
The Potential Role of Cellular Senescence in Non-Alcoholic Fatty Liver Disease



Abstract
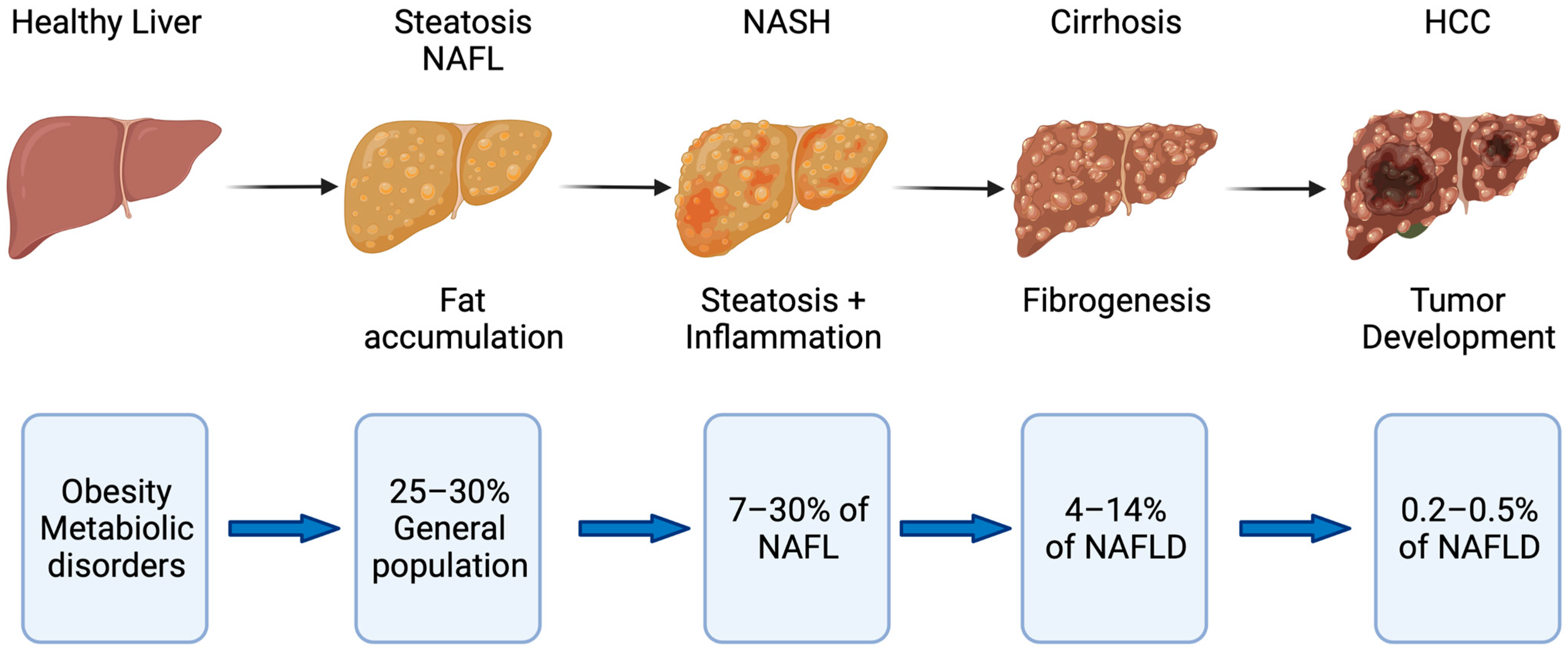
:1. Introduction
2. The Role of Cellular Senescence in Fatty Liver Disease
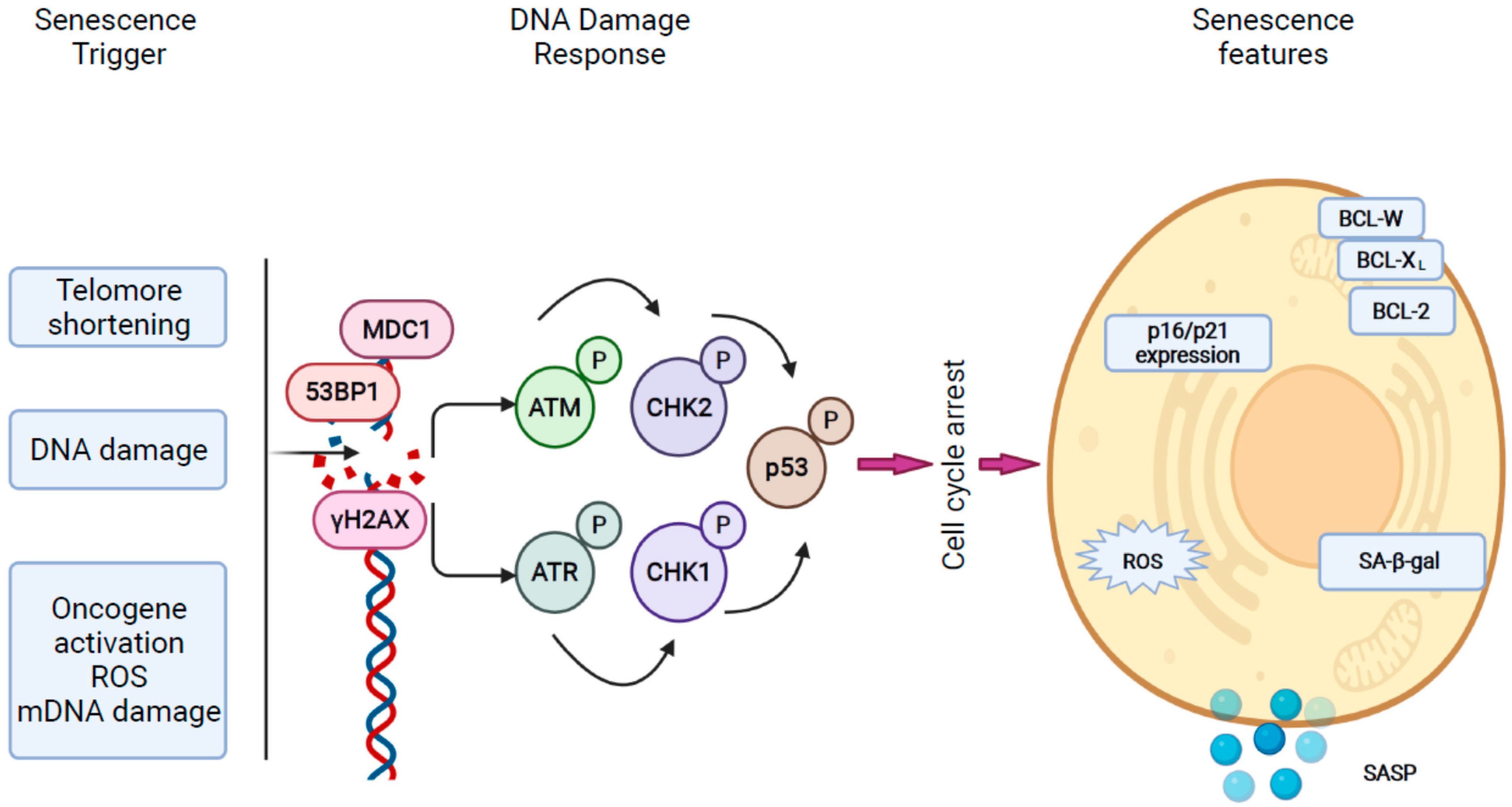
2.1. Mechanisms of Cellular Senescence in the Liver
2.2. Cellular Senescence in Humans with NAFLD
2.3. Mechanisms of Disease Progression in NAFLD
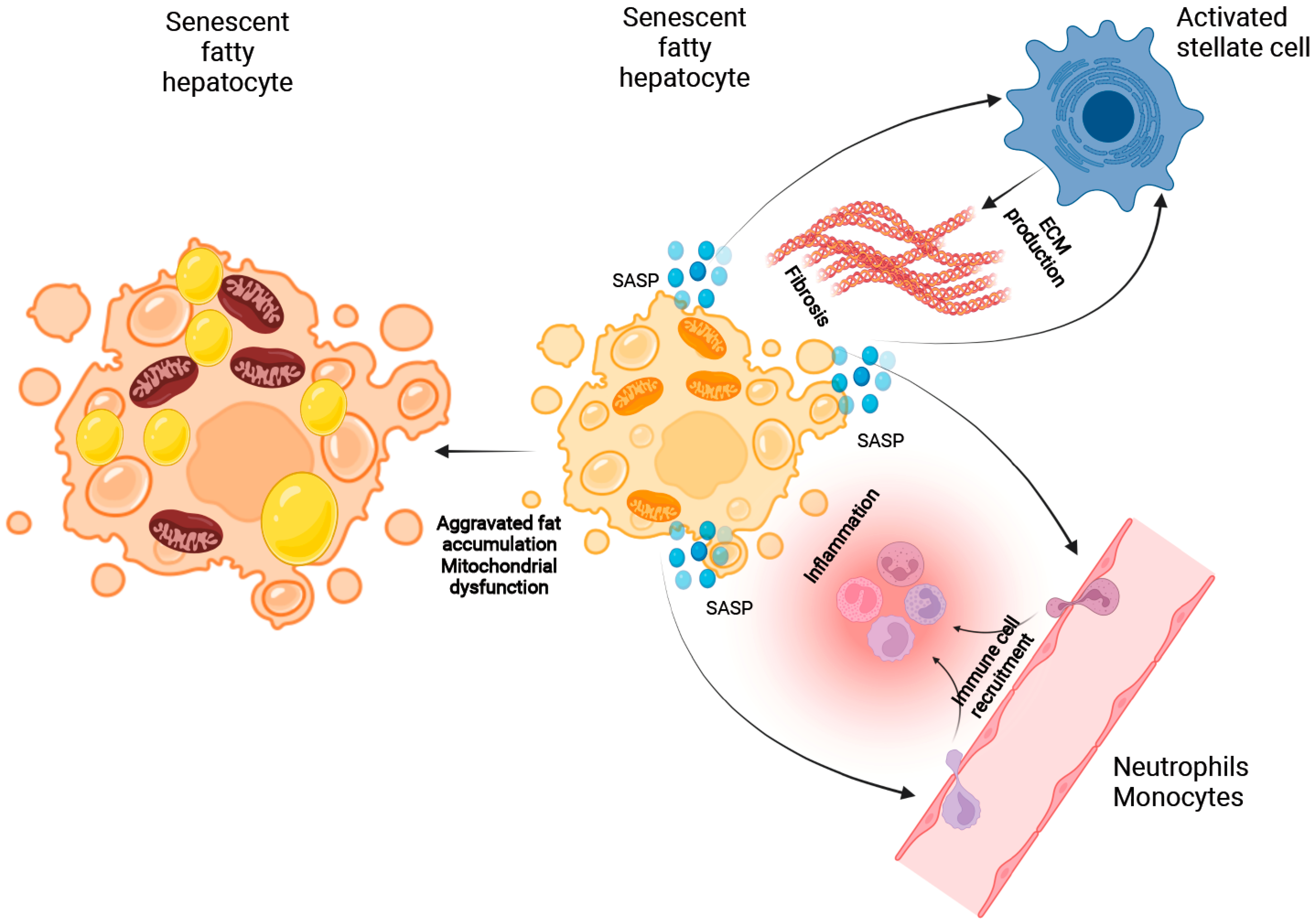
2.3.1. The Role of Cellular Senescence in Hepatocyte Fat Accumulation
2.3.2. The Putative Role of Senescence in the Process of Immune Cell Recruitment
2.3.3. The Role of Cellular Senescence in Modulating Fibrosis in NASH
3. Summary and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cotter, T.G.; Rinella, M. Nonalcoholic Fatty Liver Disease 2020, The State of the Disease. Gastroenterology 2020, 158, 1851–1864. [Google Scholar] [CrossRef]
- Haldar, D.; Kern, B.; Hodson, J.; Armstrong, M.J.; Adam, R.; Berlakovich, G.; Fritz, J.; Benedikt, F.; Popp, W.; Karam, V.; et al. Outcomes of liver transplantation for non-alcoholic steatohepatitis, A European Liver Transplant Registry study. J. Hepatol. 2019, 71, 313–322. [Google Scholar] [CrossRef] [Green Version]
- Estes, C.; Razavi, H.; Loomba, R.; Younossi, Z.; Sanyal, A.J. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology 2018, 67, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Younes, R.; Bugianesi, E. NASH in Lean Individuals. Semin. Liver Dis. 2019, 39, 86–95. [Google Scholar] [CrossRef] [Green Version]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence: Incidence; and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanwal, F.; Kramer, J.R.; Mapakshi, S.; Natarajan, Y.; Chayanupatkul, M.; Richardson, P.A.; Li, L.; Desiderio, R.; Thrift, A.P.; Asch, S.M.; et al. Risk of Hepatocellular Cancer in Patients With Non-Alcoholic Fatty Liver Disease. Gastroenterology 2018, 155, 1828–1837.e2. [Google Scholar] [CrossRef] [Green Version]
- Marchesini, G.; Bugianesi, E.; Forlani, G.; Cerrelli, F.; Lenzi, M.; Manini, R.; Natale, S.; Vanni, E.; Villanova, N.; Rizzetto, M. Nonalcoholic fatty liver; steatohepatitis; and the metabolic syndrome. Hepatology 2003, 37, 917–923. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wong, V.W.-S.; Dufour, J.-F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef]
- Younossi, Z.; Henry, L. Contribution of Alcoholic and Nonalcoholic Fatty Liver Disease to the Burden of Liver-Related Morbidity and Mortality. Gastroenterology 2016, 150, 1778–1785. [Google Scholar] [CrossRef]
- Younes, R.; Govaere, O.; Petta, S.; Miele, L.; Tiniakos, D.; Burt, A.; David, E.; Vecchio, F.M.; Maggioni, M.; Cabibi, D.; et al. Caucasian lean subjects with non-alcoholic fatty liver disease share long-term prognosis of non-lean: Time for reappraisal of BMI-driven approach? Gut 2021. [Google Scholar] [CrossRef]
- Negi, C.K.; Babica, P.; Bajard, L.; Bienertova-Vasku, J.; Tarantino, G. Insights into the molecular targets and emerging pharmacotherapeutic interventions for nonalcoholic fatty liver disease. Metabolism 2021, 126, 154925. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation; metaflammation and immunometabolic disorders. Nature 2017, 542, 177–185. [Google Scholar] [CrossRef]
- Han, J.; Kaufman, R.J. The role of ER stress in lipid metabolism and lipotoxicity. J. Lipid Res. 2016, 57, 1329–1338. [Google Scholar] [CrossRef] [Green Version]
- Puri, P.; Mirshahi, F.; Cheung, O.; Natarajan, R.; Maher, J.W.; Kellum, J.M.; Sanyal, A.J. Activation and dysregulation of the unfolded protein response in nonalcoholic fatty liver disease. Gastroenterology 2008, 134, 568–576. [Google Scholar] [CrossRef]
- Guy, C.D.; Suzuki, A.; Zdanowicz, M.; Abdelmalek, M.F.; Burchette, J.; Unalp, A.; Diehl, A.M.; Crn, F.T.N. Hedgehog pathway activation parallels histologic severity of injury and fibrosis in human nonalcoholic fatty liver disease. Hepatology 2012, 55, 1711–1721. [Google Scholar] [CrossRef] [Green Version]
- Ratziu, V.; Sanyal, A.; Harrison, S.A.; Wong, V.W.S.; Francque, S.; Goodman, Z.; Aithal, G.P.; Kowdley, K.V.; Seyedkazemi, S.; Fischer, L.; et al. Cenicriviroc Treatment for Adults with Nonalcoholic Steatohepatitis and Fibrosis, Final Analysis of the Phase 2b CENTAUR Study. Hepatology 2020, 72, 892–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henao-Mejia, J.; Elinav, E.; Jin, C.; Hao, L.; Mehal, W.Z.; Strowig, T.; Thaiss, C.A.; Kau, A.L.; Eisenbarth, S.C.; Jurczak, M.J.; et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 2012, 482, 179–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csak, T.; Ganz, M.; Pespisa, J.; Kodys, K.; Dolganiuc, A.; Szabo, G. Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology 2011, 54, 133–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.; Allen, A.M.; Wang, Z.; Prokop, L.J.; Murad, M.H.; Loomba, R. Fibrosis progression in nonalcoholic fatty liver vs. nonalcoholic steatohepatitis: A systematic review and meta-analysis of paired-biopsy studies. Clin. Gastroenterol. Hepatol. 2015, 13, 643–654.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira-Gonzalez, S.; Rodrigo-Torres, D.; Gadd, V.L.; Forbes, S.J. Cellular Senescence in Liver Disease and Regeneration. Semin. Liver Dis. 2021, 41, 50–66. [Google Scholar] [CrossRef]
- Papatheodoridi, A.M.; Chrysavgis, L.; Koutsilieris, M.; Chatzigeorgiou, A. The Role of Senescence in the Development of Nonalcoholic Fatty Liver Disease and Progression to Nonalcoholic Steatohepatitis. Hepatology 2020, 71, 363–374. [Google Scholar] [CrossRef]
- Kang, T.-W.; Yevsa, T.; Woller, N.; Hoenicke, L.; Wuestefeld, T.; Dauch, D.; Hohmeyer, A.; Gereke, M.; Rudalska, R.; Potapova, A.; et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 2011, 479, 547–551. [Google Scholar] [CrossRef]
- Chuprin, A.; Gal, H.; Biron-Shental, T.; Biran, A.; Amiel, A.; Rozenblatt, S.; Krizhanovsky, V. Cell fusion induced by ERVWE1 or measles virus causes cellular senescence. Genes Dev. 2013, 27, 2356–2366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiley, C.D.; Velarde, M.C.; Lecot, P.; Liu, S.; Sarnoski, E.A.; Freund, A.; Shirakawa, K.; Lim, H.W.; Davis, S.S.; Ramanathan, A.; et al. Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab. 2016, 23, 303–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira-Gonzalez, S.; Lu, W.Y.; Raven, A.; Dwyer, B.; Man, T.Y.; O’Duibhir, E.; Lewis, P.J.S.; Campana, L.; Kendall, T.J.; Bird, T.G.; et al. Paracrine cellular senescence exacerbates biliary injury and impairs regeneration. Nat. Commun. 2018, 9, 1020. [Google Scholar] [CrossRef] [PubMed]
- Bird, T.G.; Muller, M.; Boulter, L.; Vincent, D.F.; Ridgway, R.A.; Lopez-Guadamillas, E.; Lu, W.Y.; Jamieson, T.; Govaere, O.; Campbell, A.D.; et al. TGFbeta inhibition restores a regenerative response in acute liver injury by suppressing paracrine senescence. Sci. Transl. Med. 2018, 10, eaan1230. [Google Scholar] [CrossRef] [Green Version]
- Munoz-Espin, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef]
- Mossanen, J.C.; Kohlhepp, M.; Wehr, A.; Krenkel, O.; Liepelt, A.; Roeth, A.A.; Möckel, D.; Heymann, F.; Lammers, T.; Gassler, N.; et al. CXCR6 inhibits hepatocarcinogenesis by promoting natural killer T- and CD4+ T cell-dependent control of senescence. Gastroenterology 2019, 156, 1877–1889. [Google Scholar] [CrossRef]
- Di Micco, R.; Krizhanovsky, V.; Baker, D.; di Fagagna, F.D. Cellular senescence in ageing: From mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. 2021, 22, 75–95. [Google Scholar] [CrossRef]
- Yosef, R.; Pilpel, N.; Tokarsky-Amiel, R.; Biran, A.; Ovadya, Y.; Cohen, S.; Vadai, E.; Dassa, L.; Shahar, E.; Condiotti, R.; et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat. Commun. 2016, 7, 11190. [Google Scholar] [CrossRef]
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.-M.; DeMaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 2016, 22, 78–83. [Google Scholar] [CrossRef] [Green Version]
- Biran, A.; Zada, L.; Abou Karam, P.; Vadai, E.; Roitman, L.; Ovadya, Y.; Porat, Z.; Krizhanovsky, V. Quantitative identification of senescent cells in aging and disease. Aging Cell 2017, 16, 661–671. [Google Scholar] [CrossRef]
- Thorin-Trescases, N.; Labbe, P.; Mury, P.; Lambert, M.; Thorin, E. Angptl2 is a marker of cellular senescence: The physiological and pathophysiological impact of angptl2-related senescence. Int. J. Mol. Sci. 2021, 22, 12232. [Google Scholar] [CrossRef]
- Rodier, F.; Coppé, J.-P.; Patil, C.K.; Hoeijmakers, W.A.M.; Muñoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 2009, 11, 973–979. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; Xu, Q.; Martin, T.D.; Li, M.Z.; DeMaria, M.; Aron, L.; Lu, T.; Yankner, B.A.; Campisi, J.; Elledge, S.J. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 2015, 349, aaa5612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panasiuk, A.; Dzieciol, J.; Panasiuk, B.; Prokopowicz, D. Expression of p53; Bax and Bcl-2 proteins in hepatocytes in non-alcoholic fatty liver disease. World J. Gastroenterol. 2006, 12, 6198–6202. [Google Scholar] [CrossRef] [PubMed]
- Ping, F.; Li, Z.Y.; Lv, K.; Zhou, M.C.; Dong, Y.X.; Sun, Q.; Li, Y.X. Deoxyribonucleic acid telomere length shortening can predict the incidence of non-alcoholic fatty liver disease in patients with type 2 diabetes mellitus. J. Diabetes Investig. 2017, 8, 174–180. [Google Scholar] [CrossRef] [Green Version]
- Shampay, J.; Blackburn, E.H. Generation of telomere-length heterogeneity in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 1988, 85, 534–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laish, I.; Mannasse-Green, B.; Hadary, R.; Biron-Shental, T.; Konikoff, F.M.; Amiel, A.; Kitay-Cohen, Y. Telomere Dysfunction in Nonalcoholic Fatty Liver Disease and Cryptogenic Cirrhosis. Cytogenet Genome Res. 2016, 150, 93–99. [Google Scholar] [CrossRef]
- Aravinthan, A.; Scarpini, C.G.; Tachtatzis, P.; Verma, S.; Penrhyn-Lowe, S.; Harvey, R.; Davies, S.E.; Allison, M.; Coleman, N.; Alexander, G. Hepatocyte senescence predicts progression in non-alcohol-related fatty liver disease. J. Hepatol. 2013, 58, 549–556. [Google Scholar] [CrossRef]
- Akazawa, Y.; Nakashima, R.; Matsuda, K.; Okamaoto, K.; Hirano, R.; Kawasaki, H.; Miuma, S.; Miyaaki, H.; Malhi, H.; Abiru, S.; et al. Detection of DNA damage response in nonalcoholic fatty liver disease via p53-binding protein 1 nuclear expression. Mod. Pathol. 2019, 32, 997–1007. [Google Scholar] [CrossRef] [PubMed]
- Pirola, C.J.; Garaycoechea, M.; Flichman, D.; Castano, G.O.; Sookoian, S. Liver mitochondrial DNA damage and genetic variability of Cytochrome b—A key component of the respirasome-drive the severity of fatty liver disease. J. Intern. Med. 2021, 289, 84–96. [Google Scholar] [CrossRef]
- Ahrens, M.; Ammerpohl, O.; von Schönfels, W.; Kolarova, J.; Bens, S.; Itzel, T.; Teufel, A.; Herrmann, A.; Brosch, M.; Hinrichsen, H.; et al. DNA methylation analysis in nonalcoholic fatty liver disease suggests distinct disease-specific and remodeling signatures after bariatric surgery. Cell Metab. 2013, 18, 296–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hotta, K.; Kitamoto, A.; Kitamoto, T.; Ogawa, Y.; Honda, Y.; Kessoku, T.; Yoneda, M.; Imajo, K.; Tomeno, W.; Saito, S.; et al. Identification of differentially methylated region (DMR) networks associated with progression of nonalcoholic fatty liver disease. Sci. Rep. 2018, 8, 13567. [Google Scholar] [CrossRef] [PubMed]
- Johnson, N.D.; Wu, X.; Still, C.D.; Chu, X.; Petrick, A.T.; Gerhard, G.S.; Conneely, K.N.; DiStefano, J.K. Differential DNA methylation and changing cell-type proportions as fibrotic stage progresses in NAFLD. Clin. Epigenetics 2021, 13, 152. [Google Scholar] [CrossRef]
- Hardy, T.; Zeybel, M.; Day, C.P.; Dipper, C.; Masson, S.; McPherson, S.; Henderson, E.; Tiniakos, D.; White, S.; French, J.; et al. Plasma DNA methylation: A potential biomarker for stratification of liver fibrosis in non-alcoholic fatty liver disease. Gut 2017, 66, 1321–1328. [Google Scholar] [CrossRef]
- Aravinthan, A.; Mells, G.; Allison, M.; Leathart, J.; Kotronen, A.; Yki-Jarvinen, H.; Daly, A.K.; Day, C.P.; Anstee, Q.M. Gene polymorphisms of cellular senescence marker p21 and disease progression in non-alcohol-related fatty liver disease. Cell Cycle 2014, 13, 1489–1494. [Google Scholar] [CrossRef] [Green Version]
- Hardy, T.; Oakley, F.; Anstee, Q.M.; Day, C.P. Nonalcoholic Fatty Liver Disease, Pathogenesis and Disease Spectrum. Annu. Rev. Pathol. 2016, 11, 451–496. [Google Scholar] [CrossRef]
- Zhang, X.; Zhou, D.; Strakovsky, R.; Zhang, Y.; Pan, Y.X. Hepatic cellular senescence pathway genes are induced through histone modifications in a diet-induced obese rat model. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G558–G564. [Google Scholar] [CrossRef]
- Ogrodnik, M.; Miwa, S.; Tchkonia, T.; Tiniakos, D.; Wilson, C.L.; Lahat, A.; Day, C.P.; Burt, A.; Palmer, A.; Anstee, Q.M.; et al. Cellular senescence drives age-dependent hepatic steatosis. Nat. Commun. 2017, 8, 15691. [Google Scholar] [CrossRef]
- Wan, J.; Wu, X.; Chen, H.; Xia, X.; Song, X.; Chen, S.; Lu, X.; Jie, J.; Su, Q.; Cai, D.S.; et al. Aging-induced aberrant RAGE/PPARalpha axis promotes hepatic steatosis via dysfunctional mitochondrial beta oxidation. Aging Cell 2020, 19, e13238. [Google Scholar] [CrossRef]
- Lohr, K.; Pachl, F.; Moghaddas Gholami, A.; Geillinger, K.E.; Daniel, H.; Kuster, B.; Klingenspor, M. Reduced mitochondrial mass and function add to age-related susceptibility toward diet-induced fatty liver in C57BL/6J mice. Physiol. Rep. 2016, 4, e12988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, Y.E.; Duan, L.; He, Y.; Yuan, C.; Wang, T.; Yuan, D.; Zhang, C.C.; Liu, C.Q. Saturated Fatty Acids Promote Hepatocytic Senecence through Regulation of miR-34a/Cyclin-Dependent Kinase 6. Mol. Nutr. Food Res. 2020, 64, e2000383. [Google Scholar] [CrossRef]
- Wang, L.; Liu, X.; Nie, J.; Zhang, J.; Kimball, S.R.; Zhang, H.; Zhang, W.J.; Jefferson, L.S.; Cheng, Z.; Ji, Q.; et al. ALCAT1 controls mitochondrial etiology of fatty liver diseases; linking defective mitophagy to steatosis. Hepatology 2015, 61, 486–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinha, A.; Yen, P.M. Thyroid hormone-mediated autophagy and mitochondrial turnover in NAFLD. Cell Biosci. 2016, 6, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernansanz-Agustin, P.; Enriquez, J.A. Generation of Reactive Oxygen Species by Mitochondria. Antioxidants 2021, 10, 415. [Google Scholar] [CrossRef] [PubMed]
- Selen, E.S.; Choi, J.; Wolfgang, M.J. Discordant hepatic fatty acid oxidation and triglyceride hydrolysis leads to liver disease. JCI Insight 2021, 6, e135626. [Google Scholar] [CrossRef]
- Abate, M.; Festa, A.; Falco, M.; Lombardi, A.; Luce, A.; Grimaldi, A.; Zappavigna, S.; Sperlongano, P.; Irace, C.; Caraglia, M.; et al. Mitochondria as playmakers of apoptosis; autophagy and senescence. Semin Cell Dev. Biol. 2020, 98, 139–153. [Google Scholar] [CrossRef]
- Dabravolski, S.A.; Bezsonov, E.E.; Orekhov, A.N. The role of mitochondria dysfunction and hepatic senescence in NAFLD development and progression. Biomed. Pharmacother. 2021, 142, 112041. [Google Scholar] [CrossRef]
- Han, X.; Bao, X.; Lou, Q.; Xie, X.; Zhang, M.; Zhou, S.; Guo, H.; Jiang, G.; Shi, Q. Nicotinamide riboside exerts protective effect against aging-induced NAFLD-like hepatic dysfunction in mice. PeerJ 2019, 7, e7568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.J.; Sun, S.J.; Fu, J.T.; Ouyang, S.X.; Zhao, Q.J.; Su, L.; Ji, Q.X.; Sun, D.Y.; Zhu, J.H.; Zhang, G.Y.; et al. NAD(+)-boosting therapy alleviates nonalcoholic fatty liver disease via stimulating a novel exerkine Fndc5/irisin. Theranostics 2021, 11, 4381–4402. [Google Scholar] [CrossRef]
- Hsieh, J.Y.; Li, S.Y.; Tsai, W.C.; Liu, J.H.; Lin, C.L.; Liu, G.Y.; Hung, H.C. A small-molecule inhibitor suppresses the tumor-associated mitochondrial NAD(P)+-dependent malic enzyme (ME2) and induces cellular senescence. Oncotarget 2015, 6, 20084–20098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsyuba, E.; Mottis, A.; Zietak, M.; De Franco, F.; Van Der Velpen, V.; Gariani, K.; Ryu, D.; Cialabrini, L.; Matilainen, O.; Liscio, P.; et al. De novo NAD(+) synthesis enhances mitochondrial function and improves health. Nature 2018, 563, 354–359. [Google Scholar] [CrossRef]
- Ali, S.E.; Rychkov, G.Y.; Barritt, G.J. Targetting Ca2+ signaling in the initiation; promotion and progression of hepatocellular carcinoma. Cancers 2020, 12, 2755. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, D.V.; Vindrieux, D.; Goehrig, D.; Jaber, S.; Collin, G.; Griveau, A.; Wiel, C.; Bendrildi, N.; Ajebali, S.; Farfariello, V.; et al. Calcium channel ITPR2 and mitochondria-ER contacts promote cellular senescence and aging. Nat. Commun. 2021, 12, 720. [Google Scholar] [CrossRef] [PubMed]
- Shpilka, T.; Haynes, C.M. The mitochondrial UPR: Mechanisms; physiological functions and implications in ageing. Nat. Rev. Mol. Cell. Biol. 2018, 19, 109–120. [Google Scholar] [CrossRef]
- Archer, A.E.; Rogers, R.S.; Von Schulze, A.T.; Wheatley, J.L.; Morris, E.M.; McCoin, C.S.; Thyfault, J.P.; Geiger, P.C. Heat shock protein 72 regulates hepatic lipid accumulation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 315, R696–R707. [Google Scholar] [CrossRef] [Green Version]
- Bhaskaran, S.; Pharaoh, G.; Ranjit, R.; Murphy, A.; Matsuzaki, S.; Nair, B.C.; Forbes, B.; Gispert, S.; Auburger, G.; Humphries, K.M.; et al. Loss of mitochondrial protease ClpP protects mice from diet-induced obesity and insulin resistance. EMBO Rep. 2018, 19, e45009. [Google Scholar] [CrossRef]
- Marra, F.; Svegliati-Baroni, G. Lipotoxicity and the gut-liver axis in NASH pathogenesis. J. Hepatol. 2018, 68, 280–295. [Google Scholar] [CrossRef]
- Krenkel, O.; Tacke, F. Liver macrophages in tissue homeostasis and disease. Nat. Rev. Immunol. 2017, 17, 306–321. [Google Scholar] [CrossRef]
- Engelmann, C.; Claria, J.; Szabo, G.; Bosch, J.; Bernardi, M. Pathophysiology of decompensated cirrhosis, Portal hypertension; circulatory dysfunction; inflammation; metabolism and mitochondrial dysfunction. J. Hepatol. 2021, 75, S49–S66. [Google Scholar] [CrossRef]
- Engelmann, C.; Adebayo, D.; Oria, M.; De Chiara, F.; Novelli, S.; Habtesion, A.; Davies, N.; Andreola, F.; Jalan, R. Recombinant Alkaline Phosphatase Prevents Acute on Chronic Liver Failure. Sci. Rep. 2020, 10, 389. [Google Scholar] [CrossRef]
- Engelmann, C.; Habtesion, A.; Novelli, S.; Kerbert, A.; Davies, N.; Ferreira-Gonzalez, S.; Forbes, S.; Berg, T.; Jalan, R. Toll-like receptor 4 inhibition acts synergistically with G-CSF to prevent organ injury and to induce regeneration in acute-on-chronic liver failure. J. Hepatol. 2020, 73, 1001. [Google Scholar] [CrossRef]
- Farrell, G.C.; Larter, C.Z.; Hou, J.Y.; Zhang, R.H.; Yeh, M.M.; Williams, J.; Dela Peňa, A.; Francisco, R.; Osvath, S.R.; Brooling, J.; et al. Apoptosis in experimental NASH is associated with p53 activation and TRAIL receptor expression. J. Gastroenterol. Hepatol. 2009, 24, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhu, Y.; Hu, S.; Xu, Y.; Stroup, D.; Pan, X.; Bawa, F.C.; Chen, S.R.; Gopoju, R.; Yin, L.Y.l.; et al. Hepatocyte Nuclear Factor 4alpha Prevents the Steatosis-to-NASH Progression by Regulating p53 and Bile Acid Signaling (in mice). Hepatology 2021, 73, 2251–2265. [Google Scholar] [CrossRef]
- Tomita, K.; Teratani, T.; Suzuki, T.; Oshikawa, T.; Yokoyama, H.; Shimamura, K.; Nishiyama, K.; Mataki, N.; Irie, R.; Minamino, T.; et al. p53/p66Shc-mediated signaling contributes to the progression of non-alcoholic steatohepatitis in humans and mice. J. Hepatol. 2012, 57, 837–843. [Google Scholar] [CrossRef] [PubMed]
- Gong, D.; Shi, W.; Yi, S.J.; Chen, H.; Groffen, J.; Heisterkamp, N. TGFbeta signaling plays a critical role in promoting alternative macrophage activation. BMC Immunol. 2012, 13, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, B.; Dongiovanni, P.; Corey, K.E.; Wang, X.; Shmarakov, I.O.; Zheng, Z.; Kasikara, C.; Davra, V.; Meroni, M.; Chuang, R.; et al. Macrophage MerTK Promotes Liver Fibrosis in Nonalcoholic Steatohepatitis. Cell Metab. 2020, 31, 406–421.e7. [Google Scholar] [CrossRef]
- Tacke, F. Cenicriviroc for the treatment of non-alcoholic steatohepatitis and liver fibrosis. Expert Opin. Investig. Drugs 2018, 27, 301–311. [Google Scholar] [CrossRef]
- Daugherity, E.K.; Balmus, G.; Al Saei, A.; Moore, E.S.; Abi Abdallah, D.; Rogers, A.B.; Weiss, R.S.; Maurer, K.L. The DNA damage checkpoint protein ATM promotes hepatocellular apoptosis and fibrosis in a mouse model of non-alcoholic fatty liver disease. Cell Cycle 2012, 11, 1918–1928. [Google Scholar] [CrossRef] [Green Version]
- Kondo, Y.; Hasegawa, G.; Okada, H.; Senmaru, T.; Fukui, M.; Nakamura, N.; Sawada, M.; Kitawaki, J.; Okanoue, T.; Kishimoto, Y.; et al. Lepr(db/db) Mice with senescence marker protein-30 knockout (Lepr(db/db)Smp30(Y/-)) exhibit increases in small dense-LDL and severe fatty liver despite being fed a standard diet. PLoS ONE 2013, 8, e65698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondo, Y.; Masutomi, H.; Noda, Y.; Ozawa, Y.; Takahashi, K.; Handa, S.; Maruyama, N.; Shimizu, T.; Ishigami, A. Senescence marker protein-30/superoxide dismutase 1 double knockout mice exhibit increased oxidative stress and hepatic steatosis. FEBS Open Bio. 2014, 4, 522–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiskirchen, R.; Tacke, F. Cellular and molecular functions of hepatic stellate cells in inflammatory responses and liver immunology. Hepatobiliary Surg. Nutr. 2014, 3, 344–363. [Google Scholar] [PubMed]
- Xu, T.; Ni, M.M.; Xing, L.; Li, X.F.; Meng, X.M.; Huang, C.; Li, J. NLRC5 regulates TGF-beta1-induced proliferation and activation of hepatic stellate cells during hepatic fibrosis. Int. J. Biochem. Cell Biol. 2016, 70, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Krizhanovsky, V.; Yon, M.; Dickins, R.; Hearn, S.; Simon, J.; Miething, C.; Yee, H.; Zender, L.; Lowe, S.W. Senescence of activated stellate cells limits liver fibrosis. Cell 2008, 134, 657–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, P.; Wei, L.-L.; Zhao, S.; Sverdlov, D.Y.; Vaid, K.A.; Miyamoto, M.; Kuramitsu, K.; Lai, M.; Popov, Y.V. Hepatocyte mitochondria-derived danger signals directly activate hepatic stellate cells and drive progression of liver fibrosis. Nat. Commun. 2020, 11, 2362. [Google Scholar] [CrossRef]
- Gaul, S.; Leszczynska, A.; Alegre, F.; Kaufmann, B.; Johnson, C.D.; Adams, L.A.; Wree, A.; Damm, G.; Seehofer, D.; Calvente, C.J.; et al. Hepatocyte pyroptosis and release of inflammasome particles induce stellate cell activation and liver fibrosis. J. Hepatol. 2021, 74, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Mridha, A.R.; Wree, A.; Robertson, A.A.B.; Yeh, M.M.; Johnson, C.D.; Van Rooyen, D.M.; Haczeyni, F.; Teon, N.C.H.; Savard, C.; Ioannou, G.N.; et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J. Hepatol. 2017, 66, 1037–1046. [Google Scholar] [CrossRef]
- Nishizawa, H.; Iguchi, G.; Fukuoka, H.; Takahashi, M.; Suda, K.; Bando, H.; Matsumoto, R.; Yoshida, K.; Odake, Y.; Ogawa, W.; et al. IGF-I induces senescence of hepatic stellate cells and limits fibrosis in a p53-dependent manner. Sci. Rep. 2016, 6, 34605. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Author | Specimens | Experimental Techniques | Results |
|---|---|---|---|
| Panasiuk A et al. [37] | Liver (n = 84) | IHC 1 | Liver steatosis is associated with p53 expression and increasing pro-apoptotic BAX/BCL-2 ratio |
| Ping F et al. [38] | PBMC (blood) (n = 70) | rtPCR 2 | Telomere length in leukocytes shortened in patients with diabetes type 2 who developed NAFLD |
| Laish I et al. [40] | PBMC (blood) (NAFLD n = 22; crypt. Cirrhosis n = 20; healthy n = 20) | FISH 3, rtPCR 2 | Shorter telomere length and decreased expression of telomerase reverse transcriptase in NAFLD |
| Aravinthan A et al. [41] | Liver (NAFLD n = 70; healthy n = 60) | FISH 3, IHC 1 | NAFLD and degree of fibrosis was associated with shorter telomeres, cellular senescence (p21) and DNA damage (yH2AX) |
| Akazawa Y et al. [42] | Liver (NAFLD n = 43; healthy n = 9) | IF 4 | The number of foci with DNA double strand breaks (53BP1) increases with NAFLD and progression to NASH |
| Pirola CJ et al. [43] | Liver (NAFLD n = 252) | MT-CYB 5 sequencing; differential mtDNA 6 damage; global liver transcriptome, profiling circulating Krebs cycle metabolites; tissue levels products of lipid peroxidation and markers of oxidative stress | NASH was associated with higher MT-CYB 5 variance and changes in global liver transcriptome; liver mtDNA 6 damage, tissue levels of oxidative adducts and lipid peroxyl radicals were associated with advanced fibrosis |
| Ahrens M et al. [44] | Liver (NAFLD n = 45; healthy n = 18) | Array based DNA methylation and mRNA 7 expression | NAFLD associated with methylation differences in nine genes coding for enzymes in metabolism and insulin signaling. Methylation signatures partially reversible after bariatric surgery |
| Hotta K et al. [45] | Liver (NAFLD n = 60) | Genome-wide DNA methylation levels measured by the Illumina Infinium HumanMethylation450 BeadChip | Two differentially methylated region networks involved in NAFLD progression: 1. Genes involved in transcriptional regulation, cytoskeleton, proliferation; 2. Genes associated with metabolic pathways |
| Johnson ND et al. [46] | Liver (NAFLD n = 325) | Infinium MethylationEPIC array | DNA methylation associated with fibrosis progression with increasing proportion of natural killer cells |
| Hardy T et al. [47] | Liver and Plasma (NAFLD n = 26) | Plasma cell-free DNA methylation of PPARy 8—pyrosequencing Liver DNA methylation—laser capture microdissection and pyrosequencing | Differential DNA methylation at PPARy 8 promotor detectable in circulating cell free DNA as a non-invasive marker |
| Aravinthan A et al. [48] | PBMCs from two cohorts of NAFLD (n = 323, n = 123) | p21 polymorphisms (SNP 9)—genotyping | SNP rs762623 significantly associated with disease progression |
| Author | Species | Model | Results |
|---|---|---|---|
| Zhang G et al. [50] | Rats (Crl:CD (SD) rats) | HF diet 1 | Increasing activation of p21 and p16 pathways in livers with fat accumulation |
| Ogrodnik M et al. [51] | Mice (C57BL/6; INK-ATTAC.; Alb-Xpg) | Aged mice fed ad libitum | Cellular senescence drives hepatic steatosis and senolysis reverts the effect |
| Wan J et al. [52] | Mice (C57BL/6) | Aged mice fed with HF diet 1 or normal chow | Increasing liver fat accumulation with higher age related to upregulation of RAGE and inhibition of PPARα |
| Lohr K et al. [53] | Mice (C57BL/6) | Aged mice fed with HF diet 1 | Reduced mitochondrial mass and function foster fatty liver development |
| Qin YE et al. [54] | Mice (C57BL/6) | HF diet 1 | Liver steatosis induced hepatocyte senescence through miR-34a by targeting CDK6 |
| Han X et al. [61] | Mice (C57BL/6) | Aged mice fed with normal chow | NAD precursor nicotinamid riboside (NR) has protected from aging-induced NAFLD |
| Li DJ et al. [62] | Mice (C57BL/6; Fndc5−/−) | HF diet 1 and MCD diet 2 | Nicotinamid riboside (NR) exerts its protective effect through Fndc5/irisin upregulation |
| Archer AE et al. [68] | Wistar rats | HF diet 1 and heat treatment (41 °C) | Heat shock protein 72 improves glucose tolerance and reduces triglyceride storage |
| Bhaskaran S et al. [69] | Mice (C57BL/6; ClpP−/−) | HF diet 1 | Caseinolytic peptodase P (ClpP) regulated mitochondrial function and its deficiency protects from fat accumulation in the liver |
| Farrell GC et al. [75] | Mice (C57BL/6) | MCD diet 2 | MCD liver fat accumulation promotes p53 expression and subsequent apoptosis |
| Xu Y et al. [76] | Mice (C57BL/6; hepatocyte specific p53−/−) | HFCF diet 3 | HNF4a prevents hepatic triglyceride accumulation and promotes fatty acid oxidation but not in hepatocyte-specific p53−/− mice |
| Tomita K et al. [77] | Mice (C57BL/6; p53−/−) | MCD diet 2 | p53 promotes lipid peroxidation, apoptotic hepatocytes and progression of NADLF in a TGF-b-dependent manner |
| Daugherity EK et al. [81] | Mice (C57BL/6; Atm−/−) | HF diet 1 | Dietary liver fat induced ROS production and DNA damage which leads to apoptosis and fibrosis in a ATM dependent manner |
| Kondo Y et al. [82] | Mice (Leprdb/dbSmp30Y/−) | Normal chow (aged mice) | Senescence marker protein-30 (SMP30) increases oxidative stress and liver inflammation together with PPARa induction and fat accumulation |
| Kondo Y et al. [83] | Mice (SMP30/SOD1-DKO) | Normal chow | Dual deficiency of SMP30 and SOD1 promotes liver fat accumulation and inflammation |
| Mridha AR et al. [89] | Mice (foz/foz; C57BL/6) | MCD diet 2 | NLRP3 inflammasome stimulation promotes inflammation and fibrosis in NAFLD |
| Nishizawa H et al. [90] | Mice (C57BL/6; db/db) | MCD diet 2 | IGF-1 prevents from fibrosis in NASH by inducing cellular senescence to HSC through p53 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Engelmann, C.; Tacke, F. The Potential Role of Cellular Senescence in Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2022, 23, 652. https://doi.org/10.3390/ijms23020652
Engelmann C, Tacke F. The Potential Role of Cellular Senescence in Non-Alcoholic Fatty Liver Disease. International Journal of Molecular Sciences. 2022; 23(2):652. https://doi.org/10.3390/ijms23020652
Chicago/Turabian StyleEngelmann, Cornelius, and Frank Tacke. 2022. "The Potential Role of Cellular Senescence in Non-Alcoholic Fatty Liver Disease" International Journal of Molecular Sciences 23, no. 2: 652. https://doi.org/10.3390/ijms23020652
APA StyleEngelmann, C., & Tacke, F. (2022). The Potential Role of Cellular Senescence in Non-Alcoholic Fatty Liver Disease. International Journal of Molecular Sciences, 23(2), 652. https://doi.org/10.3390/ijms23020652