
Entailing the Next-Generation Sequencing and Metabolome for Sustainable Agriculture by Improving Plant Tolerance

 ,
,  ,
,  , , , ,
, , , ,  and
and
Abstract
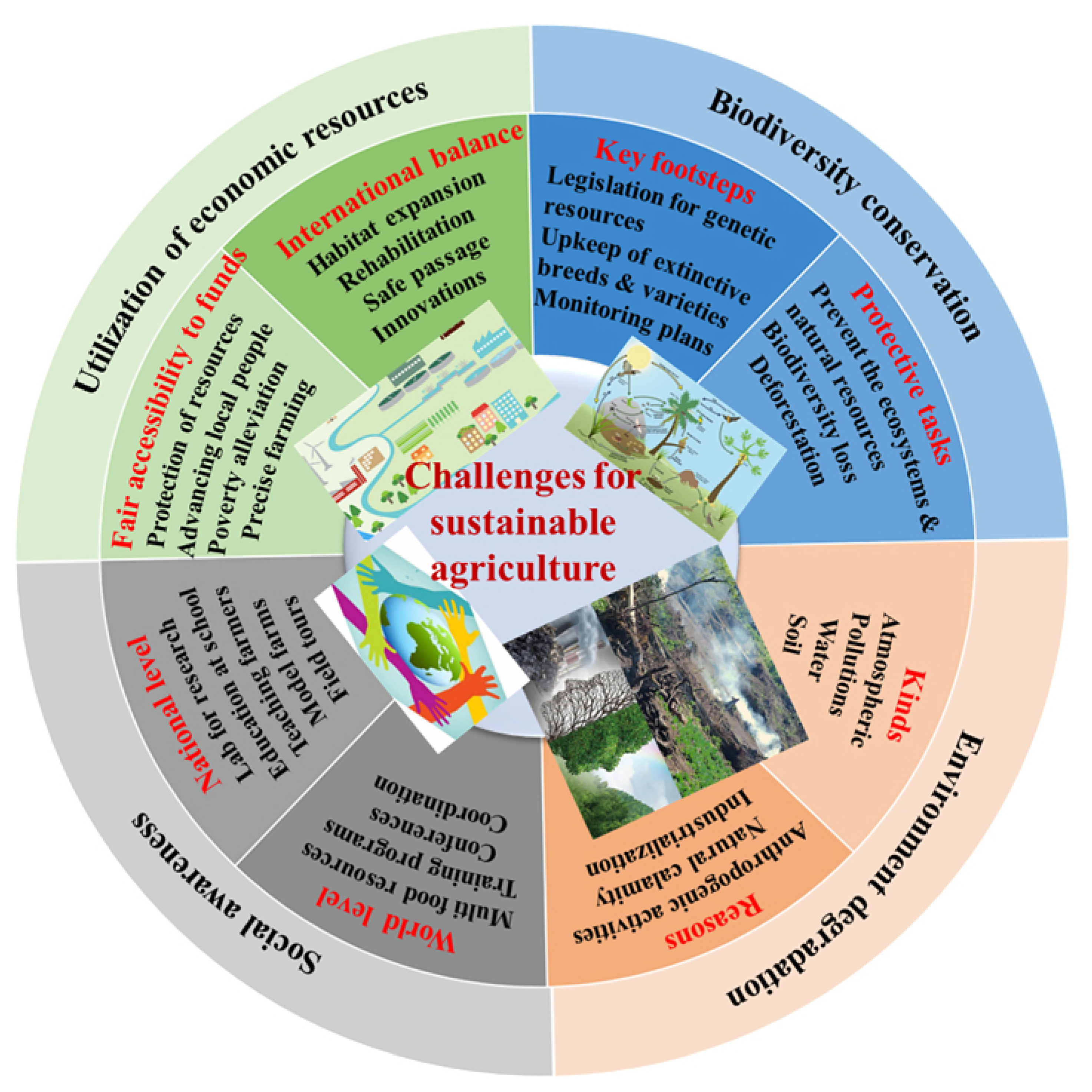
:1. Introduction
2. Progress in Sequencing
2.1. First-Generation Sequencing
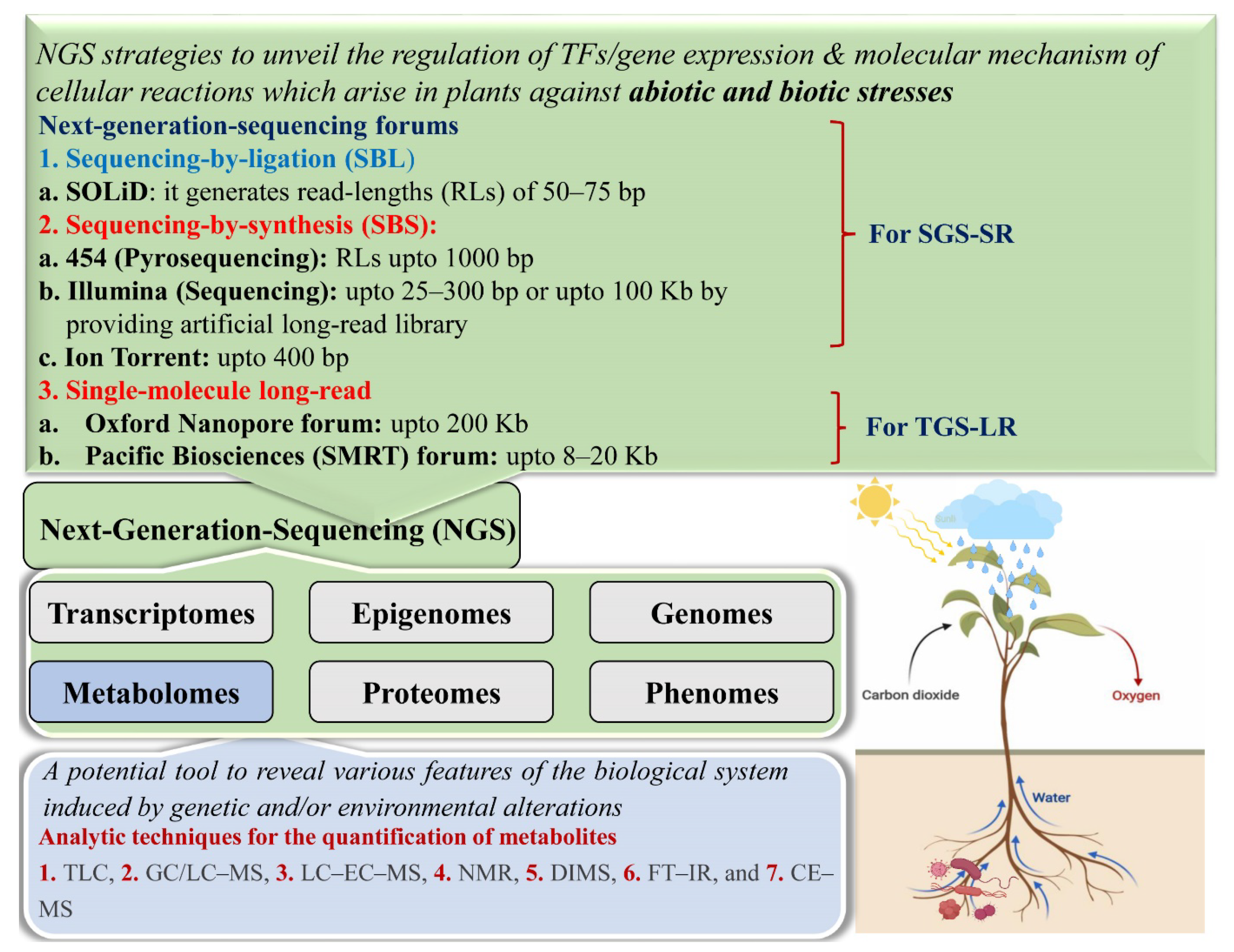
2.2. Next-Generation Sequencing
2.2.1. Second-Generation Sequencing
2.2.2. Third-Generation Sequencing
2.2.3. Challenges and Limitations of SGS and TGS Forums
3. NGS and Its Promising Aspects
Significance of the NGS
4. NGS Can Promote Sustainable Crop Production
4.1. Exploiting the Molecular Markers, Genetic Maps, and Phylogenetic Relationships Using NGS Technologies
4.2. Creating the Pan- or Super-Pan-Genome Based on NGS Technology
4.3. Sustainability by Exploiting the World Genetic Resources
4.4. Mining the Novel Genes and Regularity Pathways Using NGS to Generate Transcriptome
5. Role of Metabolomics in the Sustainable Crop Production
6. Diagnosis and Monitoring of the Disease-Causing Pathogens Using NGS and Metabolites
7. Integration between the Transcriptome and Metabolome to Achieve Crop Sustainability
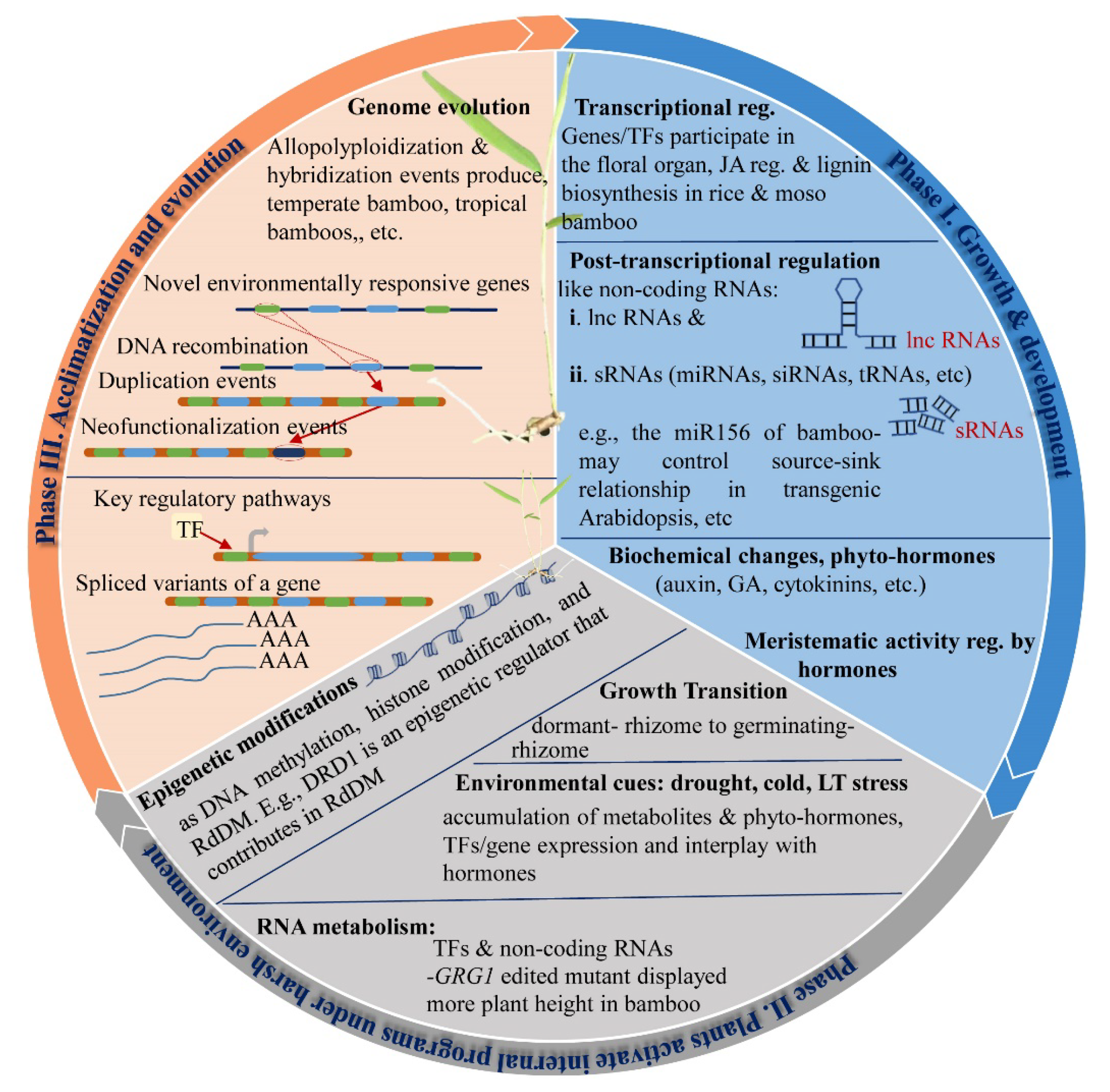
8. Understanding the Bamboos’ Tolerance Using NGS and Metabolome
9. Concluding Remarks and Promising Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Foley, J.A.; Ramankutty, N.; Brauman, K.A.; Cassidy, E.S.; Gerber, J.S.; Johnston, M.; Mueller, N.D.; O’Connell, C.; Ray, D.K.; West, P.C.; et al. Solutions for a cultivated planet. Nature 2011, 478, 337–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godfray, H.C.J.; Beddington, J.R.; Crute, I.R.; Haddad, L.; Lawrence, D.; Muir, J.F.; Pretty, J.; Robinson, S.; Thomas, S.M.; Toulmin, C. Food Security: The Challenge of Feeding 9 Billion People. Science 2010, 327, 812–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- FAO. The State of Food Security and Nutrition in the World 2020; FAO: Rome, Italy, 2019. [Google Scholar]
- Purugganan, M.D.; Jackson, S.A. Advancing crop genomics from lab to field. Nat. Genet. 2021, 53, 595–601. [Google Scholar] [CrossRef] [PubMed]
- Abubakar, M.S.; Attanda, M.L. The Concept of Sustainable Agriculture: Challenges and Prospects. IOP Conf. Series Mater. Sci. Eng. 2013, 53, 012001. [Google Scholar] [CrossRef] [Green Version]
- FAO. The State of Food and Agriculture; FAO: Rome, Italy, 2017. [Google Scholar]
- Kah, M.; Tufenkji, N.; White, J.C. Nano-enabled strategies to enhance crop nutrition and protection. Nat. Nanotechnol. 2019, 14, 532–540. [Google Scholar] [CrossRef]
- Ma, X.; Su, Z.; Ma, H. Molecular genetic analyses of abiotic stress responses during plant reproductive development. J. Exp. Bot. 2020, 71, 2870–2885. [Google Scholar] [CrossRef]
- Ashraf, M.F.; Peng, G.; Liu, Z.; Noman, A.; Alamri, S.; Hashem, M.; Qari, S.H.; Al Zoubi, O.M. Molecular Control and Application of Male Fertility for Two-Line Hybrid Rice Breeding. Int. J. Mol. Sci. 2020, 21, 7868. [Google Scholar] [CrossRef]
- Khakimov, B.; Rasmussen, M.A.; Kannangara, R.M.; Jespersen, B.M.; Munck, L.; Engelsen, S.B. From metabolome to phenotype: GC-MS metabolomics of developing mutant barley seeds reveals effects of growth, temperature and genotype. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Kaul, S.; Koo, H.L.; Jenkins, J.; Rizzo, M.; Rooney, T.; Tallon, L.J.; Feldblyum, T.; Nierman, W.; Benito, M.-I.; Lin, X.J.N. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 2000, 408, 796–815. [Google Scholar]
- Yu, J.; Hu, S.; Wang, J.; Wong, G.K.S.; Li, S.; Liu, B.; Deng, Y.; Dai, L.; Zhou, Y.; Zhang, X.; et al. A Draft Sequence of the Rice Genome (Oryza sativa L. ssp. indica). Science 2002, 296, 79–92. [Google Scholar] [CrossRef]
- Goff, S.A.; Ricke, D.; Lan, T.-H.; Presting, G.; Wang, R.; Dunn, M.; Glazebrook, J.; Sessions, A.; Oeller, P.; Varma, H.; et al. A Draft Sequence of the Rice Genome (Oryza sativa L. ssp. japonica). Science 2002, 296, 92–100. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, J.; Buell, C.R. Advances in plant genome sequencing. Plant J. 2012, 70, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Lin, F.; An, D.; Wang, W.; Huang, R. Genome Sequencing and Assembly by Long Reads in Plants. Genes 2017, 9, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bevan, M.W.; Uauy, C.; Wulff, B.; Zhou, J.; Krasileva, K.; Clark, M. Genomic innovation for crop improvement. Nature 2017, 543, 346–354. [Google Scholar] [CrossRef]
- Garcia, S.; Leitch, I.J.; Anadon-Rosell, A.; Canela, M.Á.; Gálvez, F.; Garnatje, T.; Gras, A.; Hidalgo, O.; Johnston, E.; De Xaxars, G.M.; et al. Recent updates and developments to plant genome size databases. Nucleic Acids Res. 2013, 42, D1159–D1166. [Google Scholar] [CrossRef]
- Ricroch, A. Global developments of genome editing in agriculture. Transgenic Res. 2019, 28, 45–52. [Google Scholar] [CrossRef]
- The International Wheat Genome Sequencing Consortium (IWGSC); Appels, R.; Eversole, K.; Feuillet, C.; Keller, B.; Rogers, J.; Stein, N.; Pozniak, C.J.; Choulet, F.; Distelfeld, A.; et al. Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 2018, 361, eaar7191. [Google Scholar] [CrossRef] [Green Version]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef] [PubMed]
- A Logsdon, G.; Vollger, M.R.; E Eichler, E. Long-read human genome sequencing and its applications. Nat. Rev. Genet. 2020, 21, 597. [Google Scholar] [CrossRef]
- Anandhakumar, C.; Kizaki, S.; Bando, T.; Pandian, G.; Sugiyama, H. Advancing Small-Molecule-Based Chemical Biology with Next-Generation Sequencing Technologies. ChemBioChem 2014, 16, 20–38. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Gao, Y.; Xie, W.; Gong, L.; Lu, K.; Wang, W.; Li, Y.; Liu, X.; Zhang, H.; Dong, H.; et al. Genome-wide association analyses provide genetic and biochemical insights into natural variation in rice metabolism. Nat. Genet. 2014, 46, 714–721. [Google Scholar] [CrossRef] [PubMed]
- Furbank, R.T.; Tester, M. Phenomics–technologies to relieve the phenotyping bottleneck. Trends Plant Sci. 2011, 16, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Zaidem, M.; Groen, S.C.; Purugganan, M.D. Evolutionary and ecological functional genomics, from lab to the wild. Plant J. 2019, 97, 40–55. [Google Scholar] [CrossRef] [Green Version]
- Tattaris, M.; Reynolds, M.P.; Chapman, S. A Direct Comparison of Remote Sensing Approaches for High-Throughput Phenotyping in Plant Breeding. Front. Plant Sci. 2016, 7, 1131. [Google Scholar] [CrossRef]
- Ma, C.; Zhang, H.H.; Wang, X. Machine learning for Big Data analytics in plants. Trends Plant Sci. 2014, 19, 798–808. [Google Scholar] [CrossRef]
- Esposito, S.; Carputo, D.; Cardi, T.; Tripodi, P. Applications and Trends of Machine Learning in Genomics and Phenomics for Next-Generation Breeding. Plants 2019, 9, 34. [Google Scholar] [CrossRef] [Green Version]
- Belhaj, K.; Chaparro-Garcia, A.; Kamoun, S.; Patron, N.J.; Nekrasov, V. Editing plant genomes with CRISPR/Cas. Curr. Opin. Biotechnol. 2015, 32, 76–84. [Google Scholar] [CrossRef]
- Chen, K.; Wang, Y.; Zhang, R.; Zhang, H.; Gao, C. CRISPR/Cas Genome Editing and Precision Plant Breeding in Agriculture. Annu. Rev. Plant Biol. 2019, 70, 667–697. [Google Scholar] [CrossRef]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef] [Green Version]
- Maxam, A.M.; Gilbert, W. A new method for sequencing DNA. Proc. Natl. Acad. Sci. USA 1977, 74, 560–564. [Google Scholar] [CrossRef] [Green Version]
- Devereux, H.L. Automated DNA sequencing. Methods Mol. Med. 1999, 31, 55–61. [Google Scholar] [CrossRef]
- Slatko, B.E.; Kieleczawa, J.; Ju, J.; Gardner, A.F.; Hendrickson, C.L.; Ausubel, F.M. “First Generation” Automated DNA Sequencing Technology. Curr. Protoc. Mol. Biol. 2011, 96, 7.2.1–7.2.28. [Google Scholar] [CrossRef]
- Chorley, B.N.; Wang, X.; Campbell, M.R.; Pittman, G.S.; Noureddine, M.A.; Bell, D.A. Discovery and verification of functional single nucleotide polymorphisms in regulatory genomic regions: Current and developing technologies. Mutat. Res. Mutat. Res. 2008, 659, 147–157. [Google Scholar] [CrossRef] [Green Version]
- Faber, K.; Glatting, K.-H.; Mueller, P.J.; Risch, A.; Hotz-Wagenblatt, A. Genome-wide prediction of splice-modifying SNPs in human genes using a new analysis pipeline called AASsites. BMC Bioinform. 2011, 12, S2. [Google Scholar] [CrossRef] [Green Version]
- NHGRI. The Cost of Sequencing a Human Genome; NHGRI: Bethesda, MD, USA, 2019.
- Liu, L.; Li, Y.; Li, S.; Hu, N.; He, Y.; Pong, R.; Lin, D.; Lu, L.; Law, M. Comparison of Next-Generation Sequencing Systems. J. Biomed. Biotechnol. 2012, 2012, 1–11. [Google Scholar] [CrossRef]
- Levy, S.; Sutton, G.; Ng, P.C.; Feuk, L.; Halpern, A.L.; Walenz, B.P.; Axelrod, N.; Huang, J.; Kirkness, E.F.; Denisov, G.; et al. The Diploid Genome Sequence of an Individual Human. PLoS Biol. 2007, 5, e254. [Google Scholar] [CrossRef] [Green Version]
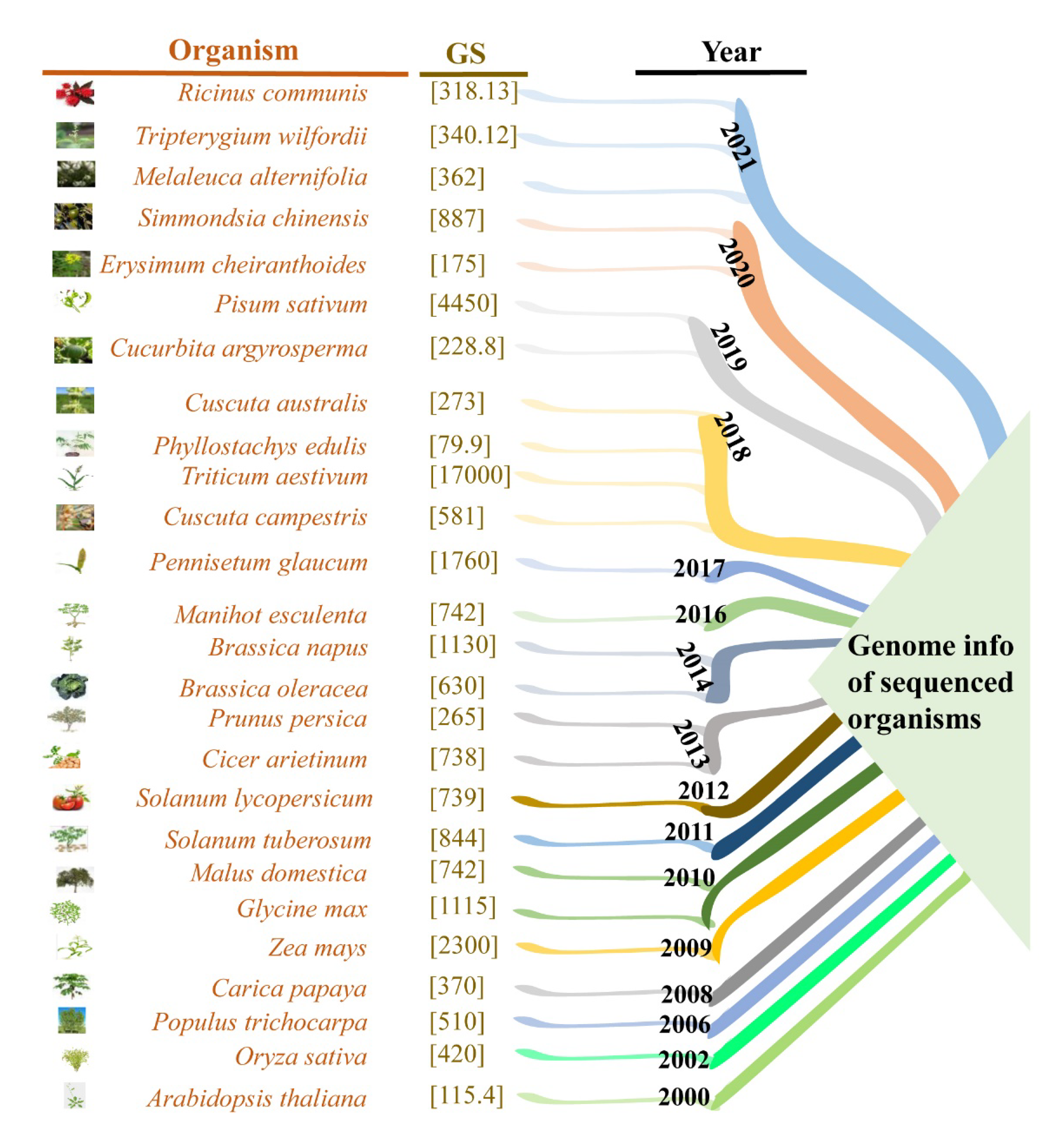
- Michael, T.P.; Jackson, S. The First 50 Plant Genomes. Plant Genome 2013, 6, 2. [Google Scholar] [CrossRef] [Green Version]
- Kiechle, F.L.; Zhang, X. The postgenomic era: Implications for the clinical laboratory. Arch. Pathol. Lab. Med. 2002, 126, 255. [Google Scholar] [CrossRef]
- Michael, M.; Savin, K.W.; Maiko, S.; Pembleton, L.W.; Cogan Noel, O.I.; Shinozuka, K.; Forster, J.W. Transcriptome sequencing of lentil based on second-generation technology permits large-scale unigene assembly and SSR marker discovery. BMC Genom. 2011, 12, 265. [Google Scholar] [CrossRef] [Green Version]
- Wheeler, D.A.; Srinivasan, M.; Egholm, M.; Shen, Y.; Chen, L.; McGuire, A.; He, W.; Chen, Y.-J.; Makhijani, V.; Roth, G.T.; et al. The complete genome of an individual by massively parallel DNA sequencing. Nature 2008, 452, 872–876. [Google Scholar] [CrossRef]
- Yuan, Y.; Bayer, P.E.; Batley, J.; Edwards, D. Current status of structural variation studies in plants. Plant Biotechnol. J. 2021, 19, 2153–2163. [Google Scholar] [CrossRef]
- Thigpen, K.G. International sequencing consortium. Environ. Heal. Perspect. 2004, 112, A406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hui, P. Next Generation Sequencing: Chemistry, Technology and Applications. Chem. Diagn. 2012, 336, 1–18. [Google Scholar] [CrossRef]
- Singh, I.; Wendeln, C.; Clark, A.W.; Cooper, J.M.; Ravoo, B.J.; Burley, G.A. Sequence-Selective Detection of Double-Stranded DNA Sequences Using Pyrrole–Imidazole Polyamide Microarrays. J. Am. Chem. Soc. 2013, 135, 3449–3457. [Google Scholar] [CrossRef]
- Park, P.J. ChIP–seq: Advantages and challenges of a maturing technology. Nat. Rev. Genet. 2009, 10, 669–680. [Google Scholar] [CrossRef] [Green Version]
- Hurd, P.J.; Nelson, C.J. Advantages of next-generation sequencing versus the microarray in epigenetic research. Brief. Funct. Genom. Proteom. 2009, 8, 174–183. [Google Scholar] [CrossRef] [Green Version]
- Unamba, C.I.N.; Nag, A.; Sharma, R.K. Next Generation Sequencing Technologies: The Doorway to the Unexplored Genomics of Non-Model Plants. Front. Plant Sci. 2015, 6, 1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fedoruk, M.J.; Vandenberg, A.; Bett, K.E. Quantitative Trait Loci Analysis of Seed Quality Characteristics in Lentil using Single Nucleotide Polymorphism Markers. Plant Genome 2013, 6. [Google Scholar] [CrossRef] [Green Version]
- Mardis, E.R. A decade’s perspective on DNA sequencing technology. Nature 2011, 470, 198–203. [Google Scholar] [CrossRef]
- Valouev, A.; Ichikawa, J.; Tonthat, T.; Stuart, J.; Ranade, S.; Peckham, H.; Zeng, K.; Malek, J.A.; Costa, G.; McKernan, K.; et al. A high-resolution, nucleosome position map of C. elegans reveals a lack of universal sequence-dictated positioning. Genome Res. 2008, 18, 1051–1063. [Google Scholar] [CrossRef] [Green Version]
- Margulies, M.; Egholm, M.; Altman, W.E.; Attiya, S.; Bader, J.S.; Bemben, L.A.; Berka, J.; Braverman, M.S.; Chen, Y.-J.; Chen, Z.J.N. Genome sequencing in microfabricated high-density picolitre reactors. Nature 2005, 437, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Shendure, J.; Porreca, G.J.; Reppas, N.B.; Lin, X.; McCutcheon, J.P.; Rosenbaum, A.M.; Wang, M.D.; Zhang, K.; Mitra, R.D.; Church, G.M. Accurate Multiplex Polony Sequencing of an Evolved Bacterial Genome. Science 2005, 309, 1728–1732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metzker, M.L. Sequencing technologies—The next generation. Nat. Rev. Genet. 2009, 11, 31–46. [Google Scholar] [CrossRef] [Green Version]
- Heather, J.M.; Chain, B. The sequence of sequencers: The history of sequencing DNA. Genomics 2016, 107, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neale, D.B.; Wegrzyn, J.L.; A Stevens, K.; Zimin, A.V.; Puiu, D.; Crepeau, M.W.; Cardeno, C.; Koriabine, M.; E Holtz-Morris, A.; Liechty, J.D.; et al. Decoding the massive genome of loblolly pine using haploid DNA and novel assembly strategies. Genome Biol. 2014, 15, R59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nystedt, B.; Street, N.R.; Wetterbom, A.; Zuccolo, A.; Lin, Y.-C.; Scofield, D.G.; Vezzi, F.; Delhomme, N.; Giacomello, S.; Alexeyenko, A.; et al. The Norway spruce genome sequence and conifer genome evolution. Nature 2013, 497, 579–584. [Google Scholar] [CrossRef] [Green Version]
- Salman-Minkov, A.; Sabath, N.; Mayrose, I. Whole-genome duplication as a key factor in crop domestication. Nat. Plants 2016, 2, 16115. [Google Scholar] [CrossRef]
- Peterson, T.W.; Nam, J.N.; Darby, A. Next gen sequencing survey. N. Am. Equity Res. 2010. [Google Scholar]
- Pei, S.; Liu, T.; Ren, X.; Li, W.; Chen, C.; Xie, Z. Benchmarking variant callers in next-generation and third-generation sequencing analysis. Brief. Bioinform. 2021, 22, 148. [Google Scholar] [CrossRef]
- Clark, T.A.; Spittle, K.E.; Turner, S.W.; Korlach, J. Direct Detection and Sequencing of Damaged DNA Bases. Genome Integr. 2011, 2, 10. [Google Scholar] [CrossRef] [Green Version]
- Harris, T.D.; Buzby, P.R.; Babcock, H.; Beer, E.; Bowers, J.; Braslavsky, I.; Causey, M.; Colonell, J.; DiMeo, J.; Efcavitch, J.W.; et al. Single-Molecule DNA Sequencing of a Viral Genome. Science 2008, 320, 106–109. [Google Scholar] [CrossRef] [Green Version]
- De Coster, W.; Weissensteiner, M.H.; Sedlazeck, F.J. Towards population-scale long-read sequencing. Nat. Rev. Genet. 2021, 22, 572–587. [Google Scholar] [CrossRef]
- Marx, V. Long road to long-read assembly. Nat. Methods 2021, 18, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Tedersoo, L.; Albertsen, M.; Anslan, S.; Callahan, B. Perspectives and Benefits of High-Throughput Long-Read Sequencing in Microbial Ecology. Appl. Environ. Microbiol. 2021, 87, 0062621. [Google Scholar] [CrossRef]
- Wang, B.; Tseng, E.; Regulski, M.; Clark, T.A.; Hon, T.; Jiao, Y.; Lu, Z.; Olson, A.; Stein, J.C.; Ware, D. Unveiling the complexity of the maize transcriptome by single-molecule long-read sequencing. Nat. Commun. 2016, 7, 11708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, T.; Chitnis, N.; Monos, D.; Dinh, A. Next-generation sequencing technologies: An overview. Hum. Immunol. 2021, 82, 801–811. [Google Scholar] [CrossRef]
- Bhat, J.A.; Yu, D.J.L.S. High-throughput NGS-based genotyping and phenotyping: Role in genomics-assisted breeding for soybean improvement. Legume Sci. 2021, 3, e81. [Google Scholar] [CrossRef]
- Bolisetty, M.T.; Rajadinakaran, G.; Graveley, B.R. Determining exon connectivity in complex mRNAs by nanopore sequencing. Genome Biol. 2015, 16, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Thanaraj, T.A. ASD: The Alternative Splicing Database. Nucleic Acids Res. 2004, 32, 64–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flint, J.; Mott, R. Finding the molecular basis of quatitative traits: Successes and pitfalls. Nat. Rev. Genet. 2001, 2, 437–445. [Google Scholar] [CrossRef]
- Abdel-Ghany, S.E.; Hamilton, M.; Jacobi, J.L.; Ngam, P.; Devitt, N.; Schilkey, F.; Ben-Hur, A.; Reddy, A.S.N. A survey of the sorghum transcriptome using single-molecule long reads. Nat. Commun. 2016, 7, 11706. [Google Scholar] [CrossRef] [Green Version]
- Steijger, T.; The RGASP Consortium; Abril, J.F.; Engström, P.; Kokocinski, F.; Hubbard, T.; Guigó, R.; Harrow, J.; Bertone, P. Assessment of transcript reconstruction methods for RNA-seq. Nat. Methods 2013, 10, 1177–1184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garalde, D.R.; Snell, E.A.; Jachimowicz, D.; Sipos, B.; Lloyd, J.H.; Bruce, M.; Pantic, N.; Admassu, T.; James, P.; Warland, A.; et al. Highly parallel direct RNA sequencing on an array of nanopores. Nat. Methods 2018, 15, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Weirather, J.L.; de Cesare, M.; Wang, Y.; Piazza, P.; Sebastiano, V.; Wang, X.-J.; Buck, D.; Au, K.F. Comprehensive comparison of Pacific Biosciences and Oxford Nanopore Technologies and their applications to transcriptome analysis. F1000Research 2017, 6, 100. [Google Scholar] [CrossRef] [PubMed]
- Montenegro, J.D.; Golicz, A.A.; Bayer, P.E.; Hurgobin, B.; Lee, H.; Chan, C.-K.K.; Visendi, P.; Lai, K.; Doležel, J.; Batley, J.; et al. The pangenome of hexaploid bread wheat. Plant J. 2017, 90, 1007–1013. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Feng, Q.; Lu, H.; Li, Y.; Wang, A.; Tian, Q.; Zhan, Q.; Lu, Y.; Zhang, L.; Huang, T.J.N.G. Pan-genome analysis highlights the extent of genomic variation in cultivated and wild rice. Nat. Genet. 2018, 50, 278–284. [Google Scholar] [CrossRef] [Green Version]
- Barabaschi, D.; Guerra, D.; Lacrima, K.; Laino, P.; Michelotti, V.; Urso, S.; Valè, G.; Cattivelli, L. Emerging Knowledge from Genome Sequencing of Crop Species. Mol. Biotechnol. 2011, 50, 250–266. [Google Scholar] [CrossRef]
- Yeh, H.-M.; Liao, M.-H.; Chu, C.-L.; Lin, Y.-H.; Sun, W.-Z.; Lai, L.-P.; Chen, P.-L. Next-generation sequencing and bioinformatics to identify genetic causes of malignant hyperthermia. J. Formos. Med Assoc. 2021, 120, 883–892. [Google Scholar] [CrossRef]
- Blätke, M.-A.; Szymanski, J.J.; Gladilin, E.; Scholz, U.; Beier, S. Editorial: Advances in Applied Bioinformatics in Crops. Front. Plant Sci. 2021, 12, 12. [Google Scholar] [CrossRef]
- Ibrahim, M.A.; Shehata, M.A.; Nasry, N.N.; Fayez, M.S.; Bishay, S.K.; Aziz, M.M.; Ratib, N.R.; Ahmed, N.K.; Ali, S.M.; Ismail, E.; et al. Bioinformatics Approaches toward Plant Breeding Programs. Asian J. Res. Rev. Agric. 2021, 3, 5–14. [Google Scholar]
- Chu, C.; Wang, S.; Rudd, J.C.; Ibrahim, A.M.; Xue, Q.; Devkota, R.N.; Baker, J.A.; Baker, S.; Simoneaux, B.; Opena, G. A New Strategy for Using Historical Imbalanced Yield Data to Conduct Genome-Wide Association Studies and Develop Genomic Prediction Models for Wheat Breeding. Mol. Breed. 2021. [Google Scholar]
- Ariyadasa, R.; Stein, N. Advances in BAC-Based Physical Mapping and Map Integration Strategies in Plants. J. Biomed. Biotechnol. 2012, 2012, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Zalapa, J.E.; Cuevas, H.; Zhu, H.; Steffan, S.; Senalik, U.; Zeldin, E.; McCown, B.; Harbut, R.; Simon, P. Using next-generation sequencing approaches to isolate simple sequence repeat (SSR) loci in the plant sciences. Am. J. Bot. 2012, 99, 193–208. [Google Scholar] [CrossRef] [Green Version]
- Gao, Q.; Yue, G.; Li, W.; Wang, J.; Xu, J.; Yin, Y. Recent Progress Using High-throughput Sequencing Technologies in Plant Molecular BreedingF. J. Integr. Plant Biol. 2012, 54, 215–227. [Google Scholar] [CrossRef]
- Paran, I.; Zamir, D. Quantitative traits in plants: Beyond the QTL. Trends Genet. 2003, 19, 303–306. [Google Scholar] [CrossRef]
- Davey, J.W.; Hohenlohe, P.A.; Etter, P.D.; Boone, J.Q.; Catchen, J.M.; Blaxter, M. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat. Rev. Genet. 2011, 12, 499–510. [Google Scholar] [CrossRef]
- Salvi, S.; Tuberosa, R. To clone or not to clone plant QTLs: Present and future challenges. Trends Plant Sci. 2005, 10, 297–304. [Google Scholar] [CrossRef]
- Liu, G.; Zhu, H.; Zhang, G.; Li, L.; Ye, G. Dynamic analysis of QTLs on tiller number in rice (Oryza sativa L.) with single segment substitution lines. Theor. Appl. Genet. 2012, 125, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, C.N.; Foerster, J.M.; Johnson, J.M.; Sekhon, R.S.; Muttoni, G.; Vaillancourt, B.; Peñagaricano, F.; Lindquist, E.; Pedraza, M.A.; Barry, K.; et al. Insights into the maize pan-genome and pan-transcriptome. Plant Cell 2014, 26, 121–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, D.; Singh, C.K.; Taunk, J.; Jadon, V.; Pal, M.; Gaikwad, K. Genome wide transcriptome analysis reveals vital role of heat responsive genes in regulatory mechanisms of lentil (Lens culinaris Medikus). Sci. Rep. 2019, 9, 1–19. [Google Scholar] [CrossRef]
- Zhou, Z.-X.; Zhang, M.-J.; Peng, X.; Takayama, Y.; Xu, X.-Y.; Huang, L.-Z.; Du, L.-L. Mapping genomic hotspots of DNA damage by a single-strand-DNA-compatible and strand-specific ChIP-seq method. Genome Res. 2013, 23, 705–715. [Google Scholar] [CrossRef] [Green Version]
- Schneeberger, K.; Weigel, D. Fast-forward genetics enabled by new sequencing technologies. Trends Plant Sci. 2011, 16, 282–288. [Google Scholar] [CrossRef]
- Hamblin, M.T.; Buckler, E.S.; Jannink, J.-L. Population genetics of genomics-based crop improvement methods. Trends Genet. 2011, 27, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Spindel, J.; Begum, H.; Akdemir, D.; Virk, P.; Collard, B.; Redoña, E.; Atlin, G.; Jannink, J.-L.; McCouch, S.R. Genomic selection and association mapping in rice (Oryza sativa): Effect of trait genetic architecture, training population composition, marker number and statistical model on accuracy of rice genomic selection in elite, tropical rice breeding lines. PLoS Genet. 2019, 2, 1004982. [Google Scholar] [CrossRef]
- Meuwissen, T.H.E.; Hayes, B.J.; Goddard, M.E. Prediction of Total Genetic Value Using Genome-Wide Dense Marker Maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [CrossRef]
- Mulder, H. Is GxE a burden or a blessing? Opportunities for genomic selection and big data. J. Anim. Breed. Genet. 2017, 134, 435–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, J.; Gupta, D.S. Prospects of next generation sequencing in lentil breeding. Mol. Biol. Rep. 2020, 47, 9043–9053. [Google Scholar] [CrossRef]
- Polanco, C.; de Miera, L.E.S.; González, A.I.; García, P.G.; Fratini, R.; Vaquero, F.; Vences, F.J.; De La Vega, M.P. Construction of a high-density interspecific (Lens culinaris x L. odemensis) genetic map based on functional markers for mapping morphological and agronomical traits, and QTLs affecting resistance to Ascochyta in lentil. PLoS ONE 2019, 14, e0214409. [Google Scholar] [CrossRef] [Green Version]
- Temel, H.Y.; Göl, D.; Akkale, H.B.K.; Kahriman, A.; Tanyolaç, M.B. Single nucleotide polymorphism discovery through Illumina-based transcriptome sequencing and mapping in lentil. Turk. J. Agric. For. 2015, 39, 470–488. [Google Scholar] [CrossRef]
- Lavin, M.; Herendeen, P.; Wojciechowski, M.F. Evolutionary Rates Analysis of Leguminosae Implicates a Rapid Diversification of Lineages during the Tertiary. Syst. Biol. 2005, 54, 575–594. [Google Scholar] [CrossRef] [Green Version]
- Sharpe, A.G.; Ramsay, L.; Sanderson, L.-A.; Fedoruk, M.J.; E Clarke, W.; Li, R.; Kagale, S.; Vijayan, P.; Vandenberg, A.; E Bett, K. Ancient orphan crop joins modern era: Gene-based SNP discovery and mapping in lentil. BMC Genom. 2013, 14, 192. [Google Scholar] [CrossRef] [Green Version]
- Brozynska, M.; Furtado, A.; Henry, R.J. Genomics of crop wild relatives: Expanding the gene pool for crop improvement. Plant Biotechnol. J. 2016, 14, 1070–1085. [Google Scholar] [CrossRef]
- Khazaei, H.; Caron, C.T.; Fedoruk, M.; Diapari, M.; Vandenberg, A.; Coyne, C.J.; McGee, R.; Bett, K.E. Genetic Diversity of Cultivated Lentil (Lens culinaris Medik.) and Its Relation to the World’s Agro-ecological Zones. Front. Plant Sci. 2016, 7, 1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horton, M.W.; Hancock, A.M.; Huang, Y.S.; Toomajian, C.; Atwell, S.; Auton, A.; Muliyati, N.W.; Platt, A.; Sperone, F.G.; Vilhjalmsson, B.J.; et al. Genome-wide patterns of genetic variation in worldwide Arabidopsis thaliana accessions from the RegMap panel. Nat. Genet. 2012, 44, 212–216. [Google Scholar] [CrossRef] [Green Version]
- Ferrero-Serrano, Á.; Assmann, S.M. Phenotypic and genome-wide association with the local environment of Arabidopsis. Nat. Ecol. Evol. 2019, 3, 274–285. [Google Scholar] [CrossRef]
- Lasky, J.R.; Marais, D.L.D.; McKay, J.K.; Richards, J.H.; Juenger, T.E.; Keitt, T.H. Data from: Characterizing genomic variation of Arabidopsis thaliana: The roles of geography and climate. Mol. Ecol. 2012, 21, 5512. [Google Scholar] [CrossRef] [PubMed]
- Gutaker, R.M.; Groen, S.C.; Bellis, E.; Choi, J.Y.; Pires, I.S.; Bocinsky, R.K.; Slayton, E.; Wilkins, O.; Castillo, C.C.; Negrão, S.; et al. Genomic history and ecology of the geographic spread of rice. Nat. Plants 2020, 6, 492–502. [Google Scholar] [CrossRef] [PubMed]
- Bilinski, P.; Albert, P.S.; Berg, J.J.; Birchler, J.A.; Grote, M.N.; Lorant, A.; Quezada, J.; Swarts, K.; Yang, J.; Ross-Ibarra, J. Parallel altitudinal clines reveal trends in adaptive evolution of genome size in Zea mays. PLoS Genet. 2018, 14, e1007162. [Google Scholar] [CrossRef]
- Lasky, J.R.; Upadhyaya, H.D.; Ramu, P.; Deshpande, S.; Hash, C.T.; Bonnette, J.; Juenger, T.E.; Hyma, K.; Acharya, C.; Mitchell, S.E.; et al. Genome-environment associations in sorghum landraces predict adaptive traits. Sci. Adv. 2015, 1, e1400218. [Google Scholar] [CrossRef] [Green Version]
- Rhoné, B.; Defrance, D.; Berthouly-Salazar, C.; Mariac, C.; Cubry, P.; Couderc, M.; Dequincey, A.; Assoumanne, A.; Kane, N.A.; Sultan, B.; et al. Pearl millet genomic vulnerability to climate change in West Africa highlights the need for regional collaboration. Nat. Commun. 2020, 11, 1–9. [Google Scholar] [CrossRef]
- Abrouk, M.; Ahmed, H.I.; Cubry, P.; Šimoníková, D.; Cauet, S.; Pailles, Y.; Bettgenhaeuser, J.; Gapa, L.; Scarcelli, N.; Couderc, M.; et al. Fonio millet genome unlocks African orphan crop diversity for agriculture in a changing climate. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Danilevicz, M.F.; Tay Fernandez, C.G.; Marsh, J.I.; Bayer, P.E.; Edwards, D. Plant pangenomics: Approaches, applications and advancements. Curr. Opin. Plant Biol. 2020, 54, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Bayer, P.; Golicz, A.; Scheben, A.; Batley, J.; Edwards, D. Plant pan-genomes are the new reference. Nat. Plants 2020, 6, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, H.; Moura, C.F.; Anderegg, W.R.L.; Ruehr, N.K.; Salmon, Y.; Allen, C.D.; Arndt, S.K.; Breshears, D.D.; Davi, H.; Galbraith, D.; et al. Research frontiers for improving our understanding of drought-induced tree and forest mortality. New Phytol. 2018, 218, 15–28. [Google Scholar] [CrossRef] [Green Version]
- Choat, B.; Brodribb, T.; Brodersen, C.R.; Duursma, R.A.; López, R.; Medlyn, B. Triggers of tree mortality under drought. Nature 2018, 558, 531–539. [Google Scholar] [CrossRef]
- Piasecka, A.; Sawikowska, A.; Kuczyńska, A.; Ogrodowicz, P.; Mikołajczak, K.; Krystkowiak, K.; Gudyś, K.; Guzy-Wróbelska, J.; Krajewski, P.; Kachlicki, P. Drought-related secondary metabolites of barley (Hordeum vulgare L.) leaves and their metabolomic quantitative trait loci. Plant J. 2017, 89, 898–913. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Song, Y.; Li, X.; Chen, J.; Mo, L.; Zhang, X.; Lin, Z.; Zhang, L. Genome sequences of horticultural plants: Past, present, and future. Hortic. Res. 2019, 6, 1–23. [Google Scholar] [CrossRef] [Green Version]
- Maroufi, A.; Van Bockstaele, E.; De Loose, M. Validation of reference genes for gene expression analysis in chicory (Cichorium intybus) using quantitative real-time PCR. BMC Mol. Biol. 2010, 11, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, research0034.1. [Google Scholar] [CrossRef] [Green Version]
- Nicot, N.; Hausman, J.-F.; Hoffmann, L.; Evers, D. Housekeeping gene selection for real-time RT-PCR normalization in potato during biotic and abiotic stress. J. Exp. Bot. 2005, 56, 2907–2914. [Google Scholar] [CrossRef] [PubMed]
- Guénin, S.; Mauriat, M.; Pelloux, J.; Van Wuytswinkel, O.; Bellini, C.; Gutierrez, L. Normalization of qRT-PCR data: The necessity of adopting a systematic, experimental conditions-specific, validation of references. J. Exp. Bot. 2009, 60, 487–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellemans, J.; Mortier, G.; De Paepe, A.; Speleman, F.; Vandesompele, J. qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol. 2007, 8, R19. [Google Scholar] [CrossRef] [Green Version]
- McHale, L.K.; Haun, W.J.; Xu, W.W.; Bhaskar, P.B.; Anderson, J.; Hyten, D.; Gerhardt, D.J.; Jeddeloh, J.A.; Stupar, R.M. Structural Variants in the Soybean Genome Localize to Clusters of Biotic Stress-Response Genes. Plant Physiol. 2012, 159, 1295–1308. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.-h.; Zhou, G.; Ma, J.; Jiang, W.; Jin, L.-g.; Zhang, Z.; Guo, Y.; Zhang, J.; Sui, Y.; Zheng, L.J.N.B. De novo assembly of soybean wild relatives for pan-genome analysis of diversity and agronomic traits. Nat. Biotechnol. 2014, 32, 1045–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schatz, M.C.; Maron, L.G.; Stein, J.C.; Wences, A.H.; Gurtowski, J.; Biggers, E.; Lee, H.; Kramer, M.; Antoniou, E.; Ghiban, E.J.G.B. Whole genome de novo assemblies of three divergent strains of rice, Oryza sativa, document novel gene space of aus and indica. Genome Biol. 2014, 15, 506. [Google Scholar]
- Tao, Y.; Zhao, X.; Mace, E.; Henry, R.; Jordan, D. Exploring and Exploiting Pan-genomics for Crop Improvement. Mol. Plant 2019, 12, 156–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lakušić, D.; Liber, Z.; Nikolić, T.; Surina, B.; Kovacic, S.; Bogdanović, S.; Stefanović, S. Molecular phylogeny of the Campanula pyramidalis species complex (Campanulaceae) inferred from chloroplast and nuclear non-coding sequences and its taxonomic implications. TAXON 2013, 62, 505–524. [Google Scholar] [CrossRef] [Green Version]
- Magdy, M.; Ou, L.; Yu, H.; Chen, R.; Zhou, Y.; Hassan, H.; Feng, B.; Taitano, N.; van der Knaap, E.; Zou, X.; et al. Pan-plastome approach empowers the assessment of genetic variation in cultivated Capsicum species. Hortic. Res. 2019, 6, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.W.; Garg, V.; Roorkiwal, M.; Golicz, A.A.; Edwards, D.; Varshney, R.K. Super-Pangenome by Integrating the Wild Side of a Species for Accelerated Crop Improvement. Trends Plant Sci. 2020, 25, 148–158. [Google Scholar] [CrossRef] [Green Version]
- Mascher, M.; Schreiber, M.; Scholz, U.; Graner, A.; Reif, J.C.; Stein, N. Genebank genomics bridges the gap between the conservation of crop diversity and plant breeding. Nat. Genet. 2019, 51, 1076–1081. [Google Scholar] [CrossRef]
- McCouch, S.; Navabi, Z.K.; Abberton, M.; Anglin, N.L.; Barbieri, R.L.; Baum, M.; Bett, K.; Booker, H.; Brown, G.L.; Bryan, G.J.; et al. Mobilizing Crop Biodiversity. Mol. Plant 2020, 13, 1341–1344. [Google Scholar] [CrossRef] [PubMed]
- Varshney, R.K.; Singh, V.K.; Kumar, A.; Powell, W.; Sorrells, M.E. Can genomics deliver climate-change ready crops? Curr. Opin. Plant Biol. 2018, 45, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Mauleon, R.; Hu, Z.; Chebotarov, D.; Tai, S.; Wu, Z.; Li, M.; Zheng, T.; Fuentes, R.R.; Zhang, F.; et al. Genomic variation in 3,010 diverse accessions of Asian cultivated rice. Nature 2018, 557, 43–49. [Google Scholar] [CrossRef]
- Wing, R.A.; Purugganan, M.D.; Zhang, Q. The rice genome revolution: From an ancient grain to Green Super Rice. Nat. Rev. Genet. 2018, 19, 505–517. [Google Scholar] [CrossRef] [PubMed]
- Whitfield, P.; Kirwan, J. Metabolomics: An Emerging Post-genomic Tool for Nutrition. Genom. Proteom. Metab. Nutraceuticals Funct. Foods 2010, 92, 271–285. [Google Scholar] [CrossRef]
- Hong, J.; Yang, L.; Zhang, D.; Shi, J. Plant Metabolomics: An Indispensable System Biology Tool for Plant Science. Int. J. Mol. Sci. 2016, 17, 767. [Google Scholar] [CrossRef]
- Kumar, R.; Bohra, A.; Pandey, A.K.; Pandey, M.K.; Kumar, A. Metabolomics for Plant Improvement: Status and Prospects. Front. Plant Sci. 2017, 8, 1302. [Google Scholar] [CrossRef] [Green Version]
- Dodds, P.N.; Rathjen, J.P. Plant immunity: Towards an integrated view of plant–pathogen interactions. Nat. Rev. Genet. 2010, 11, 539–548. [Google Scholar] [CrossRef]
- Tsuda, K.; Somssich, I.E. Transcriptional networks in plant immunity. New Phytol. 2015, 206, 932–947. [Google Scholar] [CrossRef]
- Parry, M.A.J.; Hawkesford, M. An Integrated Approach to Crop Genetic ImprovementF. J. Integr. Plant Biol. 2012, 54, 250–259. [Google Scholar] [CrossRef]
- Xavier, A.; Hall, B.; Hearst, A.A.; Cherkauer, K.A.; Rainey, K.M. Genetic Architecture of Phenomic-Enabled Canopy Coverage in Glycine max. Genetics 2017, 206, 1081–1089. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Hu, S.; Gardner, C.; Lübberstedt, T. Emerging Avenues for Utilization of Exotic Germplasm. Trends Plant Sci. 2017, 22, 624–637. [Google Scholar] [CrossRef] [Green Version]
- Turner, M.F.; Heuberger, A.L.; Kirkwood, J.S.; Collins, C.C.; Wolfrum, E.J.; Broeckling, C.D.; Prenni, J.E.; Jahn, C.E. Non-targeted Metabolomics in Diverse Sorghum Breeding Lines Indicates Primary and Secondary Metabolite Profiles Are Associated with Plant Biomass Accumulation and Photosynthesis. Front. Plant Sci. 2016, 7, 953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilu, R.; Panzeri, D.; Cassani, E.; Cerino Badone, F.; Landoni, M.; Nielsen, E. A paramutation phenomenon is involved in the genetics of maize low phytic acid1-241 (lpa1-241) trait. Heredity 2009, 102, 236–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arbona, V.; Manzi, M.; De Ollas, C.; Gómez-Cadenas, A. Metabolomics as a Tool to Investigate Abiotic Stress Tolerance in Plants. Int. J. Mol. Sci. 2013, 14, 4885–4911. [Google Scholar] [CrossRef]
- Li, X.; Lawas, L.M.; Malo, R.; Glaubitz, U.; Erban, A.; Mauleon, R.; Heuer, S.; Zuther, E.; Kopka, J.; Hincha, D.K.; et al. Metabolic and transcriptomic signatures of rice floral organs reveal sugar starvation as a factor in reproductive failure under heat and drought stress. Plant Cell Environ. 2015, 38, 2171–2192. [Google Scholar] [CrossRef] [PubMed]
- Obata, T.; Fernie, A.R. The use of metabolomics to dissect plant responses to abiotic stresses. Cell. Mol. Life Sci. 2012, 69, 3225–3243. [Google Scholar] [CrossRef] [Green Version]
- Diretto, G.; Al-Babili, S.; Tavazza, R.; Scossa, F.; Papacchioli, V.; Migliore, M.; Beyer, P.; Giuliano, G. Transcriptional-metabolic networks in beta-carotene-enriched potato tubers: The long and winding road to the Golden phenotype. Plant Physiol. 2010, 154, 899–912. [Google Scholar] [CrossRef] [Green Version]
- Paine, J.A.; Shipton, C.A.; Chaggar, S.; Howells, R.; Kennedy, M.J.; Vernon, G.; Wright, S.Y.; Hinchliffe, E.; Adams, J.L.; Silverstone, A.L.; et al. Improving the nutritional value of Golden Rice through increased pro-vitamin A content. Nat. Biotechnol. 2005, 23, 482–487. [Google Scholar] [CrossRef]
- Yang, L.; Wen, K.-S.; Ruan, X.; Zhao, Y.-X.; Wei, F.; Wang, Q. Response of Plant Secondary Metabolites to Environmental Factors. Molecules 2018, 23, 762. [Google Scholar] [CrossRef] [Green Version]
- Ramakrishna, A.; Ravishankar, G.A. Influence of abiotic stress signals on secondary metabolites in plants. Plant Signal. Behav. 2011, 6, 1720. [Google Scholar] [PubMed]
- Loskutov, I.; Shelenga, T.; Konarev, A.; Shavarda, A.; Blinova, E.; Dzubenko, N.R. The metabolomic approach to the comparative analysis of wild and cultivated species of oats (Avena L.). Russ. J. Genet. Appl. Res. 2017, 7, 501–508. [Google Scholar] [CrossRef]
- Sánchez-Martín, J.; Heald, J.; Kingston-Smith, A.; Winters, A.; Rubiales, D.; Sanz, M.; Mur, L.A.J.; Prats, E. A metabolomic study in oats (Avena sativa) highlights a drought tolerance mechanism based upon salicylate signalling pathways and the modulation of carbon, antioxidant and photo-oxidative metabolism. Plant Cell Environ. 2015, 38, 1434–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.; Rupasinghe, T.; Callahan, D.L.; Natera, S.H.; Smith, P.; Hill, C.B.; Roessner, U.; Boughton, B.S. Spatio-temporal metabolite and elemental profiling of salt stressed barley seeds during initial stages of germination by MALDI-MSI and µ-XRF spectrometry. Front. Plant Sci. 2019, 10, 1139. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zeng, X.; Xu, Q.; Mei, X.; Yuan, H.; Jiabu, D.; Sang, Z.; Nyima, T. Metabolite profiling in two contrasting Tibetan hulless barley cultivars revealed the core salt-responsive metabolome and key salt-tolerance biomarkers. AoB Plants 2019, 11, plz021. [Google Scholar] [CrossRef] [Green Version]
- Piasecka, A.; Sawikowska, A.; Krajewski, P.; Kachlicki, P. Combined mass spectrometric and chromatographic methods for in-depth analysis of phenolic secondary metabolites in barley leaves. J. Mass Spectrom. 2015, 50, 513–532. [Google Scholar] [CrossRef] [PubMed]
- Kogel, K.-H.; Voll, L.M.; Schäfer, P.; Jansen, C.; Wu, Y.; Langen, G.; Imani, J.; Hofmann, J.; Schmiedl, A.; Sonnewald, S.; et al. Transcriptome and metabolome profiling of field-grown transgenic barley lack induced differences but show cultivar-specific variances. Proc. Natl. Acad. Sci. USA 2010, 107, 6198–6203. [Google Scholar] [CrossRef] [Green Version]
- Roessner, U.; Patterson, J.H.; Forbes, M.G.; Fincher, G.B.; Langridge, P.; Bacic, A. An Investigation of Boron Toxicity in Barley Using Metabolomics. Plant Physiol. 2006, 142, 1087–1101. [Google Scholar] [CrossRef] [Green Version]
- Song, E.-H.; Jeong, J.; Park, C.Y.; Kim, H.-Y.; Kim, E.-H.; Bang, E.; Hong, Y.-S. Metabotyping of rice (Oryza sativa L.) for understanding its intrinsic physiology and potential eating quality. Food Res. Int. 2018, 111, 20–30. [Google Scholar] [CrossRef]
- Yan, S.; Huang, W.; Gao, J.; Fu, H.; Liu, J. Comparative metabolomic analysis of seed metabolites associated with seed storability in rice (Oryza sativa L.) during natural aging. Plant Physiol. Biochem. 2018, 127, 590–598. [Google Scholar] [CrossRef]
- Gayen, D.; Ghosh, S.; Paul, S.; Sarkar, S.N.; Datta, S.K.; Datta, K. Metabolic Regulation of Carotenoid-Enriched Golden Rice Line. Front. Plant Sci. 2016, 7, 1622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, C.; Shi, J.; Quan, S.; Cui, B.; Kleessen, S.; Nikoloski, Z.; Tohge, T.; Alexander, D.; Guo, L.; Lin, H.; et al. Metabolic variation between japonica and indica rice cultivars as revealed by non-targeted metabolomics. Sci. Rep. 2014, 4, 5067. [Google Scholar] [CrossRef] [Green Version]
- Zarei, I.; Luna, E.; Leach, J.E.; McClung, A.; Vilchez, S.; Koita, O.; Ryan, E.P. Comparative Rice Bran Metabolomics across Diverse Cultivars and Functional Rice Gene–Bran Metabolite Relationships. Metabolites 2018, 8, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhawi, F.; Datta, R.; Ramakrishna, W. Metabolomics, biomass and lignocellulosic total sugars analysis in foxtail millet (Setaria italica) inoculated with different combinations of plant growth promoting bacteria and mycorrhiza. Commun. Plant Sci. 2018, 8, 8. [Google Scholar] [CrossRef]
- Mareya, C.R.; Tugizimana, F.; Piater, L.A.; Madala, N.E.; Steenkamp, P.A.; Dubery, I.A. Untargeted Metabolomics Reveal Defensome-Related Metabolic Reprogramming in Sorghum bicolor against Infection by Burkholderia andropogonis. Metabolites 2019, 9, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tugizimana, F.; Djami-Tchatchou, A.T.; Steenkamp, P.A.; Piater, L.A.; Dubery, I.A. Metabolomic Analysis of Defense-Related Reprogramming in Sorghum bicolor in Response to Colletotrichum sublineolum Infection Reveals a Functional Metabolic Web of Phenylpropanoid and Flavonoid Pathways. Front. Plant Sci. 2019, 9, 1840. [Google Scholar] [CrossRef] [Green Version]
- Ogbaga, C.C.; Stępień, P.; Dyson, B.C.; Rattray, N.J.W.; Ellis, D.I.; Goodacre, R.; Johnson, G.N. Biochemical Analyses of Sorghum Varieties Reveal Differential Responses to Drought. PLoS ONE 2016, 11, e0154423. [Google Scholar] [CrossRef] [PubMed]
- Michaletti, A.; Naghavi, M.R.; Toorchi, M.; Zolla, L.; Rinalducci, S. Metabolomics and proteomics reveal drought-stress responses of leaf tissues from spring-wheat. Sci. Rep. 2018, 8, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Thomason, K.; Babar, A.; Erickson, J.E.; Mulvaney, M.; Beecher, C.; Macdonald, G. Comparative physiological and metabolomics analysis of wheat (Triticum aestivum L.) following post-anthesis heat stress. PLoS ONE 2018, 13, e0197919. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.; Zhu, A.; Jia, J.; Hu, X.; Chen, J.; Liu, W.; Ren, X.; Sun, D.; Fernie, A.R.; Cui, F.; et al. Metabolomics analysis and metabolite-agronomic trait associations using kernels of wheat (Triticum aestivum) recombinant inbred lines. Plant J. 2020, 103, 279–292. [Google Scholar] [CrossRef] [Green Version]
- Shewry, P.R.; Corol, D.I.; Jones, H.; Beale, M.H.; Ward, J.L. Defining genetic and chemical diversity in wheat grain by 1H-NMR spectroscopy of polar metabolites. Mol. Nutr. Food Res. 2017, 61, 1600807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthews, S.B.; Santra, M.; Mensack, M.M.; Wolfe, P.; Byrne, P.F.; Thompson, H.J. Metabolite Profiling of a Diverse Collection of Wheat Lines Using Ultraperformance Liquid Chromatography Coupled with Time-of-Flight Mass Spectrometry. PLoS ONE 2012, 7, e44179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Rao, R.S.P.; Zhang, Y.; Zhong, C.; Thelen, J.J. Metabolite variation in hybrid corn grain from a large-scale multisite study. Crop. J. 2016, 4, 177–187. [Google Scholar] [CrossRef] [Green Version]
- Rao, J.; Cheng, F.; Hu, C.; Quan, S.; Lin, H.; Wang, J.; Chen, G.; Zhao, X.; Alexander, D.; Guo, L.; et al. Metabolic map of mature maize kernels. Metabolomics 2014, 10, 775–787. [Google Scholar] [CrossRef]
- Barros, E.; Lezar, S.; Anttonen, M.J.; Van Dijk, J.P.; Röhlig, R.M.; Kok, E.; Engel, K.-H. Comparison of two GM maize varieties with a near-isogenic non-GM variety using transcriptomics, proteomics and metabolomics. Plant Biotechnol. J. 2010, 8, 436–451. [Google Scholar] [CrossRef]
- Lamari, N.; Zhendre, V.; Urrutia, M.; Bernillon, S.; Maucourt, M.; Deborde, C.; Prodhomme, D.; Jacob, D.; Ballias, P.; Rolin, D.; et al. Metabotyping of 30 maize hybrids under early-sowing conditions reveals potential marker-metabolites for breeding. Metabolomics 2018, 14, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Xu, G.; Cao, J.; Wang, X.; Chen, Q.; Jin, W.; Li, Z.; Tian, F. Evolutionary Metabolomics Identifies Substantial Metabolic Divergence between Maize and Its Wild Ancestor, Teosinte. Plant Cell 2019, 31, 1990–2009. [Google Scholar] [CrossRef] [Green Version]
- Jin, M.; Zhang, X.; Zhao, M.; Deng, M.; Du, Y.; Zhou, Y.; Wang, S.; Tohge, T.; Fernie, A.R.; Willmitzer, L.; et al. Integrated genomics-based mapping reveals the genetics underlying maize flavonoid biosynthesis. BMC Plant Biol. 2017, 17, 17. [Google Scholar] [CrossRef] [Green Version]
- Langridge, P.; Fleury, D. Making the most of ’omics’ for crop breeding. Trends Biotechnol. 2011, 29, 33. [Google Scholar] [CrossRef]
- Jayawardena, R.S.; Hyde, K.D.; de Farias, A.R.G.; Bhunjun, C.S.; Ferdinandez, H.S.; Manamgoda, D.S.; Udayanga, D.; Herath, I.S.; Thambugala, K.M.; Manawasinghe, I.S.; et al. What is a species in fungal plant pathogens? Fungal Divers. 2021, 109, 239–266. [Google Scholar] [CrossRef]
- Gougherty, A.V.; Davies, T.J. Towards a phylogenetic ecology of plant pests and pathogens. Philos Trans. R. Soc. Lond B Biol. Sci. 2021, 376, 20200359. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Jiang, T.; Li, Z.; Li, X.; Fan, Z.; Zhou, T. Sugarcane mosaic virus remodels multiple intracellular organelles to form genomic RNA replication sites. Arch. Virol. 2021, 166, 1921–1930. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Chen, Y.; Luo, X.; Du, Z.; Hao, K.; An, M.; Xia, Z.; Wu, Y. Recombinase Polymerase Amplification Assay for Simultaneous Detection of Maize Chlorotic Mottle Virus and Sugarcane Mosaic Virus in Maize. ACS Omega 2021, 6, 18008–18013. [Google Scholar] [CrossRef] [PubMed]
- Taş, N.; de Jong, A.E.; Li, Y.; Trubl, G.; Xue, Y.; Dove, N.C. Metagenomic tools in microbial ecology research. Curr. Opin. Biotechnol. 2021, 67, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Wuolikainen, A.; Jonsson, P.; Ahnlund, M.; Antti, H.; Marklund, S.L.; Moritz, T.; Forsgren, L.; Andersen, P.M.; Trupp, M. Multi-platform mass spectrometry analysis of the CSF and plasma metabolomes of rigorously matched amyotrophic lateral sclerosis, Parkinson’s disease and control subjects. Mol. BioSyst. 2016, 12, 1287–1298. [Google Scholar] [CrossRef]
- van Dam, N.M.; Bouwmeester, H.J. Metabolomics in the Rhizosphere: Tapping into Belowground Chemical Communication. Trends Plant Sci. 2016, 21, 256. [Google Scholar] [CrossRef]
- Fernie, A.R. Review: Metabolome characterisation in plant system analysis. Funct. Plant Biol. 2003, 30, 111–120. [Google Scholar] [CrossRef]
- Sakakibara, K.Y.; Saito, K. Review: Genetically modified plants for the promotion of human health. Biotechnol. Lett. 2006, 28, 1983–1991. [Google Scholar] [CrossRef]
- Kusano, M.; Saito, K. Role of Metabolomics in Crop Improvement. J. Plant Biochem. Biotechnol. 2012, 21, 24–31. [Google Scholar] [CrossRef]
- Butelli, E.; Titta, L.; Giorgio, M.; Mock, H.-P.; Matros, A.; Peterek, S.; Schijlen, E.G.W.M.; Hall, R.D.; Bovy, A.G.; Luo, J.; et al. Enrichment of tomato fruit with health-promoting anthocyanins by expression of select transcription factors. Nat. Biotechnol. 2008, 26, 1301–1308. [Google Scholar] [CrossRef]
- Oikawa, A.; Matsuda, F.; Kusano, M.; Okazaki, Y.; Saito, K. Rice Metabolomics. Rice 2008, 1, 63–71. [Google Scholar] [CrossRef] [Green Version]
- Fernie, A.R.; Schauer, N. Metabolomics-assisted breeding: A viable option for crop improvement? Trends Genet. 2009, 25, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Simó, C.; Ibáez, C.; Valdes, A.; Cifuentes, A.; García-Cañas, V.; Ibáñez, C. Metabolomics of Genetically Modified Crops. Int. J. Mol. Sci. 2014, 15, 18941–18966. [Google Scholar] [CrossRef] [Green Version]
- Rao, M.J.; Wang, L. CRISPR/Cas9 technology for improving agronomic traits and future prospective in agriculture. Planta 2021, 254, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Lobell, D.B.; Schlenker, W.; Costa-Roberts, J. Climate Trends and Global Crop Production Since. Science 2011, 333, 616–620. [Google Scholar] [CrossRef] [Green Version]
- Ortiz-Bobea, A.; Knippenberg, E.; Chambers, R.G. Growing climatic sensitivity of U.S. agriculture linked to technological change and regional specialization. Sci. Adv. 2018, 4, eaat4343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderegg, W.R.L.; Trugman, A.T.; Badgley, G.; Anderson, C.M.; Bartuska, A.; Ciais, P.; Cullenward, D.; Field, C.B.; Freeman, J.; Goetz, S.J.; et al. Climate-driven risks to the climate mitigation potential of forests. Science 2020, 368, 6497. [Google Scholar] [CrossRef]
- Allen, C.D.; Breshears, D.D.; McDowell, N.G. On underestimation of global vulnerability to tree mortality and forest die-off from hotter drought in the Anthropocene. Ecosphere 2015, 6, art129. [Google Scholar] [CrossRef]
- Seidl, R.; Thom, D.; Kautz, M.; Martin-Benito, D.; Peltoniemi, M.; Vacchiano, G.; Wild, J.; Ascoli, D.; Petr, M.; Honkaniemi, J.; et al. Forest disturbances under climate change. Nat. Clim. Chang. 2017, 7, 395–402. [Google Scholar] [CrossRef] [Green Version]
- Klein, T.; Hartmann, H. Climate change drives tree mortality. Science 2018, 362, 758. [Google Scholar] [CrossRef]
- Zhao, H.; Zhao, S.; Fei, B.; Liu, H.; Yang, H.; Dai, H.; Wang, D.; Jin, W.; Tang, F.; Gao, Q.; et al. Announcing the Genome Atlas of Bamboo and Rattan (GABR) project: Promoting research in evolution and in economically and ecologically beneficial plants. GigaScience 2017, 6, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canavan, S.; Richardson, D.; Visser, V.; Le Roux, J.; Vorontsova, M.S.; Wilson, J. The global distribution of bamboos: Assessing correlates of introduction and invasion. AoB Plants 2016, 9, 078. [Google Scholar] [CrossRef] [Green Version]
- Hou, D.; Zhao, Z.; Hu, Q.; Li, L.; Vasupalli, N.; Zhuo, J.; Zeng, W.; Wu, A.; Lin, X. PeSNAC-1 a NAC transcription factor from moso bamboo (Phyllostachys edulis) confers tolerance to salinity and drought stress in transgenic rice. Tree Physiol. 2020, 40, 1792–1806. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.-H.; Ma, P.-F.; Yang, G.-Q.; Hu, J.-Y.; Liu, Y.-L.; Xia, E.-H.; Zhong, M.-C.; Zhao, L.; Sun, G.-L.; Xu, Y.-X.; et al. Genome Sequences Provide Insights into the Reticulate Origin and Unique Traits of Woody Bamboos. Mol. Plant 2019, 12, 1353–1365. [Google Scholar] [CrossRef]
- Cui, K.; He, C.-Y.; Zhang, J.-G.; Duan, A.-G.; Zeng, Y.-F. Temporal and Spatial Profiling of Internode Elongation-Associated Protein Expression in Rapidly Growing Culms of Bamboo. J. Proteome Res. 2012, 11, 2492–2507. [Google Scholar] [CrossRef]
- Zhao, Z.; Hou, D.; Hu, Q.; Wei, H.; Zheng, Y.; Lin, X.J.J.A.B. Cloning and expression analysis of PeNAC047 gene from Phyllostachys edulis. Int. J. Agric. Biotechnol. 2020, 28, 58–71. [Google Scholar]
- Zhao, H.; Gao, Z.; Wang, L.; Wang, J.; Wang, S.; Fei, B.; Chen, C.; Shi, C.; Liu, X.; Zhang, H.; et al. Chromosome-level reference genome and alternative splicing atlas of moso bamboo (Phyllostachys edulis). GigaScience 2018, 7, 115. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Lu, Y.; Li, L.; Zhao, Q.; Feng, Q.; Gao, Z.; Lu, H.; Hu, T.; Yao, N.; Liu, K.; et al. The draft genome of the fast-growing non-timber forest species moso bamboo (Phyllostachys heterocycla). Nat. Genet. 2013, 45, 456–461. [Google Scholar] [CrossRef] [Green Version]
- Hou, D.; Lu, H.; Zhao, Z.; Pei, J.; Yang, H.; Wu, A.; Yu, X.; Lin, X. Integrative transcriptomic and metabolomic data provide insights into gene networks associated with lignification in postharvest Lei bamboo shoots under low temperature. Food Chem. 2021, 368, 130822. [Google Scholar] [CrossRef]
- Huang, R.; Gao, H.; Liu, J.; Li, X. WRKY transcription factors in moso bamboo that are responsive to abiotic stresses. J. Plant Biochem. Biotechnol. 2021, 1–8. [Google Scholar] [CrossRef]
- Liu, J.; Cheng, Z.; Li, X.; Xie, L.; Bai, Y.; Peng, L.; Li, J.; Gao, J. Expression Analysis and Regulation Network Identification of the CONSTANS-Like Gene Family in Moso Bamboo (Phyllostachys edulis) Under Photoperiod Treatments. DNA Cell Biol. 2019, 38, 607–626. [Google Scholar] [CrossRef]
- Li, X.; Chang, Y.; Ma, S.; Shen, J.; Hu, H.; Xiong, L. Genome-Wide Identification of SNAC1-Targeted Genes Involved in Drought Response in Rice. Front. Plant Sci. 2019, 10, 982. [Google Scholar] [CrossRef] [Green Version]
- Huang, B.; Huang, Z.; Ma, R.; Chen, J.; Zhang, Z.; Yrjälä, K. Genome-wide identification and analysis of the heat shock transcription factor family in moso bamboo (Phyllostachys edulis). Sci. Rep. 2021, 11, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Zhang, Y.; Zhang, Q.; Wu, R.; Wang, X.; Feng, S.; Chen, S.; Lu, C.; Du, L. Genome-Wide Identification and Characterization of Hexokinase Genes in Moso Bamboo (Phyllostachys edulis). Front. Plant Sci. 2020, 11, 600. [Google Scholar] [CrossRef] [PubMed]
- Shan, X.; Yang, K.; Xu, X.; Zhu, C.; Gao, Z. Genome-Wide Investigation of the NAC Gene Family and Its Potential Association with the Secondary Cell Wall in Moso Bamboo. Biomolecules 2019, 9, 609. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Chen, Z.; Wu, M.; Wang, Y.; Wang, Y.; Yan, H.; Xiang, Y. Genome-Wide Identification and Expression Analysis of the HD-Zip Gene Family in Moso Bamboo (Phyllostachys edulis). J. Plant Growth Regul. 2017, 36, 323–337. [Google Scholar] [CrossRef]
- Sun, H.; Li, L.; Lou, Y.; Zhao, H.; Gao, Z. Genome-wide identification and characterization of aquaporin gene family in moso bamboo (Phyllostachys edulis). Mol. Biol. Rep. 2016, 43, 437–450. [Google Scholar] [CrossRef]
- Wu, R.; Shi, Y.; Zhang, Q.; Zheng, W.; Chen, S.; Du, L.; Lu, C. Genome-Wide Identification and Characterization of the UBP Gene Family in Moso Bamboo (Phyllostachys edulis). Int. J. Mol. Sci. 2019, 20, 4309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Liu, H.; Wu, L.; Xiong, R.; Shi, Y.; Xiang, Y. Systematic Identification and Analysis of NAC Gene Family in Moso Bamboo (Phyllostachys edulis); Research Square: Durham, NC, USA, 2020. [Google Scholar]
- Liu, H.; Wu, M.; Zhu, D.; Pan, F.; Wang, Y.; Wang, Y.; Xiang, Y. Genome-Wide analysis of the AAAP gene family in moso bamboo (Phyllostachys edulis). BMC Plant Biol. 2017, 17, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Ma, R.; Huang, B.; Chen, J.; Huang, Z.; Yu, P.; Ruan, S.; Zhang, Z. Genome-wide identification and expression analysis of dirigent-jacalin genes from plant chimeric lectins in Moso bamboo (Phyllostachys edulis). PLoS ONE 2021, 16, e0248318. [Google Scholar] [CrossRef]
- Gao, Y.; Liu, H.; Zhang, K.; Li, F.; Wu, M.; Xiang, Y. A moso bamboo transcription factor, Phehdz1, positively regulates the drought stress response of transgenic rice. Plant Cell Rep. 2021, 40, 187–204. [Google Scholar] [CrossRef]
- Lan, Y.; Wu, L.; Wu, M.; Liu, H.; Gao, Y.; Zhang, K.; Xiang, Y. Transcriptome analysis reveals key genes regulating signaling and metabolic pathways during the growth of moso bamboo (Phyllostachys edulis) shoots. Physiol. Plant. 2021, 172, 91–105. [Google Scholar] [CrossRef]
- Li, L.; Sun, H.; Lou, Y.; Yang, Y.; Zhao, H.; Gao, Z. Cloning and expression analysis of PeLAC in Phyllostachys edulis. Plant Sci. J. 2017, 35, 252–259. [Google Scholar]
- Sun, H.; Lou, Y.; Li, L.; Zhao, H.; Gao, Z. Tissue expression pattern analysis of TIPs genes in Phyllostachys edulis. For. Res. 2016, 29, 521–528. [Google Scholar]
- Wu, M.; Zhang, K.; Xu, Y.; Wang, L.; Liu, H.; Qin, Z.; Xiang, Y. The moso bamboo WRKY transcription factor, PheWRKY86, regulates drought tolerance in transgenic plants. Plant Physiol. Biochem. 2021, 170, 180. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Ding, Y. Ultracytochemical localization of Ca2+ during the phloem ganglion development in Phyllostachys edulis. Front. Biol. China 2006, 1, 219–224. [Google Scholar] [CrossRef]
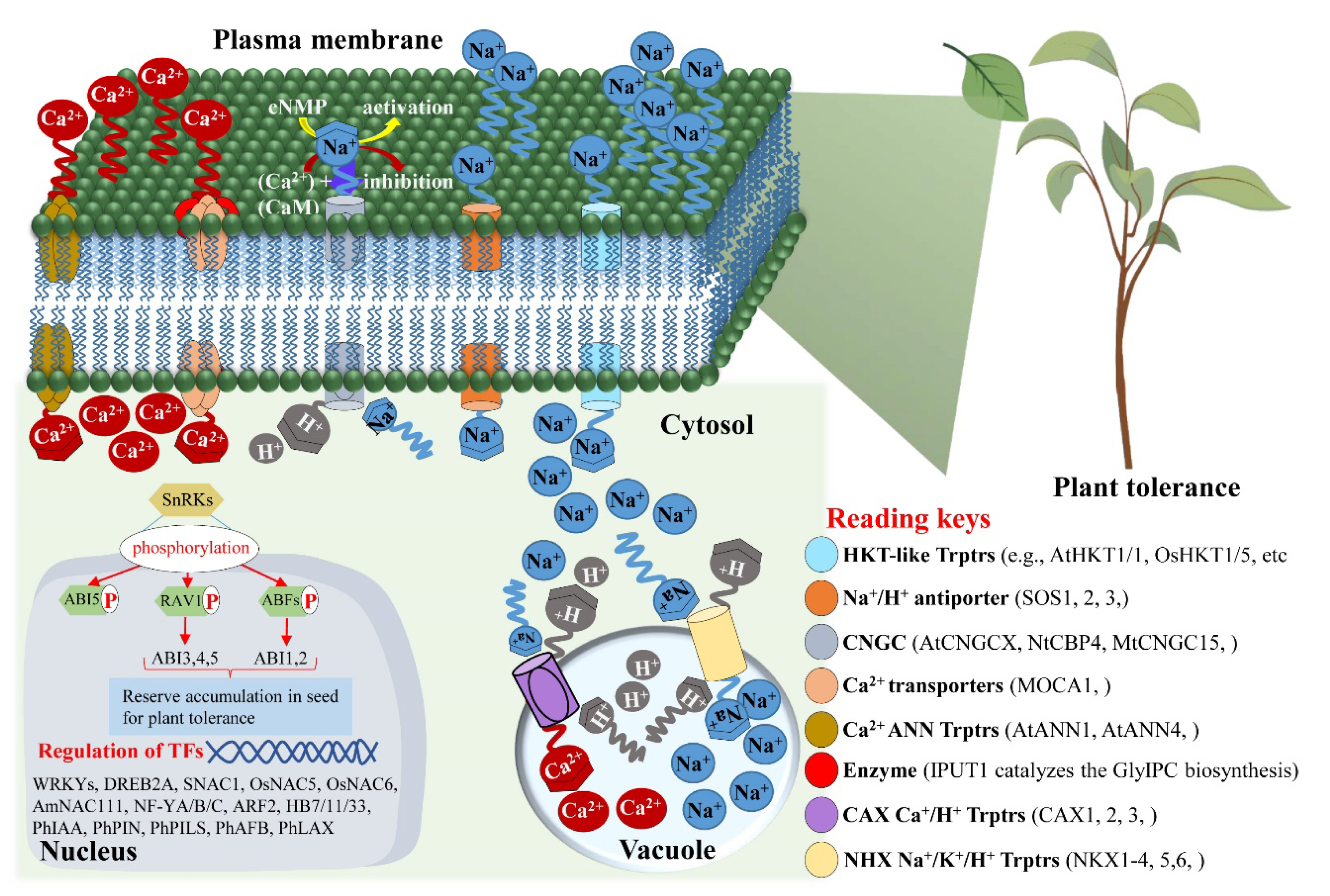
- Cushman, K.R.; Pabuayon, I.C.M.; Hinze, L.L.; Sweeney, M.E.; Reyes, B.G.D.L. Networks of Physiological Adjustments and Defenses, and Their Synergy With Sodium (Na+) Homeostasis Explain the Hidden Variation for Salinity Tolerance Across the Cultivated Gossypium hirsutum Germplasm. Front. Plant Sci. 2020, 11, 588854. [Google Scholar] [CrossRef]
- Afzal, M.Z.; Jia, Q.; Ibrahim, A.K.; Niyitanga, S.; Zhang, L. Mechanisms and Signaling Pathways of Salt Tolerance in Crops: Understanding from the Transgenic Plants. Trop. Plant Biol. 2020, 13, 297–320. [Google Scholar] [CrossRef]
- Wang, W.; Gu, L.; Ye, S.; Zhang, H.; Cai, C.; Xiang, M.; Gao, Y.; Wang, Q.; Lin, C.; Zhu, Q. Genome-wide analysis and transcriptomic profiling of the auxin biosynthesis, transport and signaling family genes in moso bamboo (Phyllostachys heterocycla). BMC Genom. 2017, 18, 870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Sun, H.Y.; Li, L.C.; Yang, Y.H.; Xu, H.; Zhao, H.; Gao, Z. Cloning and expression pattern analysis of PeDWF4 Gene in Moso bamboo (Phyllostachys edulis). For. Res. 2018, 31, 50–56. [Google Scholar] [CrossRef]
- Zhang, Z.; Yang, X.; Cheng, L.; Guo, Z.; Wang, H.; Wu, W.; Shin, K.; Zhu, J.; Zheng, X.; Bian, J.; et al. Physiological and transcriptomic analyses of brassinosteroid function in moso bamboo (Phyllostachys edulis) seedlings. Planta 2020, 252, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Sarkanen, K. Formation, structure; reactions. Classification and distribution. J. Polym. Sci. Part B Polym. Lett. 1971, 43–94. [Google Scholar]
- Shimada, N.; Munekata, N.; Tsuyama, T.; Matsushita, Y.; Fukushima, K.; Kijidani, Y.; Takabe, K.; Yazaki, K.; Kamei, I. Active Transport of Lignin Precursors into Membrane Vesicles from Lignifying Tissues of Bamboo. Plants 2021, 10, 2237. [Google Scholar] [CrossRef]
- Ishihara, A.; Hashimoto, Y.; Miyagawa, H.; Wakasa, K. Induction of serotonin accumulation by feeding of rice striped stem borer in rice leaves. Plant Signal. Behav. 2008, 3, 714–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishihara, A.; Hashimoto, Y.; Tanaka, C.; Dubouzet, J.G.; Nakao, T.; Matsuda, F.; Nishioka, T.; Miyagawa, H.; Wakasa, K. The tryptophan pathway is involved in the defense responses of rice against pathogenic infection via serotonin production. Plant J. 2008, 54, 481–495. [Google Scholar] [CrossRef] [PubMed]
- Riley, R.G.; Kolattukudy, P.E. Evidence for Covalently Attached p-Coumaric Acid and Ferulic Acid in Cutins and Suberins. Plant Physiol. 1975, 56, 650–654. [Google Scholar] [CrossRef] [Green Version]
- Bernards, M.A.; Lopez, M.L.; Zajicek, J.; Lewis, N. Hydroxycinnamic Acid-derived Polymers Constitute the Polyaromatic Domain of Suberin. J. Biol. Chem. 1995, 270, 7382–7386. [Google Scholar] [CrossRef] [Green Version]
- Rozema, J.; Broekman, R.; Blokker, P.; Meijkamp, B.B.; de Bakker, N.; van de Staaij, J.; van Beem, A.; Ariese, F.; Kars, S.M. UV-B absorbance and UV-B absorbing compounds (para-coumaric acid) in pollen and sporopollenin: The perspective to track historic UV-B levels. J. Photochem. Photobiol. B Biol. 2001, 62, 108–117. [Google Scholar] [CrossRef]
- Liu, C.-J. Biosynthesis of hydroxycinnamate conjugates: Implications for sustainable biomass and biofuel production. Biofuels 2010, 1, 745–761. [Google Scholar] [CrossRef]
- Turnbull, C.; Lillemo, M.; Hvoslef-Eide, T.A.K. Global Regulation of Genetically Modified Crops Amid the Gene Edited Crop Boom–A Review. Front. Plant Sci. 2021, 12, 630396. [Google Scholar] [CrossRef]
- Ye, S.; Chen, G.; Kohnen, M.V.; Wang, W.; Cai, C.; Ding, W.; Wu, C.; Gu, L.; Zheng, Y.; Ma, X.; et al. Robust CRISPR/Cas9 mediated genome editing and its application in manipulating plant height in the first generation of hexaploid Ma bamboo (Dendrocalamus latiflorus Munro). Plant Biotechnol. J. 2019, 18, 1501–1503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, J.-L.; Yue, J.-J.; Wu, X.-L.; Gu, X.-P. Protocol for Callus Induction and Somatic Embryogenesis in Moso Bamboo. PLoS ONE 2013, 8, e81954. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family Name | Organism | GS | NPTs | Online Accessible Links |
|---|---|---|---|---|
| Amaranthaceae | Beta vulgaris (sugar beet), spp. vulgaris var. cicla) | 604 Mbp | 34,521 | https://bvseq.boku.ac.at/ |
| Suaeda aralocaspica (shrubby sea-blite) | 467 Mbp | 29,604 | https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA428881 | |
| Arecaceae | Elaeis guineensis (African oil palm) | 1800 Mbp | 25,405 | https://www.ncbi.nlm.nih.gov/genome/?term=txid51953[orgn] |
| Phoenis dactylifera (date palm), an elite variety (Khalas) | 605.4 Mbp | ~41,660 | https://pubmed.ncbi.nlm.nih.gov/23917264/ | |
| Brassicaceae | Arabidopsis thaliana (Arabidopsis) | 125 Mbp | ~27,025 | https://www.arabidopsis.org/ and https://www.nature.com/articles/ng.807 |
| Arabidopsis lyrata (Arabidopsis) | 207 Mbp | ~32,670 | https://www.arabidopsis.org/ and https://www.nature.com/articles/ng.807 | |
| Capsella rubella (pink shepherd’s-purse) | 134.8 Mbp | ~28,447 | https://www.nature.com/articles/ng.2669 | |
| Eruca sativa (salad rocket) | ∼851 Mbp | 45,438 | https://www.frontiersin.org/articles/10.3389/fpls.2020.525102/full | |
| Eutrema salsugineum (saltwater cress) | 241 Mbp | 26,531 | https://www.frontiersin.org/articles/10.3389/fpls.2013.00046/full | |
| Cannabaceae | Cannabis sativa (hemp) | 808 Mbp | 38,828 | https://www.nature.com/articles/s41438-020-0295-3 |
| Cactaceae | Carnegiea gigantea (saguaro) | 1.40 GB | 28,292 | https://www.pnas.org/content/114/45/12003 |
| Cucurbitaceae | Cucumis melo (musk melon), doubled-haploid line DHL92 | 375 Mbp | 27,427 | https://www.pnas.org/content/109/29/11872#abstract-1 |
| Cucumis sativus (cucumber), ‘Chinese long’ inbred line 9930 | 226.2 Mbp | 26,682 | https://academic.oup.com/gigascience/article/8/6/giz072/5520540 | |
| Dioscoreaceae | Dioscorea rotundata (Yam) | 594 Mbp | 26,198 | https://bmcbiol.biomedcentral.com/articles/10.1186/s12915-017-0419-x |
| Euphorbiaceae | Manihot esculenta (cassava), domesticated KU50 | 495 Mbp | 37,592 | https://www.nature.com/articles/ncomms6110#Sec8 |
| Fabaceae | Cajanus cajan (pigeon pea) | 833.07 Mbp | 48,680 | https://www.nature.com/articles/nbt.2022 |
| Cicer arietinum (chickpea) | ∼738 Mbp | 28,269 | https://www.nature.com/articles/nbt.2491 | |
| Glycine max (soybean), cultivar Williams 82 | 969.6 Mbp | 46,430 | https://www.nature.com/articles/nature08670#Sec9 | |
| Medicago turncatula (medick or burclover) | ~330 Mbp | 50,894 | http://europepmc.org/article/MED/24767513 | |
| Vigna unguiculata (cowpea) | 640.6 Mbp | 29,773 | https://onlinelibrary.wiley.com/doi/full/10.1111/tpj.14349 | |
| Ginkgoaceae | Ginkgo biloba (ginkgo) | 10.61 Gb | 41,840 | https://gigascience.biomedcentral.com/articles/10.1186/s13742-016-0154-1 |
| Musaceae | Musa acuminata (Banana) spp. Malaccensis | 523 Mbp | 36,542 | https://www.nature.com/articles/nature11241 |
| Pinaceae | Picea abies (Norway spruce) | 20 GB | 28,354 | https://www.nature.com/articles/nature12211 |
| Poaceae | Hordeum vulgare (barley) | 5.1 GB | 26,159 | https://www.nature.com/articles/nature11543 |
| Oryza sativa (rice) | 373.2 Mbp | 3475 | https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5395016/ | |
| Phyllostachys heterocycla var. pubescens | 2.05 Gb | 31,987 | https://www.nature.com/articles/ng.2569 | |
| Phyllostachys edulis | 1.91 GB | 51,074 | https://academic.oup.com/gigascience/article/7/10/giy115/5092772 | |
| Raddia distichophylla (Schrad. ex Nees) Chase | 589 Mbp | 30,763 | https://academic.oup.com/g3journal/article/11/2/jkaa049/6066164 | |
| Sorghum bicolor (sorghum), Rio genetic material | 729.4 Mbp | 35,467 | https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-019-5734-x#Sec6 | |
| Triticum urartu (einkorn wheat), accession G1812 (PI428198) | ~4.94 GB | 34,879 | https://www.nature.com/articles/nature11997 | |
| Zea mays (maize), B73 inbred maize line | 2.3 GB | >32,000 | https://pubmed.ncbi.nlm.nih.gov/19965430/ | |
| Salicaceae | Populus tricchocarpa (poplar) | 380 Mbp | 37,238 | https://www.pnas.org/content/115/46/E10970#sec-1 |
| Solanaceae | Capsicum annuum (pepper) | 3.06 GB | 34,903 | https://www.nature.com/articles/ng.2877#Sec10 |
| Nicotiana benthamiana (tobacco) | 3.1 GB | 42,855 | https://www.biorxiv.org/content/10.1101/373506v2 | |
| Solanum lycopersicum (tomato), cv. Heinz 1706 | 799.09 Mbp | 34,384 | https://www.biorxiv.org/content/10.1101/2021.05.04.441887v1.full.pdf | |
| Solanum tuberosum (potato) | 844 Mbp | 39,031 | https://www.nature.com/articles/nature10158/ | |
| Arecaceae | Elaeis guineensis (African oil palm) | 1.8 GB | ~34,802 | https://www.nature.com/articles/nature12309 |
| Phoenis dactylifera (date palm), an elite variety (Khalas) | 605.4 Mbp | ~41,660 | https://europepmc.org/article/PMC/3741641 | |
| Rosaceae | Prumus persica (peach) | 247.33 Mbp | 26,335 | https://onlinelibrary.wiley.com/doi/10.1111/tpj.15439?af=R |
| Vitaceae | Vitis sylvestris (grape), accession of Sylvestris C1-2 | 469 Mbp | 39,031 | https://genomebiology.biomedcentral.com/articles/10.1186/s13059-020-02131-y#Sec2 |
| Plant | E | Stage and Specific Organ | Metabolites | Refs. |
|---|---|---|---|---|
| Avena sativa (oats) | E1 | Not specified using grains | PM **: malic, gluconic, and galacturonic acids, fatty acids (FAs), palmitic acid and linoleic acid. | [155] |
| E2 | Seedling stage (three weeks old) Leaves | PMS **: Ascorbate, aldarate phenylpropanoids. | [156] | |
| Hordeum vulgare (barley) | E1 | Germination using seeds | PM *: glycero(phospho)lipids, prenol lipids, sterol lipids, methylation. SM *: polyketides. | [157] |
| E2 | Two-leaf stage seedlings using leaves | PM **: organic acids (OAs), amino acids (AAs), nucleotides, and derivatives. SM *: flavonoids, absiscic acid. | [158] | |
| E3 | Three-leaf stage using leaves | SM **: chlorogenic acids, hydrocinnamic acid derivatives, and hordatines and their glycosides. | [119] | |
| E4 | Three-leaf stage and flag leaf stage using leaves | SM *: flavonoids, hydroxycinnamic acid, phenolics, glycosides, esters, and amides. | [159] | |
| E5 | During grain filling using seeds | PM *: Tricarboxylic acid (TCA), OAs, aldehydes, alcohols, polyols, FAs, carbohydrates, mevalonate. SM **: phenolic compounds, flavonoids. | [10] | |
| E6 | Four weeks old using leaves | PM *: carbohydrates, free AAs, carboxylates, phosphorylated intermediates, antioxidants, carotenoids. | [160] | |
| E7 | 1–3 weeks old using leaves and roots | PM **: AAs, sugars, OAs as fumaric acid, malic acid, glyceric acid | [161] | |
| Oryza sativa L. (rice) | E1 | Flowering and early grain filling stages using leaves, spikelets, seeds | PSM **: isoleucine, 3-cyano-alanine, phenylalanine, spermidine, polyamine, ornithine | [161] |
| E2 | At reproductive stage using leaves and grains ripe stages | PM **: saturated and unsaturated FAs, AAs, sugars, and OAs. | [162] | |
| E3 | 24 months old seeds used | PM **: sugar synthesis related compounds, AAs, free FAs, TCA cycle intermediates. | [163] | |
| E4 | Not specified using grain | PM *: aromatic AAs, carbohydrates, cofactors and vitamins, lipids, oxylipins, nucleotides. SM *: benzenoids. | [164] | |
| E5 | Maturation using mature seed | PM *: carbohydrates, lipids, cofactors, prosthetic groups, electron carriers, nucleotides. SM *: benzenoids. | [165] | |
| E6 | Maturation using mature seed | PM *: carbohydrates and lipids. SM *: α-carotene, β-carotene, and lutein. | [166] | |
| E7 | Six weeks old using leaves | PM *: AAs (arginine, ornithine, citrulline, tyrosine, phenylalanine and lysine), FAs and lipids, glutathione, carbohydrates. SM *: rutin, acetophenone, alkaloids. | [157] | |
| Setaria italica (foxtail millet) | E1 | 60 days using shoots | PM *: fructose, glucose, gluconate, formate, threonine, 4-aminobutyrate, 2-hydroxyvalerate, sarcosine, betaine, choline, isovalerate, acetate, pyruvate, TCA-OAs, and uridine. | [167] |
| E2 | 3–5 leaves stages using leaves | PM *: glycerophospholipids, AAs, OAs. SM: flavonoids, hydroxycinnamic acids, phenolamides, and vitamin-related compounds. | [167] | |
| Sorghum bicolor (sorghum) | E1 | Four-leaf stage using leaves | PM *: AAs, carboxylic acids, FAs. SM: cyanogenic glycosides, flavonoids, hydroxycinnamic acids, indoles, benzoates, phytohormones, and shikimates. | [168] |
| E2 | Four-leaf stage using leaves | SM *: 3-Deoxyanthocyanidins, phenolics, flavonoids, phytohormones, luteolinidin, apigeninidin, riboflavin. | [169] | |
| E3 | Around 26 days using roots and leaves | PM *: sugars, sugar alcohols, AAs, and OAs. | [170] | |
| E4 | Four weeks old using grain and biomass | PM **: OAs. SM **: phenylpropanoids. | [146] | |
| Triticum aestivum (wheat) | E1 | NAS using leaves | PM *: sugars, glycolysis and gluconeogenesis intermediates, AAs, nucleic acid precursors, and intermediates. SM *: chorismate, polyamines, L-pipecolate, amino-adipic acid, phenylpropanoids, terpene skeleton, and ubiquinone. | [171] |
| E2 | Physiological maturity using leaves | PM *: AAs metabolism, sugar alcohols, purine metabolism, glycerolipids, and guanine. SM *: shikimates, anthranilate, absiscic acid. | [172] | |
| E3 | Maturation using matured kernels | PM *: FAs, sugar, nucleic acids and derivatives. SM *: phenolamides, flavonoids, polyphenols, vitamins, OAs, AAs, phytohormones, and derivatives. | [173] | |
| E4 | Not specified using grain | PM *: osmolytes, glycine betaine, choline, and asparagine. | [174] | |
| E5 | Not specified using seeds | PM *: sterols, FAs, long chain FAs derivatives, glycerol (phospho) lipids. SM *: polyketides. | [175] | |
| Zea mays (maize) | E1 | R6 stage using grains | PM **: sugars, sucrose, glucose, and fructose. | [176] |
| E2 | Physiological maturity using kernels | PM *: glycolysis, TCA cycle, starch, amino acids. SM: alkaloids, benzenoids, fatty acid and sugar derivatives, flavonoids, phenylpropanoids, and terpenoids. | [177] | |
| E3 | 8 months using kernels | PM *: glucose, fructose, sucrose, tocopherol, phytosterol, inositol, asparagine, glutamic acid, pyroglutamic acid. | [178] | |
| E4 | Eight-visible-leaf stage using leaves | PM *: choline, inositol, sugars, raffinose, rhamnose, TCA cycle, AAs, trigonelline, putrescine, quinate, shikimate. SM *: flavonoids, and benzoxazinoids. | [179] | |
| E5 | Seedling stage using entire seedling | PM *: amino acids, lipids, carboxylic acid. SM *: alkaloids, terpenoids, flavonoids, alkaloids, and benzenoids. | [180] | |
| E6 | Physiological maturity using kernels | SM *: flavanones, flavones, anthocyanins, and methoxylated flavonoids. | [181] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ashraf, M.F.; Hou, D.; Hussain, Q.; Imran, M.; Pei, J.; Ali, M.; Shehzad, A.; Anwar, M.; Noman, A.; Waseem, M.; et al. Entailing the Next-Generation Sequencing and Metabolome for Sustainable Agriculture by Improving Plant Tolerance. Int. J. Mol. Sci. 2022, 23, 651. https://doi.org/10.3390/ijms23020651
Ashraf MF, Hou D, Hussain Q, Imran M, Pei J, Ali M, Shehzad A, Anwar M, Noman A, Waseem M, et al. Entailing the Next-Generation Sequencing and Metabolome for Sustainable Agriculture by Improving Plant Tolerance. International Journal of Molecular Sciences. 2022; 23(2):651. https://doi.org/10.3390/ijms23020651
Chicago/Turabian StyleAshraf, Muhammad Furqan, Dan Hou, Quaid Hussain, Muhammad Imran, Jialong Pei, Mohsin Ali, Aamar Shehzad, Muhammad Anwar, Ali Noman, Muhammad Waseem, and et al. 2022. "Entailing the Next-Generation Sequencing and Metabolome for Sustainable Agriculture by Improving Plant Tolerance" International Journal of Molecular Sciences 23, no. 2: 651. https://doi.org/10.3390/ijms23020651
APA StyleAshraf, M. F., Hou, D., Hussain, Q., Imran, M., Pei, J., Ali, M., Shehzad, A., Anwar, M., Noman, A., Waseem, M., & Lin, X. (2022). Entailing the Next-Generation Sequencing and Metabolome for Sustainable Agriculture by Improving Plant Tolerance. International Journal of Molecular Sciences, 23(2), 651. https://doi.org/10.3390/ijms23020651