
Molecular Dissection of TDP-43 as a Leading Cause of ALS/FTLD

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
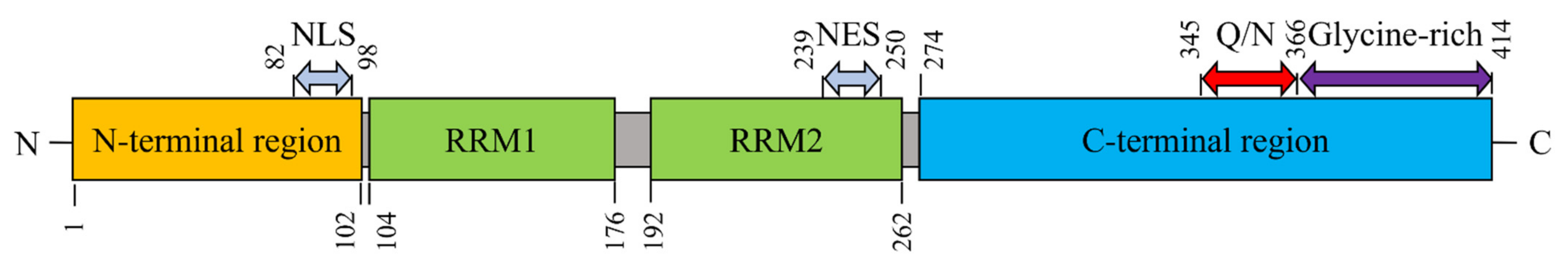
2. Physiological Structure and Functions of TDP-43
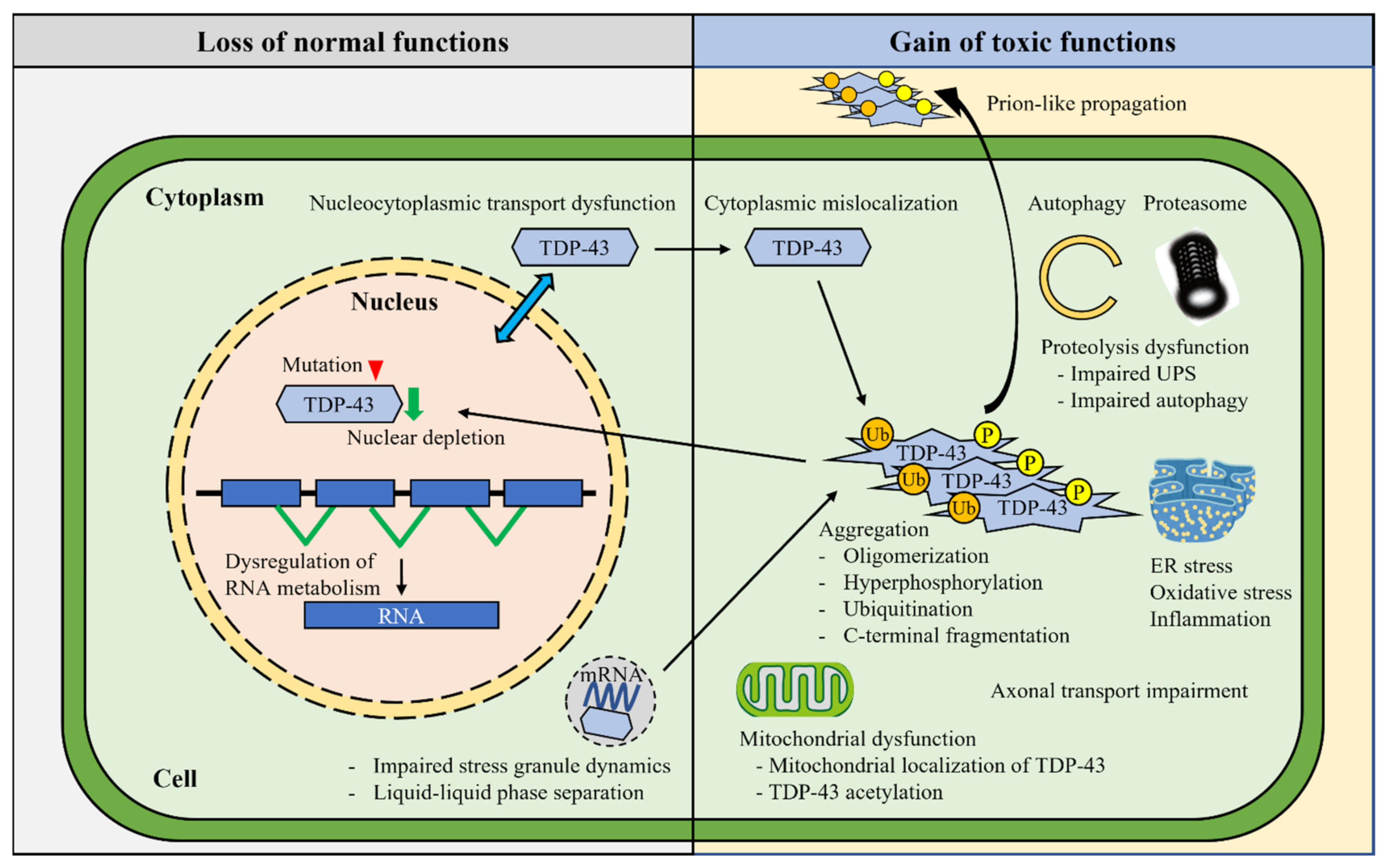
3. Pathogenic Mechanisms of TDP-43
3.1. Mutant TDP-43 Proteins
3.2. Post-Translational Modifications
3.2.1. Cleavage
3.2.2. Ubiquitination
3.2.3. Phosphorylation
3.2.4. SUMOylation
3.2.5. Acetylation
3.3. Nuclear TDP-43 Depletion
3.4. Dysregulation of RNA Metabolism
3.5. Nucleocytoplasmic Transport Dysfunction
3.6. Stress Granules
3.7. Liquid–Liquid Phase Separation
3.8. Oligomerization
3.9. Mitochondrial Dysfunction
3.10. Oxidative Stress
3.11. ER Stress
3.12. Impairment of Protein Quality Control (Ubiquitin and Autophagy Dysfunction)
3.13. Axonal Transport Impairment
3.14. Prion-like Propagation
3.15. Gliosis
4. TDP-43 Pathology in Neurodegenerative Diseases
4.1. ALS/FTLD
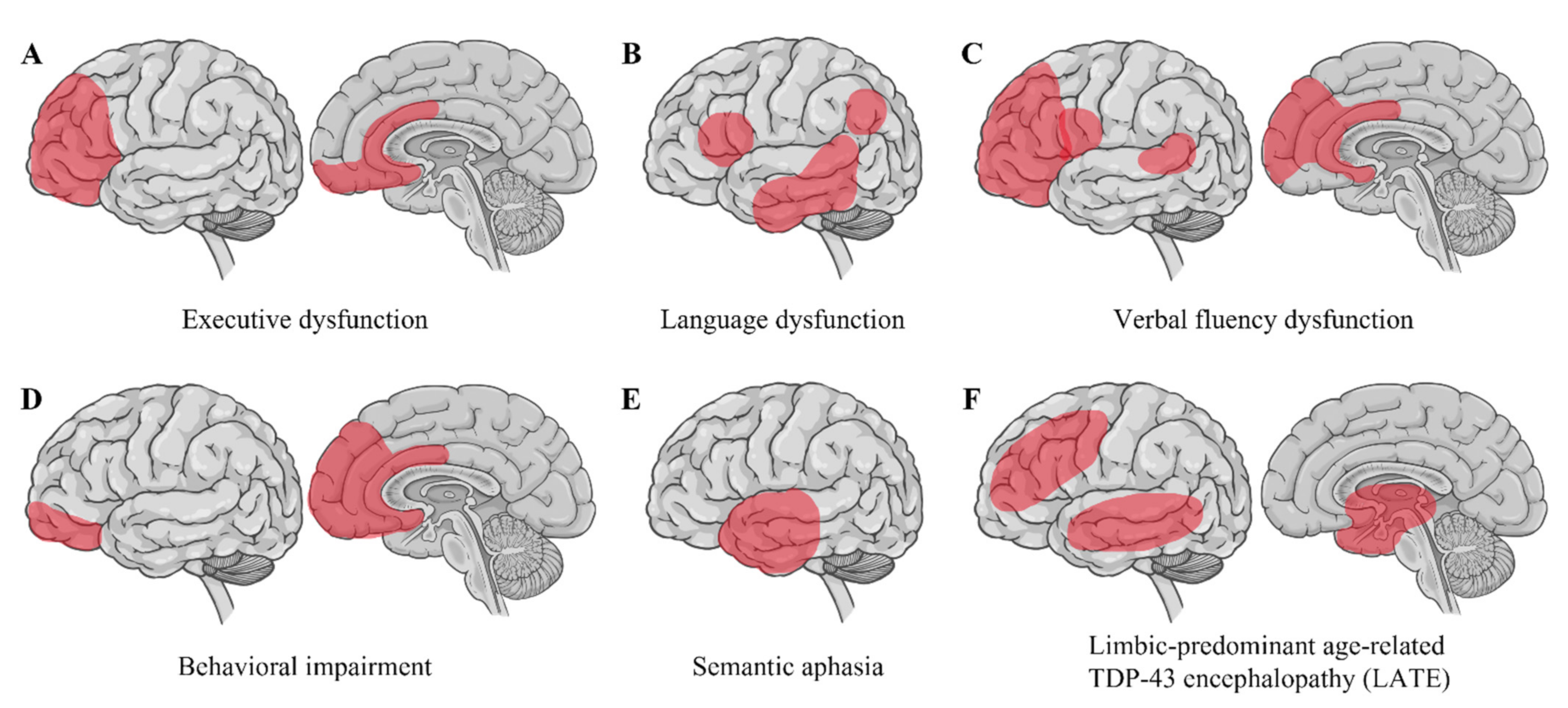
4.2. Limbic-Predominant Age-Related TDP-43 Encephalopathy (LATE)
4.3. Other Neurodegenerative Diseases
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS genetics, mechanisms, and therapeutics: Where are we now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bang, J.; Spina, S.; Miller, B.L. Frontotemporal dementia. Lancet 2015, 386, 1672–1682. [Google Scholar] [CrossRef] [Green Version]
- Burrell, J.R.; Halliday, G.M.; Kril, J.J.; Ittner, L.M.; Götz, J.; Kiernan, M.C.; Hodges, J.R. The frontotemporal dementia-motor neuron disease continuum. Lancet 2016, 388, 919–931. [Google Scholar] [CrossRef]
- Ringholz, G.M.; Appel, S.H.; Bradshaw, M.; Cooke, N.A.; Mosnik, D.M.; Schulz, P.E. Prevalence and patterns of cognitive impairment in sporadic ALS. Neurology 2005, 65, 586–590. [Google Scholar] [CrossRef] [PubMed]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [Green Version]
- Ling, S.C.; Polymenidou, M.; Cleveland, D.W. Converging mechanisms in ALS and FTD: Disrupted RNA and protein homeostasis. Neuron 2013, 79, 416–438. [Google Scholar] [CrossRef] [Green Version]
- Borroni, B.; Bonvicini, C.; Alberici, A.; Buratti, E.; Agosti, C.; Archetti, S.; Papetti, A.; Stuani, C.; Di Luca, M.; Gennarelli, M.; et al. Mutation within TARDBP leads to frontotemporal dementia without motor neuron disease. Hum. Mutat. 2009, 30, E974–E983. [Google Scholar] [CrossRef] [Green Version]
- Kabashi, E.; Valdmanis, P.N.; Dion, P.; Spiegelman, D.; McConkey, B.J.; Vande Velde, C.; Bouchard, J.P.; Lacomblez, L.; Pochigaeva, K.; Salachas, F.; et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 2008, 40, 572–574. [Google Scholar] [CrossRef]
- Kovacs, G.G.; Murrell, J.R.; Horvath, S.; Haraszti, L.; Majtenyi, K.; Molnar, M.J.; Budka, H.; Ghetti, B.; Spina, S. TARDBP variation associated with frontotemporal dementia, supranuclear gaze palsy, and chorea. Mov. Disord. 2009, 24, 1843–1847. [Google Scholar] [CrossRef]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef]
- Gao, J.; Wang, L.; Huntley, M.L.; Perry, G.; Wang, X. Pathomechanisms of TDP-43 in neurodegeneration. J. Neurochem. 2018, 146, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.T.; Dickson, D.W.; Trojanowski, J.Q.; Jack, C.R.; Boyle, P.A.; Arfanakis, K.; Rademakers, R.; Alafuzoff, I.; Attems, J.; Brayne, C.; et al. Limbic-predominant age-related TDP-43 encephalopathy (LATE): Consensus working group report. Brain 2019, 142, 1503–1527. [Google Scholar] [CrossRef] [Green Version]
- Wider, C.; Dickson, D.W.; Stoessl, A.J.; Tsuboi, Y.; Chapon, F.; Gutmann, L.; Lechevalier, B.; Calne, D.B.; Personett, D.A.; Hulihan, M.; et al. Pallidonigral TDP-43 pathology in Perry syndrome. Park. Relat. Disord. 2009, 15, 281–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Boer, E.M.J.; Orie, V.K.; Williams, T.; Baker, M.R.; De Oliveira, H.M.; Polvikoski, T.; Silsby, M.; Menon, P.; Van den Bos, M.; Halliday, G.M.; et al. TDP-43 proteinopathies: A new wave of neurodegenerative diseases. J. Neurol. Neurosurg. Psychiatry 2020, 92, 86–95. [Google Scholar] [CrossRef]
- Polymenidou, M.; Lagier-Tourenne, C.; Hutt, K.R.; Huelga, S.C.; Moran, J.; Liang, T.Y.; Ling, S.C.; Sun, E.; Wancewicz, E.; Mazur, C.; et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat. Neurosci. 2011, 14, 459–468. [Google Scholar] [CrossRef] [Green Version]
- Tollervey, J.R.; Curk, T.; Rogelj, B.; Briese, M.; Cereda, M.; Kayikci, M.; König, J.; Hortobágyi, T.; Nishimura, A.L.; Zupunski, V.; et al. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat. Neurosci. 2011, 14, 452–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riku, Y.; Seilhean, D.; Duyckaerts, C.; Boluda, S.; Iguchi, Y.; Ishigaki, S.; Iwasaki, K.; Yoshida, M.; Sobue, G.; Katsuno, M. Pathway from TDP-43-related pathology to neuronal dysfunction in amyotrophic lateral sclerosis and frontotemporal lobar degeneration. Int. J. Mol. Sci. 2021, 22, 3843. [Google Scholar] [CrossRef]
- Halliday, G.; Bigio, E.H.; Cairns, N.J.; Neumann, M.; Mackenzie, I.R.; Mann, D.M. Mechanisms of disease in frontotemporal lobar degeneration: Gain of function versus loss of function effects. Acta Neuropathol. 2012, 124, 373–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, G.; Gautier, O.; Tassoni-Tsuchida, E.; Ma, X.R.; Gitler, A.D. ALS genetics: Gains, losses, and implications for future therapies. Neuron 2020, 108, 822–842. [Google Scholar] [CrossRef] [PubMed]
- Cohen, T.J.; Lee, V.M.; Trojanowski, J.Q. TDP-43 functions and pathogenic mechanisms implicated in TDP-43 proteinopathies. Trends Mol. Med. 2011, 17, 659–667. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.L.; Zhao, J.; Yin, X.F.; He, W.T.; Yang, H.; Che, M.X.; Hu, H.Y. Two mutations G335D and Q343R within the amyloidogenic core region of TDP-43 influence its aggregation and inclusion formation. Sci. Rep. 2016, 6, 23928. [Google Scholar] [CrossRef] [Green Version]
- Kuo, P.H.; Chiang, C.H.; Wang, Y.T.; Doudeva, L.G.; Yuan, H.S. The crystal structure of TDP-43 RRM1-DNA complex reveals the specific recognition for UG- and TG-rich nucleic acids. Nucleic Acids Res. 2014, 42, 4712–4722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukavsky, P.J.; Daujotyte, D.; Tollervey, J.R.; Ule, J.; Stuani, C.; Buratti, E.; Baralle, F.E.; Damberger, F.F.; Allain, F.H. Molecular basis of UG-rich RNA recognition by the human splicing factor TDP-43. Nat. Struct. Mol. Biol. 2013, 20, 1443–1449. [Google Scholar] [CrossRef] [PubMed]
- Mompeán, M.; Romano, V.; Pantoja-Uceda, D.; Stuani, C.; Baralle, F.E.; Buratti, E.; Laurents, D.V. The TDP-43 N-terminal domain structure at high resolution. FEBS J. 2016, 283, 1242–1260. [Google Scholar] [PubMed] [Green Version]
- Qin, H.; Lim, L.Z.; Wei, Y.; Song, J. TDP-43 N terminus encodes a novel ubiquitin-like fold and its unfolded form in equilibrium that can be shifted by binding to ssDNA. Proc. Natl. Acad. Sci. USA 2014, 111, 18619–18624. [Google Scholar] [CrossRef] [Green Version]
- Shiina, Y.; Arima, K.; Tabunoki, H.; Satoh, J. TDP-43 dimerizes in human cells in culture. Cell. Mol. Neurobiol. 2010, 30, 641–652. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Caulfield, T.; Xu, Y.F.; Gendron, T.F.; Hubbard, J.; Stetler, C.; Sasaguri, H.; Whitelaw, E.C.; Cai, S.; Lee, W.C.; et al. The dual functions of the extreme N-terminus of TDP-43 in regulating its biological activity and inclusion formation. Hum. Mol. Genet. 2013, 22, 3112–3122. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.K.; Wu, T.H.; Wu, C.Y.; Chiang, M.H.; Toh, E.K.; Hsu, Y.C.; Lin, K.F.; Liao, Y.H.; Huang, T.H.; Huang, J.J. The N-terminus of TDP-43 promotes its oligomerization and enhances DNA binding affinity. Biochem. Biophys. Res. Commun. 2012, 425, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Afroz, T.; Hock, E.M.; Ernst, P.; Foglieni, C.; Jambeau, M.; Gilhespy, L.A.B.; Laferriere, F.; Maniecka, Z.; Plückthun, A.; Mittl, P.; et al. Functional and dynamic polymerization of the ALS-linked protein TDP-43 antagonizes its pathologic aggregation. Nat. Commun. 2017, 8, 45. [Google Scholar] [CrossRef]
- Wang, A.; Conicella, A.E.; Schmidt, H.B.; Martin, E.W.; Rhoads, S.N.; Reeb, A.N.; Nourse, A.; Ramirez Montero, D.; Ryan, V.H.; Rohatgi, R.; et al. A single N-terminal phosphomimic disrupts TDP-43 polymerization, phase separation, and RNA splicing. EMBO J. 2018, 37, E97452. [Google Scholar] [CrossRef] [PubMed]
- Kuo, P.H.; Doudeva, L.G.; Wang, Y.T.; Shen, C.K.; Yuan, H.S. Structural insights into TDP-43 in nucleic-acid binding and domain interactions. Nucleic Acids Res. 2009, 37, 1799–1808. [Google Scholar] [CrossRef] [PubMed]
- Shodai, A.; Ido, A.; Fujiwara, N.; Ayaki, T.; Morimura, T.; Oono, M.; Uchida, T.; Takahashi, R.; Ito, H.; Urushitani, M. Conserved acidic amino acid residues in a second RNA recognition motif regulate assembly and function of TDP-43. PLoS ONE 2012, 7, E52776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamaki, Y.; Shodai, A.; Morimura, T.; Hikiami, R.; Minamiyama, S.; Ayaki, T.; Tooyama, I.; Furukawa, Y.; Takahashi, R.; Urushitani, M. Elimination of TDP-43 inclusions linked to amyotrophic lateral sclerosis by a misfolding-specific intrabody with dual proteolytic signals. Sci. Rep. 2018, 8, 6030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lunde, B.M.; Moore, C.; Varani, G. RNA-binding proteins: Modular design for efficient function. Nat. Rev. Mol. Cell Biol. 2007, 8, 479–490. [Google Scholar] [CrossRef] [Green Version]
- Ling, J.P.; Pletnikova, O.; Troncoso, J.C.; Wong, P.C. TDP-43 repression of nonconserved cryptic exons is compromised in ALS-FTD. Science 2015, 349, 650–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koyama, A.; Sugai, A.; Kato, T.; Ishihara, T.; Shiga, A.; Toyoshima, Y.; Koyama, M.; Konno, T.; Hirokawa, S.; Yokoseki, A.; et al. Increased cytoplasmic TARDBP mRNA in affected spinal motor neurons in ALS caused by abnormal autoregulation of TDP-43. Nucleic Acids Res. 2016, 44, 5820–5836. [Google Scholar] [CrossRef] [Green Version]
- Alami, N.H.; Smith, R.B.; Carrasco, M.A.; Williams, L.A.; Winborn, C.S.; Han, S.S.W.; Kiskinis, E.; Winborn, B.; Freibaum, B.D.; Kanagaraj, A.; et al. Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron 2014, 81, 536–543. [Google Scholar] [CrossRef] [Green Version]
- Shodai, A.; Morimura, T.; Ido, A.; Uchida, T.; Ayaki, T.; Takahashi, R.; Kitazawa, S.; Suzuki, S.; Shirouzu, M.; Kigawa, T.; et al. Aberrant assembly of RNA recognition motif 1 links to pathogenic conversion of TAR DNA-binding protein of 43 kDa (TDP-43). J. Biol. Chem. 2013, 288, 14886–14905. [Google Scholar] [CrossRef] [Green Version]
- Rabdano, S.O.; Izmailov, S.A.; Luzik, D.A.; Groves, A.; Podkorytov, I.S.; Skrynnikov, N.R. Onset of disorder and protein aggregation due to oxidation-induced intermolecular disulfide bonds: Case study of RRM2 domain from TDP-43. Sci. Rep. 2017, 7, 11161. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.; Bharathi, V.; Sivalingam, V.; Girdhar, A.; Patel, B.K. Molecular mechanisms of TDP-43 misfolding and pathology in amyotrophic lateral sclerosis. Front. Mol. Neurosci. 2019, 12, 25. [Google Scholar] [CrossRef] [PubMed]
- Conicella, A.E.; Zerze, G.H.; Mittal, J.; Fawzi, N.L. ALS Mutations disrupt phase separation mediated by α-helical structure in the TDP-43 low-complexity C-terminal domain. Structure 2016, 24, 1537–1549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, B.S.; Snead, D.; Lee, J.J.; McCaffery, J.M.; Shorter, J.; Gitler, A.D. TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J. Biol. Chem. 2009, 284, 20329–20339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.; Xu, M.; Yang, M.; Yang, Y.; Li, Y.; Deng, J.; Ruan, L.; Liu, J.; Du, S.; Liu, X.; et al. An ALS-mutant TDP-43 neurotoxic peptide adopts an anti-parallel β-structure and induces TDP-43 redistribution. Hum. Mol. Genet. 2014, 23, 6863–6877. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.; Chen, Y.; Zhou, X.; Kar, A.; Ray, P.; Chen, X.; Rao, E.J.; Yang, M.; Ye, H.; Zhu, L.; et al. An ALS-associated mutation affecting TDP-43 enhances protein aggregation, fibril formation and neurotoxicity. Nat. Struct. Mol. Biol. 2011, 18, 822–830. [Google Scholar] [CrossRef] [Green Version]
- Barmada, S.J.; Skibinski, G.; Korb, E.; Rao, E.J.; Wu, J.Y.; Finkbeiner, S. Cytoplasmic mislocalization of TDP-43 is toxic to neurons and enhanced by a mutation associated with familial amyotrophic lateral sclerosis. J. Neurosci. 2010, 30, 639–649. [Google Scholar] [CrossRef] [Green Version]
- Mitsuzawa, S.; Akiyama, T.; Nishiyama, A.; Suzuki, N.; Kato, M.; Warita, H.; Izumi, R.; Osana, S.; Koyama, S.; Kato, T.; et al. TARDBP p.G376D mutation, found in rapid progressive familial ALS, induces mislocalization of TDP-43. eNeurologicalSci 2018, 11, 20–22. [Google Scholar] [CrossRef]
- Mutihac, R.; Alegre-Abarrategui, J.; Gordon, D.; Farrimond, L.; Yamasaki-Mann, M.; Talbot, K.; Wade-Martins, R. TARDBP pathogenic mutations increase cytoplasmic translocation of TDP-43 and cause reduction of endoplasmic reticulum Ca2+ signaling in motor neurons. Neurobiol. Dis. 2015, 75, 64–77. [Google Scholar] [CrossRef] [PubMed]
- Austin, J.A.; Wright, G.S.; Watanabe, S.; Grossmann, J.G.; Antonyuk, S.V.; Yamanaka, K.; Hasnain, S.S. Disease causing mutants of TDP-43 nucleic acid binding domains are resistant to aggregation and have increased stability and half-life. Proc. Natl. Acad. Sci. USA 2014, 111, 4309–4314. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, S.; Kaneko, K.; Yamanaka, K. Accelerated disease onset with stabilized familial amyotrophic lateral sclerosis (ALS)-linked mutant TDP-43 proteins. J. Biol. Chem. 2013, 288, 3641–3654. [Google Scholar] [CrossRef]
- Watanabe, S.; Oiwa, K.; Murata, Y.; Komine, O.; Sobue, A.; Endo, F.; Takahashi, E.; Yamanaka, K. ALS-linked TDP-43(M337V) knock-in mice exhibit splicing deregulation without neurodegeneration. Mol. Brain 2020, 13, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, M.A.; Kim, E.; Duffy, A.; Adalbert, R.; Phillips, B.U.; Peters, O.M.; Stephenson, J.; Yang, S.; Massenzio, F.; Lin, Z.; et al. TDP-43 gains function due to perturbed autoregulation in a Tardbp knock-in mouse model of ALS-FTD. Nat. Neurosci. 2018, 21, 552–563. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.L.; Wu, L.S.; Lee, M.; Chang, C.W.; Cheng, W.C.; Fang, Y.S.; Chen, Y.R.; Cheng, P.L.; Shen, C.J. A robust TDP-43 knock-in mouse model of ALS. Acta Neuropathol. Commun. 2020, 8, 3. [Google Scholar] [CrossRef] [Green Version]
- Santamaria, N.; Alhothali, M.; Alfonso, M.H.; Breydo, L.; Uversky, V.N. Intrinsic disorder in proteins involved in amyotrophic lateral sclerosis. Cell. Mol. Life Sci. 2017, 74, 1297–1318. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Xu, Y.F.; Cook, C.; Gendron, T.F.; Roettges, P.; Link, C.D.; Lin, W.L.; Tong, J.; Castanedes-Casey, M.; Ash, P.; et al. Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 7607–7612. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, T.; Hideyama, T.; Hachiga, K.; Teramoto, S.; Takano, J.; Iwata, N.; Saido, T.C.; Kwak, S. A role for calpain-dependent cleavage of TDP-43 in amyotrophic lateral sclerosis pathology. Nat. Commun. 2012, 3, 1307. [Google Scholar] [CrossRef] [Green Version]
- Hebron, M.L.; Lonskaya, I.; Sharpe, K.; Weerasinghe, P.P.; Algarzae, N.K.; Shekoyan, A.R.; Moussa, C.E. Parkin ubiquitinates Tar-DNA binding protein-43 (TDP-43) and promotes its cytosolic accumulation via interaction with histone deacetylase 6 (HDAC6). J. Biol. Chem. 2013, 288, 4103–4115. [Google Scholar] [CrossRef] [Green Version]
- Scotter, E.L.; Vance, C.; Nishimura, A.L.; Lee, Y.B.; Chen, H.J.; Urwin, H.; Sardone, V.; Mitchell, J.C.; Rogelj, B.; Rubinsztein, D.C.; et al. Differential roles of the ubiquitin proteasome system and autophagy in the clearance of soluble and aggregated TDP-43 species. J. Cell Sci. 2014, 127, 1263–1278. [Google Scholar] [CrossRef] [Green Version]
- Nonaka, T.; Suzuki, G.; Tanaka, Y.; Kametani, F.; Hirai, S.; Okado, H.; Miyashita, T.; Saitoe, M.; Akiyama, H.; Masai, H.; et al. Phosphorylation of TAR DNA-binding protein of 43 kDa (TDP-43) by truncated casein kinase 1δ triggers mislocalization and accumulation of TDP-43. J. Biol. Chem. 2016, 291, 5473–5483. [Google Scholar] [CrossRef] [Green Version]
- Gruijs da Silva, L.A.; Simonetti, F.; Hutten, S.; Riemenschneider, H.; Sternburg, E.L.; Pietrek, L.M.; Gebel, J.; Dötsch, V.; Edbauer, D.; Hummer, G.; et al. Disease-linked TDP-43 hyperphosphorylation suppresses TDP-43 condensation and aggregation. EMBO J. 2022, 41, E108443. [Google Scholar] [CrossRef]
- Eck, R.J.; Kraemer, B.C.; Liachko, N.F. Regulation of TDP-43 phosphorylation in aging and disease. GeroScience 2021, 43, 1605–1614. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, H.; Nonaka, T.; Yamashita, M.; Masuda-Suzukake, M.; Kametani, F.; Akiyama, H.; Mann, D.M.; Tamaoka, A.; Hasegawa, M. Epitope mapping of antibodies against TDP-43 and detection of protease-resistant fragments of pathological TDP-43 in amyotrophic lateral sclerosis and frontotemporal lobar degeneration. Biochem. Biophys. Res. Commun. 2012, 417, 116–121. [Google Scholar] [CrossRef]
- Dangoumau, A.; Veyrat-Durebex, C.; Blasco, H.; Praline, J.; Corcia, P.; Andres, C.R.; Vourc’h, P. Protein SUMOylation, an emerging pathway in amyotrophic lateral sclerosis. Int. J. Neurosci. 2013, 123, 366–374. [Google Scholar] [CrossRef]
- Maraschi, A.; Gumina, V.; Dragotto, J.; Colombrita, C.; Mompeán, M.; Buratti, E.; Silani, V.; Feligioni, M.; Ratti, A. SUMOylation regulates TDP-43 splicing activity and nucleocytoplasmic distribution. Mol. Neurobiol. 2021, 58, 5682–5702. [Google Scholar] [CrossRef]
- Irwin, D.J.; Cohen, T.J.; Grossman, M.; Arnold, S.E.; Xie, S.X.; Lee, V.M.; Trojanowski, J.Q. Acetylated tau, a novel pathological signature in Alzheimer’s disease and other tauopathies. Brain 2012, 135, 807–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, T.J.; Hwang, A.W.; Restrepo, C.R.; Yuan, C.X.; Trojanowski, J.Q.; Lee, V.M. An acetylation switch controls TDP-43 function and aggregation propensity. Nat. Commun. 2015, 6, 5845. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Wander, C.M.; Yuan, C.X.; Bereman, M.S.; Cohen, T.J. Acetylation-induced TDP-43 pathology is suppressed by an HSF1-dependent chaperone program. Nat. Commun. 2017, 8, 82. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Lu, S.; Gasior, K.; Singh, D.; Vazquez-Sanchez, S.; Tapia, O.; Toprani, D.; Beccari, M.S.; Yates, J.R.I.; Da Cruz, S.; et al. HSP70 chaperones RNA-free TDP-43 into anisotropic intranuclear liquid spherical shells. Science 2021, 371, 6529. [Google Scholar] [CrossRef]
- Wood, A.; Gurfinkel, Y.; Polain, N.; Lamont, W.; Lyn Rea, S. Molecular mechanisms underlying TDP-43 pathology in cellular and animal models of ALS and FTLD. Int. J. Mol. Sci. 2021, 22, 4705. [Google Scholar] [CrossRef]
- Ayala, Y.M.; Zago, P.; D’Ambrogio, A.; Xu, Y.F.; Petrucelli, L.; Buratti, E.; Baralle, F.E. Structural determinants of the cellular localization and shuttling of TDP-43. J. Cell Sci. 2008, 121, 3778–3785. [Google Scholar] [CrossRef]
- Lee, E.B.; Lee, V.M.; Trojanowski, J.Q. Gains or losses: Molecular mechanisms of TDP43-mediated neurodegeneration. Nat. Rev. Neurosci. 2011, 13, 38–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amlie-Wolf, A.; Ryvkin, P.; Tong, R.; Dragomir, I.; Suh, E.; Xu, Y.; Van Deerlin, V.M.; Gregory, B.D.; Kwong, L.K.; Trojanowski, J.Q.; et al. Transcriptomic changes due to cytoplasmic TDP-43 expression reveal dysregulation of histone transcripts and nuclear chromatin. PLoS ONE 2015, 10, E0141836. [Google Scholar] [CrossRef] [Green Version]
- Walker, A.K.; Spiller, K.J.; Ge, G.; Zheng, A.; Xu, Y.; Zhou, M.; Tripathy, K.; Kwong, L.K.; Trojanowski, J.Q.; Lee, V.M. Functional recovery in new mouse models of ALS/FTLD after clearance of pathological cytoplasmic TDP-43. Acta Neuropathol. 2015, 130, 643–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coyne, A.N.; Zaepfel, B.L.; Zarnescu, D.C. Failure to deliver and translate-new insights into RNA dysregulation in ALS. Front. Cell. Neurosci. 2017, 11, 243. [Google Scholar] [CrossRef] [Green Version]
- Mann, J.R.; Gleixner, A.M.; Mauna, J.C.; Gomes, E.; DeChellis-Marks, M.R.; Needham, P.G.; Copley, K.E.; Hurtle, B.; Portz, B.; Pyles, N.J.; et al. RNA binding antagonizes neurotoxic phase transitions of TDP-43. Neuron 2019, 102, 321–338. [Google Scholar]
- Hallegger, M.; Chakrabarti, A.M.; Lee, F.C.Y.; Lee, B.L.; Amalietti, A.G.; Odeh, H.M.; Copley, K.E.; Rubien, J.D.; Portz, B.; Kuret, K.; et al. TDP-43 condensation properties specify its RNA-binding and regulatory repertoire. Cell 2021, 184, 4680–4696. [Google Scholar] [CrossRef]
- Arnold, E.S.; Ling, S.C.; Huelga, S.C.; Lagier-Tourenne, C.; Polymenidou, M.; Ditsworth, D.; Kordasiewicz, H.B.; McAlonis-Downes, M.; Platoshyn, O.; Parone, P.A.; et al. ALS-linked TDP-43 mutations produce aberrant RNA splicing and adult-onset motor neuron disease without aggregation or loss of nuclear TDP-43. Proc. Natl. Acad. Sci. USA 2013, 110, E736–E745. [Google Scholar] [CrossRef] [Green Version]
- Klim, J.R.; Williams, L.A.; Limone, F.; Guerra San Juan, I.; Davis-Dusenbery, B.N.; Mordes, D.A.; Burberry, A.; Steinbaugh, M.J.; Gamage, K.K.; Kirchner, R.; et al. ALS-implicated protein TDP-43 sustains levels of STMN2, a mediator of motor neuron growth and repair. Nat. Neurosci. 2019, 22, 167–179. [Google Scholar] [CrossRef]
- Melamed, Z.; López-Erauskin, J.; Baughn, M.W.; Zhang, O.; Drenner, K.; Sun, Y.; Freyermuth, F.; McMahon, M.A.; Beccari, M.S.; Artates, J.W.; et al. Premature polyadenylation-mediated loss of stathmin-2 is a hallmark of TDP-43-dependent neurodegeneration. Nat. Neurosci. 2019, 22, 180–190. [Google Scholar] [CrossRef]
- Brown, A.L.; Wilkins, O.G.; Keuss, M.J.; Hill, S.E.; Zanovello, M.; Lee, W.C.; Bampton, A.; Lee, F.C.Y.; Masino, L.; Qi, Y.A.; et al. TDP-43 loss and ALS-risk SNPs drive mis-splicing and depletion of UNC13A. Nature 2022, 603, 131–137. [Google Scholar] [CrossRef]
- Ma, X.R.; Prudencio, M.; Koike, Y.; Vatsavayai, S.C.; Kim, G.; Harbinski, F.; Briner, A.; Rodriguez, C.M.; Guo, C.; Akiyama, T.; et al. TDP-43 represses cryptic exon inclusion in the FTD-ALS gene UNC13A. Nature 2022, 603, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Fratta, P.; Sivakumar, P.; Humphrey, J.; Lo, K.; Ricketts, T.; Oliveira, H.; Brito-Armas, J.M.; Kalmar, B.; Ule, A.; Yu, Y.; et al. Mice with endogenous TDP-43 mutations exhibit gain of splicing function and characteristics of amyotrophic lateral sclerosis. EMBO J. 2018, 37, E98684. [Google Scholar] [CrossRef] [PubMed]
- Highley, J.R.; Kirby, J.; Jansweijer, J.A.; Webb, P.S.; Hewamadduma, C.A.; Heath, P.R.; Higginbottom, A.; Raman, R.; Ferraiuolo, L.; Cooper-Knock, J.; et al. Loss of nuclear TDP-43 in amyotrophic lateral sclerosis (ALS) causes altered expression of splicing machinery and widespread dysregulation of RNA splicing in motor neurones. Neuropathol. Appl. Neurobiol. 2014, 40, 670–685. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, A.; Kimura, N.; Watanabe, Y.; Watanabe, S.; Ishihama, A. TDP-43 binds and transports G-quadruplex-containing mRNAs into neurites for local translation. Genes Cells 2016, 21, 466–481. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, A.L.; Zupunski, V.; Troakes, C.; Kathe, C.; Fratta, P.; Howell, M.; Gallo, J.M.; Hortobágyi, T.; Shaw, C.E.; Rogelj, B. Nuclear import impairment causes cytoplasmic trans-activation response DNA-binding protein accumulation and is associated with frontotemporal lobar degeneration. Brain 2010, 133, 1763–1771. [Google Scholar] [CrossRef] [Green Version]
- Guo, L.; Kim, H.J.; Wang, H.; Monaghan, J.; Freyermuth, F.; Sung, J.C.; O’Donovan, K.; Fare, C.M.; Diaz, Z.; Singh, N.; et al. Nuclear-import receptors reverse aberrant phase transitions of RNA-binding proteins with prion-like domains. Cell 2018, 173, 677–692. [Google Scholar] [CrossRef] [Green Version]
- Woerner, A.C.; Frottin, F.; Hornburg, D.; Feng, L.R.; Meissner, F.; Patra, M.; Tatzelt, J.; Mann, M.; Winklhofer, K.F.; Hartl, F.U.; et al. Cytoplasmic protein aggregates interfere with nucleocytoplasmic transport of protein and RNA. Science 2016, 351, 173–176. [Google Scholar] [CrossRef]
- Chou, C.C.; Zhang, Y.; Umoh, M.E.; Vaughan, S.W.; Lorenzini, I.; Liu, F.; Sayegh, M.; Donlin-Asp, P.G.; Chen, Y.H.; Duong, D.M.; et al. TDP-43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in ALS/FTD. Nat. Neurosci. 2018, 21, 228–239. [Google Scholar] [CrossRef] [Green Version]
- Anderson, P.; Kedersha, N. Stress granules. Curr. Biol. 2009, 19, R397–R398. [Google Scholar] [CrossRef] [Green Version]
- Aulas, A.; Vande Velde, C. Alterations in stress granule dynamics driven by TDP-43 and FUS: A link to pathological inclusions in ALS? Front. Cell. Neurosci. 2015, 9, 423. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Raphael, A.R.; LaDow, E.S.; McGurk, L.; Weber, R.A.; Trojanowski, J.Q.; Lee, V.M.; Finkbeiner, S.; Gitler, A.D.; Bonini, N.M. Therapeutic modulation of eIF2α phosphorylation rescues TDP-43 toxicity in amyotrophic lateral sclerosis disease models. Nat. Genet. 2014, 46, 152–160. [Google Scholar] [CrossRef]
- Anderson, P.; Kedersha, N. Stress granules: The Tao of RNA triage. Trends Biochem. Sci. 2008, 33, 141–150. [Google Scholar] [CrossRef]
- Colombrita, C.; Zennaro, E.; Fallini, C.; Weber, M.; Sommacal, A.; Buratti, E.; Silani, V.; Ratti, A. TDP-43 is recruited to stress granules in conditions of oxidative insult. J. Neurochem. 2009, 111, 1051–1061. [Google Scholar] [CrossRef] [PubMed]
- Dormann, D.; Haass, C. TDP-43 and FUS: A nuclear affair. Trends Neurosci. 2011, 34, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, P.; Van Hoecke, A.; Lambrechts, D.; Vanacker, P.; Bogaert, E.; van Swieten, J.; Carmeliet, P.; Van Den Bosch, L.; Robberecht, W. Progranulin functions as a neurotrophic factor to regulate neurite outgrowth and enhance neuronal survival. J. Cell Biol. 2008, 181, 37–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wójcik, C.; Rowicka, M.; Kudlicki, A.; Nowis, D.; McConnell, E.; Kujawa, M.; DeMartino, G.N. Valosin-containing protein (p97) is a regulator of endoplasmic reticulum stress and of the degradation of N-end rule and ubiquitin-fusion degradation pathway substrates in mammalian cells. Mol. Biol. Cell 2006, 17, 4606–4618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasset-Rosa, F.; Lu, S.; Yu, H.; Chen, C.; Melamed, Z.; Guo, L.; Shorter, J.; Da Cruz, S.; Cleveland, D.W. Cytoplasmic TDP-43 De-mixing independent of stress granules drives inhibition of nuclear import, loss of nuclear TDP-43, and cell death. Neuron 2019, 102, 339–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, S.; Hu, J.; Arogundade, O.A.; Goginashvili, A.; Vazquez-Sanchez, S.; Diedrich, J.K.; Gu, J.; Blum, J.; Oung, S.; Ye, Q.; et al. Heat-shock chaperone HSPB1 regulates cytoplasmic TDP-43 phase separation and liquid-to-gel transition. Nat. Cell Biol. 2022, 24, 1378–1393. [Google Scholar] [CrossRef]
- McDonald, K.K.; Aulas, A.; Destroismaisons, L.; Pickles, S.; Beleac, E.; Camu, W.; Rouleau, G.A.; Vande Velde, C. TAR DNA-binding protein 43 (TDP-43) regulates stress granule dynamics via differential regulation of G3BP and TIA-1. Hum. Mol. Genet. 2011, 20, 1400–1410. [Google Scholar] [CrossRef] [Green Version]
- Dewey, C.M.; Cenik, B.; Sephton, C.F.; Dries, D.R.; Mayer, P.I.; Good, S.K.; Johnson, B.A.; Herz, J.; Yu, G. TDP-43 is directed to stress granules by sorbitol, a novel physiological osmotic and oxidative stressor. Mol. Cell. Biol. 2011, 31, 1098–1108. [Google Scholar] [CrossRef] [Green Version]
- Liu-Yesucevitz, L.; Lin, A.Y.; Ebata, A.; Boon, J.Y.; Reid, W.; Xu, Y.F.; Kobrin, K.; Murphy, G.J.; Petrucelli, L.; Wolozin, B. ALS-linked mutations enlarge TDP-43-enriched neuronal RNA granules in the dendritic arbor. J. Neurosci. 2014, 34, 4167–4174. [Google Scholar] [CrossRef]
- Besnard-Guérin, C. Cytoplasmic localization of amyotrophic lateral sclerosis-related TDP-43 proteins modulates stress granule formation. Eur. J. Neurosci. 2020, 52, 3995–4008. [Google Scholar] [CrossRef]
- Morimoto, M.; Boerkoel, C.F. The role of nuclear bodies in gene expression and disease. Biology 2013, 2, 976–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishihara, T.; Ariizumi, Y.; Shiga, A.; Kato, T.; Tan, C.F.; Sato, T.; Miki, Y.; Yokoo, M.; Fujino, T.; Koyama, A.; et al. Decreased number of Gemini of coiled bodies and U12 snRNA level in amyotrophic lateral sclerosis. Hum. Mol. Genet. 2013, 22, 4136–4147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuiji, H.; Iguchi, Y.; Furuya, A.; Kataoka, A.; Hatsuta, H.; Atsuta, N.; Tanaka, F.; Hashizume, Y.; Akatsu, H.; Murayama, S.; et al. Spliceosome integrity is defective in the motor neuron diseases ALS and SMA. EMBO Mol. Med. 2013, 5, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Li, H.R.; Chen, T.C.; Hsiao, C.L.; Shi, L.; Chou, C.Y.; Huang, J.R. The physical forces mediating self-association and phase-separation in the C-terminal domain of TDP-43. Biochim. Biophys. Acta Proteins Proteom. 2018, 1866, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Li, H.R.; Chiang, W.C.; Chou, P.C.; Wang, W.J.; Huang, J.R. TAR DNA-binding protein 43 (TDP-43) liquid-liquid phase separation is mediated by just a few aromatic residues. J. Biol. Chem. 2018, 293, 6090–6098. [Google Scholar] [CrossRef] [Green Version]
- McGurk, L.; Gomes, E.; Guo, L.; Mojsilovic-Petrovic, J.; Tran, V.; Kalb, R.G.; Shorter, J.; Bonini, N.M. Poly(ADP-ribose) prevents pathological phase separation of TDP-43 by promoting liquid demixing and stress granule localization. Mol. Cell 2018, 71, 703–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- French, R.L.; Grese, Z.R.; Aligireddy, H.; Dhavale, D.D.; Reeb, A.N.; Kedia, N.; Kotzbauer, P.T.; Bieschke, J.; Ayala, Y.M. Detection of TAR DNA-binding protein 43 (TDP-43) oligomers as initial intermediate species during aggregate formation. J. Biol. Chem. 2019, 294, 6696–6709. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.S.; Tsai, K.J.; Chang, Y.J.; Kao, P.; Woods, R.; Kuo, P.H.; Wu, C.C.; Liao, J.Y.; Chou, S.C.; Lin, V.; et al. Full-length TDP-43 forms toxic amyloid oligomers that are present in frontotemporal lobar dementia-TDP patients. Nat. Commun. 2014, 5, 4824. [Google Scholar] [CrossRef] [Green Version]
- Davis, S.A.; Itaman, S.; Khalid-Janney, C.M.; Sherard, J.A.; Dowell, J.A.; Cairns, N.J.; Gitcho, M.A. TDP-43 interacts with mitochondrial proteins critical for mitophagy and mitochondrial dynamics. Neurosci. Lett. 2018, 678, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Kann, O.; Kovács, R. Mitochondria and neuronal activity. Am. J. Physiol. Cell Physiol. 2007, 292, C641–C657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Li, L.; Lin, W.L.; Dickson, D.W.; Petrucelli, L.; Zhang, T.; Wang, X. The ALS disease-associated mutant TDP-43 impairs mitochondrial dynamics and function in motor neurons. Hum. Mol. Genet. 2013, 22, 4706–4719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magrané, J.; Cortez, C.; Gan, W.B.; Manfredi, G. Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Hum. Mol. Genet. 2014, 23, 1413–1424. [Google Scholar] [CrossRef] [Green Version]
- Altanbyek, V.; Cha, S.J.; Kang, G.U.; Im, D.S.; Lee, S.; Kim, H.J.; Kim, K. Imbalance of mitochondrial dynamics in Drosophila models of amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2016, 481, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Braun, R.J.; Sommer, C.; Carmona-Gutierrez, D.; Khoury, C.M.; Ring, J.; Büttner, S.; Madeo, F. Neurotoxic 43-kDa TAR DNA-binding protein (TDP-43) triggers mitochondrion-dependent programmed cell death in yeast. J. Biol. Chem. 2011, 286, 19958–19972. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Wang, L.; Lu, J.; Siedlak, S.L.; Fujioka, H.; Liang, J.; Jiang, S.; Ma, X.; Jiang, Z.; da Rocha, E.L.; et al. The inhibition of TDP-43 mitochondrial localization blocks its neuronal toxicity. Nat. Med. 2016, 22, 869–878. [Google Scholar] [CrossRef]
- Zuo, X.; Zhou, J.; Li, Y.; Wu, K.; Chen, Z.; Luo, Z.; Zhang, X.; Liang, Y.; Esteban, M.A.; Zhou, Y.; et al. TDP-43 aggregation induced by oxidative stress causes global mitochondrial imbalance in ALS. Nat. Struct. Mol. Biol. 2021, 28, 132–142. [Google Scholar] [CrossRef]
- Yu, C.H.; Davidson, S.; Harapas, C.R.; Hilton, J.B.; Mlodzianoski, M.J.; Laohamonthonkul, P.; Louis, C.; Low, R.R.J.; Moecking, J.; De Nardo, D.; et al. TDP-43 Triggers Mitochondrial DNA Release via mPTP to Activate cGAS/STING in ALS. Cell 2020, 183, 636–649. [Google Scholar] [CrossRef]
- Barber, S.C.; Mead, R.J.; Shaw, P.J. Oxidative stress in ALS: A mechanism of neurodegeneration and a therapeutic target. Biochim. Biophys. Acta 2006, 1762, 1051–1067. [Google Scholar] [CrossRef] [Green Version]
- Coyle, J.T.; Puttfarcken, P. Oxidative stress, glutamate, and neurodegenerative disorders. Science 1993, 262, 689–695. [Google Scholar] [CrossRef]
- Lenaz, G.; Bovina, C.; D’Aurelio, M.; Fato, R.; Formiggini, G.; Genova, M.L.; Giuliano, G.; Merlo Pich, M.; Paolucci, U.; Parenti Castelli, G.; et al. Role of mitochondria in oxidative stress and aging. Ann. N. Y. Acad. Sci. 2002, 959, 199–213. [Google Scholar] [CrossRef] [PubMed]
- Hong, K.; Li, Y.; Duan, W.; Guo, Y.; Jiang, H.; Li, W.; Li, C. Full-length TDP-43 and its C-terminal fragments activate mitophagy in NSC34 cell line. Neurosci. Lett. 2012, 530, 144–149. [Google Scholar] [CrossRef]
- Guerrero, E.N.; Mitra, J.; Wang, H.; Rangaswamy, S.; Hegde, P.M.; Basu, P.; Rao, K.S.; Hegde, M.L. Amyotrophic lateral sclerosis-associated TDP-43 mutation Q331K prevents nuclear translocation of XRCC4-DNA ligase 4 complex and is linked to genome damage-mediated neuronal apoptosis. Hum. Mol. Genet. 2019, 28, 2459–2476. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [Green Version]
- Lautenschlaeger, J.; Prell, T.; Grosskreutz, J. Endoplasmic reticulum stress and the ER mitochondrial calcium cycle in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2012, 13, 166–177. [Google Scholar] [CrossRef] [PubMed]
- Limanaqi, F.; Biagioni, F.; Ryskalin, L.; Busceti, C.L.; Fornai, F. Molecular mechanisms linking ALS/FTD and psychiatric disorders, the potential effects of lithium. Front. Cell. Neurosci. 2019, 13, 450. [Google Scholar] [CrossRef] [Green Version]
- Stoica, R.; De Vos, K.J.; Paillusson, S.; Mueller, S.; Sancho, R.M.; Lau, K.F.; Vizcay-Barrena, G.; Lin, W.L.; Xu, Y.F.; Lewis, J.; et al. ER-mitochondria associations are regulated by the VAPB-PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat. Commun. 2014, 5, 3996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaccaro, A.; Patten, S.A.; Aggad, D.; Julien, C.; Maios, C.; Kabashi, E.; Drapeau, P.; Parker, J.A. Pharmacological reduction of ER stress protects against TDP-43 neuronal toxicity in vivo. Neurobiol. Dis. 2013, 55, 64–75. [Google Scholar] [CrossRef]
- Braun, R.J. Ubiquitin-dependent proteolysis in yeast cells expressing neurotoxic proteins. Front. Mol. Neurosci. 2015, 8, 8. [Google Scholar] [PubMed] [Green Version]
- Budini, M.; Buratti, E.; Morselli, E.; Criollo, A. Autophagy and its impact on neurodegenerative diseases: New roles for TDP-43 and C9orf72. Front. Mol. Neurosci. 2017, 10, 170. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, N.; Pandey, U.B. Autophagy dysregulation in ALS: When protein aggregates get out of hand. Front. Mol. Neurosci. 2017, 10, 263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urushitani, M.; Sato, T.; Bamba, H.; Hisa, Y.; Tooyama, I. Synergistic effect between proteasome and autophagosome in the clearance of polyubiquitinated TDP-43. J. Neurosci. Res. 2010, 88, 784–797. [Google Scholar] [CrossRef]
- Wang, X.; Fan, H.; Ying, Z.; Li, B.; Wang, H.; Wang, G. Degradation of TDP-43 and its pathogenic form by autophagy and the ubiquitin-proteasome system. Neurosci. Lett. 2010, 469, 112–116. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Gendron, T.F.; Xu, Y.F.; Ko, L.W.; Yen, S.H.; Petrucelli, L. Phosphorylation regulates proteasomal-mediated degradation and solubility of TAR DNA binding protein-43 C-terminal fragments. Mol. Neurodegener. 2010, 5, 33. [Google Scholar] [CrossRef] [Green Version]
- Dantuma, N.P.; Bott, L.C. The ubiquitin-proteasome system in neurodegenerative diseases: Precipitating factor, yet part of the solution. Front. Mol. Neurosci. 2014, 7, 70. [Google Scholar] [CrossRef] [Green Version]
- Araki, W.; Minegishi, S.; Motoki, K.; Kume, H.; Hohjoh, H.; Araki, Y.M.; Tamaoka, A. Disease-associated mutations of TDP-43 promote turnover of the protein through the proteasomal pathway. Mol. Neurobiol. 2014, 50, 1049–1058. [Google Scholar] [CrossRef]
- Van Eersel, J.; Ke, Y.D.; Gladbach, A.; Bi, M.; Götz, J.; Kril, J.J.; Ittner, L.M. Cytoplasmic accumulation and aggregation of TDP-43 upon proteasome inhibition in cultured neurons. PLoS ONE 2011, 6, E22850. [Google Scholar]
- Tashiro, Y.; Urushitani, M.; Inoue, H.; Koike, M.; Uchiyama, Y.; Komatsu, M.; Tanaka, K.; Yamazaki, M.; Abe, M.; Misawa, H.; et al. Motor neuron-specific disruption of proteasomes, but not autophagy, replicates amyotrophic lateral sclerosis. J. Biol. Chem. 2012, 287, 42984–42994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bose, J.K.; Huang, C.C.; Shen, C.K. Regulation of autophagy by neuropathological protein TDP-43. J. Biol. Chem. 2011, 286, 44441–44448. [Google Scholar] [CrossRef] [Green Version]
- Xia, Q.; Wang, H.; Hao, Z.; Fu, C.; Hu, Q.; Gao, F.; Ren, H.; Chen, D.; Han, J.; Ying, Z.; et al. TDP-43 loss of function increases TFEB activity and blocks autophagosome-lysosome fusion. EMBO J. 2016, 35, 121–142. [Google Scholar] [CrossRef] [Green Version]
- King, A.; Maekawa, S.; Bodi, I.; Troakes, C.; Al-Sarraj, S. Ubiquitinated, p62 immunopositive cerebellar cortical neuronal inclusions are evident across the spectrum of TDP-43 proteinopathies but are only rarely additionally immunopositive for phosphorylation-dependent TDP-43. Neuropathology 2011, 31, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Boland, B.; Kumar, A.; Lee, S.; Platt, F.M.; Wegiel, J.; Yu, W.H.; Nixon, R.A. Autophagy induction and autophagosome clearance in neurons: Relationship to autophagic pathology in Alzheimer’s disease. J. Neurosci. 2008, 28, 6926–6937. [Google Scholar] [CrossRef] [Green Version]
- Deng, H.X.; Chen, W.; Hong, S.T.; Boycott, K.M.; Gorrie, G.H.; Siddique, N.; Yang, Y.; Fecto, F.; Shi, Y.; Zhai, H.; et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 2011, 477, 211–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osaka, M.; Ito, D.; Suzuki, N. Disturbance of proteasomal and autophagic protein degradation pathways by amyotrophic lateral sclerosis-linked mutations in ubiquilin 2. Biochem. Biophys. Res. Commun. 2016, 472, 324–331. [Google Scholar] [CrossRef]
- Picher-Martel, V.; Renaud, L.; Bareil, C.; Julien, J.P. Neuronal expression of UBQLN2(P497H) exacerbates TDP-43 pathology in TDP-43(G348C) mice through interaction with ubiquitin. Mol. Neurobiol. 2019, 56, 4680–4696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maday, S.; Twelvetrees, A.E.; Moughamian, A.J.; Holzbaur, E.L. Axonal transport: Cargo-specific mechanisms of motility and regulation. Neuron 2014, 84, 292–309. [Google Scholar] [CrossRef] [Green Version]
- Sleigh, J.N.; Rossor, A.M.; Fellows, A.D.; Tosolini, A.P.; Schiavo, G. Axonal transport and neurological disease. Nat. Rev. Neurol. 2019, 15, 691–703. [Google Scholar] [CrossRef]
- Sleigh, J.N.; Tosolini, A.P.; Gordon, D.; Devoy, A.; Fratta, P.; Fisher, E.M.C.; Talbot, K.; Schiavo, G. Mice carrying ALS mutant TDP-43, but not mutant FUS, display in vivo defects in axonal transport of signaling endosomes. Cell Rep. 2020, 30, 3655–3662. [Google Scholar] [CrossRef] [Green Version]
- Fallini, C.; Bassell, G.J.; Rossoll, W. The ALS disease protein TDP-43 is actively transported in motor neuron axons and regulates axon outgrowth. Hum. Mol. Genet. 2012, 21, 3703–3718. [Google Scholar] [CrossRef] [Green Version]
- Brettschneider, J.; Del Tredici, K.; Lee, V.M.; Trojanowski, J.Q. Spreading of pathology in neurodegenerative diseases: A focus on human studies. Nat. Rev. Neurosci. 2015, 16, 109–120. [Google Scholar] [CrossRef] [Green Version]
- McAlary, L.; Yerbury, J.J.; Cashman, N.R. The prion-like nature of amyotrophic lateral sclerosis. Prog. Mol. Biol. Transl. Sci. 2020, 175, 261–296. [Google Scholar] [PubMed]
- Agrawal, S.; Kuo, P.H.; Chu, L.Y.; Golzarroshan, B.; Jain, M.; Yuan, H.S. RNA recognition motifs of disease-linked RNA-binding proteins contribute to amyloid formation. Sci. Rep. 2019, 9, 6171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Q.; Boyer, D.R.; Sawaya, M.R.; Ge, P.; Eisenberg, D.S. Cryo-EM structures of four polymorphic TDP-43 amyloid cores. Nat. Struct. Mol. Biol. 2019, 26, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Guenther, E.L.; Cao, Q.; Trinh, H.; Lu, J.; Sawaya, M.R.; Cascio, D.; Boyer, D.R.; Rodriguez, J.A.; Hughes, M.P.; Eisenberg, D.S. Atomic structures of TDP-43 LCD segments and insights into reversible or pathogenic aggregation. Nat. Struct. Mol. Biol. 2018, 25, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Nonaka, T.; Masuda-Suzukake, M.; Arai, T.; Hasegawa, Y.; Akatsu, H.; Obi, T.; Yoshida, M.; Murayama, S.; Mann, D.M.; Akiyama, H.; et al. Prion-like properties of pathological TDP-43 aggregates from diseased brains. Cell Rep. 2013, 4, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Porta, S.; Xu, Y.; Restrepo, C.R.; Kwong, L.K.; Zhang, B.; Brown, H.J.; Lee, E.B.; Trojanowski, J.Q.; Lee, V.M. Patient-derived frontotemporal lobar degeneration brain extracts induce formation and spreading of TDP-43 pathology in vivo. Nat. Commun. 2018, 9, 4220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smethurst, P.; Newcombe, J.; Troakes, C.; Simone, R.; Chen, Y.R.; Patani, R.; Sidle, K. In vitro prion-like behaviour of TDP-43 in ALS. Neurobiol. Dis. 2016, 96, 236–247. [Google Scholar] [CrossRef] [Green Version]
- Zeineddine, R.; Whiten, D.R.; Farrawell, N.E.; McAlary, L.; Hanspal, M.A.; Kumita, J.R.; Wilson, M.R.; Yerbury, J.J. Flow cytometric measurement of the cellular propagation of TDP-43 aggregation. Prion 2017, 11, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Feiler, M.S.; Strobel, B.; Freischmidt, A.; Helferich, A.M.; Kappel, J.; Brewer, B.M.; Li, D.; Thal, D.R.; Walther, P.; Ludolph, A.C.; et al. TDP-43 is intercellularly transmitted across axon terminals. J. Cell Biol. 2015, 211, 897–911. [Google Scholar] [CrossRef] [Green Version]
- Mishra, P.S.; Boutej, H.; Soucy, G.; Bareil, C.; Kumar, S.; Picher-Martel, V.; Dupré, N.; Kriz, J.; Julien, J.P. Transmission of ALS pathogenesis by the cerebrospinal fluid. Acta Neuropathol. Commun. 2020, 8, 65. [Google Scholar] [CrossRef] [PubMed]
- Burda, J.E.; Sofroniew, M.V. Reactive gliosis and the multicellular response to CNS damage and disease. Neuron 2014, 81, 229–248. [Google Scholar] [CrossRef] [PubMed]
- Clarke, L.E.; Barres, B.A. Emerging roles of astrocytes in neural circuit development. Nat. Rev. Neurosci. 2013, 14, 311–321. [Google Scholar] [CrossRef] [Green Version]
- Yamanaka, K.; Komine, O. The multi-dimensional roles of astrocytes in ALS. Neurosci. Res. 2018, 126, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Nagy, D.; Kato, T.; Kushner, P.D. Reactive astrocytes are widespread in the cortical gray matter of amyotrophic lateral sclerosis. J. Neurosci. Res. 1994, 38, 336–347. [Google Scholar] [CrossRef] [PubMed]
- Stallings, N.R.; Puttaparthi, K.; Luther, C.M.; Burns, D.K.; Elliott, J.L. Progressive motor weakness in transgenic mice expressing human TDP-43. Neurobiol. Dis. 2010, 40, 404–414. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.F.; Zhang, Y.J.; Lin, W.L.; Cao, X.; Stetler, C.; Dickson, D.W.; Lewis, J.; Petrucelli, L. Expression of mutant TDP-43 induces neuronal dysfunction in transgenic mice. Mol. Neurodegener. 2011, 6, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, J.; Huang, C.; Bi, F.; Wu, Q.; Huang, B.; Liu, X.; Li, F.; Zhou, H.; Xia, X.G. Expression of ALS-linked TDP-43 mutant in astrocytes causes non-cell-autonomous motor neuron death in rats. EMBO J. 2013, 32, 1917–1926. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.H.; Parsadanian, A.S.; Andreeva, A.; Snider, W.D.; Elliott, J.L. Restricted expression of G86R Cu/Zn superoxide dismutase in astrocytes results in astrocytosis but does not cause motoneuron degeneration. J. Neurosci. 2000, 20, 660–665. [Google Scholar] [CrossRef] [Green Version]
- Mackenzie, I.R.; Rademakers, R.; Neumann, M. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. 2010, 9, 995–1007. [Google Scholar] [CrossRef]
- Prater, K.E.; Latimer, C.S.; Jayadev, S. Glial TDP-43 and TDP-43 induced glial pathology, focus on neurodegenerative proteinopathy syndromes. Glia. 2022, 70, 239–255. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ho, W.Y.; Lim, K.; Feng, J.; Tucker-Kellogg, G.; Nave, K.A.; Ling, S.C. Cell-autonomous requirement of TDP-43, an ALS/FTD signature protein, for oligodendrocyte survival and myelination. Proc. Natl. Acad. Sci. USA 2018, 115, E10941–E10950. [Google Scholar] [CrossRef]
- Uchida, T.; Tamaki, Y.; Ayaki, T.; Shodai, A.; Kaji, S.; Morimura, T.; Banno, Y.; Nishitsuji, K.; Sakashita, N.; Maki, T.; et al. CUL2-mediated clearance of misfolded TDP-43 is paradoxically affected by VHL in oligodendrocytes in ALS. Sci. Rep. 2016, 6, 19118. [Google Scholar] [CrossRef] [Green Version]
- Neumann, M.; Kwong, L.K.; Lee, E.B.; Kremmer, E.; Flatley, A.; Xu, Y.; Forman, M.S.; Troost, D.; Kretzschmar, H.A.; Trojanowski, J.Q.; et al. Phosphorylation of S409/410 of TDP-43 is a consistent feature in all sporadic and familial forms of TDP-43 proteinopathies. Acta Neuropathol. 2009, 117, 137–149. [Google Scholar] [CrossRef] [Green Version]
- Igaz, L.M.; Kwong, L.K.; Xu, Y.; Truax, A.C.; Uryu, K.; Neumann, M.; Clark, C.M.; Elman, L.B.; Miller, B.L.; Grossman, M.; et al. Enrichment of C-terminal fragments in TAR DNA-binding protein-43 cytoplasmic inclusions in brain but not in spinal cord of frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Am. J. Pathol. 2008, 173, 182–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregory, J.M.; McDade, K.; Bak, T.H.; Pal, S.; Chandran, S.; Smith, C.; Abrahams, S. Executive, language and fluency dysfunction are markers of localised TDP-43 cerebral pathology in non-demented ALS. J. Neurol. Neurosurg. Psychiatry 2020, 91, 149–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prudlo, J.; König, J.; Schuster, C.; Kasper, E.; Büttner, A.; Teipel, S.; Neumann, M. TDP-43 pathology and cognition in ALS: A prospective clinicopathologic correlation study. Neurology 2016, 87, 1019–1023. [Google Scholar] [CrossRef]
- Giannini, L.A.A.; Xie, S.X.; McMillan, C.T.; Liang, M.; Williams, A.; Jester, C.; Rascovsky, K.; Wolk, D.A.; Ash, S.; Lee, E.B.; et al. Divergent patterns of TDP-43 and tau pathologies in primary progressive aphasia. Ann. Neurol. 2019, 85, 630–643. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, I.R.; Neumann, M.; Baborie, A.; Sampathu, D.M.; Du Plessis, D.; Jaros, E.; Perry, R.H.; Trojanowski, J.Q.; Mann, D.M.; Lee, V.M. A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol. 2011, 122, 111–113. [Google Scholar] [CrossRef] [Green Version]
- Van Mossevelde, S.; Engelborghs, S.; van der Zee, J.; Van Broeckhoven, C. Genotype-phenotype links in frontotemporal lobar degeneration. Nat. Rev. Neurol. 2018, 14, 363–378. [Google Scholar] [CrossRef]
- Sellami, L.; Saracino, D.; Le Ber, I. Genetic forms of frontotemporal lobar degeneration: Current diagnostic approach and new directions in therapeutic strategies. Rev. Neurol. 2020, 176, 571–581. [Google Scholar] [CrossRef]
- Boeve, B.F.; Boylan, K.B.; Graff-Radford, N.R.; DeJesus-Hernandez, M.; Knopman, D.S.; Pedraza, O.; Vemuri, P.; Jones, D.; Lowe, V.; Murray, M.E.; et al. Characterization of frontotemporal dementia and/or amyotrophic lateral sclerosis associated with the GGGGCC repeat expansion in C9ORF72. Brain 2012, 135, 765–783. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, R.M.; Caga, J.; Devenney, E.; Hsieh, S.; Bartley, L.; Highton-Williamson, E.; Ramsey, E.; Zoing, M.; Halliday, G.M.; Piguet, O.; et al. Cognition and eating behavior in amyotrophic lateral sclerosis: Effect on survival. J. Neurol. 2016, 263, 1593–1603. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, R.M.; Newcombe, R.E.; Piper, A.J.; Lewis, S.J.; Yee, B.J.; Kiernan, M.C.; Grunstein, R.R. Sleep disorders and respiratory function in amyotrophic lateral sclerosis. Sleep Med. Rev. 2016, 26, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, L.; Pradat, P.F.; Ludolph, A.C.; Loeffler, J.P. Energy metabolism in amyotrophic lateral sclerosis. Lancet Neurol. 2011, 10, 75–82. [Google Scholar] [CrossRef]
- Nakamura, R.; Kurihara, M.; Ogawa, N.; Kitamura, A.; Yamakawa, I.; Bamba, S.; Sanada, M.; Sasaki, M.; Urushitani, M. Prognostic prediction by hypermetabolism varies depending on the nutritional status in early amyotrophic lateral sclerosis. Sci. Rep. 2021, 11, 17943. [Google Scholar] [CrossRef] [PubMed]
- Gabery, S.; Ahmed, R.M.; Caga, J.; Kiernan, M.C.; Halliday, G.M.; Petersén, Å. Loss of the metabolism and sleep regulating neuronal populations expressing orexin and oxytocin in the hypothalamus in amyotrophic lateral sclerosis. Neuropathol. Appl. Neurobiol. 2021, 47, 979–989. [Google Scholar] [CrossRef]
- Araki, K.; Araki, A.; Honda, D.; Izumoto, T.; Hashizume, A.; Hijikata, Y.; Yamada, S.; Iguchi, Y.; Hara, A.; Ikumi, K.; et al. TDP-43 regulates early-phase insulin secretion via CaV1.2-mediated exocytosis in islets. J. Clin. Investig. 2019, 129, 3578–3593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurashige, T.; Morino, H.; Murao, T.; Izumi, Y.; Sugiura, T.; Kuraoka, K.; Kawakami, H.; Torii, T.; Maruyama, H. TDP-43 accumulation within intramuscular nerve bundles of patients with amyotrophic lateral sclerosis. JAMA Neurol. 2022, 79, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Vucic, S.; Tian, D.; Chong, P.S.; Cudkowicz, M.E.; Hedley-Whyte, E.T.; Cros, D. Facial onset sensory and motor neuronopathy (FOSMN syndrome): A novel syndrome in neurology. Brain 2006, 129, 3384–3390. [Google Scholar] [CrossRef] [Green Version]
- De Boer, E.M.J.; Barritt, A.W.; Elamin, M.; Anderson, S.J.; Broad, R.; Nisbet, A.; Goedee, H.S.; Vazquez Costa, J.F.; Prudlo, J.; Vedeler, C.A.; et al. Facial onset sensory and motor neuronopathy: New cases, cognitive changes, and pathophysiology. Neurol. Clin. Pract. 2021, 11, 147–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossor, A.M.; Jaunmuktane, Z.; Rossor, M.N.; Hoti, G.; Reilly, M.M. TDP43 pathology in the brain, spinal cord, and dorsal root ganglia of a patient with FOSMN. Neurology 2019, 92, E951–E956. [Google Scholar]
- Sonoda, K.; Sasaki, K.; Tateishi, T.; Yamasaki, R.; Hayashi, S.; Sakae, N.; Ohyagi, Y.; Iwaki, T.; Kira, J. TAR DNA-binding protein 43 pathology in a case clinically diagnosed with facial-onset sensory and motor neuronopathy syndrome: An autopsied case report and a review of the literature. J. Neurol. Sci. 2013, 332, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Ziso, B.; Williams, T.L.; Walters, R.J.; Jaiser, S.R.; Attems, J.; Wieshmann, U.C.; Larner, A.J.; Jacob, A. Facial onset sensory and motor neuronopathy: Further evidence for a TDP-43 proteinopathy. Case Rep. Neurol. 2015, 7, 95–100. [Google Scholar] [CrossRef]
- Higashi, S.; Iseki, E.; Yamamoto, R.; Minegishi, M.; Hino, H.; Fujisawa, K.; Togo, T.; Katsuse, O.; Uchikado, H.; Furukawa, Y.; et al. Concurrence of TDP-43, tau and alpha-synuclein pathology in brains of Alzheimer’s disease and dementia with Lewy bodies. Brain Res. 2007, 1184, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Josephs, K.A.; Murray, M.E.; Whitwell, J.L.; Parisi, J.E.; Petrucelli, L.; Jack, C.R.; Petersen, R.C.; Dickson, D.W. Staging TDP-43 pathology in Alzheimer’s disease. Acta Neuropathol. 2014, 127, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Chanson, J.B.; Echaniz-Laguna, A.; Vogel, T.; Mohr, M.; Benoilid, A.; Kaltenbach, G.; Kiesmann, M. TDP43-positive intraneuronal inclusions in a patient with motor neuron disease and Parkinson’s disease. Neurodegener. Dis. 2010, 7, 260–264. [Google Scholar] [CrossRef] [PubMed]
- Markopoulou, K.; Dickson, D.W.; McComb, R.D.; Wszolek, Z.K.; Katechalidou, L.; Avery, L.; Stansbury, M.S.; Chase, B.A. Clinical, neuropathological and genotypic variability in SNCA A53T familial Parkinson’s disease. Variability in familial Parkinson’s disease. Acta Neuropathol. 2008, 116, 25–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwab, C.; Arai, T.; Hasegawa, M.; Yu, S.; McGeer, P.L. Colocalization of transactivation-responsive DNA-binding protein 43 and huntingtin in inclusions of Huntington disease. J. Neuropathol. Exp. Neurol. 2008, 67, 1159–1165. [Google Scholar] [CrossRef] [Green Version]
- Uryu, K.; Nakashima-Yasuda, H.; Forman, M.S.; Kwong, L.K.; Clark, C.M.; Grossman, M.; Miller, B.L.; Kretzschmar, H.A.; Lee, V.M.; Trojanowski, J.Q.; et al. Concomitant TAR-DNA-binding protein 43 pathology is present in Alzheimer disease and corticobasal degeneration but not in other tauopathies. J. Neuropathol. Exp. Neurol. 2008, 67, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Koga, S.; Sanchez-Contreras, M.; Josephs, K.A.; Uitti, R.J.; Graff-Radford, N.; Van Gerpen, J.A.; Cheshire, W.P.; Wszolek, Z.K.; Rademakers, R.; Dickson, D.W. Distribution and characteristics of transactive response DNA binding protein 43 kDa pathology in progressive supranuclear palsy. Mov. Disord. 2017, 32, 246–255. [Google Scholar] [CrossRef] [Green Version]
- Yokota, O.; Davidson, Y.; Bigio, E.H.; Ishizu, H.; Terada, S.; Arai, T.; Hasegawa, M.; Akiyama, H.; Sikkink, S.; Pickering-Brown, S.; et al. Phosphorylated TDP-43 pathology and hippocampal sclerosis in progressive supranuclear palsy. Acta Neuropathol. 2010, 120, 55–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riku, Y.; Iwasaki, Y.; Ishigaki, S.; Akagi, A.; Hasegawa, M.; Nishioka, K.; Li, Y.; Riku, M.; Ikeuchi, T.; Fujioka, Y.; et al. Motor neuron TDP-43 proteinopathy in progressive supranuclear palsy and corticobasal degeneration. Brain 2022, 145, 2769–2784. [Google Scholar] [CrossRef] [PubMed]
- Geser, F.; Winton, M.J.; Kwong, L.K.; Xu, Y.; Xie, S.X.; Igaz, L.M.; Garruto, R.M.; Perl, D.P.; Galasko, D.; Lee, V.M.; et al. Pathological TDP-43 in parkinsonism-dementia complex and amyotrophic lateral sclerosis of Guam. Acta Neuropathol. 2008, 115, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M.; Arai, T.; Akiyama, H.; Nonaka, T.; Mori, H.; Hashimoto, T.; Yamazaki, M.; Oyanagi, K. TDP-43 is deposited in the Guam parkinsonism-dementia complex brains. Brain 2007, 130, 1386–1394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishima, T.; Fujioka, S.; Tomiyama, H.; Yabe, I.; Kurisaki, R.; Fujii, N.; Neshige, R.; Ross, O.A.; Farrer, M.J.; Dickson, D.W.; et al. Establishing diagnostic criteria for Perry syndrome. J. Neurol. Neurosurg. Psychiatry 2018, 89, 482–487. [Google Scholar] [CrossRef] [Green Version]
- Deshimaru, M.; Kinoshita-Kawada, M.; Kubota, K.; Watanabe, T.; Tanaka, Y.; Hirano, S.; Ishidate, F.; Hiramoto, M.; Ishikawa, M.; Uehara, Y.; et al. DCTN1 binds to TDP-43 and regulates TDP-43 aggregation. Int. J. Mol. Sci. 2021, 22, 3985. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tamaki, Y.; Urushitani, M. Molecular Dissection of TDP-43 as a Leading Cause of ALS/FTLD. Int. J. Mol. Sci. 2022, 23, 12508. https://doi.org/10.3390/ijms232012508
Tamaki Y, Urushitani M. Molecular Dissection of TDP-43 as a Leading Cause of ALS/FTLD. International Journal of Molecular Sciences. 2022; 23(20):12508. https://doi.org/10.3390/ijms232012508
Chicago/Turabian StyleTamaki, Yoshitaka, and Makoto Urushitani. 2022. "Molecular Dissection of TDP-43 as a Leading Cause of ALS/FTLD" International Journal of Molecular Sciences 23, no. 20: 12508. https://doi.org/10.3390/ijms232012508
APA StyleTamaki, Y., & Urushitani, M. (2022). Molecular Dissection of TDP-43 as a Leading Cause of ALS/FTLD. International Journal of Molecular Sciences, 23(20), 12508. https://doi.org/10.3390/ijms232012508