
Molecular Mechanisms Leading from Periodontal Disease to Cancer

 ,
,  , ,
, , 
Abstract
:1. Introduction
2. Molecular Mechanisms Linking Periodontal Disease with Cancer
2.1. Molecular Mechanisms of P. gingivalis and F. nucleatum Pathogenesis in Cancer
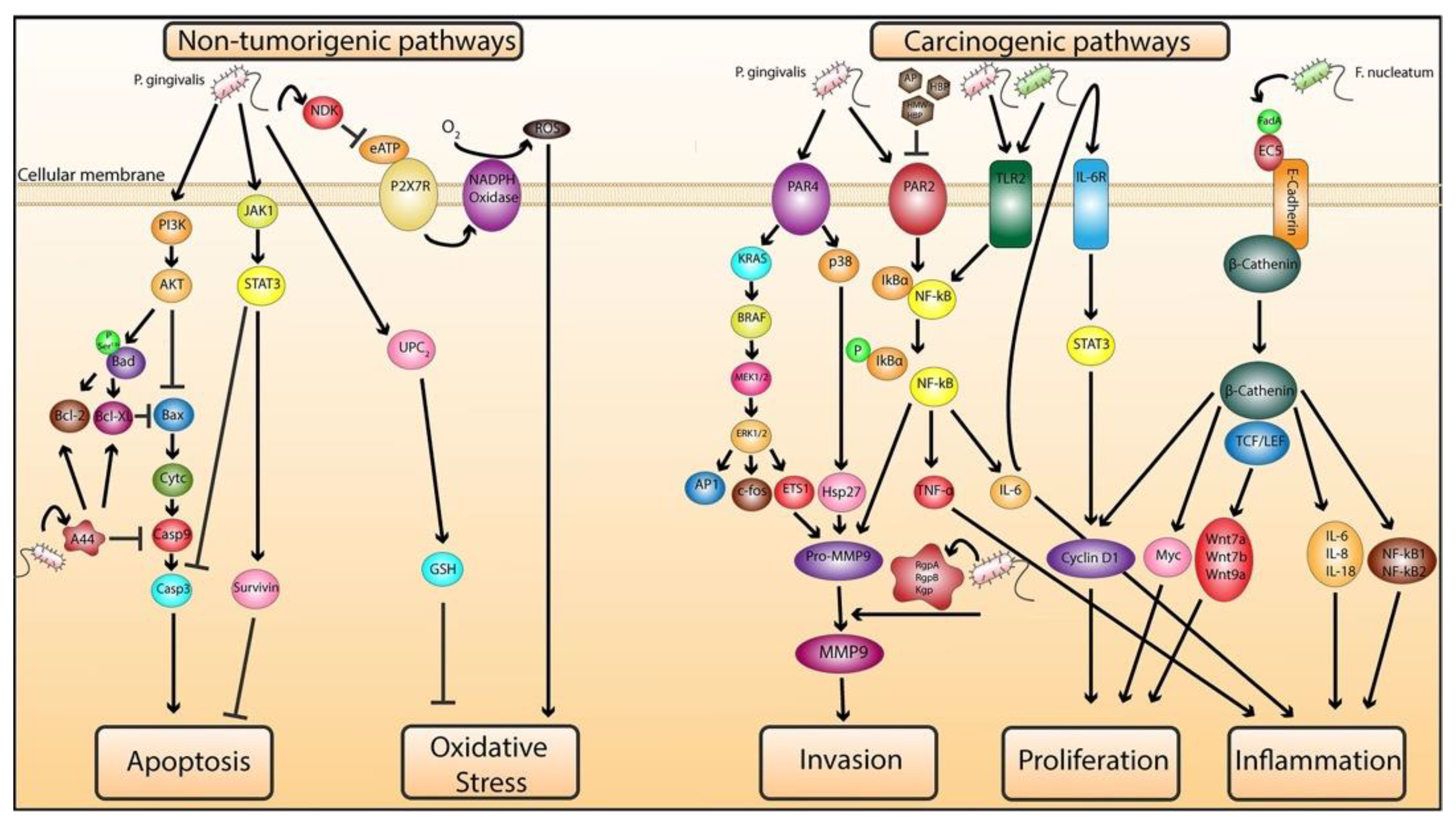
2.1.1. Role of P. gingivalis in Mediating Cellular Transformation
2.1.2. Role of P. gingivalis and F. nucleatum in Exacerbating Malignancy
2.2. The Impact of RANKL–RANK–OPG Signaling in PD
2.2.1. RANK–RANKL–OPG Axis in Bone Metabolism and Immunity
2.2.2. RANKL Isoforms and Function
2.2.3. Role of RANKL in Periodontal Disease
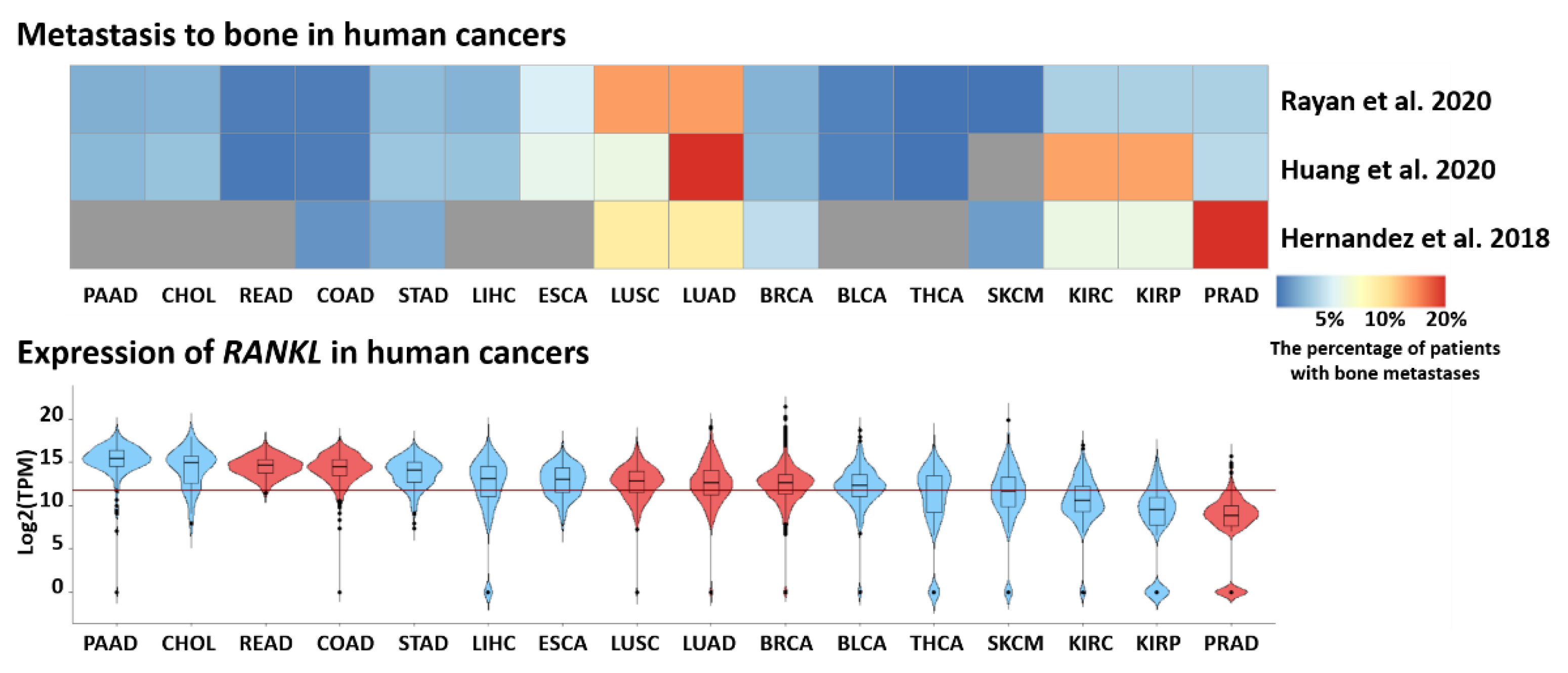
2.2.4. RANKL Links Periodontal Disease and Cancer
2.3. Periodontal Disease and Immune Response
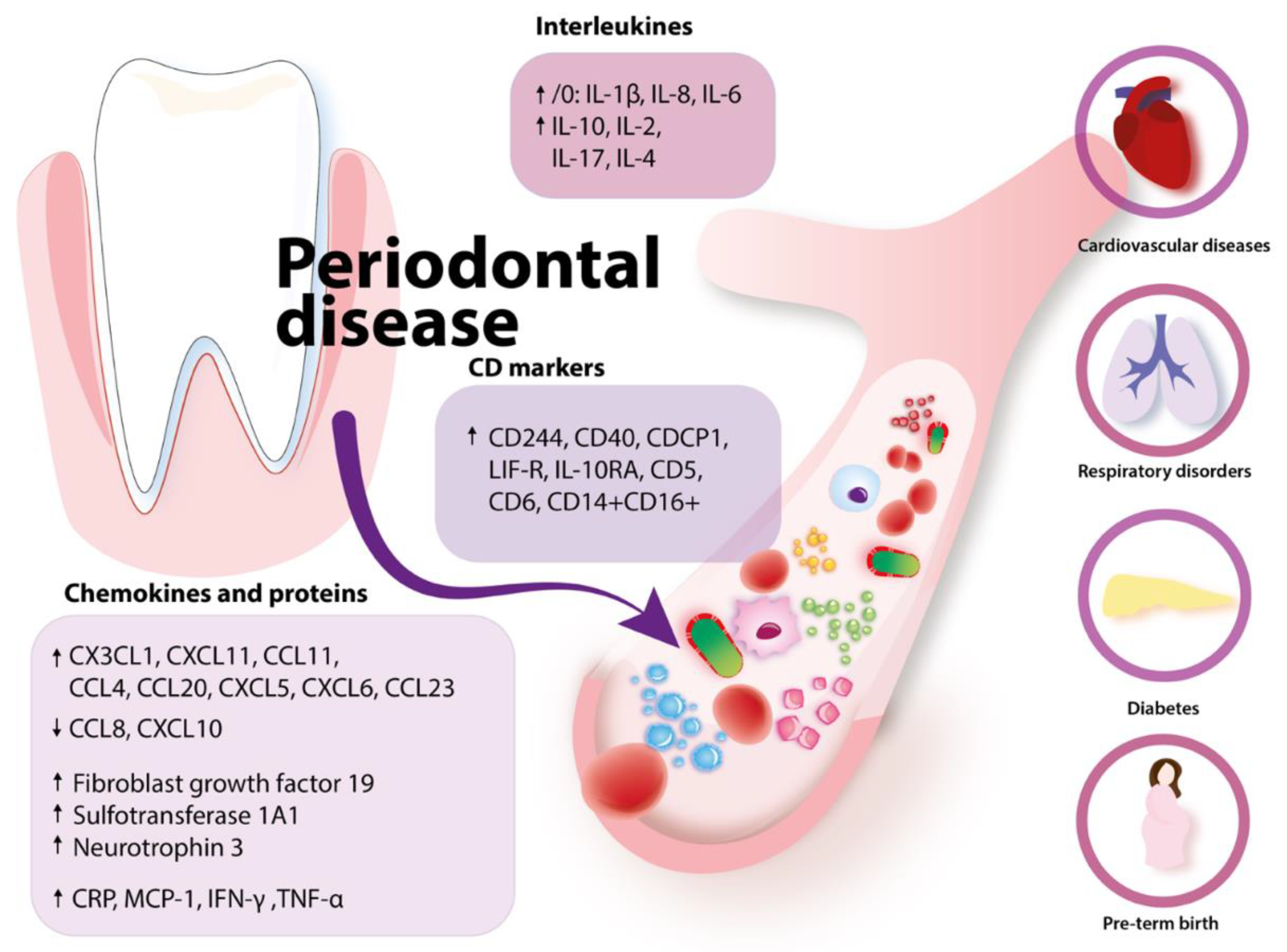
2.3.1. Periodontal Disease and Alterations in Blood: The Impact on Systemic Diseases
2.3.2. Potential Mechanisms Linking with Cancer
3. ClinicalTrials.gov Analysis
4. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- WHO Regional Office for Europe. Available online: https://www.euro.who.int/en/health-topics/disease-prevention/oral-health/data-and-statistics (accessed on 27 November 2021).
- Gopinath, D.; Menon, R.K.; Veettil, S.K.; Botelho, M.G.; Johnson, N.W. Periodontal diseases as putative risk factors for head and neck cancer: Systematic review and meta-analysis. Cancers 2020, 12, 1893. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, E.M.; Reis, C.; Manzanares-Céspedes, M.C. Chronic periodontitis, inflammatory cytokines, and interrelationship with other chronic diseases. Postgrad. Med. 2018, 130, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Mehrotra, N.; Singh, S. Periodontitis; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Curtis, M.A.; Diaz, P.I.; van Dyke, T.E. The role of the microbiota in periodontal disease. Periodontology 2000 2020, 83, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Gasner, N.S.; Schure, R.S. Periodontal Disease. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Jaramillo, A.; Arce, R.M.; Herrera, D.; Betancourth, M.; Botero, J.E.; Contreras, A. Clinical and microbiological characterization of periodontal abscesses. J. Clin. Periodontol. 2005, 32, 1213–1218. [Google Scholar] [CrossRef]
- Highfield, J. Diagnosis and Classification of Periodontal Disease. Aust. Dent. J. 2009, 54, S11–S26. [Google Scholar] [CrossRef]
- Kinane, D.F.; Stathopoulou, P.G.; Papapanou, P.N. Periodontal diseases. Nat. Rev. Dis. Prim. 2017, 3, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Hienz, S.A.; Paliwal, S.; Ivanovski, S. Mechanisms of bone resorption in periodontitis. J. Immunol. Res. 2015, 2015, 615486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez, G.A.; Miozza, V.A.; Delgado, A.; Busch, L. Salivary IL-1β and PGE2 as Biomarkers of periodontal status, before and after periodontal treatment. J. Clin. Periodontol. 2013, 40, 1112–1117. [Google Scholar] [CrossRef]
- Soutome, S.; Otsuru, M.; Kawashita, Y.; Funahara, M.; Ukai, T.; Saito, T. Effect of cancer treatment on the worsening of periodontal disease and dental caries: A preliminary, retrospective study. Oral Health Prev. Dent. 2021, 19, 399–404. [Google Scholar] [CrossRef] [PubMed]
- American Academy of Periodontology. Classification At-a-Glance; American Academy of Periodontology: Chicago, IL, USA, 2018. [Google Scholar]
- De Molon, R.S.; Rossa, C.; Thurlings, R.M.; Cirelli, J.A.; Koenders, M.I. Linkage of periodontitis and rheumatoid arthritis: Current evidence and potential biological interactions. Int. J. Mol. Sci. 2019, 20, 4541. [Google Scholar] [CrossRef] [Green Version]
- Bansal, M.; Rastogi, S.; Vineeth, N.S. Influence of periodontal disease on systemic disease: Inversion of a paradigm: A review. J. Med. Life 2013, 6, 126–130. [Google Scholar] [PubMed]
- Nwizu, N.; Wactawski-Wende, J.; Genco, R.J. Periodontal disease and cancer: Epidemiologic studies and possible mechanisms. Periodontology 2000 2020, 83, 213–233. [Google Scholar] [CrossRef] [PubMed]
- Gholizadeh, P.; Eslami, H.; Yousefi, M.; Asgharzadeh, M.; Aghazadeh, M.; Kafil, H.S. Role of oral microbiome on oral cancers, a review. Biomed. Pharmacother. 2016, 84, 552–558. [Google Scholar] [CrossRef]
- Gallimidi, A.B.; Fischman, S.; Revach, B.; Bulvik, R.; Rubinstein, A.M.; Nussbaum, G.; Elkin, M. Periodontal pathogens Porphyromonas gingivalis and Fusobacterium nucleatum promote tumor progression in an oral-specific chemical carcinogenesis model. Oncotarget 2015, 6, 22613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javed, F.; Warnakulasuriya, S. Is there a relationship between periodontal disease and oral cancer? A systematic review of currently available evidence. Crit. Rev. Oncol. Hematol. 2016, 97, 197–205. [Google Scholar] [CrossRef]
- Karmakar, S.; Kar, A.; Thakur, S.; Rao, V.U.S. Periodontitis and oral cancer-a striking link. Oral Oncol. 2020, 106, 104630. [Google Scholar] [CrossRef] [PubMed]
- Rajesh, K.S.; Thomas, D.; Hegde, S.; Kumar, M.S.A. Poor periodontal health: A cancer risk? J. Indian Soc. Periodontol. 2013, 17, 706–710. [Google Scholar] [CrossRef] [PubMed]
- Barros, S.P.; Fahimipour, F.; Tarran, R.; Kim, S.; Scarel-Caminaga, R.M.; Justice, A.; North, K. Epigenetic Reprogramming in periodontal disease: Dynamic crosstalk with potential impact in oncogenesis. Periodontology 2000 2020, 82, 157–172. [Google Scholar] [CrossRef]
- Freudenheim, J.L.; Genco, R.J.; LaMonte, M.J.; Millen, A.E.; Hovey, K.M.; Mai, X.; Nwizu, N.; Andrews, C.A.; Wactawski-Wende, J. Periodontal disease and breast cancer: Prospective cohort study of postmenopausal women. Cancer Epidemiol. Biomark. Prev. 2016, 25, 43–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michaud, D.S.; Fu, Z.; Shi, J.; Chung, M. Periodontal disease, tooth loss, and cancer risk. Epidemiol. Rev. 2017, 39, 49–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Zhu, B.L.; Wu, C.C.; Lin, R.F.; Zhang, X. Periodontal Disease and tooth loss are associated with lung cancer risk. Biomed Res. Int. 2020, 2020, 5107696. [Google Scholar] [CrossRef] [PubMed]
- Lo, C.H.; Kwon, S.; Wang, L.; Polychronidis, G.; Knudsen, M.D.; Zhong, R.; Cao, Y.; Wu, K.; Ogino, S.; Giovannucci, E.L.; et al. Periodontal disease, tooth loss, and risk of oesophageal and gastric adenocarcinoma: A prospective study. Gut 2021, 70, 620–621. [Google Scholar] [CrossRef] [PubMed]
- Lo, C.H.; Nguyen, L.H.; Wu, K.; Ogino, S.; Chan, A.T.; Giovannucci, E.L.; Song, M. Periodontal disease, tooth loss, and risk of serrated polyps and conventional adenomas. Cancer Prev. Res. 2020, 13, 699–706. [Google Scholar] [CrossRef] [PubMed]
- Momen-heravi, F.; Babic, A.; Tworoger, S.S.; Zhang, L.; Wu, K. Periodontal disease, tooth loss, and colorectal cancer risk: Results from the nurses’ health study. Int. J. Cancer 2008, 5, 2–18. [Google Scholar] [CrossRef]
- Da Silva, A.P.B.; Alluri, L.S.C.; Bissada, N.F.; Gupta, S. Association between oral pathogens and prostate cancer: Building the relationship. Am. J. Clin. Exp. Urol. 2019, 7, 1–10. [Google Scholar] [PubMed]
- Hoare, A.; Soto, C.; Rojas-Celis, V.; Bravo, D. Chronic inflammation as a link between periodontitis and carcinogenesis. Mediators Inflamm. 2019, 2019, 1029857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muthular, M.; Bálsamo, F.; Passero, P.; Jewtuchowicz, V.; Miozza, V.; Villalba, M.B.; Brusca, M.I.; Pérez, C. Effects of tamoxifen on periodontal disease and candida albicans of patients with breast cancer and other pathologies. Future Microbiol. 2019, 14, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Famili, P.; Cauley, J.A.; Greenspan, S.L. The effect of androgen deprivation therapy on periodontal disease in men with prostate cancer. J. Urol. 2007, 177, 921–924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nath, S.G.; Raveendran, R. Microbial dysbiosis in periodontitis. J. Indian Soc. Periodontol. 2013, 17, 543–545. [Google Scholar] [CrossRef]
- Kamarajan, P.; Ateia, I.; Shin, J.M.; Fenno, J.C.; Le, C.; Zhan, L.; Chang, A.; Darveau, R.; Kapila, Y.L. Periodontal pathogens promote cancer aggressivity via TLR/MyD88 triggered activation of integrin/FAK signaling that is therapeutically reversible by a probiotic bacteriocin. PLoS Pathog. 2020, 16, e1008881. [Google Scholar] [CrossRef] [PubMed]
- Makkawi, H.; Hoch, S.; Burns, E.; Hosur, K.; Hajishengallis, G.; Kirschning, C.J.; Nussbaum, G. Porphyromonas gingivalis Stimulates TLR2-PI3K Signaling to escape immune clearance and induce bone resorption independently of MyD88. Front. Cell. Infect. Microbiol. 2017, 7, 359. [Google Scholar] [CrossRef]
- Han, Y.W.; Shi, W.; Huang, G.T.J.; Kinder Haake, S.; Park, N.H.; Kuramitsu, H.; Genco, R.J. Interactions between periodontal bacteria and human oral epithelial cells: Fusobacterium nucleatum adheres to and invades epithelial cells. Infect. Immun. 2000, 68, 3140–3146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fardini, Y.; Chung, P.; Dumm, R.; Joshi, N.; Han, Y.W. Transmission of diverse oral bacteria to murine placenta: Evidence for the oral microbiome as a potential source of intrauterine infection. Infect. Immun. 2010, 78, 1789–1796. [Google Scholar] [CrossRef] [Green Version]
- Kolenbrander, P.E.; Palmer, R.J.; Periasamy, S.; Jakubovics, N.S. Oral multispecies biofilm development and the key role of cell-cell distance. Nat. Rev. Microbiol. 2010, 8, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Geng, F.; Liu, J.; Guo, Y.; Li, C.; Wang, H.; Wang, H.; Zhao, H.; Pan, Y. Persistent exposure to Porphyromonas gingivalis promotes proliferative and invasion capabilities, and tumorigenic properties of human immortalized oral epithelial cells. Front. Cell. Infect. Microbiol. 2017, 7, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, T.M.; Du, P.; Kawachi, N.; Belbin, T.J.; Wang, Y.; Schlecht, N.F.; Ow, T.J.; Keller, C.E.; Childs, G.J.; Smith, R.V.; et al. Proteomic analysis of oral cavity squamous cell carcinoma specimens identifies patient outcome-associated proteins. Arch. Pathol. Lab. Med. 2015, 139, 494–507. [Google Scholar] [CrossRef] [Green Version]
- Jeong, E.; Kim, K.; Kim, J.H.; Cha, G.S.; Kim, S.J.; Kang, H.S.; Choi, J. Porphyromonas gingivalis HSP60 peptides have distinct roles in the development of atherosclerosis. Mol. Immunol. 2015, 63, 489–496. [Google Scholar] [CrossRef]
- Tabeta, K.; Yamazaki, K.; Hotokezaka, H.; Yoshie, H.; Hara, K. Elevated humoral immune response to heat shock protein 60 (Hsp60) family in periodontitis patients. Clin. Exp. Immunol. 2000, 120, 285–293. [Google Scholar] [CrossRef]
- Chung, S.W.; Kang, H.S.; Park, H.R.; Kim, S.J.; Kim, S.J.; Choi, J.I. Immune responses to heat shock protein in Porphyromonas gingivalis-infected periodontitis and atherosclerosis patients. J. Periodontal Res. 2003, 38, 388–393. [Google Scholar] [CrossRef]
- Lee, J.Y.; Yi, N.N.; Kim, U.S.; Choi, J.S.; Kim, S.J.; Choi, J.I. Porphyromonas gingivalis heat shock protein vaccine reduces the alveolar bone loss induced by multiple periodontopathogenic bacteria. J. Periodontal Res. 2006, 41, 10–14. [Google Scholar] [CrossRef]
- Lin, F.Y.; Huang, C.Y.; Lu, H.Y.; Shih, C.M.; Tsao, N.W.; Shyue, S.K.; Lin, C.Y.; Chang, Y.J.; Tsai, C.S.; Lin, Y.W.; et al. The GroEL protein of Porphyromonas gingivalis accelerates tumor growth by enhancing endothelial progenitor cell function and neovascularization. Mol. Oral Microbiol. 2015, 30, 198–216. [Google Scholar] [CrossRef] [PubMed]
- Mao, S.; Park, Y.; Hasegawa, Y.; Tribble, G.D.; James, C.E.; Handfield, M.; Stavropoulos, M.F.; Yilmaz, Ö.; Lamont, R.J. Intrinsic apoptotic pathways of gingival epithelial cells modulated by Porphyromonas gingivalis. Cell. Microbiol. 2007, 9, 1997–2007. [Google Scholar] [CrossRef] [Green Version]
- Yilmaz, Ö.; Jungas, T.; Verbeke, P.; Ojcius, D.M. Activation of the phosphatidylinositol 3-Kinase/Akt pathway contributes to survival of primary epithelial cells infected with the periodontal pathogen Porphyromonas gingivalis. Infect. Immun. 2004, 72, 3743–3751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakhjiri, S.F.; Park, Y.; Yilmaz, O.; Chung, W.O.; Watanabe, K.; El-Sabaeny, A.; Park, K.; Lamont, R.J. Inhibition of epithelial cell apoptosis by Porphyromonas gingivalis. FEMS Microbiol. Lett. 2001, 200, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Handfield, M.; Mans, J.J.; Zheng, G.; Lopez, M.C.; Mao, S.; Progulske-Fox, A.; Narasimhan, G.; Baker, H.V.; Lamont, R.J. Distinct transcriptional profiles characterize oral epithelium-microbiota interactions. Cell. Microbiol. 2005, 7, 811–823. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Jermanus, C.; Barbetta, B.; Choi, C.; Verbeke, P.; Ojcius, D.M.; Yilmaz, Ö. Porphyromonas gingivalis infection sequesters pro-apoptotic bad through akt in primary gingival epithelial cells. Mol. Oral Microbiol. 2010, 25, 89–101. [Google Scholar] [CrossRef] [Green Version]
- Boisvert, H.; Duncan, M.J. Translocation of Porphyromonas gingivalis gingipain adhesin peptide A44 to host mitochondria prevents apoptosis. Infect. Immun. 2010, 78, 3616–3624. [Google Scholar] [CrossRef] [Green Version]
- Choi, C.H.; Spooner, R.; Deguzman, J.; Koutouzis, T.; Ojcius, D.M.; Yilmaz, Ö. Porphyromonas gingivalis-nucleoside-diphosphate-kinase inhibits ATP-induced reactive-oxygen-species via P2X7 Receptor/NADPH-oxidase signalling and contributes to persistence. Cell. Microbiol. 2013, 15, 961–976. [Google Scholar] [CrossRef] [Green Version]
- Yilmaz, Ö.; Yao, L.; Maeda, K.; Rose, T.M.; Lewis, E.L.; Duman, M.; Lamont, R.J.; Ojcius, D.M. ATP scavenging by the intracellular pathogen Porphyromonas gingivalis inhibits P2X7-mediated host-cell apoptosis. Cell. Microbiol. 2008, 10, 863–875. [Google Scholar] [CrossRef] [Green Version]
- Kuboniwa, M.; Hasegawa, Y.; Mao, S.; Shizukuishi, S.; Amano, A.; Lamont, R.J.; Yilmaz, Ö. P. gingivalis accelerates gingival epithelial cell progression through the cell cycle. Microbes Infect. 2008, 10, 122–128. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Tang, X.; Li, C.; Pan, C.; Li, Q.; Geng, F.; Pan, Y. Porphyromonas gingivalis promotes the cell cycle and inflammatory cytokine production in periodontal ligament fibroblasts. Arch. Oral Biol. 2015, 60, 1153–1161. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.; Xu, X.; Tan, L.; Lin, L.; Pan, Y. The effects of Porphyromonas gingivalis on the cell cycle progression of human gingival epithelial cells. Oral Dis. 2014, 20, 100–108. [Google Scholar] [CrossRef] [PubMed]
- O’Brien-Simpson, N.M.; Pathirana, R.D.; Walker, G.D.; Reynolds, E.C. Porphyromonas gingivalis RgpA-Kgp proteinase-adhesin complexes penetrate gingival tissue and induce proinflammatory cytokines or apoptosis in a concentration-dependent manner. Infect. Immun. 2009, 77, 1246–1261. [Google Scholar] [CrossRef] [Green Version]
- Moffatt, C.E.; Lamont, R.J. Porphyromonas gingivalis induction of MicroRNA-203 expression controls suppressor of cytokine signaling 3 in gingival epithelial cells. Infect. Immun. 2011, 79, 2632–2637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benakanakere, M.R.; Li, Q.; Eskan, M.A.; Singh, A.V.; Zhao, J.; Galicia, J.C.; Stathopoulou, P.; Knudsen, T.B.; Kinane, D.F. Modulation of TLR2 protein expression by MiR-105 in human oral keratinocytes. J. Biol. Chem. 2009, 284, 23107–23115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mu, W.; Jia, Y.; Chen, X.; Li, H.; Wang, Z.; Cheng, B. Intracellular Porphyromonas gingivalis promotes the proliferation of colorectal cancer cells via the MAPK/ERK signaling pathway. Front. Cell. Infect. Microbiol. 2020, 10, 812. [Google Scholar] [CrossRef]
- Inaba, H.; Amano, A.; Lamont, R.J.; Murakami, Y. Involvement of protease-activated receptor 4 in over-expression of matrix metalloproteinase 9 induced by Porphyromonas gingivalis. Med. Microbiol. Immunol. 2015, 204, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Inaba, H.; Tagashira, M.; Kanda, T.; Murakami, Y.; Amano, A.; Matsumoto-Nakano, M. Apple- and hop-polyphenols inhibit Porphyromonas gingivalis—Mediated precursor of matrix metalloproteinase-9 activation and invasion of oral squamous cell carcinoma cells. J. Periodontol. 2016, 87, 1103–1111. [Google Scholar] [CrossRef]
- Inaba, H.; Sugita, H.; Kuboniwa, M.; Iwai, S.; Hamada, M.; Noda, T.; Morisaki, I.; Lamont, R.J.; Amano, A. Porphyromonas gingivalis promotes invasion of oral squamous cell carcinoma through induction of ProMMP9 and its activation. Cell. Microbiol. 2014, 16, 131–145. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.S.; Zheng, M.; Zhang, M.; Pang, X.; Li, L.; Wang, S.S.; Yang, X.; Wu, J.B.; Tang, Y.J.; Tang, Y.L.; et al. Porphyromonas gingivalis promotes 4-nitroquinoline-1-oxide-induced oral carcinogenesis with an alteration of fatty acid metabolism. Front. Microbiol. 2018, 9, 1–13. [Google Scholar] [CrossRef]
- Polak, D.; Wilensky, A.; Shapira, L.; Halabi, A.; Goldstein, D.; Weiss, E.I.; Houri-Haddad, Y. Mouse model of experimental periodontitis induced by Porphyromonas gingivalis/Fusobacterium nucleatum infection: Bone loss and host response. J. Clin. Periodontol. 2009, 36, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Woo, B.H.; Kim, D.J.; Choi, J.I.; Kim, S.J.; Park, B.S.; Song, J.M.; Lee, J.H.; Park, H.R. Oral cancer cells sustainedly infected with Porphyromonas gingivalis exhibit resistance to taxol and have higher metastatic potential. Oncotarget 2017, 8, 46981–46992. [Google Scholar] [CrossRef] [Green Version]
- Ha, N.H.; Park, D.G.; Woo, B.H.; Kim, D.J.; Choi, J.I.; Park, B.S.; Kim, Y.D.; Lee, J.H.; Park, H.R. Porphyromonas gingivalis increases the invasiveness of oral cancer cells by upregulating IL-8 and MMPs. Cytokine 2016, 86, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Ha, N.H.; Woo, B.H.; Kim, D.J.; Ha, E.S.; Choi, J.I.; Kim, S.J.; Park, B.S.; Lee, J.H.; Park, H.R. Prolonged and repetitive exposure to Porphyromonas gingivalis increases aggressiveness of oral cancer cells by promoting acquisition of cancer stem cell properties. Tumor Biol. 2015, 36, 9947–9960. [Google Scholar] [CrossRef] [PubMed]
- Fernando, R.I.; Castillo, M.D.; Litzinger, M.; Hamilton, D.H.; Palena, C. IL-8 Signaling plays a critical role in the epithelial-mesenchymal transition of human carcinoma cells. Cancer Res. 2011, 71, 5296–5306. [Google Scholar] [CrossRef] [Green Version]
- Song, J.M.; Woo, B.H.; Lee, J.H.; Yoon, S.; Cho, Y.; Kim, Y.D.; Park, H.R. Oral administration of Porphyromonas gingivalis, a major pathogen of chronic periodontitis, promotes resistance to paclitaxel in mouse xenografts of oral squamous cell carcinoma. Int. J. Mol. Sci. 2019, 20, 2494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, T.J.; Wee, S.W.; Woo, V.H.; Choi, J.I.; Kim, S.J.; Shin, H.I.; Lee, J.H.; Park, H.R. Porphyromonas gingivalis-induced autophagy suppresses cell proliferation through G1 arrest in oral cancer cells. Arch. Oral Biol. 2014, 59, 370–378. [Google Scholar] [CrossRef]
- Rubinstein, M.R.; Wang, X.; Liu, W.; Hao, Y.; Cai, G.; Han, Y.W. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-Cadherin/β-Catenin Signaling via its FadA adhesin. Cell Host Microbe 2013, 14, 195–206. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Xie, M.; Xie, Y.; Mei, F.; Lu, X.; Li, X.; Chen, L. The roles of osteocytes in alveolar bone destruction in periodontitis. J. Transl. Med. 2020, 18, 1–15. [Google Scholar] [CrossRef]
- Yasuda, H.; Shima, N.; Nakagawa, N.; Mochizuki, S.I.; Yano, K.; Fujise, N.; Sato, Y.; Goto, M.; Yamaguchi, K.; Kuriyama, M.; et al. Identity of osteoclastogenesis inhibitory factor (OCIF) and osteoprotegerin (OPG): A mechanism by which OPG/OCIF inhibits osteoclastogenesis in vitro. Endocrinology 1998, 139, 1329–1337. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.; Lacey, D.L.; Dunstan, C.R.; Solovyev, I.; Colombero, A.; Timms, E.; Tan, H.L.; Elliott, G.; Kelley, M.J.; Sarosi, I.; et al. Tumor necrosis factor receptor family member RANK mediates osteoclast differentiation and activation induced by osteoprotegerin ligand. Proc. Natl. Acad. Sci. USA 1999, 96, 3540–3545. [Google Scholar] [CrossRef] [Green Version]
- Lacey, D.L.; Timms, E.; Tan, H.L.; Kelley, M.J.; Dunstan, C.R.; Burgess, T.; Elliott, R.; Colombero, A.; Elliott, G.; Scully, S.; et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell 1998, 93, 165–176. [Google Scholar] [CrossRef] [Green Version]
- Lloyd, S.A.J.; Yuan, Y.Y.; Kostenuik, P.J.; Ominsky, M.S.; Lau, A.G.; Morony, S.; Stolina, M.; Asuncion, F.J.; Bateman, T.A. Soluble RANKL Induces high bone turnover and decreases bone volume, density, and strength in mice. Calcif. Tissue Int. 2008, 82, 361–372. [Google Scholar] [CrossRef]
- Li, J.; Sarosi, I.; Yan, X.Q.; Morony, S.; Capparelli, C.; Tan, H.L.; McCabe, S.; Elliott, R.; Scully, S.; Van, G.; et al. RANK is the intrinsic hematopoietic cell surface receptor that controls osteoclastogenesis and regulation of bone mass and calcium metabolism. Proc. Natl. Acad. Sci. USA 2000, 97, 1566–1571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, Y.; Yoshida, H.; Sarosi, I.; Tan, H.; Timms, E.; Capparelli, C.; Morony, S.; Oliveira-dos-santos, A.J.; Van, G.; Itie, A.; et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature 1999, 397, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Bucay, N.; Sarosi, I.; Dunstan, C.R.; Morony, S.; Tarpley, J.; Capparelli, C.; Scully, S.; Tan, H.L.; Xu, W.; Lacey, D.L.; et al. Osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev. 1998, 12, 1260–1268. [Google Scholar] [CrossRef] [PubMed]
- Boyce, B.F.; Xing, L. Functions of RANKL/RANK/OPG in bone modeling and remodeling. Arch. Biochem. Biophys. 2008, 473, 139–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, C.A. Control of RANKL gene expression. Bone 2010, 46, 911–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosa, J.T.; Laizé, V.; Gavaia, P.J.; Cancela, M.L. Fish models of induced osteoporosis. Front. Cell Dev. Biol. 2021, 9, 1924. [Google Scholar] [CrossRef]
- Dougall, W.C.; Glaccum, M.; Charrier, K.; Rohrbach, K.; Brasel, K.; De Smedt, T.; Daro, E.; Smith, J.; Tometsko, M.E.; Maliszewski, C.R.; et al. RANK is essential for osteoclast and lymph node development. Genes Dev. 1999, 13, 2412–2424. [Google Scholar] [CrossRef]
- Kung, Y.Y.; Felge, U.; Sarosi, I.; Bolon, B.; Taturi, A.; Morony, S.; Capparelli, C.; Li, J.; Elliott, R.; McCabe, S.; et al. Activated t cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature 1999, 402, 304–309. [Google Scholar] [CrossRef]
- Irla, M. RANK signaling in the differentiation and regeneration of thymic epithelial cells. Front. Immunol. 2021, 11, 3592. [Google Scholar] [CrossRef]
- Lopes, N.; Vachon, H.; Marie, J.; Irla, M. Administration of RANKL Boosts thymic regeneration upon bone marrow transplantation. EMBO Mol. Med. 2017, 9, 835–851. [Google Scholar] [CrossRef] [PubMed]
- Sobacchi, C.; Menale, C.; Villa, A. The RANKL-RANK axis: A bone to thymus round trip. Front. Immunol. 2019, 10, 629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, D.M.; Maraskovsky, E.; Billingsley, W.L.; Dougall, W.C.; Tometsko, M.E.; Roux, E.R.; Teepe, M.C.; DuBose, R.F.; Cosman, D.; Galibert, L. A Homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature 1997, 390, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Wong, B.R.; Rho, J.; Arron, J.; Robinson, E.; Orlinick, J.; Chao, M.; Kalachikov, S.; Cayani, E.; Bartlett, F.S.; Frankel, W.N.; et al. TRANCE is a novel ligand of the tumor necrosis factor receptor family that activates c-jun n-terminal kinase in t cells. J. Biol. Chem. 1997, 272, 25190–25194. [Google Scholar] [CrossRef] [Green Version]
- Dong, C.; Yang, D.D.; Tournler, C.; Whitmarsh, A.J.; Xu, J.; Davis, R.J.; Flavell, R.A. JNK Is required for effector T-cell function but not for T-cell activation. Nature 2000, 405, 91–94. [Google Scholar] [CrossRef]
- Kawai, T.; Matsuyama, T.; Hosokawa, Y.; Makihira, S.; Seki, M.; Karimbux, N.Y.; Goncalves, R.B.; Valverde, P.; Dibart, S.; Li, Y.P.; et al. B and T lymphocytes are the primary sources of RANKL in the bone resorptive lesion of periodontal disease. Am. J. Pathol. 2006, 169, 987–998. [Google Scholar] [CrossRef] [Green Version]
- Meednu, N.; Zhang, H.; Owen, T.; Sun, W.; Wang, V.; Cistrone, C.; Rangel-Moreno, J.; Xing, L.; Anolik, J.H. Production of RANKL by memory B cells: A link between B cells and bone erosion in rheumatoid arthritis. Arthritis Rheumatol. 2016, 68, 805–816. [Google Scholar] [CrossRef]
- Page, G.; Miossec, P. RANK and RANKL Expression as markers of dendritic cell-T cell interactions in paired samples of rheumatoid synovium and lymph nodes. Arthritis Rheum. 2005, 52, 2307–2312. [Google Scholar] [CrossRef]
- Ono, T.; Hayashi, M.; Sasaki, F.; Nakashima, T. RANKL biology: Bone metabolism, the immune system, and beyond. Inflamm. Regen. 2020, 40, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elango, J.; Bao, B.; Wu, W. The hidden secrets of soluble RANKL in bone biology. Cytokine 2021, 144, 155559. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.R.; Branstetter, D.; Manna, E.; Dougall, W.C.; Bussiere, J.; Johnson, C.W. Distribution of rank and rank ligand in normal human tissues as determined by an optimized immunohistochemical method. Appl. Immunohistochem. Mol. Morphol. 2017, 25, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Willard-Mack, C.L. Normal structure, function, and histology of lymph nodes. Toxicol. Pathol. 2006, 34, 409–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeda, T.; Kasai, M.; Utsuyama, M.; Hirokawa, K. Determination of three isoforms of the receptor activator of nuclear factor-ΚB ligand and their differential expression in bone and thymus. Endocrinology 2001, 142, 1419–1426. [Google Scholar] [CrossRef] [PubMed]
- Buckle, C.H.; de Leenheer, E.; Lawson, M.A.; Yong, K.; Rabin, N.; Perry, M.; Vanderkerken, K.; Croucher, P.I. Soluble Rank ligand produced by myeloma cells causes generalised bone loss in multiple myeloma. PLoS ONE 2012, 7, e41127. [Google Scholar] [CrossRef] [Green Version]
- Hikita, A.; Yana, I.; Wakeyama, H.; Nakamura, M.; Kadono, Y.; Oshima, Y.; Nakamura, K.; Seiki, M.; Tanaka, S. Negative regulation of osteoclastogenesis by ectodomain shedding of receptor activator of NF-ΚB ligand. J. Biol. Chem. 2006, 281, 36846–36855. [Google Scholar] [CrossRef] [Green Version]
- Mosheimer, B.A.; Kaneider, N.C.; Feistritzer, C.; Sturn, D.H.; Wiedermann, C.J. Expression and function of RANK in human monocyte chemotaxis. Arthritis Rheum. 2004, 50, 2309–2316. [Google Scholar] [CrossRef]
- Henriksen, K.; Karsdal, M.; Delaissé, J.M.; Engsig, M.T. RANKL and vascular endothelial growth factor (VEGF) induce osteoclast chemotaxis through an ERK1/2-dependent mechanism. J. Biol. Chem. 2003, 278, 48745–48753. [Google Scholar] [CrossRef] [Green Version]
- Francisconi, C.F.; Vieira, A.E.; Azevedo, M.C.S.; Tabanez, A.P.; Fonseca, A.C.; Trombone, A.P.F.; Letra, A.; Silva, R.M.; Sfeir, C.S.; Little, S.R.; et al. RANKL triggers treg-mediated immunoregulation in inflammatory osteolysis. J. Dent. Res. 2018, 97, 917–927. [Google Scholar] [CrossRef]
- Tsukasaki, M.; Komatsu, N.; Nagashima, K.; Nitta, T.; Pluemsakunthai, W.; Shukunami, C.; Iwakura, Y.; Nakashima, T.; Okamoto, K.; Takayanagi, H. Host defense against oral microbiota by bone-damaging T cells. Nat. Commun. 2018, 9, 701. [Google Scholar] [CrossRef]
- Pan, W.; Wang, Q.; Chen, Q. The cytokine network involved in the host immune response to periodontitis. Int. J. Oral Sci. 2019, 11, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groeger, S.; Meyle, J. Oral mucosal epithelial cells. Front. Immunol. 2019, 10, 208. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, C.; Suliman, S.; Almarhoumi, R.; Vega, M.E.; Rojas, C.; Monasterio, G.; Galindo, M.; Vernal, R.; Kantarci, A. Regulatory T cell phenotype and anti-osteoclastogenic function in experimental periodontitis. Sci. Rep. 2020, 10, 19018. [Google Scholar] [CrossRef]
- Tsukasaki, M. RANKL and osteoimmunology in periodontitis. J. Bone Miner. Metab. 2021, 39, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Yuce, H.B.; Gokturk, O.; Turkal, H.A.; Inanir, A.; Benli, I.; Demir, O. Assessment of local and systemic 25-hydroxy-vitamin D, RANKL, OPG, and TNF levels in patients with rheumatoid arthritis and periodontitis. J. Oral Sci. 2017, 59, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Asif, S.; Ahmad, B.; Hamza, S.A.; Taib, H.; Kassim, N.K.; Zainuddin, S.L.A. Investigation of salivary RANKL and OPG levels in periodontitis patients at hospital universiti sains malaysia. Eur. J. Dent. 2021. [Google Scholar] [CrossRef]
- Teodorescu, A.C.; Martu, I.; Teslaru, S.; Kappenberg-Nitescu, D.C.; Goriuc, A.; Luchian, I.; Martu, M.A.; Solomon, S.M.; Mârțu, S. Assessment of Salivary Levels of RANKL and OPG in Aggressive versus Chronic Periodontitis. J. Immunol. Res. 2019, 2019, 195258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanzaki, H.; Makihira, S.; Suzuki, M.; Ishii, T.; Movila, A.; Hirschfeld, J.; Mawardi, H.; Lin, X.; Han, X.; Taubman, M.A.; et al. Soluble RANKL cleaved from activated lymphocytes by TNF-α–converting enzyme contributes to osteoclastogenesis in periodontitis. J. Immunol. 2016, 197, 3871–3883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohan, M.J.; Seaton, T.; Mitchell, J.; Howe, A.; Blackburn, K.; Burkhart, W.; Moyer, M.; Patel, I.; Waitt, G.M.; Becherer, J.D.; et al. The tumor necrosis factor-α converting enzyme (TACE): A unique metalloproteinase with highly defined substrate selectivity. Biochemistry 2002, 41, 9462–9469. [Google Scholar] [CrossRef] [PubMed]
- Bostanci, N.; Emingil, G.; Afacan, B.; Han, B.; Ilgenli, T.; Atilla, G.; Hughes, F.J.; Belibasakis, G.N. Tumor Necrosis Factor-alpha-Converting Enzyme (TACE) Levels. J. Dent. Res. 2008, 87, 273–277. [Google Scholar] [CrossRef] [PubMed]
- De Almeida Guardiola, C.J.; Clemente-Napimoga, J.T.; Martinez, E.F.; Abdalla, H.B.; Peruzzo, D.C.; Joly, J.C.; Napimoga, M.H. Dc-stamp and tace levels are higher in patients with periodontitis. Braz. Dent. J. 2020, 31, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.J.; Yang, B.E.; Yoo, D.M.; Kim, S.J.; Choi, H.G.; Byun, S.H. Analysis of the relationship between periodontitis and osteoporosis/fractures: A cross-sectional study. BMC Oral Health 2021, 21, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Burska, A.N.; El-Jawhari, J.J.; Wu, J.; Wakefield, R.J.; Marzo-Ortega, H.; Conaghan, P.G.; Emery, P.; Ponchel, F.; Freeston, J.E. Receptor activator of nuclear factor kappa-β ligand (RANKL) serum levels are associated with progression to seropositive/negative rheumatoid arthritis. Clin. Exp. Rheumatol. 2021, 39, 456–462. [Google Scholar]
- Mohammadpour, A.H.; Shamsara, J.; Nazemi, S.; Ghadirzadeh, S.; Shahsavand, S.; Ramezani, M. Evaluation of RANKL/OPG serum concentration ratio as a new biomarker for coronary artery calcification: A pilot study. Thrombosis 2012, 2012, 306263. [Google Scholar] [CrossRef] [PubMed]
- Werner de Castro, G.R.; Buss, Z.D.S.; Rosa, J.S.; Facchin, B.M.; Fröde, T.S. Evaluation of bone metabolism biomarkers in paget’s disease of bone. Cureus 2019, 11, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Goranova-Marinova, V.; Goranov, S.; Pavlov, P.; Tzvetkova, T. Serum levels of OPG, RANKL and RANKL/OPG ratios in newly-diagnosed patients with multiple myeloma. Clinical correlations. Haematologica 2007, 92, 1000–1001. [Google Scholar] [CrossRef] [Green Version]
- Granchi, D.; Garaventa, A.; Amato, I.; Paolucci, P.; Baldini, N. Plasma Levels of receptor activator of nuclear factor-ΚB ligand and osteoprotegerin in patients with neuroblastoma. Int. J. Cancer 2006, 119, 146–151. [Google Scholar] [CrossRef]
- Li, X.; Liu, Y.; Wu, B.; Dong, Z.; Wang, Y.; Lu, J.; Shi, P.; Bai, W.; Wang, Z. Potential Role of the OPG/RANK/RANKL Axis in prostate cancer invasion and bone metastasis. Oncol. Rep. 2014, 32, 2605–2611. [Google Scholar] [CrossRef] [Green Version]
- Kiechl, S.; Schramek, D.; Widschwendter, M.; Fourkala, E.O.; Zaikin, A.; Jones, A.; Jaeger, B.; Rack, B.; Janni, W.; Scholz, C.; et al. Aberrant regulation of RANKL/OPG in women at high risk of developing breast cancer. Oncotarget 2017, 8, 3811–3825. [Google Scholar] [CrossRef] [Green Version]
- Owen, S.; Ye, L.; Sanders, A.J.; Mason, M.D.; Jiang, W.G. Expression profile of receptor activator of nuclear-Κb (Rank), rank ligand (rankl) and osteoprotegerin (OPG) in breast cancer. Anticancer Res. 2013, 33, 199–206. [Google Scholar]
- Chen, G.; Sircar, K.; Aprikian, A.; Potti, A.; Goltzman, D.; Rabbani, S.A. Expression of RANKL/RANK/OPG in primary and metastatic human prostate cancer as markers of disease stage and functional regulation. Cancer 2006, 107, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Meignin, V.; Quillard, J.; Meduri, G.; Guiochon-Mantel, A.; Fermand, J.P.; Milgrom, E.; Mariette, X. RANK (receptor activator of nuclear factor-ΚB) and RANKL expression in multiple myeloma. Br. J. Haematol. 2002, 117, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Casimiro, S.; Mohammad, K.S.; Pires, R.; Tato-Costa, J.; Alho, I.; Teixeira, R.; Carvalho, A.; Ribeiro, S.; Lipton, A.; Guise, T.A.; et al. RANKL/RANK/MMP-1 Molecular triad contributes to the metastatic phenotype of breast and prostate cancer cells in vitro. PLoS ONE 2013, 8, e63153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roodman, G.D.; Dougall, W.C. RANK Ligand as a therapeutic target for bone metastases and multiple myeloma. Cancer Treat. Rev. 2008, 34, 92–101. [Google Scholar] [CrossRef]
- Kiechl, S.; Wittmann, J.; Giaccari, A.; Knoflach, M.; Willeit, P.; Bozec, A.; Moschen, A.R.; Muscogiuri, G.; Sorice, G.P.; Kireva, T.; et al. Blockade of receptor activator of nuclear factor-ΚB (RANKL) signaling improves hepatic insulin resistance and prevents development of diabetes mellitus. Nat. Med. 2013, 19, 358–363. [Google Scholar] [CrossRef]
- Sarink, D.; Schock, H.; Johnson, T.; Overvad, K.; Holm, M.; Tjønneland, A.; Boutron-Ruault, M.C.; His, M.; Kvaskoff, M.; Boeing, H.; et al. Circulating RANKL and RANKL/OPG and breast cancer risk by ER and PR subtype: Results from the EPIC cohort. Cancer Prev. Res. 2017, 10, 525–534. [Google Scholar] [CrossRef] [Green Version]
- Shao, J.; Wu, L.; Leng, W.D.; Fang, C.; Zhu, Y.J.; Jin, Y.H.; Zeng, X.T. Periodontal disease and breast cancer: A meta-analysis of 173,162 participants. Front. Oncol. 2018, 8, 601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, G.W.; Kim, Y.S.; Lee, S.H.; Park, S.G.; Kim, D.H.; Cho, J.Y.; Hahm, K.B.; Hong, S.P.; Yoo, J.H. Periodontitis is associated with an increased risk for proximal colorectal neoplasms. Sci. Rep. 2019, 9, 7528. [Google Scholar] [CrossRef]
- Xuan, K.; Jha, A.R.; Zhao, T.; Uy, J.P.; Sun, C. Is periodontal disease associated with increased risk of colorectal cancer? A meta-analysis. Int. J. Dent. Hyg. 2021, 19, 50–61. [Google Scholar] [CrossRef]
- Sisay, M.; Mengistu, G.; Edessa, D. The RANK/RANKL/OPG System in tumorigenesis and metastasis of cancer stem cell: Potential targets for anticancer therapy. Onco. Targets. Ther. 2017, 10, 3801–3810. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Wang, Y.; Lu, Y.; Zhu, Q.; Xie, W.; Tang, N.; Huang, L.; An, T.; Zhang, D.; Yan, A.; et al. RANK Promotes colorectal cancer migration and invasion by activating the Ca2+-Calcineurin/NFATC1-ACP5 axis. Cell Death Dis. 2021, 12, 336. [Google Scholar] [CrossRef] [PubMed]
- Ming, J.; Cronin, S.J.F.; Penninger, J.M. Targeting the RANKL/RANK/OPG Axis for Cancer Therapy. Front. Oncol. 2020, 10, 1283. [Google Scholar] [CrossRef]
- Wu, X.; Li, F.; Dang, L.; Liang, C.; Lu, A.; Zhang, G. RANKL/RANK System-Based mechanism for breast cancer bone metastasis and related therapeutic strategies. Front. Cell Dev. Biol. 2020, 8, 76. [Google Scholar] [CrossRef] [PubMed]
- Ryan, C.; Stoltzfus, K.C.; Horn, S.; Chen, H.; Louie, A.V.; Lehrer, E.J.; Trifiletti, D.M.; Fox, E.J.; Abraham, J.A.; Zaorsky, N.G. Epidemiology of bone metastases. Bone 2020, 1, 115783. [Google Scholar] [CrossRef]
- Huang, J.-F.; Shen, J.; Li, X.; Rengan, R.; Silvestris, N.; Wang, M.; Derosa, L.; Zheng, X.; Belli, A.; Zhang, X.-L.; et al. Incidence of patients with bone metastases at diagnosis of solid tumors in adults: A large population-based study. Ann. Transl. Med. 2020, 8, 482. [Google Scholar] [CrossRef]
- Hernandez, R.K.; Wade, S.W.; Reich, A.; Pirolli, M.; Liede, A.; Lyman, G.H. Incidence of bone metastases in patients with solid tumors: Analysis of oncology electronic medical records in the United States. BMC Cancer 2018, 18, 44. [Google Scholar] [CrossRef] [Green Version]
- Togashi, Y.; Shitara, K.; Nishikawa, H. Regulatory T Cells in cancer immunosuppression—Implications for anticancer therapy. Nat. Rev. Clin. Oncol. 2019, 16, 356–371. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, Y.; Zhu, B. T-Cell exhaustion in the tumor microenvironment. Cell Death Dis. 2015, 6, e1792. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.S.; Yang, F.H.; Li, Y.; Pei, F.; Kulkarni, A.B.; Chen, Z.; Zhang, L. The Expression of PD-1 and LAG-3 in periapical lesions. Am. J. Transl. Res. 2018, 10, 2677–2684. [Google Scholar]
- Cheng, R.; Billet, S.; Liu, C.; Haldar, S.; Choudhury, D.; Tripathi, M.; Hav, M.; Merchant, A.; Hu, T.; Huang, H.; et al. Periodontal inflammation recruits distant metastatic breast cancer cells by increasing myeloid-derived suppressor cells. Oncogene 2020, 39, 1543–1556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Croucher, P.I.; McDonald, M.M.; Martin, T.J. Bone metastasis: The importance of the neighbourhood. Nat. Rev. Cancer 2016, 16, 373–386. [Google Scholar] [CrossRef] [PubMed]
- Esposito, M.; Guise, T.; Kang, Y. The biology of bone metastasis. Cold Spring Harb. Perspect. Med. 2018, 8, a031252. [Google Scholar] [CrossRef]
- Asano, T.; Okamoto, K.; Nakai, Y.; Tsutsumi, M.; Muro, R.; Suematsu, A.; Hashimoto, K.; Okamura, T.; Ehata, S.; Nitta, T.; et al. Soluble RANKL is physiologically dispensable but accelerates tumour metastasis to bone. Nat. Metab. 2019, 1, 868–875. [Google Scholar] [CrossRef]
- Tan, W.; Zhang, W.; Strasner, A.; Grivennikov, S.; Cheng, J.Q.; Hoffman, R.M.; Karin, M. Tumour-infiltrating regulatory T cells stimulate mammary cancermetastasis through RANKL-RANK signalling. Nature 2011, 470, 548–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Zhou, L.; Liu, X.; Wen, X.; Li, H.; Li, W. Meta-analysis of clinical trials to assess denosumab over zoledronic acid in bone metastasis. Int. J. Clin. Pharm. 2021, 43, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Terpos, E.; Raje, N.; Croucher, P.; Garcia-Sanz, R.; Leleu, X.; Pasteiner, W.; Wang, Y.; Glennane, A.; Canon, J.; Pawlyn, C. Denosumab compared with zoledronic acid on PFS in multiple myeloma: Exploratory results of an international phase 3 study. Blood Adv. 2021, 5, 725–736. [Google Scholar] [CrossRef]
- Okuma, S.; Matsuda, Y.; Nariai, Y.; Karino, M.; Suzuki, R.; Kanno, T. A Retrospective observational study of risk factors for denosumab-related osteonecrosis of the jaw in patients with bone metastases from solid cancers. Cancers 2020, 12, 1209. [Google Scholar] [CrossRef]
- Forner, L.; Larsen, T.; Kilian, M.; Holmstrup, P. Incidence of bacteremia after chewing, tooth brushing and scaling in individuals with periodontal inflammation. J. Clin. Periodontol. 2006, 33, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Miyajima, S.I.; Naruse, K.; Kobayashi, Y.; Nakamura, N.; Nishikawa, T.; Adachi, K.; Suzuki, Y.; Kikuchi, T.; Mitani, A.; Mizutani, M.; et al. Periodontitis-activated monocytes/macrophages cause aortic inflammation. Sci. Rep. 2014, 4, 5171. [Google Scholar] [CrossRef] [Green Version]
- Aarabi, G.; Eberhard, J.; Reissmann, D.R.; Heydecke, G.; Seedorf, U. Interaction between periodontal disease and atherosclerotic vascular disease–Fact or fiction? Atherosclerosis 2015, 241, 555–560. [Google Scholar] [CrossRef]
- Eberhard, J.; Grote, K.; Luchtefeld, M.; Heuer, W.; Schuett, H.; Divchev, D.; Scherer, R.; Schmitz-Streit, R.; Langfeldt, D.; Stumpp, N.; et al. Experimental gingivitis induces systemic inflammatory markers in young healthy individuals: A single-subject interventional study. PLoS ONE 2013, 8, e55265. [Google Scholar] [CrossRef] [PubMed]
- Hegde, R.; Awan, K.H. Effects of periodontal disease on systemic health. Disease-a-Month 2019, 65, 185–192. [Google Scholar] [CrossRef]
- Jagannathan, R.; Vamsi Lavu, S.R.R. Comparison of the proportion of non classical (CD14+ CD16+) monocytes/macrophages in peripheral blood and gingiva of healthy individuals and chronic periodontitis patients. J. Periodontol. 2013, 85, 852–858. [Google Scholar] [CrossRef] [PubMed]
- Duarte, P.M.; da Rocha, M.; Sampaio, E.; Mestnik, M.J.; Feres, M.; Figueiredo, L.C.; Bastos, M.F.; Faveri, M. Serum levels of cytokines in subjects with generalized chronic and aggressive periodontitis before and after non-surgical periodontal therapy: A pilot study. J. Periodontol. 2010, 81, 1056–1063. [Google Scholar] [CrossRef] [PubMed]
- Kampits, C.; Montenegro, M.M.; Ribeiro, I.W.J.; Furtado, M.V.; Polanczyk, C.A.; Rösing, C.K.; Haas, A.N. Periodontal disease and inflammatory blood cytokines in patients with stable coronary artery disease. J. Appl. Oral Sci. 2016, 24, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Escobar-Arregoces, F.; Latorre-Uriza, C.; Velosa-Porras, J.; Roa-Molina, N.; Ruiz, A.J.; Silva, J.; Arias, E.; Echeverri, J. Respuesta inflamatoria en mujer embarazada con alto riesgo de parto prematuro y su relacion con problemas periodontal. Acta Odontol. Latinoam. 2018, 31, 53–57. [Google Scholar] [PubMed]
- Panezai, J.; Ghaffar, A.; Altamash, M.; Sundqvist, K.G.; Engström, P.E.; Larsson, A. Correlation of serum cytokines, chemokines, growth factors and enzymes with periodontal disease parameters. PLoS ONE 2017, 12, e0188945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palomino, D.C.; Arolin., T.; Marti, L.C.; Avalheir. Chemokines and immunity. Einstein 2015, 13, 469–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zlotnik, A. Perspective: Insights on the nomenclature of cytokines and chemokines. Front. Immunol. 2020, 11, 908. [Google Scholar] [CrossRef] [PubMed]
- Holmlund, A.; Holm, G.; Lind, L. Severity of Periodontal disease and number of remaining teeth are related to the prevalence of myocardial infarction and hypertension in a study based on 4254 subjects. J. Periodontol. 2006, 77, 1173–1178. [Google Scholar] [CrossRef]
- Zhu, X.; Du, L.; Feng, J.; Ling, Y.; Xu, S. Clinicopathological and prognostic significance of serum cytokine levels in breast cancer. Clin. Lab. 2014, 60, 1145–1151. [Google Scholar] [CrossRef]
- Pan, E.Y.; Merl, M.Y.; Lin, K. The impact of corticosteroid use during anti-PD1 treatment. J. Oncol. Pharm. Pract. Off. Publ. Int. Soc. Oncol. Pharm. Pract. 2020, 26, 814–822. [Google Scholar] [CrossRef]
- Khosravi, N.; Stoner, L.; Farajivafa, V.; Hanson, E.D. Exercise training, circulating cytokine levels and immune function in cancer survivors: A meta-analysis. Brain. Behav. Immun. 2019, 81, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Seruga, B.; Zhang, H.; Bernstein, L.J.; Tannock, I.F. Cytokines and their relationship to the symptoms and outcome of cancer. Nat. Rev. Cancer 2008, 8, 887–899. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, J.; Ślebioda, T.; Kmieć, Z.; Zaucha, R. Changes in systemic immune response after stereotactic ablative radiotherapy. preliminary results of a prospective study in patients with early lung cancer. Polish Arch. Intern. Med. 2017, 127, 245–253. [Google Scholar] [CrossRef] [Green Version]
- Gazdic, M.; Simovic Markovic, B.; Jovicic, N.; Misirkic-Marjanovic, M.; Djonov, V.; Jakovljevic, V.; Arsenijevic, N.; Lukic, M.L.; Volarevic, V. Mesenchymal stem cells promote metastasis of lung cancer cells by downregulating systemic antitumor immune response. Stem Cells Int. 2017, 2017, 6294717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Liu, S.; Parajuli, K.R.; Zhang, W.; Zhang, K.; Mo, Z.; Liu, J.; Chen, Z.; Yang, S.; Wang, A.R.; et al. Interleukin-17 promotes prostate cancer via MMP7-induced epithelial-to-mesenchymal transition. Oncogene 2017, 36, 687–699. [Google Scholar] [CrossRef] [Green Version]
- Casadó-Llombart, S.; de Andrés, M.V.; Català, C.; Leyton-Pereira, A.; Lozano, F.; Bosch, E. Contribution of evolutionary selected immune gene polymorphism to immune-related disorders: The case of lymphocyte scavenger receptors Cd5 and Cd6. Int. J. Mol. Sci. 2021, 22, 5315. [Google Scholar] [CrossRef] [PubMed]
- Potrony, M.; Carreras, E.; Aranda, F.; Zimmer, L.; Puig-Butille, J.A.; Tell-Martí, G.; Armiger, N.; Sucker, A.; Giménez-Xavier, P.; Martínez-Florensa, M.; et al. Inherited functional variants of the lymphocyte receptor CD5 influence melanoma survival. Int. J. Cancer 2016, 139, 1297–1302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velasco-de Andrés, M.; Casadó-Llombart, S.; Català, C.; Leyton-Pereira, A.; Lozano, F.; Aranda, F. Soluble CD5 and CD6: Lymphocytic class i scavenger receptors as immunotherapeutic agents. Cells 2020, 9, 2589. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Xu, Q.; Zhang, P.; Michalek, S.M.; Katz, J. Phenotype and Function of Myeloidderived Suppressor Cells Induced by Porphyromonas gingivalis Infection. Infect. Immun. 2017, 85, e00213-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Health Problems Associated with Peridontal Disease | Mechanism |
|---|---|
| Cardiovascular | Elevated acute phase proteins (CRP, haptoglobin, alfa1-antitrypsin, fibrinogen) due to periodontitis Elevated CRP is associated with a higher risk of myocardial infarction and peripheral artery disease |
| Diabetes | Progression of diabetes: IL-1β and TNF-α increase insulin Endotoxins or LPS → inflammation |
| Respiratory | Aspiration of oral bacteria Enzymes secreted with saliva in periodontal disease may change mucosa and lead to higher adhesion and colonization of respiratory microbes Enzymes secreted in periodontal disease by P. gingivalis degrade salivary elements that bind pathogens and preclude them from mucosal adhesion Cytokines secreted in periodontal disease may modify respiratory epithelium |
| Problems with pregnancy | Low birth weight Preterm birth: elevated LPS stimulates placenta calls to secrete IL-1β and PGE-2 |
| Rheumatoid arthritis | P. gingivalis synthetizes citrullinated proteins causing the organism to produce anti-citrullinated proteins antibodies |
| Title of Project | ClinicalTrials.gov Identifier/Current Status | Condition | Number of Particiapnts (n) | Intervention | Primary Outcome | Secondary Outcome | Country |
|---|---|---|---|---|---|---|---|
| The Link Between Periodontitis, Smoking and Oral Cancer | NCT04047212/not yet recruiting |
| 200 |
| Periodontitis: occurrence of periodontitis or increase in the grade of an already existing case of periodontitis | Oral cancer: occurrence of a lesion of oral cancer or a premalignant lesion | Egypt |
| Postradiation Dental Disease Amongst Head and Neck Cancer Patients | NCT03703648/recruiting |
| 215 | Radiation: radiotherapy (curative) for head and neck cancer | Dental caries: the mean number of carious teeth amongst head and neck cancer patients postradiotherapy |
| UK |
| Cytokine Profiles in Breast Cancer Patients Undergoing Chemotherapy | NCT03244943/completed |
| 40 | Procedure: nonsurgical periodontal treatment | Cytokines: cytokine levels and changes before and after posttreatment | Correlation of cytokines: cytokine levels between parameters clinical | Brazil |
| Oral Health in Breast Cancer Survivors on Aromatase Inhibitors | NCT01693731/completed |
| 300 | ND | Periodontal diseases |
| USA |
| Towards a Viral Etiology of Periodontal Disease in Relation to Radiotherapy Treatment of Head and Neck Cancers | NCT02180932/completed | Periodontal disease | 25 | Biological: periodontal pocket samples | Measure of level of EBV nucleic acids Measure of level of EBV nucleic acids | NA | France |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sobocki, B.K.; Basset, C.A.; Bruhn-Olszewska, B.; Olszewski, P.; Szot, O.; Kaźmierczak-Siedlecka, K.; Guziak, M.; Nibali, L.; Leone, A. Molecular Mechanisms Leading from Periodontal Disease to Cancer. Int. J. Mol. Sci. 2022, 23, 970. https://doi.org/10.3390/ijms23020970
Sobocki BK, Basset CA, Bruhn-Olszewska B, Olszewski P, Szot O, Kaźmierczak-Siedlecka K, Guziak M, Nibali L, Leone A. Molecular Mechanisms Leading from Periodontal Disease to Cancer. International Journal of Molecular Sciences. 2022; 23(2):970. https://doi.org/10.3390/ijms23020970
Chicago/Turabian StyleSobocki, Bartosz Kamil, Charbel A. Basset, Bożena Bruhn-Olszewska, Paweł Olszewski, Olga Szot, Karolina Kaźmierczak-Siedlecka, Mateusz Guziak, Luigi Nibali, and Angelo Leone. 2022. "Molecular Mechanisms Leading from Periodontal Disease to Cancer" International Journal of Molecular Sciences 23, no. 2: 970. https://doi.org/10.3390/ijms23020970
APA StyleSobocki, B. K., Basset, C. A., Bruhn-Olszewska, B., Olszewski, P., Szot, O., Kaźmierczak-Siedlecka, K., Guziak, M., Nibali, L., & Leone, A. (2022). Molecular Mechanisms Leading from Periodontal Disease to Cancer. International Journal of Molecular Sciences, 23(2), 970. https://doi.org/10.3390/ijms23020970