
Paving Therapeutic Avenues for FOXG1 Syndrome: Untangling Genotypes and Phenotypes from a Molecular Perspective


{kind=link}
{kind=link}
{kind=link}
Abstract
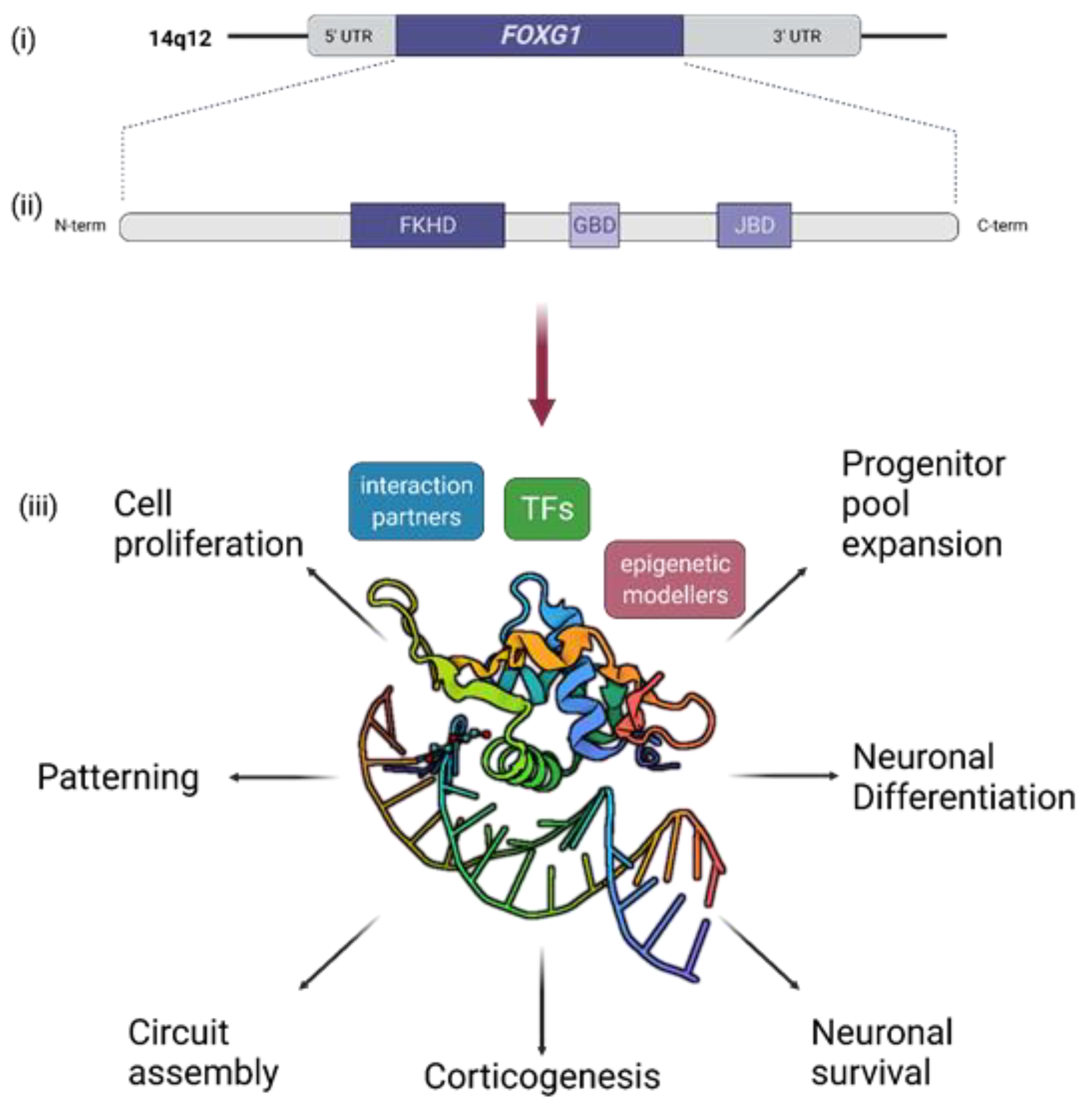
:1. Introduction
2. Clinical Manifestations of FOXG1 Syndrome
3. Short Recapitulation of FOXG1 Functions in Brain Development and Function
4. Human Cell-Derived Models of FOXG1 Syndrome and Function
5. Future of FOXG1 Syndrome Modelling, Therapeutic Prospects, and Limitations
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hou, P.-S.; hAilín, D.Ó.; Vogel, T.; Hanashima, C. Transcription and Beyond: Delineating FOXG1 Function in Cortical Development and Disorders. Front. Cell. Neurosci. 2020, 14, 35. [Google Scholar] [CrossRef] [PubMed]
- Tao, W.; Lai, E. Telencephalon-Restricted Expression of BF-1, a New Member of the HNF-3/Fork Head Gene Family, in the Developing Rat Brain. Neuron 1992, 8, 957–966. [Google Scholar] [CrossRef]
- Chahrour, M.; Zoghbi, H.Y. The Story of Rett Syndrome: From Clinic to Neurobiology. Neuron 2007, 56, 422–437. [Google Scholar] [CrossRef] [Green Version]
- Kortum, F.; Das, S.; Flindt, M.; Morris-Rosendahl, D.J.; Stefanova, I.; Goldstein, A.; Horn, D.; Klopocki, E.; Kluger, G.; Martin, P.; et al. The Core FOXG1 Syndrome Phenotype Consists of Postnatal Microcephaly, Severe Mental Retardation, Absent Language, Dyskinesia, and Corpus Callosum Hypogenesis. J. Med. Genet. 2011, 48, 396–406. [Google Scholar] [CrossRef] [PubMed]
- Mitter, D.; Pringsheim, M.; Kaulisch, M.; Plümacher, K.S.; Schröder, S.; Warthemann, R.; Abou Jamra, R.; Baethmann, M.; Bast, T.; Büttel, H.-M.; et al. FOXG1 Syndrome: Genotype–Phenotype Association in 83 Patients with FOXG1 Variants. Genet. Med. 2018, 20, 98–108. [Google Scholar] [CrossRef] [Green Version]
- Mariani, J.; Coppola, G.; Zhang, P.; Abyzov, A.; Provini, L.; Tomasini, L.; Amenduni, M.; Szekely, A.; Palejev, D.; Wilson, M.; et al. FOXG1-Dependent Dysregulation of GABA/Glutamate Neuron Differentiation in Autism Spectrum Disorders. Cell 2015, 162, 375–390. [Google Scholar] [CrossRef] [Green Version]
- What Is FOXG1 Syndrome? Available online: https://foxg1research.org/foxg1syndrome (accessed on 23 November 2021).
- Ellaway, C.J.; Ho, G.; Bettella, E.; Knapman, A.; Collins, F.; Hackett, A.; McKenzie, F.; Darmanian, A.; Peters, G.B.; Fagan, K.; et al. 14q12 Microdeletions Excluding FOXG1 Give Rise to a Congenital Variant Rett Syndrome-like Phenotype. Eur. J. Hum. Genet. 2013, 21, 522–527. [Google Scholar] [CrossRef] [Green Version]
- Takagi, M.; Sasaki, G.; Mitsui, T.; Honda, M.; Tanaka, Y.; Hasegawa, T. A 2.0 Mb Microdeletion in Proximal Chromosome 14q12, Involving Regulatory Elements of FOXG1, with the Coding Region of FOXG1 Being Unaffected, Results in Severe Developmental Delay, Microcephaly, and Hypoplasia of the Corpus Callosum. Eur. J. Med. Genet. 2013, 56, 526–528. [Google Scholar] [CrossRef]
- Brunetti-Pierri, N.; Paciorkowski, A.R.; Ciccone, R.; Mina, E.D.; Bonaglia, M.C.; Borgatti, R.; Schaaf, C.P.; Sutton, V.R.; Xia, Z.; Jelluma, N.; et al. Duplications of FOXG1 in 14q12 Are Associated with Developmental Epilepsy, Mental Retardation, and Severe Speech Impairment. Eur. J. Hum. Genet. EJHG 2011, 19, 102–107. [Google Scholar] [CrossRef] [Green Version]
- Vegas, N.; Cavallin, M.; Maillard, C.; Boddaert, N.; Toulouse, J.; Schaefer, E.; Lerman-Sagie, T.; Lev, D.; Magalie, B.; Moutton, S.; et al. Delineating FOXG1 Syndrome: From Congenital Microcephaly to Hyperkinetic Encephalopathy. Neurol. Genet. 2018, 4, e281. [Google Scholar] [CrossRef] [Green Version]
- Wong, L.-C.; Wu, Y.-T.; Hsu, C.-J.; Weng, W.-C.; Tsai, W.-C.; Lee, W.-T. Cognition and Evolution of Movement Disorders of FOXG1-Related Syndrome. Front. Neurol. 2019, 10, 641. [Google Scholar] [CrossRef]
- Shoichet, S.A.; Kunde, S.-A.; Viertel, P.; Schell-Apacik, C.; von Voss, H.; Tommerup, N.; Ropers, H.-H.; Kalscheuer, V.M. Haploinsufficiency of Novel FOXG1B Variants in a Patient with Severe Mental Retardation, Brain Malformations and Microcephaly. Hum. Genet. 2005, 117, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Ariani, F.; Hayek, G.; Rondinella, D.; Artuso, R.; Mencarelli, M.A.; Spanhol-Rosseto, A.; Pollazzon, M.; Buoni, S.; Spiga, O.; Ricciardi, S.; et al. FOXG1 Is Responsible for the Congenital Variant of Rett Syndrome. Am. J. Hum. Genet. 2008, 83, 89–93. [Google Scholar] [CrossRef] [Green Version]
- Byun, C.K.; Lee, J.S.; Lim, B.C.; Kim, K.J.; Hwang, Y.S.; Chae, J.-H. FOXG1 Mutation Is a Low-Incidence Genetic Cause in Atypical Rett Syndrome. Child Neurol. Open 2015, 2, 2329048X14568151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bredenkamp, N.; Seoighe, C.; Illing, N. Comparative Evolutionary Analysis of the FoxG1 Transcription Factor from Diverse Vertebrates Identifies Conserved Recognition Sites for MicroRNA Regulation. Dev. Genes Evol. 2007, 217, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Wiese, S.; Murphy, D.B.; Schlung, A.; Burfeind, P.; Schmundt, D.; Schnülle, V.; Mattei, M.G.; Thies, U. The Genes for Human Brain Factor 1 and 2, Members of the Fork Head Gene Family, Are Clustered on Chromosome 14q. Biochim. Biophys Acta 1995, 1262, 105–112. [Google Scholar] [CrossRef]
- Hettige, N.C.; Ernst, C. FOXG1 Dose in Brain Development. Front. Pediatr. 2019, 7, 482. [Google Scholar] [CrossRef] [Green Version]
- Kumamoto, T.; Hanashima, C. Evolutionary Conservation and Conversion of Foxg1 Function in Brain Development. Dev. Growth Differ. 2017, 59, 258–269. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Dai, S.; Chen, X.; Liang, X.; Qu, L.; Jiang, L.; Guo, M.; Zhou, Z.; Wei, H.; Zhang, H.; et al. Mechanism of Forkhead Transcription Factors Binding to a Novel Palindromic DNA Site. Nucleic Acids Res. 2021, 49, 3573–3583. [Google Scholar] [CrossRef]
- Dai, S.; Qu, L.; Li, J.; Chen, Y. Toward a Mechanistic Understanding of DNA Binding by Forkhead Transcription Factors and Its Perturbation by Pathogenic Mutations. Nucleic Acids Res. 2021, 49, 10235–10249. [Google Scholar] [CrossRef]
- Dai, S.; Li, J.; Zhang, H.; Chen, X.; Guo, M.; Chen, Z.; Chen, Y. Structural Basis for DNA Recognition by FOXG1 and the Characterization of Disease-Causing FOXG1 Mutations. J. Mol. Biol. 2020, 432, 6146–6156. [Google Scholar] [CrossRef]
- Caporali, C.; Signorini, S.; De Giorgis, V.; Pichiecchio, A.; Zuffardi, O.; Orcesi, S. Early-Onset Movement Disorder as Diagnostic Marker in Genetic Syndromes: Three Cases of FOXG1-Related Syndrome. Eur. J. Paediatr. Neurol. 2018, 22, 336–339. [Google Scholar] [CrossRef]
- Christodoulou, J.; Grimm, A.; Maher, T.; Bennetts, B. RettBASE: The IRSA MECP2 Variation Database-a New Mutation Database in Evolution. Hum. Mutat. 2003, 21, 466–472. [Google Scholar] [CrossRef]
- Krishnaraj, R.; Ho, G.; Christodoulou, J. RettBASE: Rett Syndrome Database Update. Hum. Mutat. 2017, 38, 922–931. [Google Scholar] [CrossRef]
- Seltzer, L.E.; Ma, M.; Ahmed, S.; Bertrand, M.; Dobyns, W.B.; Wheless, J.; Paciorkowski, A.R. Epilepsy and Outcome in FOXG1-Related Disorders. Epilepsia 2014, 55, 1292–1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bębenek, A.; Ziuzia-Graczyk, I. Fidelity of DNA Replication—A Matter of Proofreading. Curr. Genet. 2018, 64, 985–996. [Google Scholar] [CrossRef] [Green Version]
- Mehrjouy, M.M.; Fonseca, A.C.S.; Ehmke, N.; Paskulin, G.; Novelli, A.; Benedicenti, F.; Mencarelli, M.A.; Renieri, A.; Busa, T.; Missirian, C.; et al. Regulatory Variants of FOXG1 in the Context of Its Topological Domain Organisation. Eur. J. Hum. Genet. 2018, 26, 186–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papandreou, A.; Schneider, R.B.; Augustine, E.F.; Ng, J.; Mankad, K.; Meyer, E.; McTague, A.; Ngoh, A.; Hemingway, C.; Robinson, R.; et al. Delineation of the Movement Disorders Associated with FOXG1 Mutations. Neurology 2016, 86, 1794–1800. [Google Scholar] [CrossRef] [Green Version]
- Vignoli, A.; Savini, M.N.; Nowbut, M.S.; Peron, A.; Turner, K.; La Briola, F.; Canevini, M.P. Effectiveness and Tolerability of Antiepileptic Drugs in 104 Girls with Rett Syndrome. Epilepsy Behav. 2017, 66, 27–33. [Google Scholar] [CrossRef]
- FOXG1 Research Projects. Available online: https://foxg1research.org/foxg1-research-projects (accessed on 22 December 2021).
- Xuan, S.; Baptista, C.A.; Balas, G.; Tao, W.; Soares, V.C.; Lai, E. Winged Helix Transcription Factor BF-1 Is Essential for the Development of the Cerebral Hemispheres. Neuron 1995, 14, 1141–1152. [Google Scholar] [CrossRef] [Green Version]
- Hanashima, C.; Shen, L.; Li, S.C.; Lai, E. Brain Factor-1 Controls the Proliferation and Differentiation of Neocortical Progenitor Cells through Independent Mechanisms. J. Neurosci. 2002, 22, 6526–6536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanashima, C.; Fernandes, M.; Hebert, J.M.; Fishell, G. The Role of Foxg1 and Dorsal Midline Signaling in the Generation of Cajal-Retzius Subtypes. J. Neurosci. 2007, 27, 11103–11111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyoshi, G.; Fishell, G. Dynamic FoxG1 Expression Coordinates the Integration of Multipolar Pyramidal Neuron Precursors into the Cortical Plate. Neuron 2012, 74, 1045–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanashima, C. Foxg1 Suppresses Early Cortical Cell Fate. Science 2004, 303, 56–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumamoto, T.; Toma, K.; Gunadi; McKenna, W.; Kasukawa, T.; Katzman, S.; Chen, B.; Hanashima, C. Foxg1 Coordinates the Switch from Non-Radially to Radially Migrating Glutamatergic Subtypes in the Neocortex through Spatiotemporal Repression. Cell Rep. 2013, 3, 931–945. [Google Scholar] [CrossRef] [Green Version]
- Hou, P.-S.; Miyoshi, G.; Hanashima, C. Sensory Cortex Wiring Requires Preselection of Short- and Long-Range Projection Neurons through an Egr-Foxg1-COUP-TFI Network. Nat. Commun. 2019, 10, 3581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cargnin, F.; Kwon, J.-S.; Katzman, S.; Chen, B.; Lee, J.W.; Lee, S.-K. FOXG1 Orchestrates Neocortical Organization and Cortico-Cortical Connections. Neuron 2018, 100, 1083–1096.e5. [Google Scholar] [CrossRef] [Green Version]
- Hébert, J.M.; McConnell, S.K. Targeting of Cre to the Foxg1 (BF-1) Locus Mediates LoxP Recombination in the Telencephalon and Other Developing Head Structures. Dev. Biol. 2000, 222, 296–306. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.; Nam, H.-S.; Song, P.; Moore, H.; Anderson, S.A. FoxG1 Haploinsufficiency Results in Impaired Neurogenesis in the Postnatal Hippocampus and Contextual Memory Deficits. Hippocampus 2006, 16, 875–890. [Google Scholar] [CrossRef]
- Eagleson, K.L.; Schlueter McFadyen-Ketchum, L.J.; Ahrens, E.T.; Mills, P.H.; Does, M.D.; Nickols, J.; Levitt, P. Disruption of Foxg1 Expression by Knock-in of Cre Recombinase: Effects on the Development of the Mouse Telencephalon. Neuroscience 2007, 148, 385–399. [Google Scholar] [CrossRef] [Green Version]
- Siegenthaler, J.A.; Tremper-Wells, B.A.; Miller, M.W. Foxg1 Haploinsufficiency Reduces the Population of Cortical Intermediate Progenitor Cells: Effect of Increased P21 Expression. Cereb. Cortex 2008, 18, 1865–1875. [Google Scholar] [CrossRef] [Green Version]
- Dou, C.; Lee, J.; Liu, B.; Liu, F.; Massague, J.; Xuan, S.; Lai, E. BF-1 Interferes with Transforming Growth Factor Beta Signaling by Associating with Smad Partners. Mol. Cell Biol. 2000, 20, 6201–6211. [Google Scholar] [CrossRef]
- Seoane, J.; Le, H.-V.; Shen, L.; Anderson, S.A.; Massagué, J. Integration of Smad and Forkhead Pathways in the Control of Neuroepithelial and Glioblastoma Cell Proliferation. Cell 2004, 117, 211–223. [Google Scholar] [CrossRef] [Green Version]
- Vezzali, R.; Weise, S.C.; Hellbach, N.; Heidrich, S.; Vogel, T. The FOXG1/FOXO/SMAD Network Balances Proliferation and Differentiation of Cortical Progenitors and Activates Kcnh3 Expression in Mature Neurons. Oncotarget 2016, 7, 37436–37455. [Google Scholar] [CrossRef] [Green Version]
- McConnell, S.K. Constructing the Cerebral Cortex: Neurogenesis and Fate Determination. Neuron 1995, 15, 761–768. [Google Scholar] [CrossRef] [Green Version]
- Agirman, G.; Broix, L.; Nguyen, L. Cerebral Cortex Development: An Outside-in Perspective. FEBS Lett. 2017, 591, 3978–3992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cadwell, C.R.; Bhaduri, A.; Mostajo-Radji, M.A.; Keefe, M.G.; Nowakowski, T.J. Development and Arealization of the Cerebral Cortex. Neuron 2019, 103, 980–1004. [Google Scholar] [CrossRef] [PubMed]
- Fame, R.M.; MacDonald, J.L.; Macklis, J.D. Development, Specification, and Diversity of Callosal Projection Neurons. Trends Neurosci. 2011, 34, 41–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Testa, G.; Mainardi, M.; Olimpico, F.; Pancrazi, L.; Cattaneo, A.; Caleo, M.; Costa, M. A Triheptanoin-Supplemented Diet Rescues Hippocampal Hyperexcitability and Seizure Susceptibility in FoxG1+/− Mice. Neuropharmacology 2019, 148, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Testa, G.; Olimpico, F.; Pancrazi, L.; Borello, U.; Cattaneo, A.; Caleo, M.; Costa, M.; Mainardi, M. Cortical Seizures in FoxG1+/− Mice Are Accompanied by Akt/S6 Overactivation, Excitation/Inhibition Imbalance and Impaired Synaptic Transmission. IJMS 2019, 20, 4127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pancrazi, L.; Di Benedetto, G.; Colombaioni, L.; Della Sala, G.; Testa, G.; Olimpico, F.; Reyes, A.; Zeviani, M.; Pozzan, T.; Costa, M. Foxg1 Localizes to Mitochondria and Coordinates Cell Differentiation and Bioenergetics. Proc. Natl. Acad. Sci. USA 2015, 112, 13910–13915. [Google Scholar] [CrossRef] [Green Version]
- Liebhaber, G.M.; Riemann, E.; Baumeister, F.A.M. Ketogenic Diet in Rett Syndrome. J. Child Neurol. 2003, 18, 74–75. [Google Scholar] [CrossRef]
- Mantis, J.G.; Centeno, N.A.; Todorova, M.T.; McGowan, R.; Seyfried, T.N. Management of Multifactorial Idiopathic Epilepsy in EL Mice with Caloric Restriction and the Ketogenic Diet: Role of Glucose and Ketone Bodies. Nutr. Metab. 2004, 1, 11. [Google Scholar] [CrossRef] [Green Version]
- Park, M.J.; Aja, S.; Li, Q.; Degano, A.L.; Penati, J.; Zhuo, J.; Roe, C.R.; Ronnett, G.V. Anaplerotic Triheptanoin Diet Enhances Mitochondrial Substrate Use to Remodel the Metabolome and Improve Lifespan, Motor Function, and Sociability in MeCP2-Null Mice. PLoS ONE 2014, 9, e109527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patriarchi, T.; Amabile, S.; Frullanti, E.; Landucci, E.; Lo Rizzo, C.; Ariani, F.; Costa, M.; Olimpico, F.; Hell, J.W.; Vaccarino, F.M.; et al. Imbalance of Excitatory/Inhibitory Synaptic Protein Expression in IPSC-Derived Neurons from FOXG1(+/−) Patients and in Foxg1(+/−) Mice. Eur. J. Hum. Genet. EJHG 2016, 24, 871–880. [Google Scholar] [CrossRef] [Green Version]
- Weise, S.C.; Arumugam, G.; Villarreal, A.; Videm, P.; Heidrich, S.; Nebel, N.; Dumit, V.I.; Sananbenesi, F.; Reimann, V.; Craske, M.; et al. FOXG1 Regulates PRKAR2B Transcriptionally and Posttranscriptionally via MiR200 in the Adult Hippocampus. Mol. Neurobiol. 2019, 56, 5188–5201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, A.R.; Fernandes, T.G.; Cabral, J.M.S.; Diogo, M.M. Modeling Rett Syndrome with Human Pluripotent Stem Cells: Mechanistic Outcomes and Future Clinical Perspectives. Int. J. Mol. Sci. 2021, 22, 3751. [Google Scholar] [CrossRef] [PubMed]
- Marchetto, M.C.N.; Carromeu, C.; Acab, A.; Yu, D.; Yeo, G.W.; Mu, Y.; Chen, G.; Gage, F.H.; Muotri, A.R. A Model for Neural Development and Treatment of Rett Syndrome Using Human Induced Pluripotent Stem Cells. Cell 2010, 143, 527–539. [Google Scholar] [CrossRef] [Green Version]
- Livide, G.; Patriarchi, T.; Amenduni, M.; Amabile, S.; Yasui, D.; Calcagno, E.; Lo Rizzo, C.; De Falco, G.; Ulivieri, C.; Ariani, F.; et al. GluD1 Is a Common Altered Player in Neuronal Differentiation from Both MECP2-Mutated and CDKL5-Mutated IPS Cells. Eur. J. Hum. Genet. EJHG 2015, 23, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Croci, S.; Carriero, M.L.; Capitani, K.; Daga, S.; Donati, F.; Papa, F.T.; Frullanti, E.; Lopergolo, D.; Lamacchia, V.; Tita, R.; et al. AAV-Mediated FOXG1 Gene Editing in Human Rett Primary Cells. Eur. J. Hum. Genet. EJHG 2020, 28, 1446–1458. [Google Scholar] [CrossRef] [PubMed]
- Waite, A.J.; Millar, D.; Clarke, A. The Generation of an Induced Pluripotent Stem Cell Line (DCGi001-A) from an Individual with FOXG1 Syndrome Carrying the c.460dupG (p.Glu154fs) Variation in the FOXG1 Gene. Stem Cell Res. 2020, 49, 102018. [Google Scholar] [CrossRef]
- Sampieri, K.; Meloni, I.; Scala, E.; Ariani, F.; Caselli, R.; Pescucci, C.; Longo, I.; Artuso, R.; Bruttini, M.; Mencarelli, M.A.; et al. Italian Rett Database and Biobank. Hum. Mutat. 2007, 28, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Meng, X.; Liu, Y.; Song, D.; Jiang, C.; Cai, J. Applications of Brain Organoids in Neurodevelopment and Neurological Diseases. J. Biomed. Sci. 2021, 28, 30. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Knoblich, J.A. Generation of Cerebral Organoids from Human Pluripotent Stem Cells. Nat. Protoc. 2014, 9, 2329–2340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, W.; Zhang, B.; Li, M.; Mo, F.; Mi, T.; Wu, Y.; Teng, Z.; Zhou, Q.; Li, W.; Hu, B. Precisely Controlling Endogenous Protein Dosage in HPSCs and Derivatives to Model FOXG1 Syndrome. Nat. Commun. 2019, 10, 928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, A.R.; Fernandes, T.G.; Vaz, S.H.; Silva, T.P.; Bekman, E.P.; Xapelli, S.; Duarte, S.; Ghazvini, M.; Gribnau, J.; Muotri, A.R.; et al. Modeling Rett Syndrome With Human Patient-Specific Forebrain Organoids. Front. Cell Dev. Biol. 2020, 8, 610427. [Google Scholar] [CrossRef] [PubMed]
- Samarasinghe, R.A.; Miranda, O.A.; Buth, J.E.; Mitchell, S.; Ferando, I.; Watanabe, M.; Allison, T.F.; Kurdian, A.; Fotion, N.N.; Gandal, M.J.; et al. Identification of Neural Oscillations and Epileptiform Changes in Human Brain Organoids. Nat. Neurosci. 2021, 24, 1488–1500. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, C.A.; Adams, J.W.; Negraes, P.D.; Carromeu, C.; Tejwani, L.; Acab, A.; Tsuda, B.; Thomas, C.A.; Sodhi, N.; Fichter, K.M.; et al. Pharmacological Reversal of Synaptic and Network Pathology in Human MECP2 -KO Neurons and Cortical Organoids. EMBO Mol. Med. 2021, 13, e12523. [Google Scholar] [CrossRef]
- Mellios, N.; Feldman, D.A.; Sheridan, S.D.; Ip, J.P.K.; Kwok, S.; Amoah, S.K.; Rosen, B.; Rodriguez, B.A.; Crawford, B.; Swaminathan, R.; et al. MeCP2-Regulated MiRNAs Control Early Human Neurogenesis through Differential Effects on ERK and AKT Signaling. Mol. Psychiatry 2018, 23, 1051–1065. [Google Scholar] [CrossRef] [Green Version]
- Nakashima, H.; Tsujimura, K.; Irie, K.; Imamura, T.; Trujillo, C.A.; Ishizu, M.; Uesaka, M.; Pan, M.; Noguchi, H.; Okada, K.; et al. MeCP2 Controls Neural Stem Cell Fate Specification through MiR-199a-Mediated Inhibition of BMP-Smad Signaling. Cell Rep. 2021, 35, 109124. [Google Scholar] [CrossRef]
- Takahashi, R.-U.; Prieto-Vila, M.; Kohama, I.; Ochiya, T. Development of MiRNA-Based Therapeutic Approaches for Cancer Patients. Cancer Sci. 2019, 110, 1140–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, D.; Evans, L.; Perrone, F.; Sokleva, V.; Lim, K.; Rezakhani, S.; Lutolf, M.; Zilbauer, M.; Rawlins, E.L. A Functional Genetic Toolbox for Human Tissue-Derived Organoids. Elife 2021, 10, e67886. [Google Scholar] [CrossRef]
- Pham, M.T.; Pollock, K.M.; Rose, M.D.; Cary, W.A.; Stewart, H.R.; Zhou, P.; Nolta, J.A.; Waldau, B. Generation of Human Vascularized Brain Organoids. Neuroreport 2018, 29, 588–593. [Google Scholar] [CrossRef]
- Shi, Y.; Sun, L.; Wang, M.; Liu, J.; Zhong, S.; Li, R.; Li, P.; Guo, L.; Fang, A.; Chen, R.; et al. Vascularized Human Cortical Organoids (VOrganoids) Model Cortical Development in Vivo. PLOS Biol. 2020, 18, e3000705. [Google Scholar] [CrossRef] [PubMed]
- Ahn, Y.; An, J.-H.; Yang, H.-J.; Lee, D.G.; Kim, J.; Koh, H.; Park, Y.-H.; Song, B.-S.; Sim, B.-W.; Lee, H.J.; et al. Human Blood Vessel Organoids Penetrate Human Cerebral Organoids and Form a Vessel-Like System. Cells 2021, 10, 2036. [Google Scholar] [CrossRef]
- Bergmann, S.; Lawler, S.E.; Qu, Y.; Fadzen, C.M.; Wolfe, J.M.; Regan, M.S.; Pentelute, B.L.; Agar, N.Y.R.; Cho, C.-F. Blood-Brain-Barrier Organoids for Investigating the Permeability of CNS Therapeutics. Nat. Protoc. 2018, 13, 2827–2843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagley, J.A.; Reumann, D.; Bian, S.; Lévi-Strauss, J.; Knoblich, J.A. Fused Cerebral Organoids Model Interactions between Brain Regions. Nat. Methods 2017, 14, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, E.; Albanna, W.; Pasquini, G.; Ramani, A.; Josipovic, N.; Mariappan, A.; Schinzel, F.; Karch, C.M.; Bao, G.; Gottardo, M.; et al. Human Brain Organoids Assemble Functionally Integrated Bilateral Optic Vesicles. Cell Stem Cell 2021, 28, 1740–1757.e8. [Google Scholar] [CrossRef]
- Pereira, J.D.; DuBreuil, D.M.; Devlin, A.-C.; Held, A.; Sapir, Y.; Berezovski, E.; Hawrot, J.; Dorfman, K.; Chander, V.; Wainger, B.J. Human Sensorimotor Organoids Derived from Healthy and Amyotrophic Lateral Sclerosis Stem Cells Form Neuromuscular Junctions. Nat. Commun. 2021, 12, 4744. [Google Scholar] [CrossRef]
- Faustino Martins, J.-M.; Fischer, C.; Urzi, A.; Vidal, R.; Kunz, S.; Ruffault, P.-L.; Kabuss, L.; Hube, I.; Gazzerro, E.; Birchmeier, C.; et al. Self-Organizing 3D Human Trunk Neuromuscular Organoids. Cell Stem Cell 2020, 26, 172–186.e6. [Google Scholar] [CrossRef]
- Lenin, S.; Ponthier, E.; Scheer, K.G.; Yeo, E.C.F.; Tea, M.N.; Ebert, L.M.; Oksdath Mansilla, M.; Poonnoose, S.; Baumgartner, U.; Day, B.W.; et al. A Drug Screening Pipeline Using 2D and 3D Patient-Derived In Vitro Models for Pre-Clinical Analysis of Therapy Response in Glioblastoma. Int. J. Mol. Sci. 2021, 22, 4322. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tang, P.; Cai, S.; Peng, J.; Hua, G. Organoid Based Personalized Medicine: From Bench to Bedside. Cell Regen. 2020, 9, 21. [Google Scholar] [CrossRef] [PubMed]
- Azar, J.; Bahmad, H.F.; Daher, D.; Moubarak, M.M.; Hadadeh, O.; Monzer, A.; Al Bitar, S.; Jamal, M.; Al-Sayegh, M.; Abou-Kheir, W. The Use of Stem Cell-Derived Organoids in Disease Modeling: An Update. Int. J. Mol. Sci. 2021, 22, 7667. [Google Scholar] [CrossRef] [PubMed]
- Lavazza, A.; Massimini, M. Cerebral Organoids: Ethical Issues and Consciousness Assessment. J. Med. Ethics 2018, 44, 606–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akol, I.; Gather, F.; Vogel, T. Paving Therapeutic Avenues for FOXG1 Syndrome: Untangling Genotypes and Phenotypes from a Molecular Perspective. Int. J. Mol. Sci. 2022, 23, 954. https://doi.org/10.3390/ijms23020954
Akol I, Gather F, Vogel T. Paving Therapeutic Avenues for FOXG1 Syndrome: Untangling Genotypes and Phenotypes from a Molecular Perspective. International Journal of Molecular Sciences. 2022; 23(2):954. https://doi.org/10.3390/ijms23020954
Chicago/Turabian StyleAkol, Ipek, Fabian Gather, and Tanja Vogel. 2022. "Paving Therapeutic Avenues for FOXG1 Syndrome: Untangling Genotypes and Phenotypes from a Molecular Perspective" International Journal of Molecular Sciences 23, no. 2: 954. https://doi.org/10.3390/ijms23020954
APA StyleAkol, I., Gather, F., & Vogel, T. (2022). Paving Therapeutic Avenues for FOXG1 Syndrome: Untangling Genotypes and Phenotypes from a Molecular Perspective. International Journal of Molecular Sciences, 23(2), 954. https://doi.org/10.3390/ijms23020954