
Three Microbial Musketeers of the Seas: Shewanella baltica, Aliivibrio fischeri and Vibrio harveyi, and Their Adaptation to Different Salinity Probed by a Proteomic Approach

 , , , , , ,
, , , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
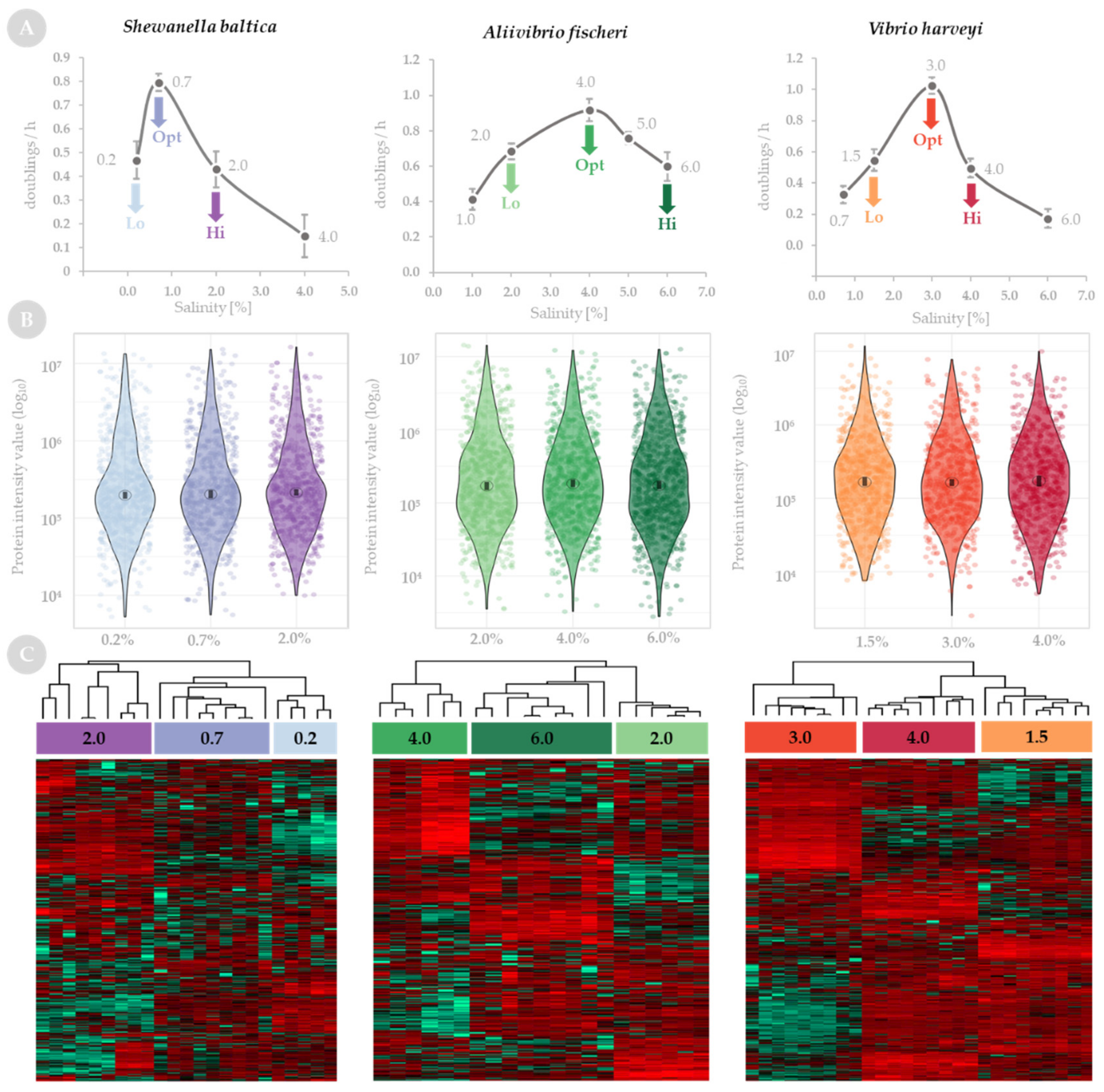
2.1. Quantitative Analysis of Global Proteome Response to Salinity Challenge
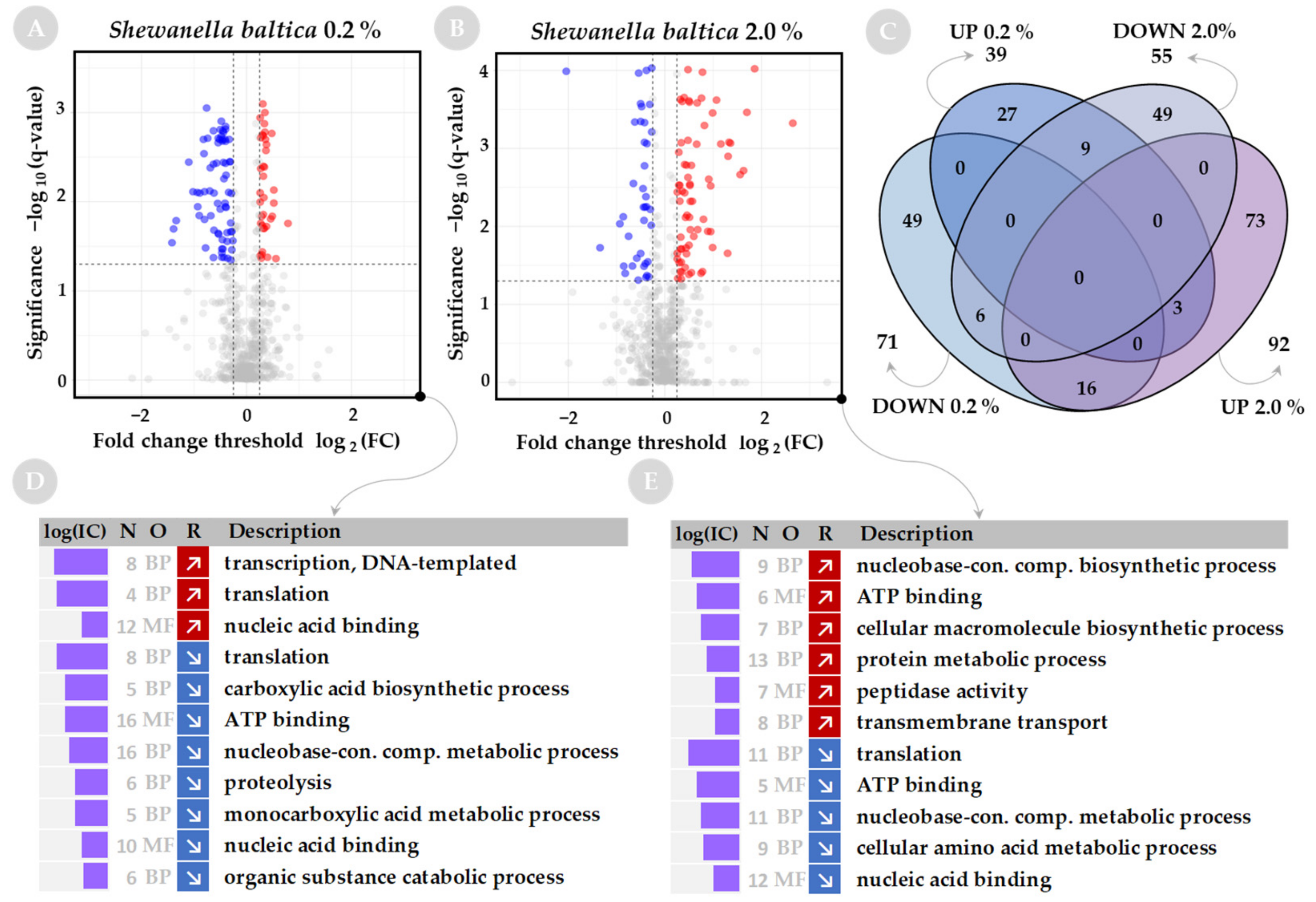
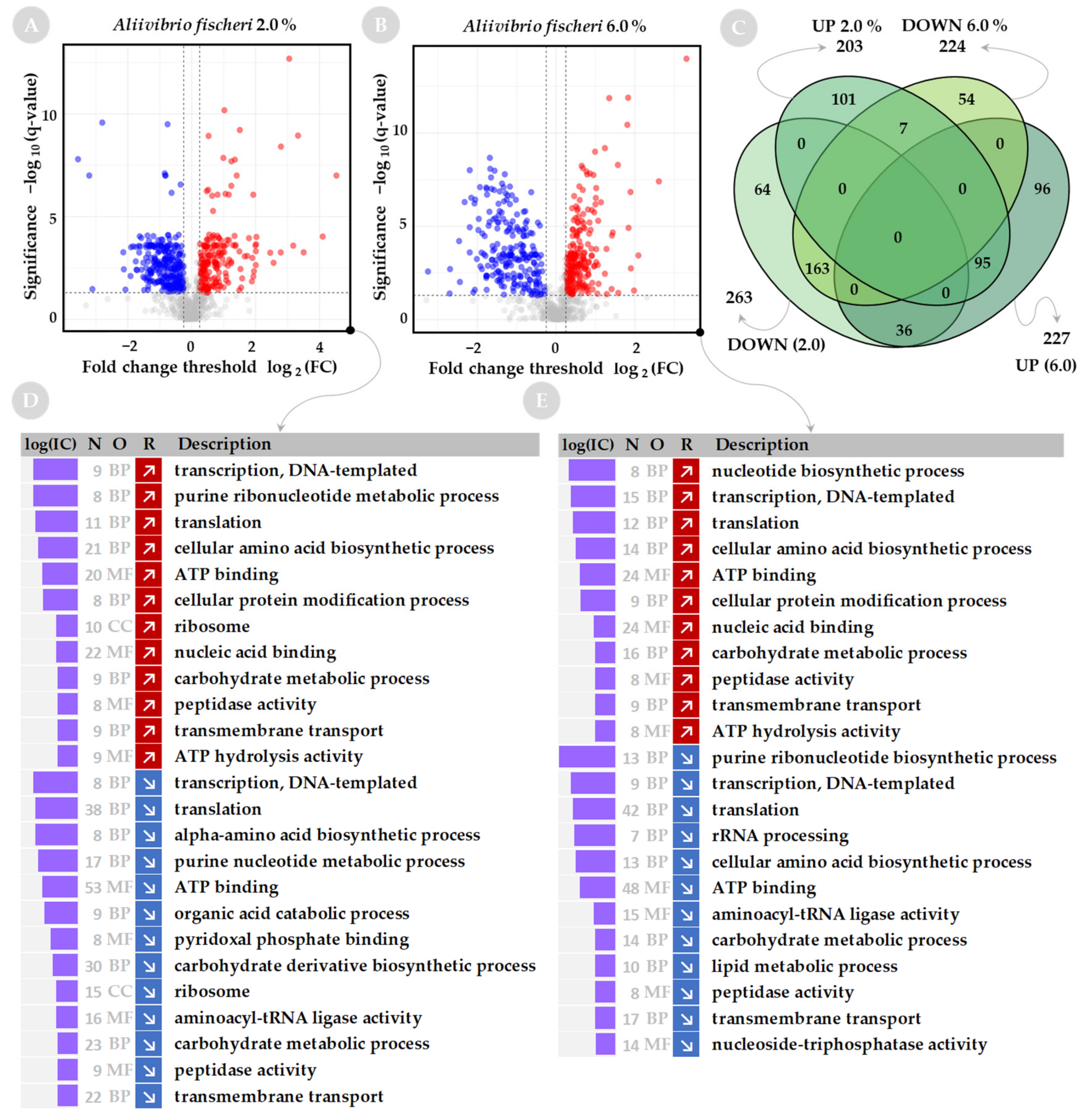
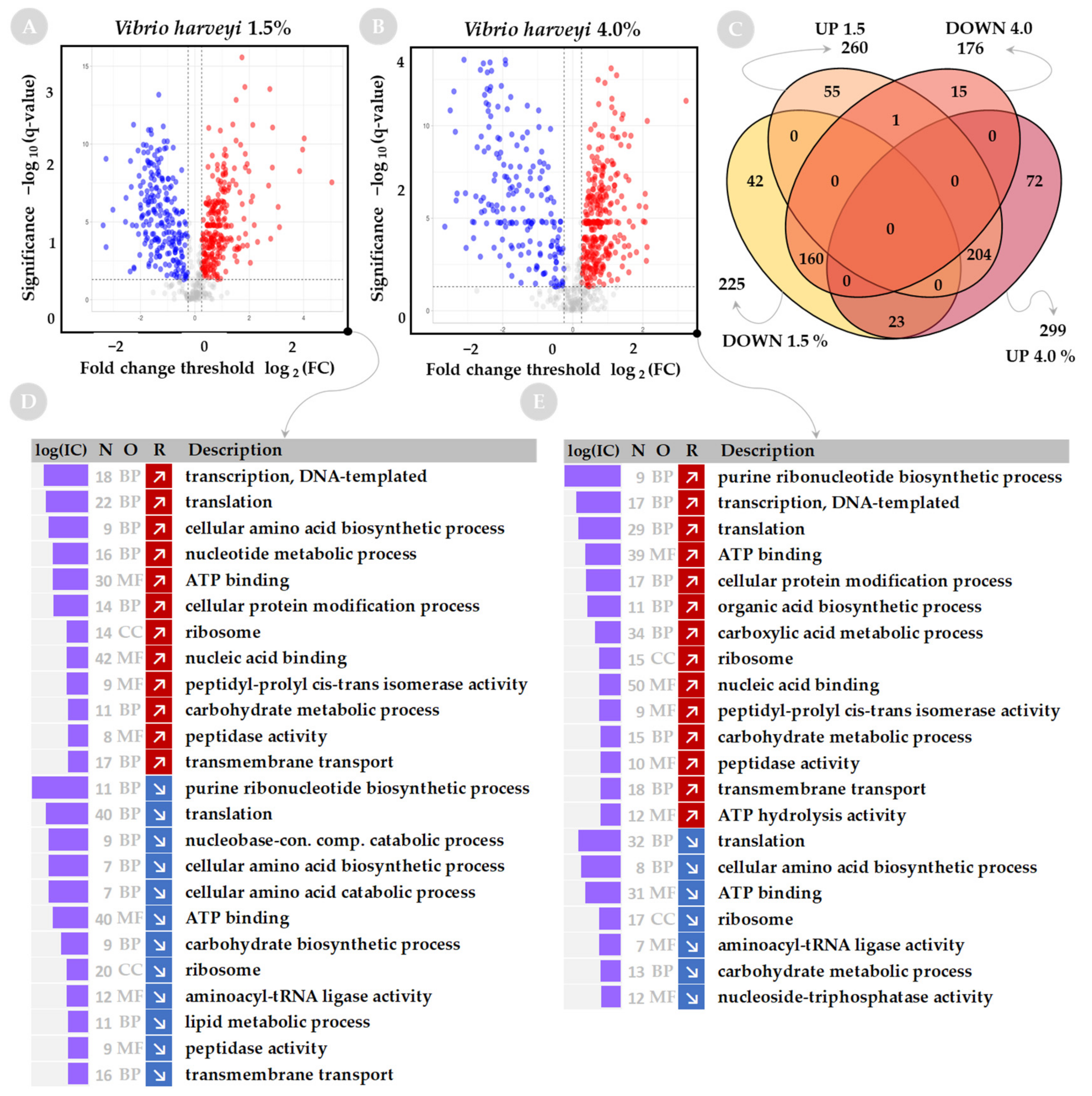
2.2. Functional Analysis of Global Proteome in Response to Salinity Challenge
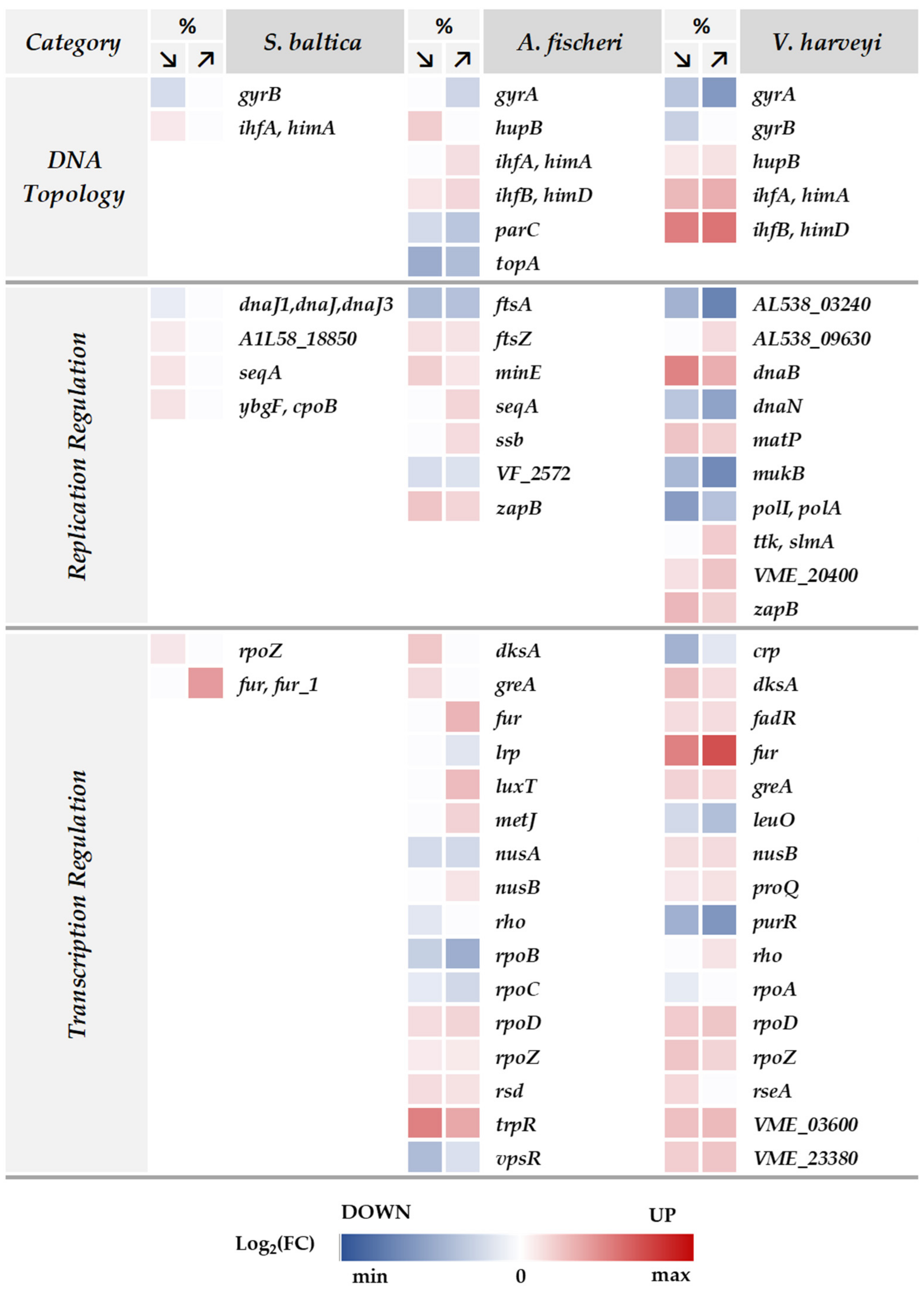
2.3. Changes in Salinity Affect the Nucleic Acid-Binding Proteins
2.4. Reprogramming of the Transcription Machinery as an Adjustment to Osmotic Changes
2.5. The Effect of Osmotic Changes on Translation-Related Proteins
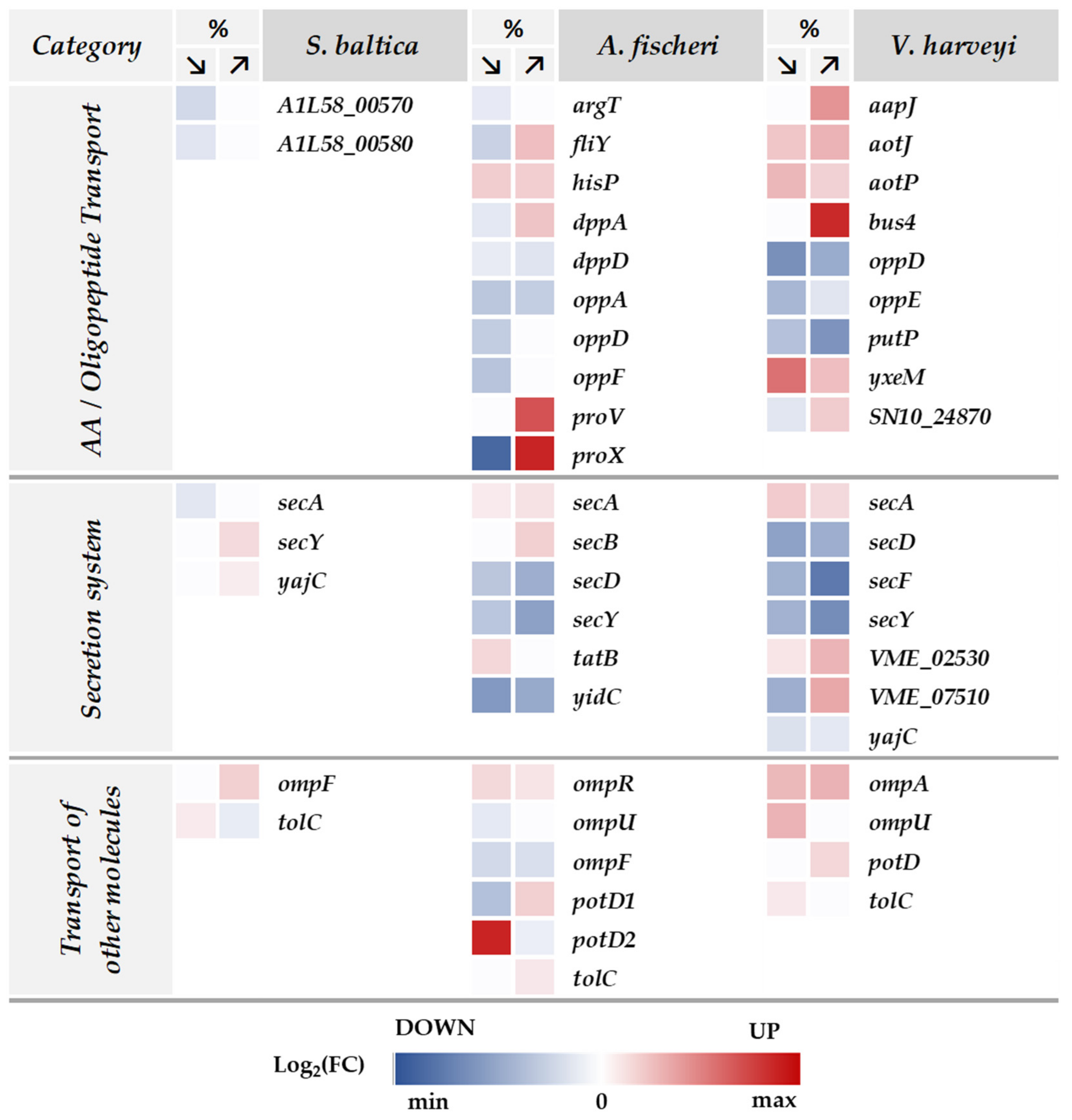
2.6. Transmembrane Transport in Response to Salinity Stress
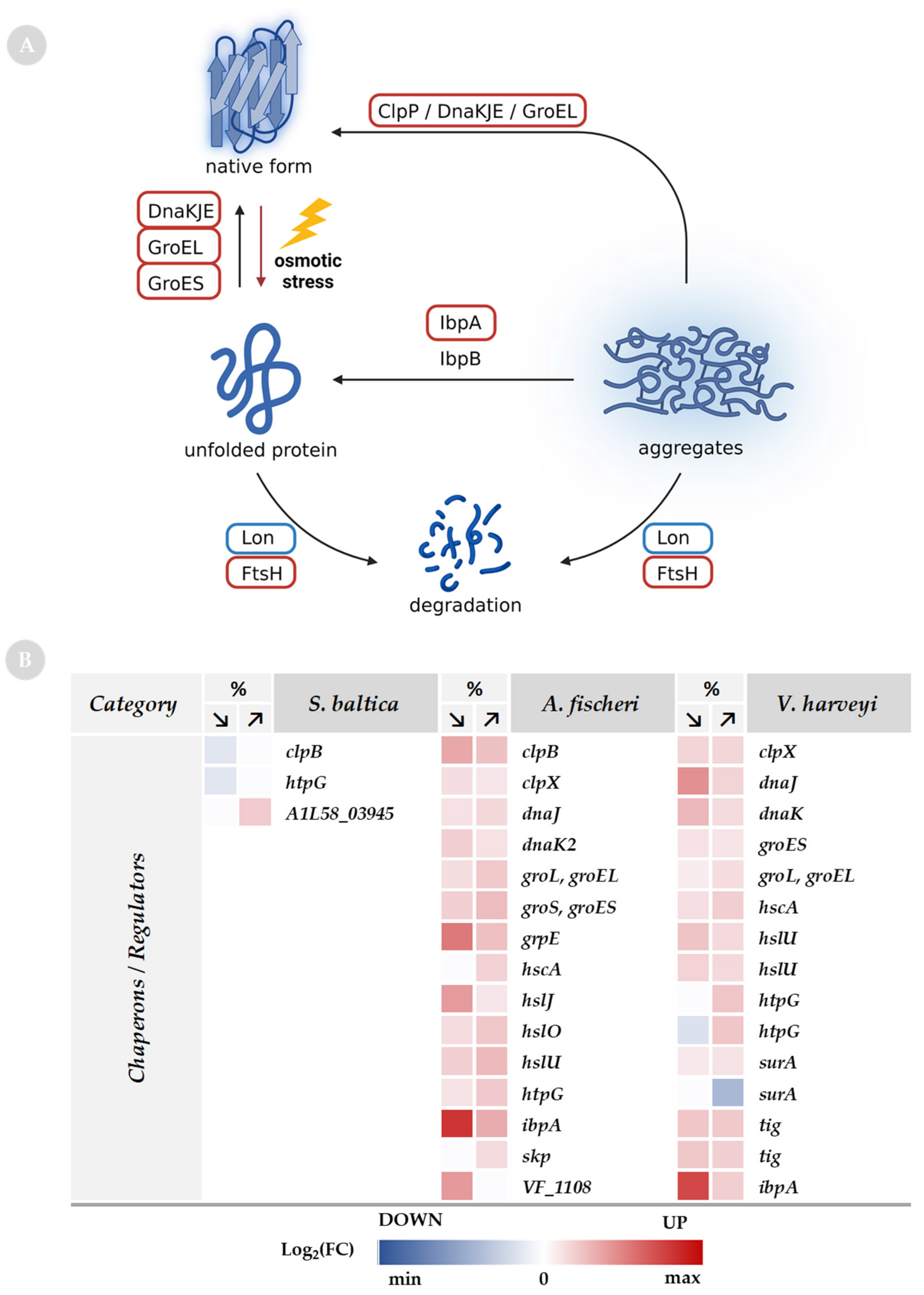
2.7. Chaperones as a Response to Osmolarity Alterations
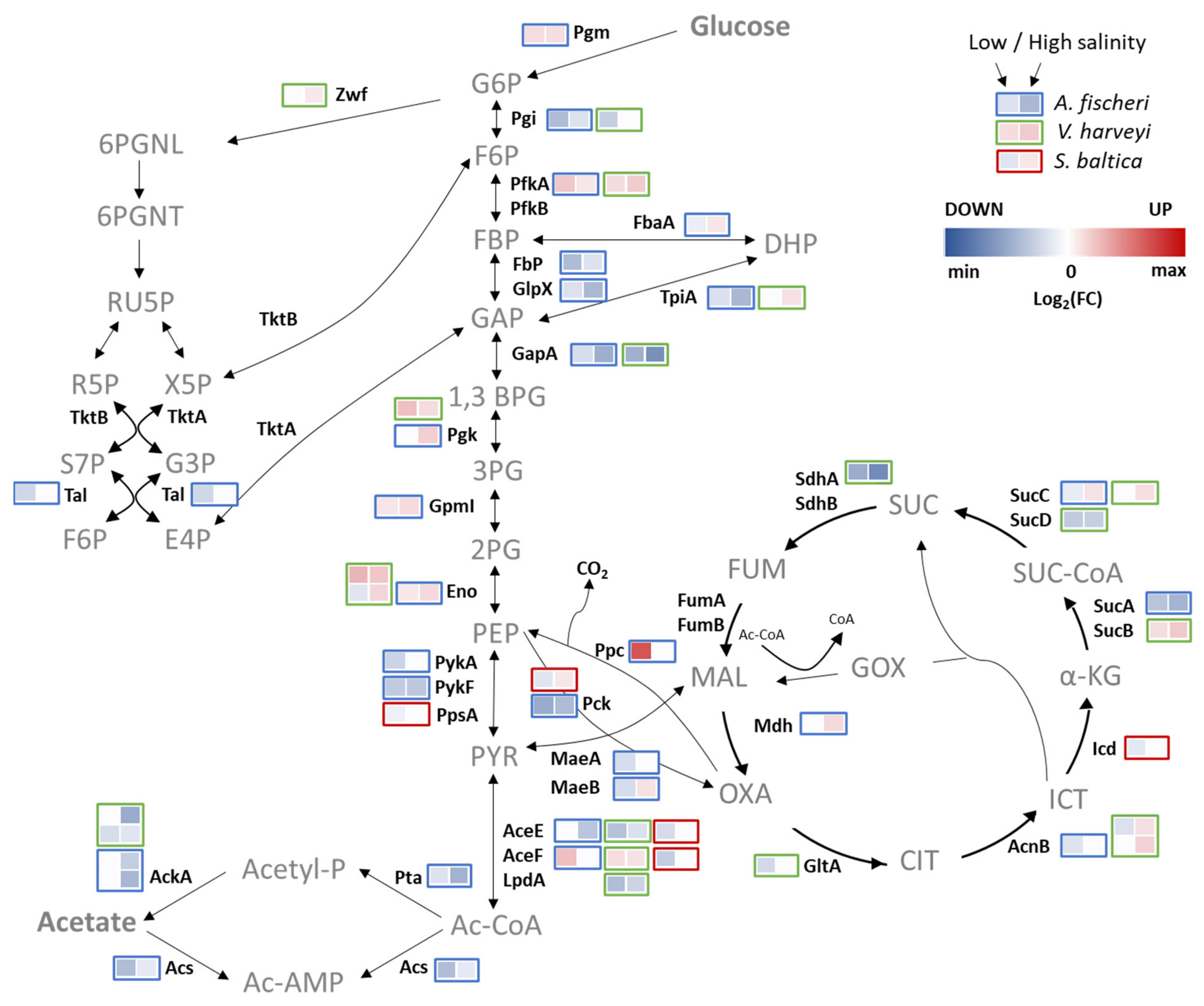
2.8. Metabolic Adjustment to Salinity Changes
2.9. The “All-Hands-on-Board” to Protect the Nucleoid Hypothesis
3. Materials and Methods
3.1. Bacterial Strains and Growth Conditions
3.2. Protein Digestion
3.3. Mass Spectrometry Measurements
3.4. Data Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yancey, P.H. Organic osmolytes as compatible, metabolic and counteracting cytoprotectants in high osmolarity and other stresses. J. Exp. Biol. 2005, 208, 2819–2830. [Google Scholar] [CrossRef] [Green Version]
- Record, M.T.J.; Courtenay, E.S.; Cayley, D.S.; Guttman, H.J. Responses of E. coli to osmotic stress: Large changes in amounts of cytoplasmic solutes and water. Trends Biochem. Sci. 1998, 23, 143–148. [Google Scholar] [CrossRef]
- Lang, F. Mechanisms and significance of cell volume regulation. J. Am. Coll. Nutr. 2007, 26, 613S–623S. [Google Scholar] [CrossRef]
- Bourque, C.W. Central mechanisms of osmosensation and systemic osmoregulation. Nat. Rev. Neurosci. 2008, 9, 519–531. [Google Scholar] [CrossRef]
- Wood, J.M. Bacterial osmoregulation: A paradigm for the study of cellular homeostasis. Annu. Rev. Microbiol. 2011, 65, 215–238. [Google Scholar] [CrossRef] [Green Version]
- Bolen, D.W. Protein stabilization by naturally occurring osmolytes. Methods Mol. Biol. 2001, 168, 17–36. [Google Scholar] [CrossRef]
- Altendorf, K.; Booth, I.R.; Gralla, J.; Greie, J.-C.; Rosenthal, A.Z.; Wood, J.M. Osmotic Stress. EcoSal Plus 2009, 3. [Google Scholar] [CrossRef]
- Kempf, B.; Bremer, E. Uptake and synthesis of compatible solutes as microbial stress responses to high-osmolality environments. Arch. Microbiol. 1998, 170, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Poolman, B.; Blount, P.; Folgering, J.H.A.; Friesen, R.H.E.; Moe, P.C.; van der Heide, T. How do membrane proteins sense water stress? Mol. Microbiol. 2002, 44, 889–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, J.M. Bacterial responses to osmotic challenges. J. Gen. Physiol. 2015, 145, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Schlösser, A.; Meldorf, M.; Stumpe, S.; Bakker, E.P.; Epstein, W. TrkH and its homolog, TrkG, determine the specificity and kinetics of cation transport by the Trk system of Escherichia coli. J. Bacteriol. 1995, 177, 1908–1910. [Google Scholar] [CrossRef] [Green Version]
- Gralla, J.D.; Vargas, D.R. Potassium glutamate as a transcriptional inhibitor during bacterial osmoregulation. EMBO J. 2006, 25, 1515–1521. [Google Scholar] [CrossRef] [Green Version]
- Wood, J.M. Osmosensing by bacteria. Sci. STKE 2006, 2006, pe43. [Google Scholar] [CrossRef]
- Shabala, L.; Bowman, J.; Brown, J.; Ross, T.; McMeekin, T.; Shabala, S. Ion transport and osmotic adjustment in Escherichia coli in response to ionic and non-ionic osmotica. Environ. Microbiol. 2009, 11, 137–148. [Google Scholar] [CrossRef]
- Whatmore, A.M.; Reed, R.H. Determination of turgor pressure in Bacillus subtilis: A possible role for K+ in turgor regulation. J. Gen. Microbiol. 1990, 136, 2521–2526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poolman, B.; Glaasker, E. Regulation of compatible solute accumulation in bacteria. Mol. Microbiol. 1998, 29, 397–407. [Google Scholar] [CrossRef] [Green Version]
- Sleator, R.D.; Hill, C. Bacterial osmoadaptation: The role of osmolytes in bacterial stress and virulence. FEMS Microbiol. Rev. 2002, 26, 49–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sukharev, S.I.; Blount, P.; Martinac, B.; Blattner, F.R.; Kung, C. A large-conductance mechanosensitive channel in E. coli encoded by mscL alone. Nature 1994, 368, 265–268. [Google Scholar] [CrossRef]
- Levina, N.; Tötemeyer, S.; Stokes, N.R.; Louis, P.; Jones, M.A.; Booth, I.R. Protection of Escherichia coli cells against extreme turgor by activation of MscS and MscL mechanosensitive channels: Identification of genes required for MscS activity. EMBO J. 1999, 18, 1730–1737. [Google Scholar] [CrossRef]
- Krämer, R. Bacterial stimulus perception and signal transduction: Response to osmotic stress. Chem. Rec. 2010, 10, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Naughton, L.M.; Blumerman, S.L.; Carlberg, M.; Boyd, E.F. Osmoadaptation among Vibrio species and unique genomic features and physiological responses of Vibrio parahaemolyticus. Appl. Environ. Microbiol. 2009, 75, 2802–2810. [Google Scholar] [CrossRef] [Green Version]
- Penn, K.; Jensen, P.R. Comparative genomics reveals evidence of marine adaptation in Salinispora species. BMC Genom. 2012, 13, 86. [Google Scholar] [CrossRef] [Green Version]
- Rao, N.V.; Shashidhar, R.; Bandekar, J.R. Comparative analysis of induction of osmotic-stress-dependent genes in Vibrio vulnificus exposed to hyper- and hypo-osmotic stress. Can. J. Microbiol. 2013, 59, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Kohler, C.; Lourenço, R.F.; Bernhardt, J.; Albrecht, D.; Schüler, J.; Hecker, M.; Gomes, S.L. A comprehensive genomic, transcriptomic and proteomic analysis of a hyperosmotic stress sensitive α-proteobacterium. BMC Microbiol. 2015, 15, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ongagna-Yhombi, S.Y.; McDonald, N.D.; Boyd, E.F. Deciphering the role of multiple betaine-carnitine-choline transporters in the Halophile Vibrio parahaemolyticus. Appl. Environ. Microbiol. 2015, 81, 351–363. [Google Scholar] [CrossRef] [Green Version]
- Gregory, G.J.; Boyd, E.F. Stressed out: Bacterial response to high salinity using compatible solute biosynthesis and uptake systems, lessons from Vibrionaceae. Comput. Struct. Biotechnol. J. 2021, 19, 1014–1027. [Google Scholar] [CrossRef] [PubMed]
- Fonnesbech Vogel, B.; Venkateswaran, K.; Satomi, M.; Gram, L. Identification of Shewanella baltica as the most important H2S-producing species during iced storage of Danish marine fish. Appl. Environ. Microbiol. 2005, 71, 6689–6697. [Google Scholar] [CrossRef] [Green Version]
- Ge, Y.; Zhu, J.; Ye, X.; Yang, Y. Spoilage potential characterization of Shewanella and Pseudomonas isolated from spoiled large yellow croaker (Pseudosciaena crocea). Lett. Appl. Microbiol. 2017, 64, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Kloska, A.; Cech, G.M.; Sadowska, M.; Krause, K.; Szalewska-Pałasz, A.; Olszewski, P. Adaptation of the Marine Bacterium Shewanella baltica to Low Temperature Stress. Int. J. Mol. Sci. 2020, 21, 4338. [Google Scholar] [CrossRef] [PubMed]
- Harris, H.W.; Sánchez-Andrea, I.; McLean, J.S.; Salas, E.C.; Tran, W.; El-Naggar, M.Y.; Nealson, K.H. Redox Sensing within the Genus Shewanella. Front. Microbiol. 2017, 8, 2568. [Google Scholar] [CrossRef]
- Deng, J.; Brettar, I.; Luo, C.; Auchtung, J.; Konstantinidis, K.T.; Rodrigues, J.L.M.; Hofle, M.; Tiedje, J.M. Stability, genotypic and phenotypic diversity of Shewanella baltica in the redox transition zone of the Baltic Sea. Environ. Microbiol. 2014, 16, 1854–1866. [Google Scholar] [CrossRef]
- Milewska, K.; Krause, K.; Szalewska-Palasz, A. The stringent response of marine bacteria—Assessment of (p)ppGpp accumulation upon stress conditions. J. Appl. Genet. 2020, 61, 123–130. [Google Scholar] [CrossRef]
- Ma, S.; Kim, A.; Lee, W.; Kim, S.; Lee, S.; Yoon, D.; Bae, J.-S.; Park, C.-I.; Kim, S. Vibrio harveyi Infection Significantly Alters Amino Acid and Carbohydrate Metabolism in Whiteleg Shrimp, Litopenaeus vannamei. Metabolites 2020, 10, 265. [Google Scholar] [CrossRef]
- Zhang, X.-H.; He, X.; Austin, B. Vibrio harveyi: A serious pathogen of fish and invertebrates in mariculture. Mar. life Sci. Technol. 2020, 3, 231–245. [Google Scholar] [CrossRef] [Green Version]
- Owens, L.; Busico-Salcedo, N. Vibrio harveyi: Pretty Problems in Paradise. In The Biology of Vibrios; Thompson, F.L., Austin, B., Swings, J., Eds.; ASM Press: Washington, DC, USA, 2006; pp. 266–280. [Google Scholar]
- Waters, C.M.; Bassler, B.L. Quorum sensing: Cell-to-cell communication in bacteria. Annu. Rev. Cell Dev. Biol. 2005, 21, 319–346. [Google Scholar] [CrossRef] [Green Version]
- Montánchez, I.; Kaberdin, V.R. Vibrio harveyi: A brief survey of general characteristics and recent epidemiological traits associated with climate change. Mar. Environ. Res. 2020, 154, 104850. [Google Scholar] [CrossRef] [PubMed]
- Ruby, E.G.; Urbanowski, M.; Campbell, J.; Dunn, A.; Faini, M.; Gunsalus, R.; Lostroh, P.; Lupp, C.; McCann, J.; Millikan, D.; et al. Complete genome sequence of Vibrio fischeri: A symbiotic bacterium with pathogenic congeners. Proc. Natl. Acad. Sci. USA 2005, 102, 3004–3009. [Google Scholar] [CrossRef] [Green Version]
- Morris, A.R.; Visick, K.L. Control of biofilm formation and colonization in Vibrio fischeri: A role for partner switching? Environ. Microbiol. 2010, 12, 2051–2059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graf, J.; Ruby, E.G. Host-derived amino acids support the proliferation of symbiotic bacteria. Proc. Natl. Acad. Sci. USA 1998, 95, 1818–1822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Dufour, Y.S.; Carlson, H.K.; Donohue, T.J.; Marletta, M.A.; Ruby, E.G. H-NOX-mediated nitric oxide sensing modulates symbiotic colonization by Vibrio fischeri. Proc. Natl. Acad. Sci. USA 2010, 107, 8375–8380. [Google Scholar] [CrossRef] [Green Version]
- Jeong, H.S.; Kim, S.M.; Lim, M.S.; Kim, K.S.; Choi, S.H. Direct interaction between quorum-sensing regulator SmcR and RNA polymerase is mediated by integration host factor to activate vvpE encoding elastase in Vibrio vulnificus. J. Biol. Chem. 2010, 285, 9357–9366. [Google Scholar] [CrossRef] [Green Version]
- Chaparian, R.R.; Olney, S.G.; Hustmyer, C.M.; Rowe-Magnus, D.A.; van Kessel, J.C. Integration host factor and LuxR synergistically bind DNA to coactivate quorum-sensing genes in Vibrio harveyi. Mol. Microbiol. 2016, 101, 823–840. [Google Scholar] [CrossRef]
- Learman, D.R.; Yi, H.; Brown, S.D.; Martin, S.L.; Geesey, G.G.; Stevens, A.M.; Hochella, M.F., Jr. Involvement of Shewanella oneidensis MR-1 LuxS in biofilm development and sulfur metabolism. Appl. Environ. Microbiol. 2009, 75, 1301–1307. [Google Scholar] [CrossRef] [Green Version]
- Broyles, S.S.; Pettijohn, D.E. Interaction of the Escherichia coli HU protein with DNA. Evidence for formation of nucleosome-like structures with altered DNA helical pitch. J. Mol. Biol. 1986, 187, 47–60. [Google Scholar] [CrossRef]
- Roy, S.; Dimitriadis, E.K.; Kar, S.; Geanacopoulos, M.; Lewis, M.S.; Adhya, S. Gal repressor-operator-HU ternary complex: Pathway of repressosome formation. Biochemistry 2005, 44, 5373–5380. [Google Scholar] [CrossRef]
- Oberto, J.; Nabti, S.; Jooste, V.; Mignot, H.; Rouviere-Yaniv, J. The HU regulon is composed of genes responding to anaerobiosis, acid stress, high osmolarity and SOS induction. PLoS ONE 2009, 4, e4367. [Google Scholar] [CrossRef] [Green Version]
- Floc’h, K.; Lacroix, F.; Servant, P.; Wong, Y.-S.; Kleman, J.-P.; Bourgeois, D.; Timmins, J. Cell morphology and nucleoid dynamics in dividing Deinococcus radiodurans. Nat. Commun. 2019, 10, 3815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balandina, A.; Kamashev, D.; Rouviere-Yaniv, J. The bacterial histone-like protein HU specifically recognizes similar structures in all nucleic acids. DNA, RNA, and their hybrids. J. Biol. Chem. 2002, 277, 27622–27628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamashev, D.; Rouviere-Yaniv, J. The histone-like protein HU binds specifically to DNA recombination and repair intermediates. EMBO J. 2000, 19, 6527–6535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cech, G.M.; Szalewska-Pałasz, A.; Kubiak, K.; Malabirade, A.; Grange, W.; Arluison, V.; Węgrzyn, G. The Escherichia coli Hfq Protein: An Unattended DNA-Transactions Regulator. Front. Mol. Biosci. 2016, 3, 36. [Google Scholar] [CrossRef] [Green Version]
- Sheng, H.; Stauffer, W.T.; Hussein, R.; Lin, C.; Lim, H.N. Nucleoid and cytoplasmic localization of small RNAs in Escherichia coli. Nucleic Acids Res. 2017, 45, 2919–2934. [Google Scholar] [CrossRef]
- Malabirade, A.; Jiang, K.; Kubiak, K.; Diaz-Mendoza, A.; Liu, F.; van Kan, J.A.; Berret, J.-F.; Arluison, V.; van der Maarel, J.R.C. Compaction and condensation of DNA mediated by the C-terminal domain of Hfq. Nucleic Acids Res. 2017, 45, 7299–7308. [Google Scholar] [CrossRef] [PubMed]
- Wien, F.; Martinez, D.; Le Brun, E.; Jones, N.C.; Vrønning Hoffmann, S.; Waeytens, J.; Berbon, M.; Habenstein, B.; Arluison, V. The Bacterial Amyloid-Like Hfq Promotes In Vitro DNA Alignment. Microorganisms 2019, 7, 639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tehei, M.; Franzetti, B.; Maurel, M.-C.; Vergne, J.; Hountondji, C.; Zaccai, G. The search for traces of life: The protective effect of salt on biological macromolecules. Extremophiles 2002, 6, 427–430. [Google Scholar] [CrossRef] [PubMed]
- Saenger, W.; Hunter, W.N.; Kennard, O. DNA conformation is determined by economics in the hydration of phosphate groups. Nature 1986, 324, 385–388. [Google Scholar] [CrossRef]
- Chitra, S.R.; Ramalakshmi, N.; Arunkumar, S.; Manimegalai, P. A Comprehensive Review on DNA Gyrase Inhibitors. Infect. Disord.—Drug Targets 2021, 20, 765–777. [Google Scholar] [CrossRef] [PubMed]
- Bush, N.G.; Evans-Roberts, K.; Maxwell, A. DNA Topoisomerases. EcoSal Plus 2015, 6, 2. [Google Scholar] [CrossRef]
- Matthews, L.A.; Simmons, L.A. Regulation of DNA Binding and High-Order Oligomerization of the DnaB Helicase Loader. J. Bacteriol. 2020, 202, e00286-20. [Google Scholar] [CrossRef]
- Zhang, W.; Carneiro, M.J.V.M.; Turner, I.J.; Allen, S.; Roberts, C.J.; Soultanas, P. The Bacillus subtilis DnaD and DnaB Proteins Exhibit Different DNA Remodelling Activities. J. Mol. Biol. 2005, 351, 66–75. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, M.A.; Zeng, W. Structures of the nucleoid occlusion protein SlmA bound to DNA and the C-terminal domain of the cytoskeletal protein FtsZ. Proc. Natl. Acad. Sci. USA 2016, 113, 4988–4993. [Google Scholar] [CrossRef] [Green Version]
- Kurkela, J.; Fredman, J.; Salminen, T.A.; Tyystjärvi, T. Revealing secrets of the enigmatic omega subunit of bacterial RNA polymerase. Mol. Microbiol. 2021, 115, 1–11. [Google Scholar] [CrossRef]
- Zenkin, N.; Yuzenkova, Y. New Insights into the Functions of Transcription Factors that Bind the RNA Polymerase Secondary Channel. Biomolecules 2015, 5, 1195–1209. [Google Scholar] [CrossRef] [Green Version]
- Potrykus, K.; Vinella, D.; Murphy, H.; Szalewska-Palasz, A.; D’Ari, R.; Cashel, M. Antagonistic regulation of Escherichia coli ribosomal RNA rrnB P1 promoter activity by GreA and DksA. J. Biol. Chem. 2006, 281, 15238–15248. [Google Scholar] [CrossRef] [Green Version]
- Rutherford, S.T.; Lemke, J.J.; Vrentas, C.E.; Gaal, T.; Ross, W.; Gourse, R.L. Effects of DksA, GreA, and GreB on transcription initiation: Insights into the mechanisms of factors that bind in the secondary channel of RNA polymerase. J. Mol. Biol. 2007, 366, 1243–1257. [Google Scholar] [CrossRef] [Green Version]
- Paul, B.J.; Berkmen, M.B.; Gourse, R.L. DksA potentiates direct activation of amino acid promoters by ppGpp. Proc. Natl. Acad. Sci. USA 2005, 102, 7823–7828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, B.J.; Barker, M.M.; Ross, W.; Schneider, D.A.; Webb, C.; Foster, J.W.; Gourse, R.L. DksA: A critical component of the transcription initiation machinery that potentiates the regulation of rRNA promoters by ppGpp and the initiating NTP. Cell 2004, 118, 311–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potrykus, K.; Cashel, M. (p)ppGpp: Still magical? Annu. Rev. Microbiol. 2008, 62, 35–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinnbier, U.; Limpinsel, E.; Schmid, R.; Bakker, E.P. Transient accumulation of potassium glutamate and its replacement by trehalose during adaptation of growing cells of Escherichia coli K-12 to elevated sodium chloride concentrations. Arch. Microbiol. 1988, 150, 348–357. [Google Scholar] [CrossRef]
- Brown, L.; Gentry, D.; Elliott, T.; Cashel, M. DksA Affects ppGpp Induction of RpoS at a Translational Level. J. Bacteriol. 2002, 184, 4455–4465. [Google Scholar] [CrossRef] [Green Version]
- Yamanaka, K.; Zheng, W.; Crooke, E.; Wang, Y.H.; Inouye, M. CspD, a novel DNA replication inhibitor induced during the stationary phase in Escherichia coli. Mol. Microbiol. 2001, 39, 1572–1584. [Google Scholar] [CrossRef]
- Crozat, E.; Tardin, C.; Salhi, M.; Rousseau, P.; Lablaine, A.; Bertoni, T.; Holcman, D.; Sclavi, B.; Cicuta, P.; Cornet, F. Post-replicative pairing of sister ter regions in Escherichia coli involves multiple activities of MatP. Nat. Commun. 2020, 11, 3796. [Google Scholar] [CrossRef]
- LaMonte, B.L.; Hughes, J.A. In vivo hydrolysis of S-adenosylmethionine induces the met regulon of Escherichia coli. Microbiology 2006, 152, 1451–1459. [Google Scholar] [CrossRef] [Green Version]
- Argandoña, M.; Nieto, J.J.; Iglesias-Guerra, F.; Calderón, M.I.; García-Estepa, R.; Vargas, C. Interplay between iron homeostasis and the osmotic stress response in the halophilic bacterium Chromohalobacter salexigens. Appl. Environ. Microbiol. 2010, 76, 3575–3589. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, T.; Schütz, A.; Brosius, M.; Völker, A.; Völker, U.; Bremer, E. High-salinity-induced iron limitation in Bacillus subtilis. J. Bacteriol. 2002, 184, 718–727. [Google Scholar] [CrossRef] [Green Version]
- Steil, L.; Hoffmann, T.; Budde, I.; Völker, U.; Bremer, E. Genome-wide transcriptional profiling analysis of adaptation of Bacillus subtilis to high salinity. J. Bacteriol. 2003, 185, 6358–6370. [Google Scholar] [CrossRef] [Green Version]
- Du, H.; Sheng, X.; Zhang, H.; Zou, X.; Ni, B.; Xu, S.; Zhu, X.; Xu, H.; Huang, X. RpoE may promote flagellar gene expression in Salmonella enterica serovar typhi under hyperosmotic stress. Curr. Microbiol. 2011, 62, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Selven, S.; Philip, R. Salinity a significant environmental factor for Vibrio harveyi virulence in Fenneropenaeus indicus. Aquac. Res. 2013, 44, 747–759. [Google Scholar] [CrossRef]
- Weber, A.; Jung, K. Profiling early osmostress-dependent gene expression in Escherichia coli using DNA macroarrays. J. Bacteriol. 2002, 184, 5502–5507. [Google Scholar] [CrossRef] [Green Version]
- Bessaiah, H.; Pokharel, P.; Loucif, H.; Kulbay, M.; Sasseville, C.; Habouria, H.; Houle, S.; Bernier, J.; Massé, É.; Van Grevenynghe, J.; et al. The RyfA small RNA regulates oxidative and osmotic stress responses and virulence in uropathogenic Escherichia coli. PLoS Pathog. 2021, 17, e1009617. [Google Scholar] [CrossRef] [PubMed]
- Garre, E.; Romero-Santacreu, L.; Barneo-Muñoz, M.; Miguel, A.; Pérez-Ortín, J.E.; Alepuz, P. Nonsense-mediated mRNA decay controls the changes in yeast ribosomal protein pre-mRNAs levels upon osmotic stress. PLoS ONE 2013, 8, e61240. [Google Scholar] [CrossRef] [PubMed]
- Pircher, A.; Bakowska-Zywicka, K.; Schneider, L.; Zywicki, M.; Polacek, N. An mRNA-derived noncoding RNA targets and regulates the ribosome. Mol. Cell 2014, 54, 147–155. [Google Scholar] [CrossRef] [Green Version]
- Dai, X.; Zhu, M.; Warren, M.; Balakrishnan, R.; Patsalo, V.; Okano, H.; Williamson, J.R.; Fredrick, K.; Wang, Y.-P.; Hwa, T. Reduction of translating ribosomes enables Escherichia coli to maintain elongation rates during slow growth. Nat. Microbiol. 2016, 2, 16231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bremer, H.; Dennis, P.P. Modulation of Chemical Composition and Other Parameters of the Cell at Different Exponential Growth Rates. EcoSal Plus 2008, 3, 1. [Google Scholar] [CrossRef] [PubMed]
- Bremer, E.; Krämer, R. Responses of Microorganisms to Osmotic Stress. Annu. Rev. Microbiol. 2019, 73, 313–334. [Google Scholar] [CrossRef]
- Gevrekci, A.Ö. The roles of polyamines in microorganisms. World J. Microbiol. Biotechnol. 2017, 33, 204. [Google Scholar] [CrossRef]
- Michael, A.J. Polyamines in Eukaryotes, Bacteria, and Archaea. J. Biol. Chem. 2016, 291, 14896–14903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller-Fleming, L.; Olin-Sandoval, V.; Campbell, K.; Ralser, M. Remaining Mysteries of Molecular Biology: The Role of Polyamines in the Cell. J. Mol. Biol. 2015, 427, 3389–3406. [Google Scholar] [CrossRef] [PubMed]
- Mou, X.; Vila-Costa, M.; Sun, S.; Zhao, W.; Sharma, S.; Moran, M.A. Metatranscriptomic signature of exogenous polyamine utilization by coastal bacterioplankton. Environ. Microbiol. Rep. 2011, 3, 798–806. [Google Scholar] [CrossRef]
- Kalamaki, M.S.; Merkouropoulos, G.; Kanellis, A.K. Can ornithine accumulation modulate abiotic stress tolerance in Arabidopsis? Plant Signal. Behav. 2009, 4, 1099–1101. [Google Scholar] [CrossRef] [Green Version]
- Frain, K.M.; Robinson, C.; van Dijl, J.M. Transport of Folded Proteins by the Tat System. Protein J. 2019, 38, 377–388. [Google Scholar] [CrossRef] [Green Version]
- Den Brave, F.; Cairo, L.V.; Jagadeesan, C.; Ruger-Herreros, C.; Mogk, A.; Bukau, B.; Jentsch, S. Chaperone-Mediated Protein Disaggregation Triggers Proteolytic Clearance of Intra-nuclear Protein Inclusions. Cell Rep. 2020, 31, 107680. [Google Scholar] [CrossRef] [PubMed]
- Mogk, A.; Deuerling, E.; Vorderwülbecke, S.; Vierling, E.; Bukau, B. Small heat shock proteins, ClpB and the DnaK system form a functional triade in reversing protein aggregation. Mol. Microbiol. 2003, 50, 585–595. [Google Scholar] [CrossRef]
- Langklotz, S.; Baumann, U.; Narberhaus, F. Structure and function of the bacterial AAA protease FtsH. Biochim. Biophys. Acta 2012, 1823, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Tomoyasu, T.; Gamer, J.; Bukau, B.; Kanemori, M.; Mori, H.; Rutman, A.J.; Oppenheim, A.B.; Yura, T.; Yamanaka, K.; Niki, H. Escherichia coli FtsH is a membrane-bound, ATP-dependent protease which degrades the heat-shock transcription factor sigma 32. EMBO J. 1995, 14, 2551–2560. [Google Scholar] [CrossRef] [PubMed]
- Bruckner, R.C.; Gunyuzlu, P.L.; Stein, R.L. Coupled kinetics of ATP and peptide hydrolysis by Escherichia coli FtsH protease. Biochemistry 2003, 42, 10843–10852. [Google Scholar] [CrossRef] [PubMed]
- Kohlstedt, M.; Sappa, P.K.; Meyer, H.; Maaß, S.; Zaprasis, A.; Hoffmann, T.; Becker, J.; Steil, L.; Hecker, M.; van Dijl, J.M.; et al. Adaptation of Bacillus subtilis carbon core metabolism to simultaneous nutrient limitation and osmotic challenge: A multi-omics perspective. Environ. Microbiol. 2014, 16, 1898–1917. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, Y.; Zhu, Y.; Mao, S.; Li, Y. Proteomic analyses to reveal the protective role of glutathione in resistance of Lactococcus lactis to osmotic stress. Appl. Environ. Microbiol. 2010, 76, 3177–3186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceylan, S.; Yilan, G.; Akbulut, B.S.; Poli, A.; Kazan, D. Interplay of adaptive capabilities of Halomonas sp. AAD12 under salt stress. J. Biosci. Bioeng. 2012, 114, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Cánovas, M.; Bernal, V.; Sevilla, A.; Torroglosa, T.; Iborra, J.L. Salt stress effects on the central and carnitine metabolisms of Escherichia coli. Biotechnol. Bioeng. 2007, 96, 722–737. [Google Scholar] [CrossRef]
- Klein, A.H.; Shulla, A.; Reimann, S.A.; Keating, D.H.; Wolfe, A.J. The intracellular concentration of acetyl phosphate in Escherichia coli is sufficient for direct phosphorylation of two-component response regulators. J. Bacteriol. 2007, 189, 5574–5581. [Google Scholar] [CrossRef] [Green Version]
- Joyeux, M. Bacterial Nucleoid: Interplay of DNA Demixing and Supercoiling. Biophys. J. 2020, 118, 2141–2150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iacomino, G.; Picariello, G.; D’Agostino, L. DNA and nuclear aggregates of polyamines. Biochim. Biophys. Acta—Mol. Cell Res. 2012, 1823, 1745–1755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karczewska-Golec, J.; Strapagiel, D.; Sadowska, M.; Szalewska-Pałasz, A.; Golec, P. Draft Genome Sequence of Shewanella baltica M1 Isolated from Brackish Surface Water of the Gulf of Gdańsk. Genome Announc. 2016, 4, e00611-16. [Google Scholar] [CrossRef] [Green Version]
- Czyz, A.; Jasiecki, J.; Bogdan, A.; Szpilewska, H.; Wegrzyn, G. Genetically modified Vibrio harveyi strains as potential bioindicators of mutagenic pollution of marine environments. Appl. Environ. Microbiol. 2000, 66, 599–605. [Google Scholar] [CrossRef] [Green Version]
- Graf, J.; Dunlap, P.V.; Ruby, E.G. Effect of transposon-induced motility mutations on colonization of the host light organ by Vibrio fischeri. J. Bacteriol. 1994, 176, 6986–6991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiśniewski, J.R. Quantitative Evaluation of Filter Aided Sample Preparation (FASP) and Multienzyme Digestion FASP Protocols. Anal. Chem. 2016, 88, 5438–5443. [Google Scholar] [CrossRef] [Green Version]
- Rappsilber, J.; Mann, M.; Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2007, 2, 1896–1906. [Google Scholar] [CrossRef]
- Gillet, L.C.; Navarro, P.; Tate, S.; Röst, H.; Selevsek, N.; Reiter, L.; Bonner, R.; Aebersold, R. Targeted Data Extraction of the MS/MS Spectra Generated by Data-independent Acquisition: A New Concept for Consistent and Accurate Proteome Analysis. Mol. Cell. Proteomics 2012, 11, O111.016717. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Bilbao, A.; Bruderer, T.; Luban, J.; Strambio-De-Castillia, C.; Lisacek, F.; Hopfgartner, G.; Varesio, E. The Use of Variable Q1 Isolation Windows Improves Selectivity in LC–SWATH–MS Acquisition. J. Proteome Res. 2015, 14, 4359–4371. [Google Scholar] [CrossRef]
- Deutsch, E.W.; Csordas, A.; Sun, Z.; Jarnuczak, A.; Perez-Riverol, Y.; Ternent, T.; Campbell, D.S.; Bernal-Llinares, M.; Okuda, S.; Kawano, S.; et al. The ProteomeXchange consortium in 2017: Supporting the cultural change in proteomics public data deposition. Nucleic Acids Res. 2017, 45, D1100–D1106. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef]
- Lewandowska, A.E.; Macur, K.; Czaplewska, P.; Liss, J.; Łukaszuk, K.; Ołdziej, S. Human follicular fluid proteomic and peptidomic composition quantitative studies by SWATH-MS methodology. Applicability of high pH RP-HPLC fractionation. J. Proteomics 2019, 191, 131–142. [Google Scholar] [CrossRef]
- Bateman, A.; Martin, M.-J.; Orchard, S.; Magrane, M.; Agivetova, R.; Ahmad, S.; Alpi, E.; Bowler-Barnett, E.H.; Britto, R.; Bursteinas, B.; et al. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Postma, M.; Goedhart, J. PlotsOfData—A web app for visualizing data together with their summaries. PLoS Biol. 2019, 17, e3000202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goedhart, J.; Luijsterburg, M.S. VolcaNoseR is a web app for creating, exploring, labeling and sharing volcano plots. Sci. Rep. 2020, 10, 20560. [Google Scholar] [CrossRef]
- Heberle, H.; Meirelles, G.V.; da Silva, F.R.; Telles, G.P.; Minghim, R. InteractiVenn: A web-based tool for the analysis of sets through Venn diagrams. BMC Bioinform. 2015, 16, 169. [Google Scholar] [CrossRef] [PubMed]
- Ayllon-Benitez, A.; Bourqui, R.; Thébault, P.; Mougin, F. GSAn: An alternative to enrichment analysis for annotating gene sets. NAR Genom. Bioinform. 2020, 2, lqaa017. [Google Scholar] [CrossRef] [Green Version]
- UniProt Consortium, T. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2018, 46, 2699. [Google Scholar] [CrossRef] [Green Version]
- Binns, D.; Dimmer, E.; Huntley, R.; Barrell, D.; O’Donovan, C.; Apweiler, R. QuickGO: A web-based tool for Gene Ontology searching. Bioinformatics 2009, 25, 3045–3046. [Google Scholar] [CrossRef] [Green Version]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kloska, A.; Cech, G.M.; Nowicki, D.; Maciąg-Dorszyńska, M.; Bogucka, A.E.; Markert, S.; Becher, D.; Potrykus, K.; Czaplewska, P.; Szalewska-Pałasz, A. Three Microbial Musketeers of the Seas: Shewanella baltica, Aliivibrio fischeri and Vibrio harveyi, and Their Adaptation to Different Salinity Probed by a Proteomic Approach. Int. J. Mol. Sci. 2022, 23, 619. https://doi.org/10.3390/ijms23020619
Kloska A, Cech GM, Nowicki D, Maciąg-Dorszyńska M, Bogucka AE, Markert S, Becher D, Potrykus K, Czaplewska P, Szalewska-Pałasz A. Three Microbial Musketeers of the Seas: Shewanella baltica, Aliivibrio fischeri and Vibrio harveyi, and Their Adaptation to Different Salinity Probed by a Proteomic Approach. International Journal of Molecular Sciences. 2022; 23(2):619. https://doi.org/10.3390/ijms23020619
Chicago/Turabian StyleKloska, Anna, Grzegorz M. Cech, Dariusz Nowicki, Monika Maciąg-Dorszyńska, Aleksandra E. Bogucka, Stephanie Markert, Dörte Becher, Katarzyna Potrykus, Paulina Czaplewska, and Agnieszka Szalewska-Pałasz. 2022. "Three Microbial Musketeers of the Seas: Shewanella baltica, Aliivibrio fischeri and Vibrio harveyi, and Their Adaptation to Different Salinity Probed by a Proteomic Approach" International Journal of Molecular Sciences 23, no. 2: 619. https://doi.org/10.3390/ijms23020619
APA StyleKloska, A., Cech, G. M., Nowicki, D., Maciąg-Dorszyńska, M., Bogucka, A. E., Markert, S., Becher, D., Potrykus, K., Czaplewska, P., & Szalewska-Pałasz, A. (2022). Three Microbial Musketeers of the Seas: Shewanella baltica, Aliivibrio fischeri and Vibrio harveyi, and Their Adaptation to Different Salinity Probed by a Proteomic Approach. International Journal of Molecular Sciences, 23(2), 619. https://doi.org/10.3390/ijms23020619