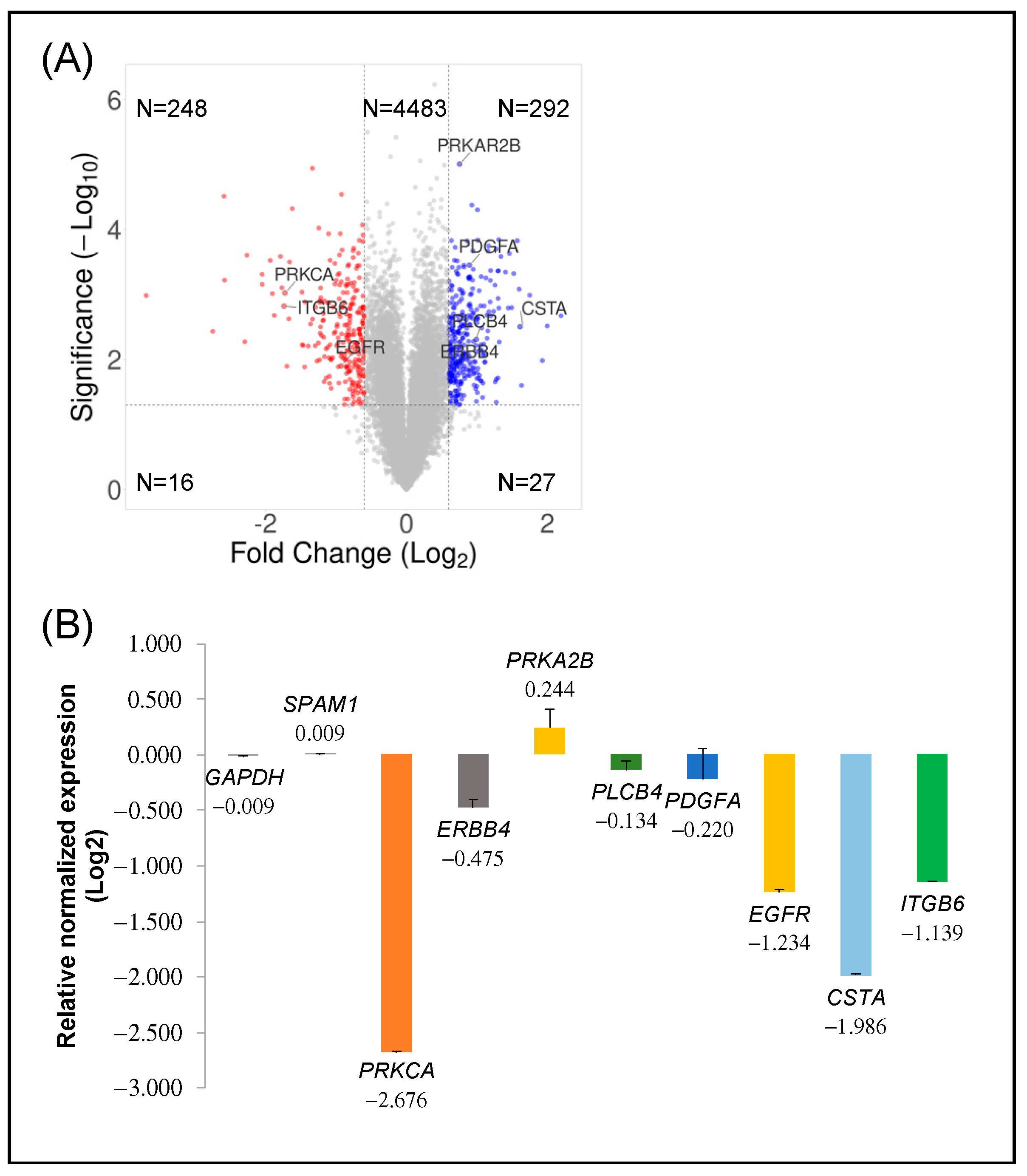
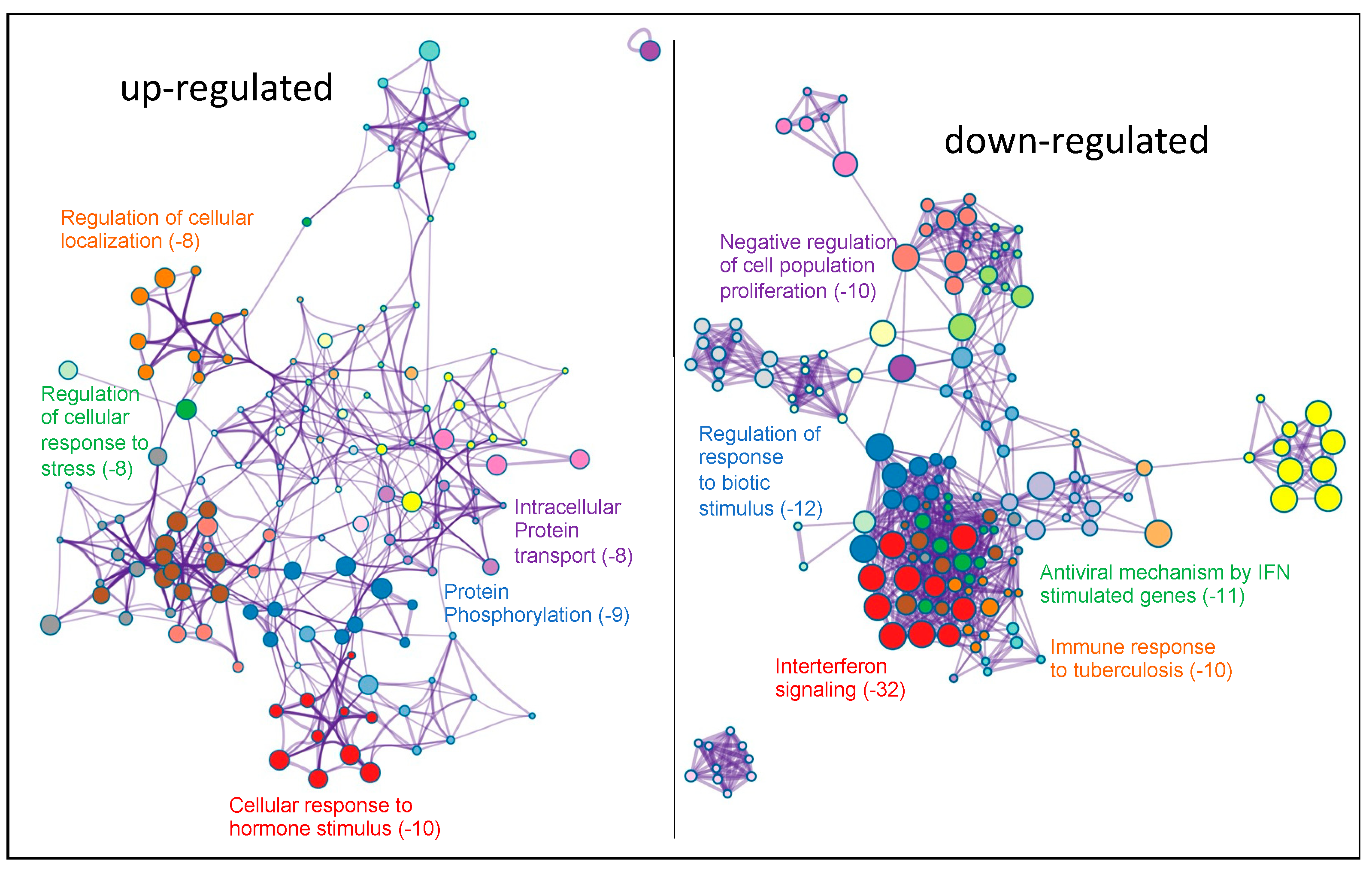
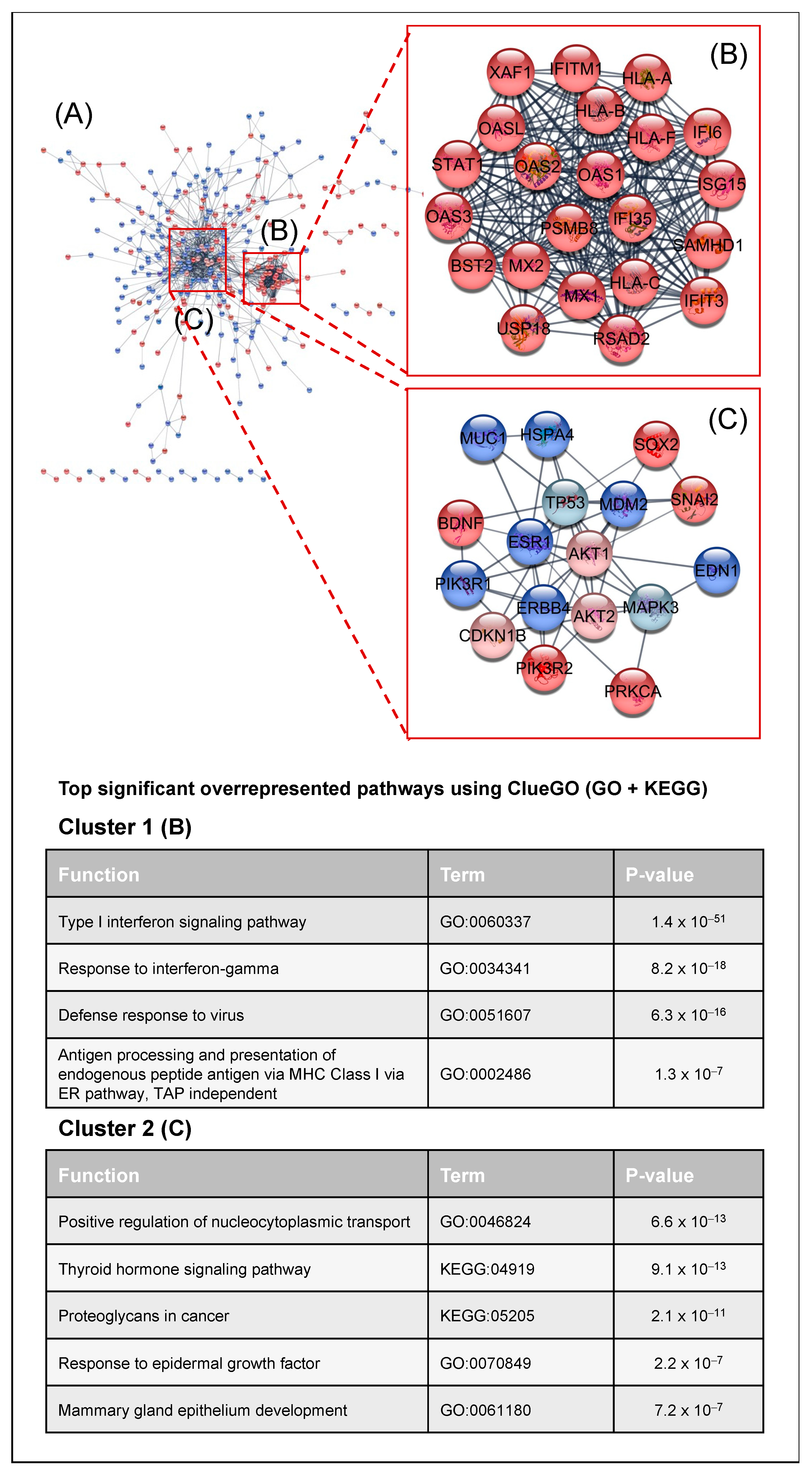
2.2. Transcriptomic Analysis of PKCα Knock-Down MCF-7 Cells Reveals Essential Alterations in Signaling Pathways
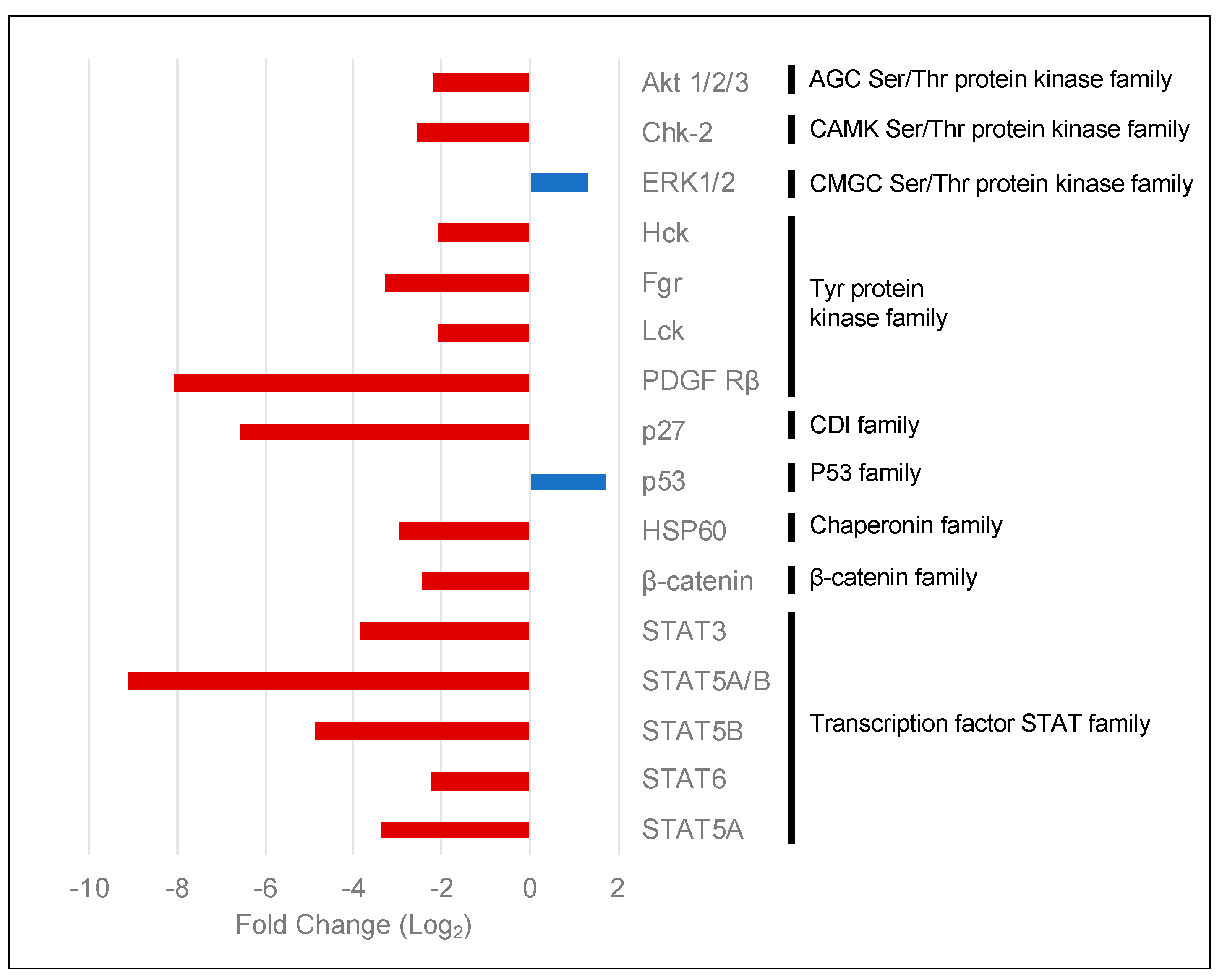
2.3. PKCα Controls the Phosphorylation of Essential Ser/Thr and Tyr Kinases and Transcription Factors
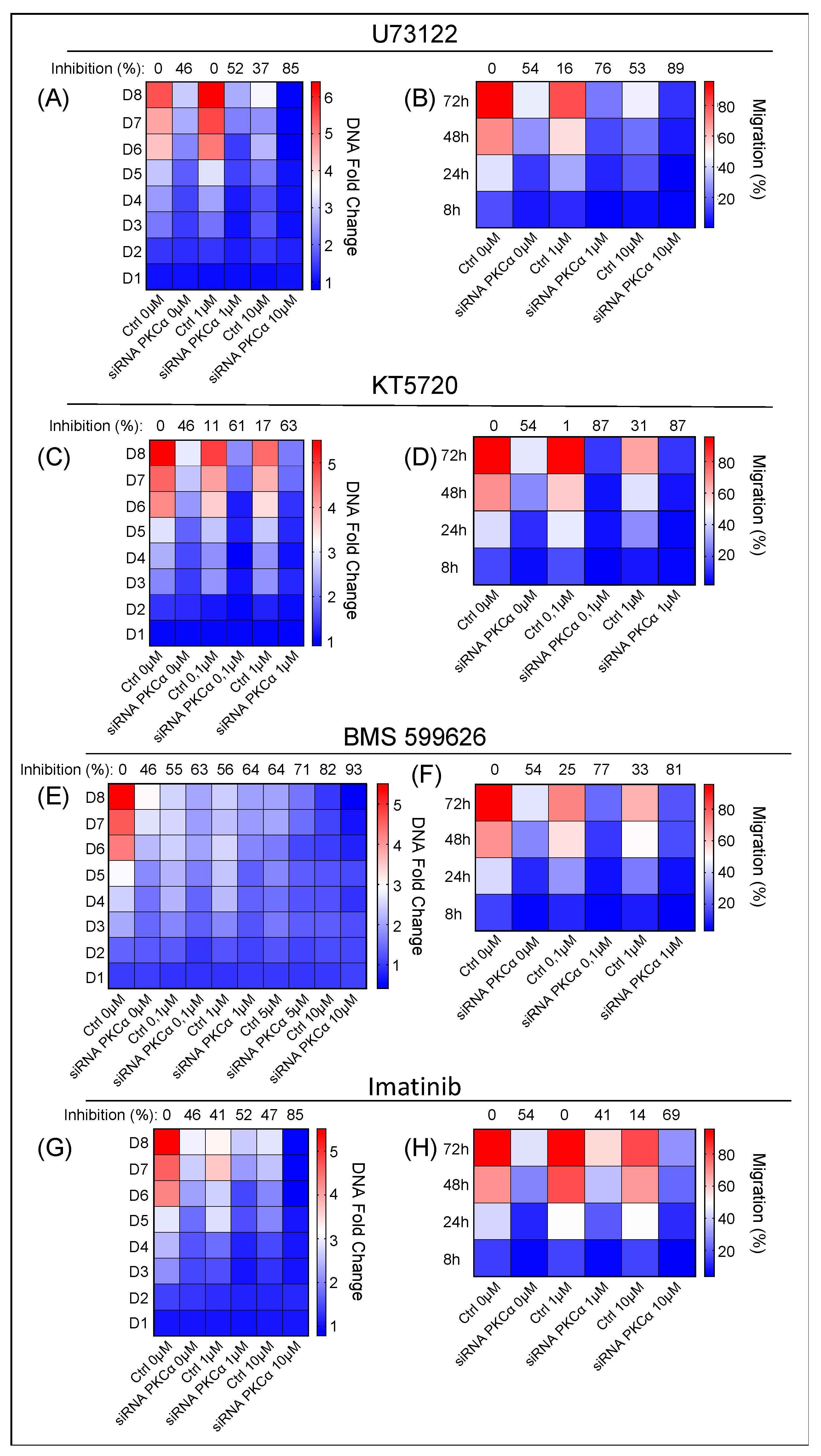
2.4. Potential Targeted Therapy by Using Specific Inhibitors of Key Signaling Pathways
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lei, S.; Zheng, R.; Zhang, S.; Wang, S.; Chen, R.; Sun, K.; Zeng, H.; Zhou, J.; Wei, W. Global Patterns of Breast Cancer Incidence and Mortality: A Population-Based Cancer Registry Data Analysis from 2000 to 2020. Cancer Commun. 2021, 41, 1183–1194. [Google Scholar] [CrossRef] [PubMed]
- Lønne, G.K.; Cornmark, L.; Zahirovic, I.O.; Landberg, G.; Jirström, K.; Larsson, C. PKCα Expression Is a Marker for Breast Cancer Aggressiveness. Mol. Cancer 2010, 9, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, M.; Kishimoto, A.; Takai, Y.; Nishizuka, Y. Studies on a Cyclic Nucleotide-Independent Protein Kinase and Its Proenzyme in Mammalian Tissues. II. Proenzyme and Its Activation by Calcium-Dependent Protease from Rat Brain. J. Biol. Chem. 1977, 252, 7610–7616. [Google Scholar] [CrossRef]
- Castagna, M.; Takai, Y.; Kaibuchi, K.; Sano, K.; Kikkawa, U.; Nishizuka, Y. Direct Activation of Calcium-Activated, Phospholipid-Dependent Protein Kinase by Tumor-Promoting Phorbol Esters. J. Biol. Chem. 1982, 257, 7847–7851. [Google Scholar] [CrossRef]
- Cooke, M.; Magimaidas, A.; Casado-Medrano, V.; Kazanietz, M.G. Protein Kinase C in Cancer: The Top Five Unanswered Questions. Mol. Carcinog. 2017, 56, 1531–1542. [Google Scholar] [CrossRef]
- Masso-Welch, P.A.; Verstovsek, G.; Ip, M.M. Alterations in the Expression and Localization of Protein Kinase C Isoforms during Mammary Gland Differentiation. Eur. J. Cell Biol. 1999, 78, 497–510. [Google Scholar] [CrossRef]
- Urtreger, A.J.; Kazanietz, M.G.; Bal De Kier Joffé, E.D. Contribution of Individual PKC Isoforms to Breast Cancer Progression. IUBMB Life 2012, 64, 18–26. [Google Scholar] [CrossRef]
- Lahn, M.; Köhler, G.; Sundell, K.; Su, C.; Li, S.; Paterson, B.M.; Bumol, T.F. Protein Kinase C Alpha Expression in Breast and Ovarian Cancer. Oncology 2004, 67, 1–10. [Google Scholar] [CrossRef]
- Tan, M.; Li, P.; Sun, M.; Yin, G.; Yu, D. Upregulation and Activation of PKC Alpha by ErbB2 through Src Promotes Breast Cancer Cell Invasion That Can Be Blocked by Combined Treatment with PKC Alpha and Src Inhibitors. Oncogene 2006, 25, 3286–3295. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the CBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The CBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griner, E.M.; Kazanietz, M.G. Protein Kinase C and Other Diacylglycerol Effectors in Cancer. Nat. Rev. Cancer 2007, 7, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.-H. Protein Kinase C (PKC) Isozymes and Cancer. New J. Sci. 2014, 8, 168–173. [Google Scholar] [CrossRef] [Green Version]
- Newton, A.C.; Brognard, J. Reversing the Paradigm: Protein Kinase C as a Tumor Suppressor. Trends Pharmacol. Sci. 2017, 38, 438–447. [Google Scholar] [CrossRef] [Green Version]
- Tonetti, D.A.; Chisamore, M.J.; Grdina, W.; Schurz, H.; Jordan, V.C. Stable Transfection of Protein Kinase C Alpha CDNA in Hormone-Dependent Breast Cancer Cell Lines. Br. J. Cancer 2000, 83, 782. [Google Scholar] [CrossRef] [Green Version]
- Tonetti, D.A.; Morrow, M.; Kidwai, N.; Gupta, A.; Badve, S. Elevated Protein Kinase C Alpha Expression May Be Predictive of Tamoxifen Treatment Failure. Br. J. Cancer 2003, 88, 1400. [Google Scholar] [CrossRef] [Green Version]
- Correction: Novel Antitumor Effect of Estradiol in Athymic Mice Injected with a T47D Breast Cancer Cell Line Overexpressing Protein Kinase C Alpha. Clin. Cancer Res. 2016, 22, 4756. [CrossRef] [Green Version]
- Hsu, Y.-H.; Yao, J.; Chan, L.-C.; Wu, T.-J.; Hsu, J.L.; Fang, Y.-F.; Wei, Y.; Wu, Y.; Huang, W.-C.; Liu, C.-L.; et al. Definition of PKC-, CDK6, and MET as Therapeutic Targets in Triple-Negative Breast Cancer. Cancer Res. 2014, 74, 4822–4835. [Google Scholar] [CrossRef] [Green Version]
- Tam, W.L.; Lu, H.; Buikhuisen, J.; Soh, B.S.; Lim, E.; Reinhardt, F.; Wu, Z.J.; Krall, J.A.; Bierie, B.; Guo, W.; et al. Protein Kinase C α Is a Central Signaling Node and Therapeutic Target for Breast Cancer Stem Cells. Cancer Cell 2013, 24, 347–364. [Google Scholar] [CrossRef] [Green Version]
- Kawano, T.; Inokuchi, J.; Eto, M.; Murata, M.; Kang, J.H. Activators and Inhibitors of Protein Kinase C (PKC): Their Applications in Clinical Trials. Pharmaceutics 2021, 13, 1748. [Google Scholar] [CrossRef]
- Kannaiyan, R.; Mahadevan, D. A Comprehensive Review of Protein Kinase Inhibitors for Cancer Therapy. Expert Rev. Anticancer Ther. 2018, 18, 1249–1270. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.; Cross, D.; Jänne, P.A. Kinase Drug Discovery 20 Years after Imatinib: Progress and Future Directions. Nat. Rev. Drug Discov. 2021, 20, 551–569. [Google Scholar] [CrossRef] [PubMed]
- Bhullar, K.S.; Lagarón, N.O.; Mcgowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-Targeted Cancer Therapies: Progress, Challenges and Future Directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaoka, T.; Kusumoto, S.; Ando, K.; Ohba, M.; Ohmori, T. Receptor Tyrosine Kinase-Targeted Cancer Therapy. Int. J. Mol. Sci. 2018, 19, 3491. [Google Scholar] [CrossRef] [Green Version]
- Alharbi, K.S.; Javed Shaikh, M.A.; Afzal, O.; Alfawaz Altamimi, A.S.; Almalki, W.H.; Alzarea, S.I.; Kazmi, I.; Al-Abbasi, F.A.; Singh, S.K.; Dua, K.; et al. An Overview of Epithelial Growth Factor Receptor (EGFR) Inhibitors in Cancer Therapy. Chem. Biol. Interact. 2022, 366, 110108. [Google Scholar] [CrossRef]
- Alzahrani, A.S. PI3K/Akt/MTOR Inhibitors in Cancer: At the Bench and Bedside. Semin. Cancer Biol. 2019, 59, 125–132. [Google Scholar] [CrossRef]
- Anwar, S.; Mohammad, T.; Shamsi, A.; Queen, A.; Parveen, S.; Luqman, S.; Hasan, G.M.; Alamry, K.A.; Azum, N.; Asiri, A.M.; et al. Discovery of Hordenine as a Potential Inhibitor of Pyruvate Dehydrogenase Kinase 3: Implication in Lung Cancer Therapy. Biomedicines 2020, 8, 119. [Google Scholar] [CrossRef]
- Fassl, A.; Geng, Y.; Sicinski, P. CDK4 and CDK6 Kinases: From Basic Science to Cancer Therapy. Science 2022, 375, eabc1495. [Google Scholar] [CrossRef]
- Du, R.; Huang, C.; Liu, K.; Li, X.; Dong, Z. Targeting AURKA in Cancer: Molecular Mechanisms and Opportunities for Cancer Therapy. Mol. Cancer 2021, 20, 1–27. [Google Scholar] [CrossRef]
- Yesilkanal, A.E.; Johnson, G.L.; Ramos, A.F.; Rosner, M.R. New Strategies for Targeting Kinase Networks in Cancer. J. Biol. Chem. 2021, 297, 101128. [Google Scholar] [CrossRef]
- Moradi Kashkooli, F.; Soltani, M. Evaluation of Solid Tumor Response to Sequential Treatment Cycles via a New Computational Hybrid Approach. Sci. Rep. 2021, 11, 21475. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape Provides a Biologist-Oriented Resource for the Analysis of Systems-Level Datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING Database in 2021: Customizable Protein-Protein Networks, and Functional Characterization of User-Uploaded Gene/Measurement Sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Bader, G.D.; Hogue, C.W.V. An Automated Method for Finding Molecular Complexes in Large Protein Interaction Networks. BMC Bioinform. 2003, 4, 2. [Google Scholar] [CrossRef] [Green Version]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pagès, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape Plug-in to Decipher Functionally Grouped Gene Ontology and Pathway Annotation Networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [Green Version]
- Burgdorf, C.; Schäfer, U.; Richardt, G.; Kurz, T. U73122, an Aminosteroid Phospholipase C Inhibitor, Is a Potent Inhibitor of Cardiac Phospholipase D by a PIP2-Dependent Mechanism. J. Cardiovasc. Pharmacol. 2010, 55, 555–559. [Google Scholar] [CrossRef]
- Parker, P.J.; Brown, S.J.; Calleja, V.; Chakravarty, P.; Cobbaut, M.; Linch, M.; Marshall, J.J.T.; Martini, S.; McDonald, N.Q.; Soliman, T.; et al. Equivocal, Explicit and Emergent Actions of PKC Isoforms in Cancer. Nat. Rev. Cancer 2021, 21, 51–63. [Google Scholar] [CrossRef]
- Black, A.R.; Black, J.D. The Complexities of PKCα Signaling in Cancer. Adv. Biol. Regul. 2021, 80, 100769. [Google Scholar] [CrossRef]
- Isakov, N. Protein Kinase C (PKC) Isoforms in Cancer, Tumor Promotion and Tumor Suppression. Semin. Cancer Biol. 2018, 48, 36–52. [Google Scholar] [CrossRef]
- Caiazza, F.; Elliott, L.; Fennelly, D.; Sheahan, K.; Doherty, G.A.; Ryan, E.J. Targeting EGFR in Metastatic Colorectal Cancer beyond the Limitations of KRAS Status: Alternative Biomarkers and Therapeutic Strategies. Biomark. Med. 2015, 9, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Caiazza, F.; Harvey, B.J.; Thomas, W. Cytosolic Phospholipase A 2 Activation Correlates with HER2 Overexpression and Mediates Estrogen-Dependent Breast Cancer Cell Growth. Mol. Endocrinol. 2010, 24, 953–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cubas, R.; Zhang, S.; Li, M.; Chen, C.; Yao, Q. Trop2 Expression Contributes to Tumor Pathogenesis by Activating the ERK MAPK Pathway. Mol. Cancer 2010, 9, 253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wieczorek, M.; Abualrous, E.T.; Sticht, J.; Álvaro-Benito, M.; Stolzenberg, S.; Noé, F.; Freund, C. Major Histocompatibility Complex (MHC) Class I and MHC Class II Proteins: Conformational Plasticity in Antigen Presentation. Front. Immunol. 2017, 8, 292. [Google Scholar] [CrossRef] [Green Version]
- Pishesha, N.; Harmand, T.J.; Ploegh, H.L. A Guide to Antigen Processing and Presentation. Nat. Rev. Immunol. 2022, 26, 1–14. [Google Scholar] [CrossRef]
- Dersh, D.; Hollý, J.; Yewdell, J.W. A Few Good Peptides: MHC Class I-Based Cancer Immunosurveillance and Immunoevasion. Nat. Rev. Immunol. 2021, 21, 116–128. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, X.; Cheng, M.; Guan, J.; Gai, J.; Fu, L.; Zhang, R.; Du, T.; Li, Q. Expression of IFN-Induced 2′-5′-Oligoadenylate Synthetases Correlates with Immune Infiltration, Revealing Potential Targets and New Biomarkers for Basal-like Breast Cancer Prognosis. Int. Immunopharmacol. 2020, 88, 106916. [Google Scholar] [CrossRef]
- Zhang, Y.; Yu, C. Prognostic Characterization of OAS1/OAS2/OAS3/OASL in Breast Cancer. BMC Cancer 2020, 20, 575. [Google Scholar] [CrossRef]
- Cooper, J.; Giancotti, F.G. Cancer Cell Review Integrin Signaling in Cancer: Mechanotransduction, Stemness, Epithelial Plasticity, and Therapeutic Resistance. Cancer Cell 2019, 35, 347–367. [Google Scholar] [CrossRef]
- Karantza, V. Keratins in Health and Cancer: More than Mere Epithelial Cell Markers. Oncogene 2010, 30, 127–138. [Google Scholar] [CrossRef]
- Heo, S.H.; Lee, J.Y.; Yang, K.M.; Park, K.S. ELK3 Expression Correlates with Cell Migration, Invasion, and Membrane Type 1-Matrix Metalloproteinase Expression in MDA-MB-231 Breast Cancer Cells. Gene Expr. 2015, 16, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Hoxhaj, G.; Manning, B.D. The PI3K–AKT Network at the Interface of Oncogenic Signalling and Cancer Metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Miricescu, D.; Totan, A.; Stanescu-Spinu, I.I.; Badoiu, S.C.; Stefani, C.; Greabu, M. PI3K/AKT/MTOR Signaling Pathway in Breast Cancer: From Molecular Landscape to Clinical Aspects. Int. J. Mol. Sci. 2021, 22, 173. [Google Scholar] [CrossRef] [PubMed]
- Costa, R.L.B.; Han, H.S.; Gradishar, W.J. Targeting the PI3K/AKT/MTOR Pathway in Triple-Negative Breast Cancer: A Review. Breast Cancer Res. Treat. 2018, 169, 397–406. [Google Scholar] [CrossRef]
- Thorpe, L.M.; Yuzugullu, H.; Zhao, J.J. PI3K in Cancer: Divergent Roles of Isoforms, Modes of Activation and Therapeutic Targeting. Nat. Rev. Cancer 2015, 15, 7–24. [Google Scholar] [CrossRef] [Green Version]
- Rathinaswamy, M.K.; Burke, J.E. Advances in Biological Regulation Class I Phosphoinositide 3-Kinase (PI3K) Regulatory Subunits and Their Roles in Signaling and Disease. Adv. Biol. Regul. 2020, 75, 100657. [Google Scholar] [CrossRef]
- Taniguchi, C.M.; Winnay, J.; Kondo, T.; Bronson, R.T.; Guimaraes, A.R.; Alemán, J.O.; Luo, J.; Stephanopoulos, G.; Weissleder, R.; Cantley, L.C.; et al. The Phosphoinositide 3-Kinase Regulatory Subunit P85α Can Exert Tumor Suppressor Properties through Negative Regulation of Growth Factor Signaling. Cancer Res. 2010, 70, 5305–5315. [Google Scholar] [CrossRef] [Green Version]
- Cizkova, M.; Vacher, S.; Meseure, D.; Trassard, M.; Susini, A.; Mlcuchova, D.; Callens, C.; Rouleau, E.; Spyratos, F.; Lidereau, R.; et al. PIK3R1 Underexpression Is an Independent Prognostic Marker in Breast Cancer. BMC Cancer 2013, 13, 545. [Google Scholar] [CrossRef] [Green Version]
- Cortés, I.; Sánchez-Ruíz, J.; Zuluaga, S.; Calvanese, V.; Marqués, M.; Hernández, C.; Rivera, T.; Kremer, L.; González-García, A.; Carrera, A.C. P85β Phosphoinositide 3-Kinase Subunit Regulates Tumor Progression. Proc. Natl. Acad. Sci. USA 2012, 109, 11318–11323. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Liu, G.; Wang, X.; Sethi, S.; Ali-Fehmi, R.; Abrams, J.; Zheng, Z.; Zhang, K.; Ethier, S.; Yang, Z.Q. ERLIN2 Promotes Breast Cancer Cell Survival by Modulating Endoplasmic Reticulum Stress Pathways. BMC Cancer 2012, 12, 225. [Google Scholar] [CrossRef] [Green Version]
- Ivashkiv, L.B. IFNγ: Signalling, Epigenetics and Roles in Immunity, Metabolism, Disease and Cancer Immunotherapy. Nat. Rev. Immunol. 2018, 18, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Barrat, F.J.; Crow, M.K.; Ivashkiv, L.B. Interferon Target-Gene Expression and Epigenomic Signatures in Health and Disease. Nat. Immunol. 2019, 20, 1574–1583. [Google Scholar] [CrossRef] [PubMed]
- Van der Vorst, E.P.C.; Theodorou, K.; Wu, Y.; Hoeksema, M.A.; Goossens, P.; Bursill, C.A.; Aliyev, T.; Huitema, L.F.A.; Tas, S.W.; Wolfs, I.M.J.; et al. High-Density Lipoproteins Exert Pro-Inflammatory Effects on Macrophages via Passive Cholesterol Depletion and PKC-NF-ΚB/STAT1-IRF1 Signaling. Cell Metab. 2017, 25, 197–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, X.; Li, A.S.; Zheng, H.; Zhao, D.; Guan, D.; Zou, H. Different Associations of CD45 Isoforms with STAT3, PKC and ERK Regulate IL-6-Induced Proliferation in Myeloma. PLoS ONE 2015, 10, e0119780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhoeven, Y.; Tilborghs, S.; Jacobs, J.; De Waele, J.; Quatannens, D.; Deben, C.; Prenen, H.; Pauwels, P.; Trinh, X.B.; Wouters, A.; et al. The Potential and Controversy of Targeting STAT Family Members in Cancer. Semin. Cancer Biol. 2020, 60, 41–56. [Google Scholar] [CrossRef]
- Loh, C.Y.; Arya, A.; Naema, A.F.; Wong, W.F.; Sethi, G.; Looi, C.Y. Signal Transducer and Activator of Transcription (STATs) Proteins in Cancer and Inflammation: Functions and Therapeutic Implication. Front. Oncol. 2019, 9, 48. [Google Scholar] [CrossRef] [Green Version]
- Rah, B.; Rather, R.A.; Bhat, G.R.; Baba, A.B.; Mushtaq, I.; Farooq, M.; Yousuf, T.; Dar, S.B.; Parveen, S.; Hassan, R.; et al. JAK/STAT Signaling: Molecular Targets, Therapeutic Opportunities, and Limitations of Targeted Inhibitions in Solid Malignancies. Front. Pharmacol. 2022, 13, 821344. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; Kundu, M.; Das, S.; Jena, B.C.; Mandal, M. Understanding the Function and Regulation of Sox2 for Its Therapeutic Potential in Breast Cancer. Biochim. Biophys. Acta Rev. Cancer 2022, 1877, 188692. [Google Scholar] [CrossRef] [PubMed]
- Alves, C.L.; Elias, D.; Lyng, M.B.; Bak, M.; Ditzel, H.J. SNAI2 Upregulation Is Associated with an Aggressive Phenotype in Fulvestrant-Resistant Breast Cancer Cells and Is an Indicator of Poor Response to Endocrine Therapy in Estrogen Receptor-Positive Metastatic Breast Cancer. Breast Cancer Res. 2018, 20, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obr, A.E.; Kumar, S.; Chang, Y.J.; Bulatowicz, J.J.; Barnes, B.J.; Birge, R.B.; Lazzarino, D.A.; Gallagher, E.; Leroith, D.; Wood, T.L. Insulin-like Growth Factor Receptor Signaling in Breast Tumor Epithelium Protects Cells from Endoplasmic Reticulum Stress and Regulates the Tumor Microenvironment 11 Medical and Health Sciences 1112 Oncology and Carcinogenesis. Breast Cancer Res. 2018, 20, 138. [Google Scholar] [CrossRef]
- Opyrchal, M.; Gil, M.; Salisbury, J.L.; Goetz, M.P.; Suman, V.; Degnim, A.; McCubrey, J.; Haddad, T.; Iankov, I.; Kurokawa, C.B.; et al. Molecular Targeting of the Aurora-A/SMAD5 Oncogenic Axis Restores Chemosensitivity in Human Breast Cancer Cells. Oncotarget 2017, 8, 91803–91816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavoie, H.; Gagnon, J.; Therrien, M. ERK Signalling: A Master Regulator of Cell Behaviour, Life and Fate. Nat. Rev. Mol. Cell Biol. 2020, 21, 607–632. [Google Scholar] [CrossRef] [PubMed]
- Kyriakis, J.M.; Avruch, J. MAP Kinase Pathways. In Compendium of Inflammatory Diseases; Springer: Berlin/Heidelberg, Germany, 2016; pp. 892–908. [Google Scholar] [CrossRef]
- Khojasteh Poor, F.; Keivan, M.; Ramazii, M.; Ghaedrahmati, F.; Anbiyaiee, A.; Panahandeh, S.; Khoshnam, S.E.; Farzaneh, M. Mini Review: The FDA-Approved Prescription Drugs That Target the MAPK Signaling Pathway in Women with Breast Cancer. Breast Dis. 2021, 40, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, D.A.J.; Negm, O.H.; Alabdullah, M.L.; Mirza, S.; Hamed, M.R.; Band, V.; Green, A.R.; Ellis, I.O.; Rakha, E.A. Clinicopathological and Prognostic Significance of Mitogen-Activated Protein Kinases (MAPK) in Breast Cancers. Breast Cancer Res. Treat. 2016, 159, 457–467. [Google Scholar] [CrossRef]
- Lin, R.; Bao, X.; Wang, H.; Zhu, S.; Liu, Z.; Chen, Q.; Ai, K.; Shi, B. TRPM2 Promotes Pancreatic Cancer by PKC/MAPK Pathway. Cell Death Dis. 2021, 12, 585. [Google Scholar] [CrossRef]
- Kim, S.J.; Pham, T.H.; Bak, Y.; Ryu, H.W.; Oh, S.R.; Yoon, D.Y. Orientin Inhibits Invasion by Suppressing MMP-9 and IL-8 Expression via the PKCα/ ERK/AP-1/STAT3-Mediated Signaling Pathways in TPA-Treated MCF-7 Breast Cancer Cells. Phytomedicine 2018, 50, 35–42. [Google Scholar] [CrossRef]
- Huang, C.; Cao, Z.; Ma, J.; Shen, Y.; Bu, Y.; Khoshaba, R.; Shi, G.; Huang, D.; Liao, D.F.; Ji, H.; et al. AKR1B10 Activates Diacylglycerol (DAG) Second Messenger in Breast Cancer Cells. Mol. Carcinog. 2018, 57, 1300–1310. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.T.; Chang, F.; Lehmann, B.; Terrian, D.M.; Milella, M.; Tafuri, A.; et al. Roles of the Raf/MEK/ERK Pathway in Cell Growth, Malignant Transformation and Drug Resistance. Biochim. Biophys. Acta-Mol. Cell Res. 2007, 1773, 1263–1284. [Google Scholar] [CrossRef] [Green Version]
- Kruiswijk, F.; Labuschagne, C.F.; Vousden, K.H. P53 in Survival, Death and Metabolic Health: A Lifeguard with a Licence to Kill. Nat. Rev. Mol. Cell Biol. 2015, 16, 393–405. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, C.; Hu, W.; Feng, Z. Tumor Suppressor P53 and Metabolism. J. Mol. Cell Biol. 2019, 11, 284–292. [Google Scholar] [CrossRef] [Green Version]
- Roos, W.P.; Thomas, A.D.; Kaina, B. DNA Damage and the Balance between Survival and Death in Cancer Biology. Nat. Rev. Cancer 2016, 16, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Boehme, K.A.; Kulikov, R.; Blattner, C. P53 Stabilization in Response to DNA Damage Requires Akt/PKB and DNA-PK. Proc. Natl. Acad. Sci. USA 2008, 105, 7785–7790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, T.T.; Hsieh, Y.H.; Hsieh, Y.S.; Liu, J.Y. Reduction of PKCα Decreases Cell Proliferation, Migration, and Invasion of Human Malignant Hepatocellular Carcinoma. J. Cell. Biochem. 2008, 103, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.D.; Enge, M.; Bao, W.; Thullberg, M.; Costa, T.D.F.; Olofsson, H.; Gashi, B.; Selivanova, G.; Strömblad, S. Protein Kinase Cα (PKCα) Regulates P53 Localization and Melanoma Cell Survival Downstream of Integrin Av in Three-Dimensional Collagen and in Vivo. J. Biol. Chem. 2012, 287, 29336–29347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brufsky, A.M.; Dickler, M.N. Estrogen Receptor-Positive Breast Cancer: Exploiting Signaling Pathways Implicated in Endocrine Resistance. Oncologist 2018, 23, 528–539. [Google Scholar] [CrossRef] [Green Version]
- Lei, J.T.; Gou, X.; Seker, S.; Ellis, M.J. ESR1 Alterations and Metastasis in Estrogen Receptor Positive Breast Cancer. J. Cancer Metastasis Treat. 2019, 5, 38. [Google Scholar] [CrossRef] [Green Version]
- Toska, E.; Osmanbeyoglu, H.U.; Castel, P.; Chan, C.; Hendrickson, R.C.; Elkabets, M.; Dickler, M.N.; Scaltriti, M.; Leslie, C.S.; Armstrong, S.A.; et al. PI3K Pathway Regulates ER-Dependent Transcription in Breast Cancer through the Epigenetic Regulator KMT2D. Science 2017, 355, 1324–1330. [Google Scholar] [CrossRef] [Green Version]
- Bosch, A.; Li, Z.; Bergamaschi, A.; Ellis, H.; Toska, E.; Prat, A.; Tao, J.J.; Spratt, D.E.; Viola-Villegas, N.T.; Castel, P.; et al. PI3K Inhibition Results in Enhanced Estrogen Receptor Function and Dependence in Hormone Receptor-Positive Breast Cancer. Sci. Transl. Med. 2015, 7, 283ra51. [Google Scholar] [CrossRef] [Green Version]
- Guardiola, S.; Varese, M.; Sánchez-Navarro, M.; Giralt, E. A Third Shot at EGFR: New Opportunities in Cancer Therapy. Trends Pharmacol. Sci. 2019, 40, 941–955. [Google Scholar] [CrossRef]
- Harris, R.C.; Chung, E.; Coffey, R.J. EGF Receptor Ligands. Exp. Cell Res. 2003, 284, 2–13. [Google Scholar] [CrossRef]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging Functions of the EGFR in Cancer. Mol. Oncol. 2018, 12, 3–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Gamal, M.I.; Mewafi, N.H.; Abdelmotteleb, N.E.; Emara, M.A.; Tarazi, H.; Sbenati, R.M.; Madkour, M.M.; Zaraei, S.O.; Shahin, A.I.; Anbar, H.S. A Review of HER4 (ErbB4) Kinase, Its Impact on Cancer, and Its Inhibitors. Molecules 2021, 26, 7376. [Google Scholar] [CrossRef] [PubMed]
- Segers, V.F.M.; Dugaucquier, L.; Feyen, E.; Shakeri, H.; De Keulenaer, G.W. The Role of ErbB4 in Cancer. Cell. Oncol. 2020, 43, 335–352. [Google Scholar] [CrossRef]
- Lucas, L.M.; Dwivedi, V.; Senfeld, J.I.; Cullum, R.L.; Mill, C.P.; Piazza, J.T.; Bryant, I.N.; Cook, L.J.; Miller, S.T.; Lott, J.H.; et al. The Yin and Yang of ERBB4: Tumor Suppressor and Oncoprotein. Pharmacol. Rev. 2022, 74, 18–47. [Google Scholar] [CrossRef] [PubMed]
- Avraham, R.; Yarden, Y. Feedback Regulation of EGFR Signalling: Decision Making by Early and Delayed Loops. Nat. Rev. Mol. Cell Biol. 2011, 12, 104–117. [Google Scholar] [CrossRef]
- Fernández-Ulibarri, I.; Vilella, M.; Lázaro-Diéguez, F.; Sarri, E.; Martínez, S.E.; Jiménez, N.; Claro, E.; Mérida, I.; Burger, K.N.J.; Egea, G. Diacylglycerol Is Required for the Formation of COPI Vesicles in the Golgi-to-ER Transport Pathway. Mol. Biol. Cell 2007, 18, 3250–3263. [Google Scholar] [CrossRef] [Green Version]
- Lonic, A.; Gehling, F.; Belle, L.; Li, X.; Schieber, N.L.; Nguyen, E.V.; Goodall, G.J.; Parton, R.G.; Daly, R.J.; Khew-Goodall, Y. Phosphorylation of PKCδ by FER Tips the Balance from EGFR Degradation to Recycling. J. Cell Biol. 2021, 220, e201902073. [Google Scholar] [CrossRef]
- Liu, M.; Idkowiak-Baldys, J.; Roddy, P.L.; Baldys, A.; Raymond, J.; Clarke, C.J.; Hannun, Y.A. Sustained Activation of Protein Kinase C Induces Delayed Phosphorylation and Regulates the Fate of Epidermal Growth Factor Receptor. PLoS ONE 2013, 8, e80721. [Google Scholar] [CrossRef] [Green Version]
- Heckman, C.A.; Biswas, T.; Dimick, D.M.; Cayer, M.L. Activated Protein Kinase C (PKC) Is Persistently Trafficked with Epidermal Growth Factor (EGF) Receptor. Biomolecules 2020, 10, 1288. [Google Scholar] [CrossRef]
- Bao, J.; Alroy, I.; Waterman, H.; Schejter, E.D.; Brodie, C.; Gruenberg, J.; Yarden, Y. Threonine Phosphorylation Diverts Internalized Epidermal Growth Factor Receptors from a Degradative Pathway to the Recycling Endosome. J. Biol. Chem. 2000, 275, 26178–26186. [Google Scholar] [CrossRef] [Green Version]
- Dietrich, M.; Malik, M.S.; Skeie, M.; Bertelsen, V.; Stang, E. Protein Kinase C Regulates ErbB3 Turnover. Exp. Cell Res. 2019, 382, 111473. [Google Scholar] [CrossRef] [PubMed]
- Basso, F.; Rocchetti, F.; Rodriguez, S.; Nesterova, M.; Cormier, F.; Stratakis, C.A.; Ragazzon, B.; Bertherat, J.; Rizk-Rabin, M. Comparison of the Effects of PRKAR1A and PRKAR2B Depletion on Signaling Pathways, Cell Growth, and Cell Cycle Control of Adrenocortical Cells. Horm. Metab. Res. 2014, 46, 883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saidy, B.; Kotecha, S.; Butler, A.; Rakha, E.A.; Ellis, I.O.; Green, A.R.; Martin, S.G.; Storr, S.J. PP1, PKA and DARPP-32 in Breast Cancer: A Retrospective Assessment of Protein and MRNA Expression. J. Cell. Mol. Med. 2021, 25, 5015. [Google Scholar] [CrossRef] [PubMed]
- Li, C.-F.; Liu, T.-T.; Chuang, I.-C.; Chen, Y.-Y.; Fang, F.-M.; Chan, T.-C.; Li, W.-S.; Huang, H.-Y. PLCB4 Copy Gain and PLCß4 Overexpression in Primary Gastrointestinal Stromal Tumors: Integrative Characterization of a Lipid-Catabolizing Enzyme Associated with Worse Disease-Free Survival Network; PLCB4, Phospholipase C-SS4. Oncotarget 2017, 8, 19997–20010. [Google Scholar] [CrossRef] [Green Version]
- Han, N.; Zhang, Y.Y.; Zhang, Z.M.; Zhang, F.; Zeng, T.Y.; Zhang, Y.B.; Zhao, W.C. High Expression of PDGFA Predicts Poor Prognosis of Esophageal Squamous Cell Carcinoma. Medicine 2021, 100, e25932. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of Image Analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Ho, J.; Tumkaya, T.; Aryal, S.; Choi, H.; Claridge-Chang, A. Moving beyond P Values: Data Analysis with Estimation Graphics. Nat. Methods 2019, 16, 565–566. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serrano-López, E.M.; Coronado-Parra, T.; Marín-Vicente, C.; Szallasi, Z.; Gómez-Abellán, V.; López-Andreo, M.J.; Gragera, M.; Gómez-Fernández, J.C.; López-Nicolás, R.; Corbalán-García, S. Deciphering the Role and Signaling Pathways of PKCα in Luminal A Breast Cancer Cells. Int. J. Mol. Sci. 2022, 23, 14023. https://doi.org/10.3390/ijms232214023
Serrano-López EM, Coronado-Parra T, Marín-Vicente C, Szallasi Z, Gómez-Abellán V, López-Andreo MJ, Gragera M, Gómez-Fernández JC, López-Nicolás R, Corbalán-García S. Deciphering the Role and Signaling Pathways of PKCα in Luminal A Breast Cancer Cells. International Journal of Molecular Sciences. 2022; 23(22):14023. https://doi.org/10.3390/ijms232214023
Chicago/Turabian StyleSerrano-López, Emilio M., Teresa Coronado-Parra, Consuelo Marín-Vicente, Zoltan Szallasi, Victoria Gómez-Abellán, María José López-Andreo, Marcos Gragera, Juan C. Gómez-Fernández, Rubén López-Nicolás, and Senena Corbalán-García. 2022. "Deciphering the Role and Signaling Pathways of PKCα in Luminal A Breast Cancer Cells" International Journal of Molecular Sciences 23, no. 22: 14023. https://doi.org/10.3390/ijms232214023
APA StyleSerrano-López, E. M., Coronado-Parra, T., Marín-Vicente, C., Szallasi, Z., Gómez-Abellán, V., López-Andreo, M. J., Gragera, M., Gómez-Fernández, J. C., López-Nicolás, R., & Corbalán-García, S. (2022). Deciphering the Role and Signaling Pathways of PKCα in Luminal A Breast Cancer Cells. International Journal of Molecular Sciences, 23(22), 14023. https://doi.org/10.3390/ijms232214023