
Pyroptosis-Mediated Periodontal Disease

 ,
,  and
and
Abstract
:1. Introduction
2. Methods
3. Results
4. Discussion
4.1. Part 1—Updated Knowledge of Pyroptosis Inflammatory Pathways
4.1.1. Innate Immune System and Inflammation
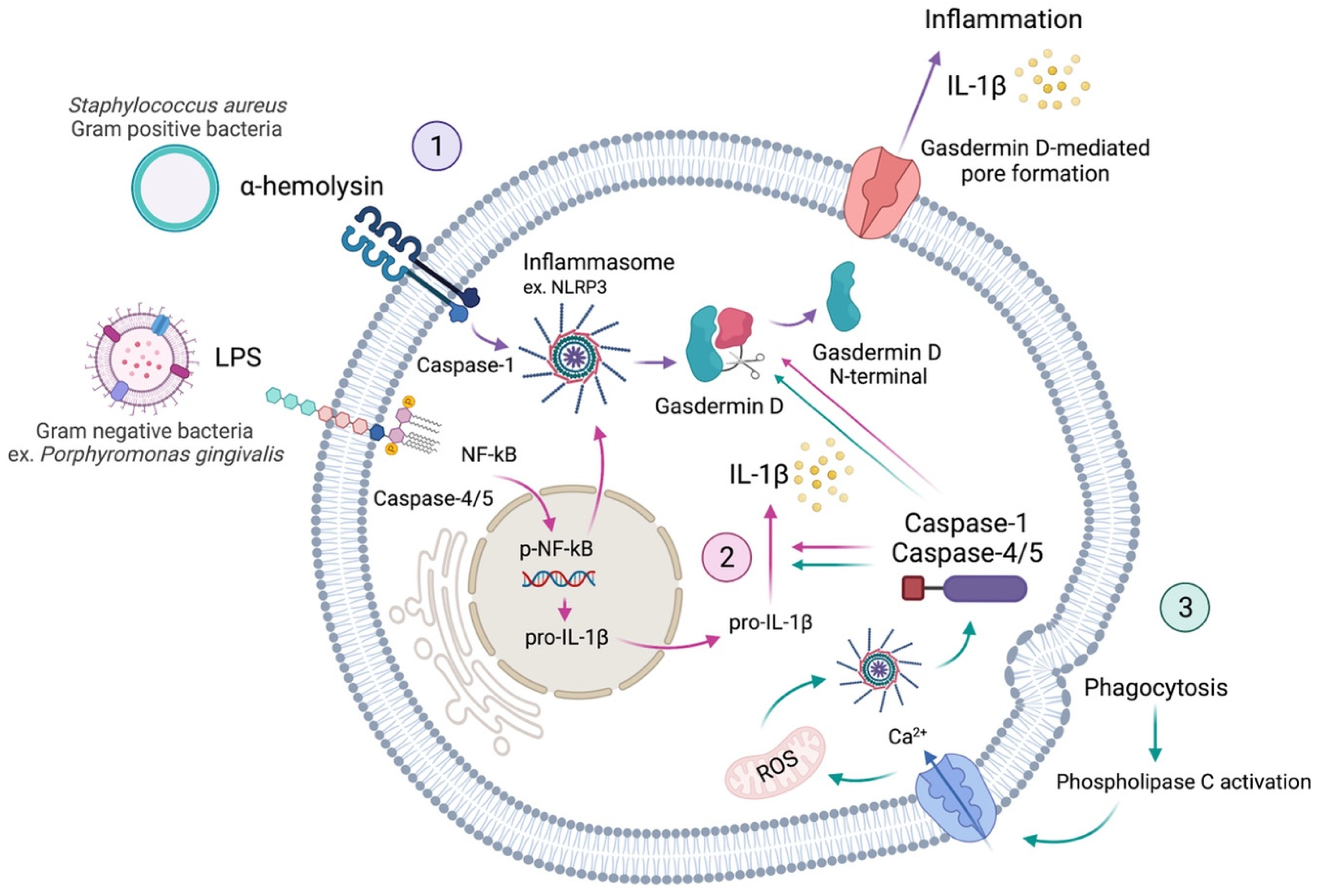
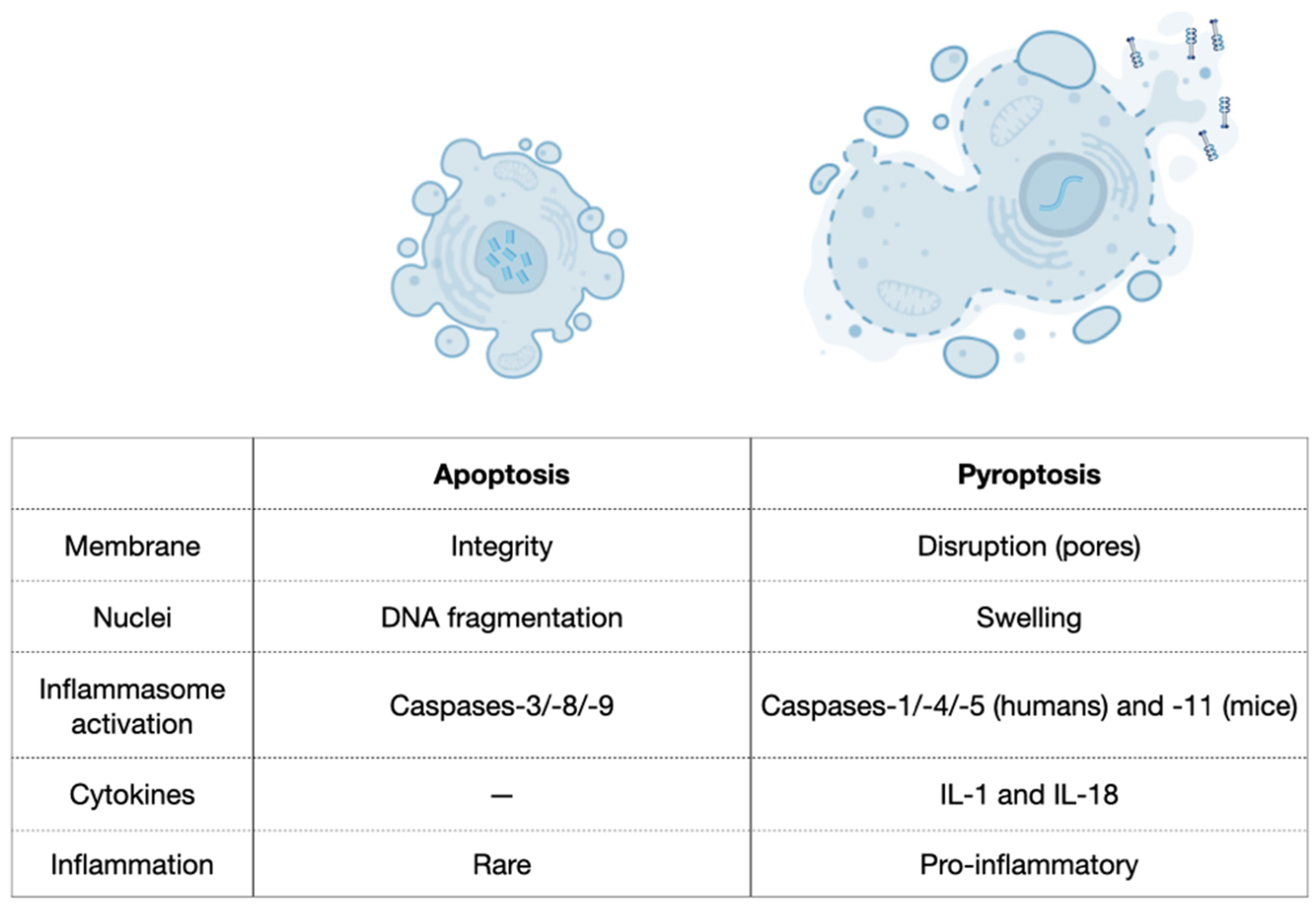
4.1.2. Pyroptosis
4.1.3. Virulence Factors
4.1.4. Inflammasomes
4.1.5. Caspases
4.1.6. Gasdermin D
4.1.7. Interleukins
4.1.8. Clinical Relevance
4.2. Part 2—Pyroptosis on the Periodontal Diseases and Periodontal Therapy
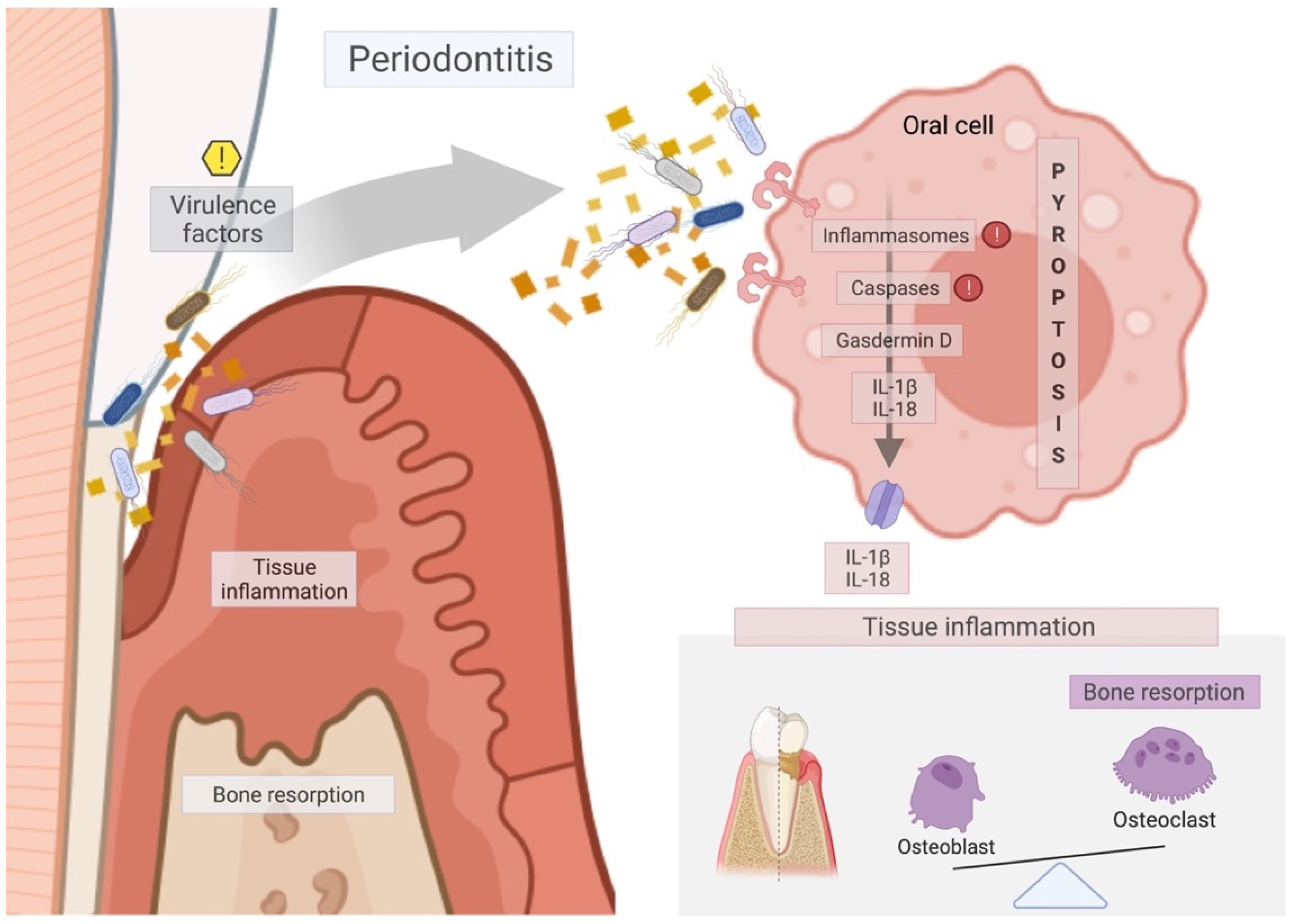
4.2.1. Clinical and In Vivo Pieces of Evidence of Pyroptosis on the Periodontal Tissues
4.2.2. In Vitro Research on Pyroptosis on the Periodontal Disease
4.2.3. Virulence Factors Associated with Pyroptosis on the Periodontal Disease
4.2.4. Systemic Disorders Associated with the Periodontal Disease through Pyroptosis
4.2.5. Therapeutic Approaches for Pyroptosis-Related Periodontal Disease
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Oka, S.; Li, X.; Sato, F.; Zhang, F.; Tewari, N.; Kim, I.-S.; Zhong, L.; Hamada, N.; Makishima, M.; Liu, Y.; et al. A Deficiency of Dec2 Triggers Periodontal Inflammation and Pyroptosis. J. Periodontal Res. 2021, 56, 492–500. [Google Scholar] [CrossRef]
- Chen, Q.; Liu, X.; Wang, D.; Zheng, J.; Chen, L.; Xie, Q.; Liu, X.; Niu, S.; Qu, G.; Lan, J.; et al. Periodontal Inflammation-Triggered by Periodontal Ligament Stem Cell Pyroptosis Exacerbates Periodontitis. Front. Cell Dev. Biol. 2021, 9, 663037. [Google Scholar] [CrossRef]
- Cheng, R.; Feng, Y.; Zhang, R.; Liu, W.; Lei, L.; Hu, T. The Extent of Pyroptosis Varies in Different Stages of Apical Periodontitis. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 226–237. [Google Scholar] [CrossRef]
- Zhang, X.; He, S.; Lu, W.; Lin, L.; Xiao, H. Glycogen Synthase Kinase-3β (GSK-3β) Deficiency Inactivates the NLRP3 Inflammasome-Mediated Cell Pyroptosis in LPS-Treated Periodontal Ligament Cells (PDLCs). In Vitro Cell. Dev. Biol. Anim. 2021, 57, 404–414. [Google Scholar] [CrossRef]
- Yu, C.; Zhang, C.; Kuang, Z.; Zheng, Q. The Role of NLRP3 Inflammasome Activities in Bone Diseases and Vascular Calcification. Inflammation 2021, 44, 434–449. [Google Scholar] [CrossRef]
- Lu, W.L.; Song, D.Z.; Yue, J.L.; Wang, T.T.; Zhou, X.D.; Zhang, P.; Zhang, L.; Huang, D.M. NLRP3 Inflammasome May Regulate Inflammatory Response of Human Periodontal Ligament Fibroblasts in an Apoptosis-Associated Speck-like Protein Containing a CARD (ASC)-Dependent Manner. Int. Endod. J. 2017, 50, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Domon, H.; Takahashi, N.; Honda, T.; Nakajima, T.; Tabeta, K.; Abiko, Y.; Yamazaki, K. Up-Regulation of the Endoplasmic Reticulum Stress-Response in Periodontal Disease. Clin. Chim. Acta 2009, 401, 134–140. [Google Scholar] [CrossRef] [PubMed]
- De Andrade, K.Q.; Almeida-da-Silva, C.L.C.; Coutinho-Silva, R. Immunological Pathways Triggered by Porphyromonas gingivalis and Fusobacterium nucleatum: Therapeutic Possibilities? Mediat. Inflamm. 2019, 2019, 7241312. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Huang, X. Methodology for Comprehensive Detection of Pyroptosis. Methods Mol. Biol. 2021, 2255, 149–157. [Google Scholar] [CrossRef]
- Liu, S.; Du, J.; Li, D.; Yang, P.; Kou, Y.; Li, C.; Zhou, Q.; Lu, Y.; Hasegawa, T.; Li, M. Oxidative Stress Induced Pyroptosis Leads to Osteogenic Dysfunction of MG63 Cells. J. Mol. Histol. 2020, 51, 221–232. [Google Scholar] [CrossRef]
- Li, J.; Wang, X.; Mei, K.-C.; Chang, C.H.; Jiang, J.; Liu, X.; Liu, Q.; Guiney, L.M.; Hersam, M.C.; Liao, Y.-P.; et al. Lateral Size of Graphene Oxide Determines Differential Cellular Uptake and Cell Death Pathways in Kupffer Cells, LSECs, and Hepatocytes. Nano Today 2021, 37, 101061. [Google Scholar] [CrossRef] [PubMed]
- Spel, L.; Martinon, F. Inflammasomes Contributing to Inflammation in Arthritis. Immunol. Rev. 2020, 294, 48–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Cao, M.; Ge, H. Knockdown of MALAT1 Inhibits the Progression of Chronic Periodontitis via Targeting MiR-769-5p/HIF3A Axis. BioMed Res. Int. 2021, 2021, 8899863. [Google Scholar] [CrossRef]
- Cheng, R.; Liu, W.; Zhang, R.; Feng, Y.; Bhowmick, N.A.; Hu, T. Porphyromonas Gingivalis-Derived Lipopolysaccharide Combines Hypoxia to Induce Caspase-1 Activation in Periodontitis. Front. Cell. Infect. Microbiol. 2017, 7, 474. [Google Scholar] [CrossRef] [PubMed]
- Cecil, J.D.; O’Brien-Simpson, N.M.; Lenzo, J.C.; Holden, J.A.; Singleton, W.; Perez-Gonzalez, A.; Mansell, A.; Reynolds, E.C. Outer Membrane Vesicles Prime and Activate Macrophage Inflammasomes and Cytokine Secretion In Vitro and In Vivo. Front. Immunol. 2017, 8, 1017. [Google Scholar] [CrossRef] [Green Version]
- Fleetwood, A.J.; Lee, M.K.S.; Singleton, W.; Achuthan, A.; Lee, M.-C.; O’Brien-Simpson, N.M.; Cook, A.D.; Murphy, A.J.; Dashper, S.G.; Reynolds, E.C.; et al. Metabolic Remodeling, Inflammasome Activation, and Pyroptosis in Macrophages Stimulated by Porphyromonas gingivalis and Its Outer Membrane Vesicles. Front. Cell. Infect. Microbiol. 2017, 7, 351. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.M.; Kennedy, D.J.; Morton, R.E.; Febbraio, M. CD36/SR-B2-TLR2 Dependent Pathways Enhance Porphyromonas gingivalis Mediated Atherosclerosis in the Ldlr KO Mouse Model. PLoS ONE 2015, 10, e0125126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taxman, D.J.; Swanson, K.V.; Broglie, P.M.; Wen, H.; Holley-Guthrie, E.; Huang, M.T.-H.; Callaway, J.B.; Eitas, T.K.; Duncan, J.A.; Ting, J.P.Y. Porphyromonas gingivalis Mediates Inflammasome Repression in Polymicrobial Cultures through a Novel Mechanism Involving Reduced Endocytosis. J. Biol. Chem. 2012, 287, 32791–32799. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Du, C.; Xu, L. Circ_0081572 Inhibits the Progression of Periodontitis through Regulating the MiR-378h/RORA Axis. Arch. Oral Biol. 2021, 124, 105053. [Google Scholar] [CrossRef]
- Liu, P.; Cui, L.; Shen, L. Knockdown of TRIM52 Alleviates LPS-Induced Inflammatory Injury in Human Periodontal Ligament Cells through the TLR4/NF-ΚB Pathway. Biosci. Rep. 2020, 40, BSR20201223. [Google Scholar] [CrossRef]
- Zhang, K.; He, S.; Dai, Z.; Cao, L.; Yue, S.; Bai, Y.; Zheng, M. Axin 1 Knockdown Inhibits Osteoblastic Apoptosis Induced by Porphyromonas gingivalis Lipopolysaccharide. Arch. Oral Biol. 2020, 112, 104667. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, H.; Zhang, G.; He, Y.; Zhang, P.; Sun, Z.; Gao, Y.; Tan, Y. Calcitonin Gene-related Peptide Reduces Porphyromonas gingivalis LPS-induced TNF-α Release and Apoptosis in Osteoblasts. Mol. Med. Rep. 2018, 17, 3246–3254. [Google Scholar] [CrossRef] [Green Version]
- Shirasugi, M.; Nishioka, K.; Yamamoto, T.; Nakaya, T.; Kanamura, N. Normal Human Gingival Fibroblasts Undergo Cytostasis and Apoptosis after Long-Term Exposure to Butyric Acid. Biochem. Biophys. Res. Commun. 2017, 482, 1122–1128. [Google Scholar] [CrossRef]
- Zhu, X.; Lu, W.; Chen, Y.; Cheng, X.; Qiu, J.; Xu, Y.; Sun, Y. Effects of Porphyromonas gingivalis LipopolysaccharideTolerized Monocytes on Inflammatory Responses in Neutrophils. PLoS ONE 2016, 11, e0161482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deepak, V.; Kasonga, A.; Kruger, M.C.; Coetzee, M. Carvacrol Inhibits Osteoclastogenesis and Negatively Regulates the Survival of Mature Osteoclasts. Biol. Pharm. Bull. 2016, 39, 1150–1158. [Google Scholar] [CrossRef] [Green Version]
- Jönsson, D.; Nilsson, B.-O. The Antimicrobial Peptide LL-37 Is Anti-Inflammatory and Proapoptotic in Human Periodontal Ligament Cells. J. Periodontal Res. 2012, 47, 330–335. [Google Scholar] [CrossRef]
- Zaric, S.; Shelburne, C.; Darveau, R.; Quinn, D.J.; Weldon, S.; Taggart, C.C.; Coulter, W.A. Impaired Immune Tolerance to Porphyromonas gingivalis Lipopolysaccharide Promotes Neutrophil Migration and Decreased Apoptosis. Infect. Immun. 2010, 78, 4151–4156. [Google Scholar] [CrossRef] [Green Version]
- Thammasitboon, K.; Goldring, S.R.; Boch, J.A. Role of Macrophages in LPS-Induced Osteoblast and PDL Cell Apoptosis. Bone 2006, 38, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Bateman, G.; Hill, B.; Knight, R.; Boucher, D. Great Balls of Fire: Activation and Signalling of Inflammatory Caspases. Biochem. Soc. Trans. 2021, 49, BST20200986. [Google Scholar] [CrossRef]
- Dahlen, G.; Basic, A.; Bylund, J. Importance of Virulence Factors for the Persistence of Oral Bacteria in the Inflamed Gingival Crevice and in the Pathogenesis of Periodontal Disease. J. Clin. Med. 2019, 8, 1339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, H.; Kawai, T.; Akira, S. Pathogen Recognition by the Innate Immune System. Int. Rev. Immunol. 2011, 30, 16–34. [Google Scholar] [CrossRef]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of Assembly, Regulation and Signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Wang, L.; Sharif, H.; Vora, S.M.; Zheng, Y.; Wu, H. Structures and Functions of the Inflammasome Engine. J. Allergy Clin. Immunol. 2021, 147, 2021–2029. [Google Scholar] [CrossRef] [PubMed]
- Miao, E.A.; Leaf, I.A.; Treuting, P.M.; Mao, D.P.; Dors, M.; Sarkar, A.; Warren, S.E.; Wewers, M.D.; Aderem, A. Caspase-1-Induced Pyroptosis Is an Innate Immune Effector Mechanism against Intracellular Bacteria. Nat. Immunol. 2010, 11, 1136–1142. [Google Scholar] [CrossRef] [PubMed]
- Miao, E.A.; Rajan, J.V.; Aderem, A. Caspase-1 Induced Pyroptotic Cell Death. Immunol. Rev. 2011, 243, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Duprez, L.; Wirawan, E.; Vanden Berghe, T.; Vandenabeele, P. Major Cell Death Pathways at a Glance. Microbes Infect. 2009, 11, 1050–1062. [Google Scholar] [CrossRef] [PubMed]
- Guan, R.; Chen, Y.; Zeng, L.; Rees, T.W.; Jin, C.; Huang, J.; Chen, Z.-S.; Ji, L.; Chao, H. Oncosis-Inducing Cyclometalated Iridium(Iii) Complexes. Chem. Sci. 2018, 9, 5183–5190. [Google Scholar] [CrossRef] [Green Version]
- Declercq, W.; Vanden Berghe, T.; Vandenabeele, P. RIP Kinases at the Crossroads of Cell Death and Survival. Cell 2009, 138, 229–232. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; He, W.-T.; Hu, L.; Li, J.; Fang, Y.; Wang, X.; Xu, X.; Wang, Z.; Huang, K.; Han, J. Pyroptosis Is Driven by Non-Selective Gasdermin-D Pore and Its Morphology Is Different from MLKL Channel-Mediated Necroptosis. Cell Res. 2016, 26, 1007–1020. [Google Scholar] [CrossRef]
- Remijsen, Q.; Kuijpers, T.W.; Wirawan, E.; Lippens, S.; Vandenabeele, P.; Vanden Berghe, T. Dying for a Cause: NETosis, Mechanisms behind an Antimicrobial Cell Death Modality. Cell Death Differ. 2011, 18, 581–588. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Non-Apoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host Cell Death and Inflammation. Nat. Rev. Microbiol. 2009, 7, 99–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fink, S.L.; Cookson, B.T. Caspase-1-Dependent Pore Formation during Pyroptosis Leads to Osmotic Lysis of Infected Host Macrophages. Cell. Microbiol. 2006, 8, 1812–1825. [Google Scholar] [CrossRef]
- Zychlinsky, A.; Prevost, M.C.; Sansonetti, P.J. Shigella flexneri Induces Apoptosis in Infected Macrophages. Nature 1992, 358, 167–169. [Google Scholar] [CrossRef]
- Miao, E.A.; Scherer, C.A.; Tsolis, R.M.; Kingsley, R.A.; Adams, L.G.; Bäumler, A.J.; Miller, S.I. Salmonella typhimurium Leucine-Rich Repeat Proteins Are Targeted to the SPI1 and SPI2 Type III Secretion Systems. Mol. Microbiol. 1999, 34, 850–864. [Google Scholar] [CrossRef] [Green Version]
- Cookson, B.T.; Brennan, M.A. Pro-Inflammatory Programmed Cell Death. Trends Microbiol. 2001, 9, 113–114. [Google Scholar] [CrossRef]
- Liu, W.; Liu, J.; Wang, W.; Wang, Y.; Ouyang, X. NLRP6 Induces Pyroptosis by Activation of Caspase-1 in Gingival Fibroblasts. J. Dent. Res. 2018, 97, 1391–1398. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, Y.; Liu, Q.; Zheng, Q.; Dong, X.; Liu, X.; Gao, W.; Bai, X.; Li, Z. Caspase-1-Dependent Pyroptosis of Peripheral Blood Mononuclear Cells Is Associated with the Severity and Mortality of Septic Patients. BioMed Res. Int. 2020, 2020, 9152140. [Google Scholar] [CrossRef] [Green Version]
- Fantuzzi, G.; Dinarello, C.A. Interleukin-18 and Interleukin-1 Beta: Two Cytokine Substrates for ICE (Caspase-1). J. Clin. Immunol. 1999, 19, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Watson, P.R.; Gautier, A.V.; Paulin, S.M.; Bland, A.P.; Jones, P.W.; Wallis, T.S. Salmonella enterica Serovars Typhimurium and Dublin Can Lyse Macrophages by a Mechanism Distinct from Apoptosis. Infect. Immun. 2000, 68, 3744–3747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sendler, M.; Mayerle, J.; Lerch, M.M. Necrosis, Apoptosis, Necroptosis, Pyroptosis: It Matters How Acinar Cells Die During Pancreatitis. Cell. Mol. Gastroenterol. Hepatol. 2016, 2, 407–408. [Google Scholar] [CrossRef] [Green Version]
- Lamkanfi, M.; Dixit, V.M. Inflammasomes and Their Roles in Health and Disease. Annu. Rev. Cell Dev. Biol. 2012, 28, 137–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincent, W.J.B.; Freisinger, C.M.; Lam, P.-Y.; Huttenlocher, A.; Sauer, J.-D. Macrophages Mediate Flagellin Induced Inflammasome Activation and Host Defense in Zebrafish. Cell. Microbiol. 2016, 18, 591–604. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Zhou, W.; Wang, H.; Liang, S. Roles of Porphyromonas gingivalis and Its Virulence Factors in Periodontitis. Adv. Protein Chem. Struct. Biol. 2020, 120, 45–84. [Google Scholar] [CrossRef]
- Bostanci, N.; Emingil, G.; Saygan, B.; Turkoglu, O.; Atilla, G.; Curtis, M.A.; Belibasakis, G.N. Expression and Regulation of the NALP3 Inflammasome Complex in Periodontal Diseases. Clin. Exp. Immunol. 2009, 157, 415–422. [Google Scholar] [CrossRef] [Green Version]
- Muñoz-Planillo, R.; Franchi, L.; Miller, L.S.; Núñez, G. A Critical Role for Hemolysins and Bacterial Lipoproteins in Staphylococcus aureus-Induced Activation of the Nlrp3 Inflammasome. J. Immunol. 2009, 183, 3942–3948. [Google Scholar] [CrossRef] [Green Version]
- Soong, G.; Chun, J.; Parker, D.; Prince, A. Staphylococcus aureus Activation of Caspase 1/Calpain Signaling Mediates Invasion through Human Keratinocytes. J. Infect. Dis. 2012, 205, 1571–1579. [Google Scholar] [CrossRef] [Green Version]
- Hruz, P.; Zinkernagel, A.S.; Jenikova, G.; Botwin, G.J.; Hugot, J.-P.; Karin, M.; Nizet, V.; Eckmann, L. NOD2 Contributes to Cutaneous Defense against Staphylococcus aureus through Alpha-Toxin-Dependent Innate Immune Activation. Proc. Natl. Acad. Sci. USA 2009, 106, 12873–12878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, A.D.; Bubeck Wardenburg, J.; Gardner, D.J.; Long, D.; Whitney, A.R.; Braughton, K.R.; Schneewind, O.; DeLeo, F.R. Targeting of Alpha-Hemolysin by Active or Passive Immunization Decreases Severity of USA300 Skin Infection in a Mouse Model. J. Infect. Dis. 2010, 202, 1050–1058. [Google Scholar] [CrossRef] [Green Version]
- Miller, L.S.; Pietras, E.M.; Uricchio, L.H.; Hirano, K.; Rao, S.; Lin, H.; O’Connell, R.M.; Iwakura, Y.; Cheung, A.L.; Cheng, G.; et al. Inflammasome-Mediated Production of IL-1beta Is Required for Neutrophil Recruitment against Staphylococcus aureus In Vivo. J. Immunol. 2007, 179, 6933–6942. [Google Scholar] [CrossRef] [Green Version]
- Muñoz-Planillo, R.; Kuffa, P.; Martínez-Colón, G.; Smith, B.L.; Rajendiran, T.M.; Núñez, G. K+ Efflux Is the Common Trigger of NLRP3 Inflammasome Activation by Bacterial Toxins and Particulate Matter. Immunity 2013, 38, 1142–1153. [Google Scholar] [CrossRef] [Green Version]
- Pressman, B.C. Biological Applications of Ionophores. Annu. Rev. Biochem. 1976, 45, 501–530. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Weiss, D.S.; Newton, K.; McBride, J.; O’Rourke, K.; Roose-Girma, M.; Lee, W.P.; Weinrauch, Y.; Monack, D.M.; Dixit, V.M. Cryopyrin Activates the Inflammasome in Response to Toxins and ATP. Nature 2006, 440, 228–232. [Google Scholar] [CrossRef]
- He, W.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.-H.; Zhong, C.-Q.; Han, J. Gasdermin D Is an Executor of Pyroptosis and Required for Interleukin-1β Secretion. Cell Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by Inflammatory Caspases Determines Pyroptotic Cell Death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 Cleaves Gasdermin D for Non-Canonical Inflammasome Signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Pihlstrom, B.L.; Michalowicz, B.S.; Johnson, N.W. Periodontal Diseases. Lancet 2005, 366, 1809–1820. [Google Scholar] [CrossRef] [Green Version]
- Hajishengallis, G. Periodontitis: From Microbial Immune Subversion to Systemic Inflammation. Nat. Rev. Immunol. 2015, 15, 30–44. [Google Scholar] [CrossRef] [PubMed]
- Rocha, F.R.G.; Delitto, A.E.; de Souza, J.A.C.; González-Maldonado, L.A.; Wallet, S.M.; Rossa Junior, C. Relevance of Caspase-1 and Nlrp3 Inflammasome on Inflammatory Bone Resorption in A Murine Model of Periodontitis. Sci. Rep. 2020, 10, 7823. [Google Scholar] [CrossRef]
- Bullon, P.; Navarro, J.M. Inflammasome as a Key Pathogenic Mechanism in Endometriosis. Curr. Drug Targets 2017, 18, 997–1002. [Google Scholar] [CrossRef]
- Lu, A.; Magupalli, V.G.; Ruan, J.; Yin, Q.; Atianand, M.K.; Vos, M.R.; Schröder, G.F.; Fitzgerald, K.A.; Wu, H.; Egelman, E.H. Unified Polymerization Mechanism for the Assembly of ASC-Dependent Inflammasomes. Cell 2014, 156, 1193–1206. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, M.; Moehring, D.; Muñoz-Planillo, R.; Núñez, G.; Callaway, J.; Ting, J.; Scurria, M.; Ugo, T.; Bernad, L.; Cali, J.; et al. A Bioluminescent Caspase-1 Activity Assay Rapidly Monitors Inflammasome Activation in Cells. J. Immunol. Methods 2017, 447, 1–13. [Google Scholar] [CrossRef]
- Hörauf, J.-A.; Kany, S.; Janicova, A.; Xu, B.; Vrdoljak, T.; Sturm, R.; Dunay, I.R.; Martin, L.; Relja, B. Short Exposure to Ethanol Diminishes Caspase-1 and ASC Activation in Human HepG2 Cells In Vitro. Int. J. Mol. Sci. 2020, 21, 3196. [Google Scholar] [CrossRef]
- Yang, K.; Xu, S.; Zhao, H.; Liu, L.; Lv, X.; Hu, F.; Wang, L.; Ji, Q. Hypoxia and Porphyromonas gingivalis-Lipopolysaccharide Synergistically Induce NLRP3 Inflammasome Activation in Human Gingival Fibroblasts. Int. Immunopharmacol. 2021, 94, 107456. [Google Scholar] [CrossRef]
- Kovacs, S.B.; Miao, E.A. Gasdermins: Effectors of Pyroptosis. Trends Cell Biol. 2017, 27, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Delaleu, N.; Bickel, M. Interleukin-1 Beta and Interleukin-18: Regulation and Activity in Local Inflammation. Periodontol. 2000 2004, 35, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Walle, L.V.; Van Opdenbosch, N.; Jacques, P.; Fossoul, A.; Verheugen, E.; Vogel, P.; Beyaert, R.; Elewaut, D.; Kanneganti, T.-D.; van Loo, G.; et al. Negative Regulation of the NLRP3 Inflammasome by A20 Protects against Arthritis. Nature 2014, 512, 69–73. [Google Scholar] [CrossRef] [Green Version]
- Nakanishi, K.; Yoshimoto, T.; Tsutsui, H.; Okamura, H. Interleukin-18 Regulates Both Th1 and Th2 Responses. Annu. Rev. Immunol. 2001, 19, 423–474. [Google Scholar] [CrossRef] [PubMed]
- Bui, F.Q.; Johnson, L.; Roberts, J.; Hung, S.-C.; Lee, J.; Atanasova, K.R.; Huang, P.-R.; Yilmaz, Ö.; Ojcius, D.M. Fusobacterium nucleatum Infection of Gingival Epithelial Cells Leads to NLRP3 Inflammasome-Dependent Secretion of IL-1β and the Danger Signals ASC and HMGB1. Cell. Microbiol. 2016, 18, 970–981. [Google Scholar] [CrossRef] [Green Version]
- Simon, A.; van der Meer, J.W.M. Pathogenesis of Familial Periodic Fever Syndromes or Hereditary Autoinflammatory Syndromes. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R86–R98. [Google Scholar] [CrossRef] [Green Version]
- Olsen, I. Porphyromonas gingivalis-Induced Neuroinflammation in Alzheimer’s Disease. Front. Neurosci. 2021, 15, 691016. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhao, Y.; Zhang, J.; Yang, G. Mechanisms of NLRP3 Inflammasome Activation: Its Role in the Treatment of Alzheimer’s Disease. Neurochem. Res. 2020, 45, 2560–2572. [Google Scholar] [CrossRef]
- Schenkein, H.A.; Papapanou, P.N.; Genco, R.; Sanz, M. Mechanisms Underlying the Association between Periodontitis and Atherosclerotic Disease. Periodontol. 2000 2020, 83, 90–106. [Google Scholar] [CrossRef] [PubMed]
- Kong, R.; Sun, L.; Li, H.; Wang, D. The Role of NLRP3 Inflammasome in the Pathogenesis of Rheumatic Disease. Autoimmunity 2021, 1–7. [Google Scholar] [CrossRef]
- Imai, J.; Ichikawa, H.; Kitamoto, S.; Golob, J.L.; Kaneko, M.; Nagata, J.; Takahashi, M.; Gillilland, M.G.; Tanaka, R.; Nagao-Kitamoto, H.; et al. A Potential Pathogenic Association between Periodontal Disease and Crohn’s Disease. JCI Insight 2021, 6, e148543. [Google Scholar] [CrossRef] [PubMed]
- Cobelli, N.; Scharf, B.; Crisi, G.M.; Hardin, J.; Santambrogio, L. Mediators of the Inflammatory Response to Joint Replacement Devices. Nat. Rev. Rheumatol. 2011, 7, 600–608. [Google Scholar] [CrossRef] [PubMed]
- Chadha, S.; Behl, T.; Bungau, S.; Kumar, A.; Arora, R.; Gupta, A.; Uddin, M.S.; Zengin, G.; Aleya, L.; Setia, D.; et al. Mechanistic Insights into the Role of Pyroptosis in Rheumatoid Arthritis. Curr. Res. Transl. Med. 2020, 68, 151–158. [Google Scholar] [CrossRef]
- Burska, A.; Boissinot, M.; Ponchel, F. Cytokines as Biomarkers in Rheumatoid Arthritis. Mediat. Inflamm. 2014, 2014, 545493. [Google Scholar] [CrossRef]
- Tan, G.; Huang, C.; Chen, J.; Chen, B.; Zhi, F. Gasdermin-E-Mediated Pyroptosis Participates in the Pathogenesis of Crohn’s Disease by Promoting Intestinal Inflammation. Cell Rep. 2021, 35, 109265. [Google Scholar] [CrossRef]
- Zhou, L.; Liu, T.; Huang, B.; Luo, M.; Chen, Z.; Zhao, Z.; Wang, J.; Leung, D.; Yang, X.; Chan, K.W.; et al. Excessive Deubiquitination of NLRP3-R779C Variant Contributes to Very-Early-Onset Inflammatory Bowel Disease Development. J. Allergy Clin. Immunol. 2021, 147, 267–279. [Google Scholar] [CrossRef]
- Xue, F.; Shu, R.; Xie, Y. The Expression of NLRP3, NLRP1 and AIM2 in the Gingival Tissue of Periodontitis Patients: RT-PCR Study and Immunohistochemistry. Arch. Oral Biol. 2015, 60, 948–958. [Google Scholar] [CrossRef]
- Li, Y.; Li, B.; Liu, Y.; Wang, H.; He, M.; Liu, Y.; Sun, Y.; Meng, W. Porphyromonas gingivalis Lipopolysaccharide Affects Oral Epithelial Connections via Pyroptosis. J. Dent. Sci. 2021, 16, 1255–1263. [Google Scholar] [CrossRef]
- Jun, H.-K.; Jung, Y.-J.; Ji, S.; An, S.-J.; Choi, B.-K. Caspase-4 Activation by a Bacterial Surface Protein Is Mediated by Cathepsin G in Human Gingival Fibroblasts. Cell Death Differ. 2018, 25, 380–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Chen, K.; Wan, X.; Wang, F.; Guo, Z.; Mo, Z. NLRP3 Inflammasome Activation in Mesenchymal Stem Cells Inhibits Osteogenic Differentiation and Enhances Adipogenic Differentiation. Biochem. Biophys. Res. Commun. 2017, 484, 871–877. [Google Scholar] [CrossRef] [PubMed]
- Ji, S.; Choi, Y. Microbial and Host Factors That Affect Bacterial Invasion of the Gingiva. J. Dent. Res. 2020, 99, 1013–1020. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Kurita-Ochiai, T.; Kobayashi, R.; Suzuki, T.; Ando, T. Activation of the NLRP3 Inflammasome in Porphyromonas gingivalis-Accelerated Atherosclerosis. Pathog. Dis. 2015, 73, ftv011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.K.; Dhasmana, N.; Dubey, N.; Kumar, N.; Gangwal, A.; Gupta, M.; Singh, Y. Bacterial Virulence Factors: Secreted for Survival. Indian J. Microbiol. 2017, 57, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Bostanci, N.; Belibasakis, G.N. Porphyromonas gingivalis: An Invasive and Evasive Opportunistic Oral Pathogen. FEMS Microbiol. Lett. 2012, 333, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Thay, B.; Damm, A.; Kufer, T.A.; Wai, S.N.; Oscarsson, J. Aggregatibacter actinomycetemcomitans Outer Membrane Vesicles Are Internalized in Human Host Cells and Trigger NOD1- and NOD2-Dependent NF-ΚB Activation. Infect. Immun. 2014, 82, 4034–4046. [Google Scholar] [CrossRef] [Green Version]
- Kieselbach, T.; Zijnge, V.; Granström, E.; Oscarsson, J. Proteomics of Aggregatibacter actinomycetemcomitans Outer Membrane Vesicles. PLoS ONE 2015, 10, e0138591. [Google Scholar] [CrossRef] [Green Version]
- Vanaja, S.K.; Russo, A.J.; Behl, B.; Banerjee, I.; Yankova, M.; Deshmukh, S.D.; Rathinam, V.A.K. Bacterial Outer Membrane Vesicles Mediate Cytosolic Localization of LPS and Caspase-11 Activation. Cell 2016, 165, 1106–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demirel, I.; Persson, A.; Brauner, A.; Särndahl, E.; Kruse, R.; Persson, K. Activation of NLRP3 by Uropathogenic Escherichia coli Is Associated with IL-1β Release and Regulation of Antimicrobial Properties in Human Neutrophils. Sci. Rep. 2020, 10, 21837. [Google Scholar] [CrossRef]
- Okano, T.; Ashida, H.; Suzuki, S.; Shoji, M.; Nakayama, K.; Suzuki, T. Porphyromonas gingivalis Triggers NLRP3-Mediated Inflammasome Activation in Macrophages in a Bacterial Gingipains-Independent Manner. Eur. J. Immunol. 2018, 48, 1965–1974. [Google Scholar] [CrossRef] [Green Version]
- Belibasakis, G.N.; Maula, T.; Bao, K.; Lindholm, M.; Bostanci, N.; Oscarsson, J.; Ihalin, R.; Johansson, A. Virulence and Pathogenicity Properties of Aggregatibacter actinomycetemcomitans. Pathogens 2019, 8, 222. [Google Scholar] [CrossRef] [Green Version]
- Kelk, P.; Johansson, A.; Claesson, R.; Hänström, L.; Kalfas, S. Caspase 1 Involvement in Human Monocyte Lysis Induced by Actinobacillus actinomycetemcomitans Leukotoxin. Infect. Immun. 2003, 71, 4448–4455. [Google Scholar] [CrossRef] [Green Version]
- Belibasakis, G.N.; Johansson, A. Aggregatibacter actinomycetemcomitans Targets NLRP3 and NLRP6 Inflammasome Expression in Human Mononuclear Leukocytes. Cytokine 2012, 59, 124–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belibasakis, G.N.; Guggenheim, B.; Bostanci, N. Down-Regulation of NLRP3 Inflammasome in Gingival Fibroblasts by Subgingival Biofilms: Involvement of Porphyromonas gingivalis. Innate Immun. 2013, 19, 3–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibata, K. Historical Aspects of Studies on Roles of the Inflammasome in the Pathogenesis of Periodontal Diseases. Mol. Oral Microbiol. 2018, 33, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Apaza-Bedoya, K.; Tarce, M.; Benfatti, C.A.M.; Henriques, B.; Mathew, M.T.; Teughels, W.; Souza, J.C.M. Synergistic Interactions between Corrosion and Wear at Titanium-Based Dental Implant Connections: A Scoping Review. J. Periodontal Res. 2017, 52, 946–954. [Google Scholar] [CrossRef] [PubMed]
- Burton, L.; Paget, D.; Binder, N.B.; Bohnert, K.; Nestor, B.J.; Sculco, T.P.; Santambrogio, L.; Ross, F.P.; Goldring, S.R.; Purdue, P.E. Orthopedic Wear Debris Mediated Inflammatory Osteolysis Is Mediated in Part by NALP3 Inflammasome Activation. J. Orthop. Res. Off. Publ. Orthop. Res. Soc. 2013, 31, 73–80. [Google Scholar] [CrossRef]
- Jämsen, E.; Pajarinen, J.; Kouri, V.-P.; Rahikkala, A.; Goodman, S.B.; Manninen, M.; Nordström, D.C.; Eklund, K.K.; Nurmi, K. Tumor Necrosis Factor Primes and Metal Particles Activate the NLRP3 Inflammasome in Human Primary Macrophages. Acta Biomater. 2020, 108, 347–357. [Google Scholar] [CrossRef]
- Polak, D.; Shapira, L. An Update on the Evidence for Pathogenic Mechanisms That May Link Periodontitis and Diabetes. J. Clin. Periodontol. 2018, 45, 150–166. [Google Scholar] [CrossRef]
- Aoyama, N.; Kure, K.; Minabe, M.; Izumi, Y. Increased Heart Failure Prevalence in Patients with a High Antibody Level against Periodontal Pathogen. Int. Heart. J. 2019, 60, 1142–1146. [Google Scholar] [CrossRef] [Green Version]
- Di Spirito, F.; Toti, P.; Pilone, V.; Carinci, F.; Lauritano, D.; Sbordone, L. The Association between Periodontitis and Human Colorectal Cancer: Genetic and Pathogenic Linkage. Life 2020, 10, 211. [Google Scholar] [CrossRef]
- Dominy, S.S.; Lynch, C.; Ermini, F.; Benedyk, M.; Marczyk, A.; Konradi, A.; Nguyen, M.; Haditsch, U.; Raha, D.; Griffin, C.; et al. Porphyromonas gingivalis in Alzheimer’s Disease Brains: Evidence for Disease Causation and Treatment with Small-Molecule Inhibitors. Sci. Adv. 2019, 5, eaau3333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Spirito, F.; La Rocca, M.; De Bernardo, M.; Rosa, N.; Sbordone, C.; Sbordone, L. Possible Association of Periodontal Disease and Macular Degeneration: A Case-Control Study. Dent. J. 2020, 9, 1. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Sun, X.; Liu, S.; Tang, Y.; Shi, Y.; Bai, Y.; Wang, Y.; Yang, Q.; Yang, Q.; Jiang, W.; et al. Caspase-11-Gasdermin D-Mediated Pyroptosis Is Involved in the Pathogenesis of Atherosclerosis. Front. Pharmacol. 2021, 12, 657486. [Google Scholar] [CrossRef]
- Zeng, W.; Wu, D.; Sun, Y.; Suo, Y.; Yu, Q.; Zeng, M.; Gao, Q.; Yu, B.; Jiang, X.; Wang, Y. The Selective NLRP3 Inhibitor MCC950 Hinders Atherosclerosis Development by Attenuating Inflammation and Pyroptosis in Macrophages. Sci. Rep. 2021, 11, 19305. [Google Scholar] [CrossRef] [PubMed]
- Kabeerdoss, J.; Jayakanthan, P.; Pugazhendhi, S.; Ramakrishna, B.S. Alterations of Mucosal Microbiota in the Colon of Patients with Inflammatory Bowel Disease Revealed by Real Time Polymerase Chain Reaction Amplification of 16S Ribosomal Ribonucleic Acid. Indian J. Med. Res. 2015, 142, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Mao, H.; Pan, Y.; Li, H.; Lei, L. Cyclin-Dependent Kinase 9 Inhibition Suppresses Necroptosis and Pyroptosis in the Progress of Endotoxemia. Inflammation 2020, 43, 2061–2074. [Google Scholar] [CrossRef]
- Huang, C.; Zhang, C.; Yang, P.; Chao, R.; Yue, Z.; Li, C.; Guo, J.; Li, M. Eldecalcitol Inhibits LPS-Induced NLRP3 Inflammasome-Dependent Pyroptosis in Human Gingival Fibroblasts by Activating the Nrf2/HO-1 Signaling Pathway. Drug Des. Dev. Ther. 2020, 14, 4901–4913. [Google Scholar] [CrossRef] [PubMed]
- Sholapurkar, A.; Sharma, D.; Glass, B.; Miller, C.; Nimmo, A.; Jennings, E. Professionally Delivered Local Antimicrobials in the Treatment of Patients with Periodontitis—A Narrative Review. Dent. J. 2021, 9, 2. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Authors | Year | Journal | Study Type | Methods | Outcomes | Title and Reference | |
|---|---|---|---|---|---|---|---|
| 1 | Zhang X, He S, Lu W, Lin L, Xiao H. | 2021 | In Vitro Cellular & Developmental Biololy-Animal | In Vitro | PDLCs were stimulated with E. coli LPS (1, 5, and 10 µg/mL for 6 h and 12 h). GCF were collected from periodontitis patients and healthy volunteers. | LPS suppressed PDLCs viability and led to production and secretion of IL-1β, IL-18, IL-6, and TNF-α in a time- and concentration-dependent manner. LPS activated NLRP3 and GSDMD, cleaved caspase-1, and upregulated GSK-3β. Blockage of GSK-3β restrained NLRP3-mediated pyroptosis. Pro-inflammatory cytokines were upregulated in periodontal patients’ GCF but not in healthy volunteers. | Glycogen synthase kinase-3β (GSK-3β) deficiency inactivates the NLRP3 inflammasome-mediated cell pyroptosis in LPS-treated periodontal ligament cells (PDLCs) [4] |
| 2 | Oka S, Li X, Sato F, Zhang F, Tewari N, Kim I-S, Zhong L, Hamada N, et al. | 2021 | Journal of Periodontal Research | In vitro and in vivo | HGFs and PDLCs were stimulated with P. gingivalis LPS (10 µg/mL for 24 h). Mouse experimental periodontitis model (WT and differentiated embryo chondrocyte 2 (Dec2) KO) was established. | LPS activated caspase-1, caspase-11, and NF-κB. Dec2 KO upregulated LPS-induced pyroptosis, resulting in IL-1β release. The inhibition of Dec2 led to the activation of caspase-1 and GSDMD, reduced the phosphorylation and translocation of NF-κB, decreased IL-1β expression, reducing pyroptosis. | A deficiency of Dec2 triggers periodontal inflammation and pyroptosis [1] |
| 3 | Chen Q, Cao M, Ge H. | 2021 | BioMed Research International | In Vitro | PDLCs were treated with P. gingivalis LPS (100 ng/mL for 72 h). The expression of metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) and miR-769-5p in gingival tissues of patients with periodontitis and LPS-treated PDLCs was evaluated. | MALAT1 KO promoted cell viability and inhibited inflammation and pyroptosis. The expression of MALAT1 and hypoxia-inducible factor 3A (HIF3A) was enhanced, and the expression of miR-769-5p was reduced in gingival tissues of patients with periodontitis and LPS-treated PDLCs. | Knockdown of MALAT1 inhibits the progression of chronic periodontitis via targeting miR-769-5p/HIF3A axis [13] |
| 4 | Liu S, Du J, Li D, Yang P, Kou Y, Li C, Zhou Q, Lu Y, et al. | 2020 | Journal of Molecular Histology | In Vitro | Human osteoblast-like cells were exposed to E. coli LPS (0.5, 1, or 2 μg/mL) for 24 h and 48 h. N-acetyl-L-cysteine (NAC) was used to decrease the intracellular ROS level and MCC950 was used to inhibit pyroptosis. | LPS led to NLRP3-mediated pyroptosis in a time- and dose-dependent manner. The inhibition of ROS with NAC attenuated oxidative stress-mediated pyroptosis. The inhibition of pyroptosis with MCC950 restored the expression of osteogenic differentiation-related proteins of osteoblasts. | Oxidative stress induced pyroptosis leads to osteogenic dysfunction of MG63 cells [10] |
| 5 | Cheng R, Feng Y, Zhang R, Liu W, Lei L, Hu T. | 2018 | Biochimica et Biophysica Acta—Molecular Basis of Disease | In vitro and in vivo | PDLCs were stimulated with E. coli LPS (1 μg/mL) or P. gingivalis LPS (10 μg/mL) for 24 h. Rat experimental periodontitis model was established. VX765 caspase-1 inhibitor was used to block pyroptosis. | VX765 inhibited the expressions of IL-1β, monocyte chemoattractant protein-1 (MCP-1), IL-6, and IL-8 In vitro, decreasing inflammatory responses during periodontitis. VX765 suppressed bone loss in vivo, linking pyroptosis to bone resorption in acute apical periodontitis. | The extent of pyroptosis varies in different stages of apical periodontitis [3] |
| 6 | Chen R, Liu W, Zhang R, Feng Y, Bhowmick NA, Hu T. | 2017 | Frontiers in Cellular and Infection Microbiology | In vitro and in vivo | HGFs were stimulated with E. coli LPS (1 μg/mL) or P. gingivalis LPS (10 μg/mL) at 2% or 20% O2 for 6 h. Mouse experimental periodontitis model was established. | P. gingivalis LPS slightly decreased the level of NLRP3 and IL-1β under normoxia. Hypoxia reversed the effects of P. gingivalis LPS, promoting caspase-1 activation and IL-1β maturation. E. coli LPS enhanced IL-1β maturation in both normoxia and hypoxia, and turned normoxia into hypoxia in the periodontitis model, suggesting to increase the inflammatory effect of P. gingivalis LPS. | Porphyromonas gingivalis-derived lipopolysaccharide combines hypoxia to induce caspase-1 activation in periodontitis [14] |
| 7 | Cecil JD, O’Brien-Simpson NM, Lenzo JC, Holden JA, Singleton W, Perez-Gonzalez A, Mansell A, Reynolds EC. | 2017 | Frontiers in Immunology | In vitro and in vivo | THP-1 (monocytes) and macrophages extracted from C57BL/6 J mice (ex vivo and in vivo) were treated with intraperitoneal injections of P. gingivalis, T. denticola, and T. forsythia OMVs (100 ng protein/mL) for 4 h. Cells were stimulated with nigericin (10 µM), silica (125 mg/mL), or transfected with poly(dAdT) (250 ng/mL) using lipofectamine LTX for 6 h. | OMVs interacted with monocytes and macrophages, inducing phagocytosis, NF-κB activation, IL-1β secretion, and cell death via NLRP3 activation. The immune stimulatory effects of P. gingivalis OMVs are suggested to dysregulate the host immune response and initiate the disease, while the pro-inflammatory effects of T. denticola and T. forsythia OMVs are suggested to promote disease progression. | Outer membrane vesicles prime and activate macrophage inflammasomes and cytokine secretion In vitro and in vivo [15] |
| 8 | Fleetwood AJ, Lee MKS, Singleton W, Achutan A, Lee M-C, O’Brien-Simpson NM, Cook AD, Murphy AJ, et al. | 2017 | Frontiers in Cellular and Infection Microbiology | In vitro and in vivo | C57BL/6 mouse and human macrophages were treated with viable P. gingivalis, heat-killed P. gingivalis, OMVs, or heat-inactivated OMVs at a MOI of 10:1, 25:1 (protein concentration of about 3.0 μg/mL) or 100:1 bacilli or OMVs/cell for 2 h. | P. gingivalis did not lead to the activation of NLRP3 while P. gingivalis OMVs activated caspase-1, produced large amounts of IL-1β and IL-18, released lactate dehydrogenase (LDH), and were positive for 7-amino actinomycin D (7-AAD) staining, thus indicating of pyroptosis. | Metabolic remodeling, inflammasome activation, and pyroptosis in macrophages stimulated by Porphyromonas gingivalis and its outer membrane vesicles [16] |
| 9 | Lu WL, Song DZ, Yue JL, Wang TT, Zhou XD, Zhang P, Zhang L, Huang DM. | 2017 | International Endodontic Journal | In Vitro | PDLCs were stimulated with MDP (10 μg/mL) for 0, 1, 3, 8, 14 or 24 h; E. coli LPS (0.5 μg/mL) for 0, 4, 8 or 24 h; or MDP and LPS in combination for 0, 4, 8 or 24 h. | MDP, LPS, or MDP in combination with LPS promoted the expression of NLRP3, caspase-1, and induced IL-1β secretion. MDP exhibited synergistic or additive effects with LPS to upregulate the expression of NLRP3, ASC and caspase-1. | NLRP3 inflammasome may regulate inflammatory response of human periodontal ligament fibroblasts in an apoptosis-associated speck-like protein containing a CARD (ASC)-dependent manner [6] |
| 10 | Brown PM, Kennedy DJ, Morton RE, Febbraio M. | 2015 | PLoS ONE | In Vivo | Cd36/Ldlr and Ldlr mice were derived from a cross between Cd36° and Ldlr mice. P. gingivalis (~2 × 109 CFU/mL) were resuspended in saline containing 2% carboxymethylcellulose (as a thickener to promote adherence) prior to oral inoculation of mice. | An increase of 225% (females) and 175% (males) was found in periodontal lesions compared to uninfected mice. This increase was CD36/SR-B2-dependent since there was no significant change in lesion burden between infected and uninfected Cd36°/Ldlr mice. Activation of the NLRP3 by P. gingivalis is mediated by CD36/SR-B2 and TLR2, leading to systemic release of IL-1β and inducing pyroptosis. | CD36/SR-B2-TLR2 dependent pathways enhance Porphyromonas gingivalis mediated atherosclerosis in the Ldlr KO mouse model [17] |
| 11 | Taxman DJ, Swanson KV, Broglie PM, Wen H, Holley-Guthrie E, Huang MT-H, Callaway JB, Eitas TK, et al. | 2012 | Journal of Biological Chemistry | In vitro and in vivo | MyD88−/−, Nlrp3−/−, Asc−/−, and Casp1−/− mice macrophages were infected with P. gingivalis. Macrophages were stimulated with E. coli LPS (1 μg/mL) for 3 h, followed by ATP (2 mM) for 0.5 h, nigericin (20 μM) for 0.5 h, monosodium urate (200 μg/mL) for 6 h, alum crystals (400 μg/mL) for 6 h, or S. aureus peptidoglycan (20 μg/mL) for 14-16 h. | P. gingivalis lacks signaling capability for the NLRP3 activation and can suppress NLRP3 activation by F. nucleatum, thus repressing IL-1β and IL-18 release and cell death. P. gingivalis can repress NLRP3 activation by E. coli, and by DAMPs and PAMPs that mediate activation through endocytosis, but cannot suppress NLRP3 activation by ATP or nigericin, suggesting that P. gingivalis preferentially suppress endocytic pathways towards NLRP3 activation. | Porphyromonas gingivalis mediates inflammasome repression in polymicrobial cultures through a novel mechanism involving reduced endocytosis [18] |
| 12 | Domon H, Takahashi N, Honda T, Nakajima T, Tabeta K, Abiko Y, Yamazaki K. | 2009 | Clinica Chimica Acta | In Vitro | Cells were obtained from human periodontitis patients. Macrophages were stimulated with E. coli LPS (1 µg/mL), P. gingivalis LPS (1 µg/mL), IFN-γ (100 or 500 U), or tunicamycin (1 μg/mL) for 1, 3, 6, 12, or 24 h. The expression of unfolded protein response (UPR) was analysed. | The expression of UPR-related genes was higher in periodontitis than in gingivitis lesions. P. gingivalis LPS (but not E. coli LPS or IFN-γ) failed to up-regulate gene expressions. Macrophages stimulated with E. coli LPS or IFN-γ expressed IL-β and caspase-4 at the gene level while tunicamycin did not. | Up-regulation of the endoplasmic reticulum stress-response in periodontal disease [7] |
| Authors | Year | Journal | Main Findings | Title and Reference |
|---|---|---|---|---|
| De Andrade KQ, Almeida-da-Silva CLC, Coutinho-Silva R. | 2017 | Mediators of Inflammation | Inflammasomes are involved in the pathogenesis of periodontitis; however, it is necessary to determine which inflammasomes, others than the typical NLRP3, contribute to the pathogenesis of periodontitis induced by P. gingivalis and F. nucleatum. With more solid literature on the signaling pathways and immune responses during infection with these bacteria, more effective treatments for periodontitis may appear. | Immunological pathways triggered by Porphyromonas gingivalis and Fusobacterium nucleatum: therapeutic possibilities? [8] |
| Authors | Year | Journal | Type of Study | Reason of Exclusion | Title and Reference | |
|---|---|---|---|---|---|---|
| 1 | Wang J, Du, C, Xu L. | 2021 | Archives of Oral Biology | In Vitro | Studied apoptosis | Circ_0081572 inhibits the progression of periodontitis through regulating the miR-378h/RorA axis [19] |
| 2 | Liu P, Cui, L, Shen L. | 2020 | Bioscience Reports | In Vitro | Studied apoptosis | Knockdown of TRIM52 alleviates LPS-induced inflammatory injury in human periodontal ligament cells through the TLR4/NF-κB pathway [20] |
| 3 | Zhang K, He S, Dai Z, Cao L, Yue S, Bai Y, Zheng M. | 2020 | Archives of Oral Biology | In Vitro | Studied apoptosis | Axin 1 knockdown inhibits osteoblastic apoptosis induced by Porphyromonas gingivalis lipopolysaccharide [21] |
| 4 | Zhou Y, Zhang H, Zhang G, He Y, Zhang P, Sun Z, Gao Y, Tan Y. | 2018 | Molecular Medicine Reports | In Vitro | Studied apoptosis | Calcitonin gene-related peptide reduces Porphyromonas gingivalis LPS-induced TNF-α release and apoptosis in osteoblasts [22] |
| 5 | Shirasugi M, Nishioka K, Yamamoto T, Nakaya T, Kanamura N. | 2017 | Biochemical and Biophysical Research Communications | In Vitro | Studied apoptosis and cytostasis | Normal human gingival fibroblasts undergo cytostasis and apoptosis after long-term exposure to butyric acid [23] |
| 6 | Zhu X, Lu W, Chen Y, Cheng X, Qiu J, Xu Y, Sun Y. | 2016 | PLoS ONE | In Vitro | Studied apoptosis | Effects of Porphyromonas gingivalis Lipopolysaccharide olerized monocytes on inflammatory responses in neutrophils [24] |
| 7 | Deepak V, Kasonga A, Kruger MC, Coetzee M. | 2016 | Biological and Pharmaceutical Bulletin | In Vitro | Studied apoptosis | Carvacrol inhibits osteoclastogenesis and negatively regulates the survival of mature osteoclasts [25] |
| 8 | Jönsson D, Nilsson B-O. | 2012 | Journal of Periodontal Research | In Vitro | Studied apoptosis | The antimicrobial peptide LL-37 is anti-inflammatory and proapoptotic in human periodontal ligament cells [26] |
| 9 | Zaric, S, Shelburne C, Darveau R, Quinn DJ, Weldon S, Taggart CC, Coulter WA. | 2010 | Infection and Immunity | In Vitro | Studied apoptosis | Impaired immune tolerance to Porphyromonas gingivalis lipopolysaccharide promotes neutrophil migration and decreased apoptosis [27] |
| 10 | Thammasitboon K, Goldring SR, Boch JA. | 2006 | Bone | In Vitro | Studied apoptosis | Role of macrophages in LPS-induced osteoblast and PDL cell apoptosis [28] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sordi, M.B.; Magini, R.d.S.; Panahipour, L.; Gruber, R. Pyroptosis-Mediated Periodontal Disease. Int. J. Mol. Sci. 2022, 23, 372. https://doi.org/10.3390/ijms23010372
Sordi MB, Magini RdS, Panahipour L, Gruber R. Pyroptosis-Mediated Periodontal Disease. International Journal of Molecular Sciences. 2022; 23(1):372. https://doi.org/10.3390/ijms23010372
Chicago/Turabian StyleSordi, Mariane Beatriz, Ricardo de Souza Magini, Layla Panahipour, and Reinhard Gruber. 2022. "Pyroptosis-Mediated Periodontal Disease" International Journal of Molecular Sciences 23, no. 1: 372. https://doi.org/10.3390/ijms23010372
APA StyleSordi, M. B., Magini, R. d. S., Panahipour, L., & Gruber, R. (2022). Pyroptosis-Mediated Periodontal Disease. International Journal of Molecular Sciences, 23(1), 372. https://doi.org/10.3390/ijms23010372