2.2. Case Study
To gain experience with the basic functionalities of GA_v2, we recommend the users to consult the technical details described in the additional files of GA_v1.19 [
5] and also the Manual.pdf available in the docs folder. All of the scripts are available in the folder scripts and are open source as GA_v2. In order to make the most of GA_v2, we created four .mp4 videos with Kazam application (
https://launchpad.net/kazam; accessed on 31 August 2022), which are available as
Supplementary Materials (Video S1–S4).
Although the results of a JQ alignment may be reported from left to right in the GUI either in the red-blue or blue-red order (depending on the orientation of the insertion), we will further use the expression “blue-red” for both alternatives. The TGN represents the first blue nucleotide next to a red NT nucleotide at the genome–transposon junction border/juxtaposition, and it is highlighted in green.
The nucleotide sequences uploaded in the genome database (GD) or in the transposon database (TD) of GA_v2 are indexed as reference sequences; therefore, any query sequence may be aligned against each of these references. Whenever one introduces in the GD a collection of contigs standing for a new sequenced genome, each of the reads/contigs is indexed as a reference sequence. Similarly, if the canonical release of a model genome (such as r6.47 of D. melanogaster) is deposited in the GD, each of the chromosome sequences is implicitly converted into an indexed reference sequence. To avoid confusions between the canonical reference assembly of a model genome and the genome assembly generated in a new sequencing project, we will further use the terms canonical reference genome (CRG) and alternative reference genome (ARG) to describe the workflows and performances of GA_v2.
The loading time (see
Video S2: Loading_Genomes.mp4) of a reference genome in the GD varies from a few seconds (for a bacterial genome) to a few minutes for a eukaryotic genome of moderate size (such as of
D. melanogaster). Obviously, for a mammalian genome, the loading time could rise to even a few tens of minutes. We recommend using a computer equipped with at least 16 Gb of RAM, 500 Gb SSD and a processor able to work at around 3.5–4.00 GHz. It may be noticed that the mid-range laptops fulfill these requirements; the performance of GA_v2 is significantly increased when it is run on a more powerful computing machine. The mapping results we present herein are obtained by working with regular computers, equipped with Linux Mint or Ubuntu OS, 16–64 Gb of RAM, Intel or AMD processors of maximum 4.5 GHz, and no more than 1 Tb of SSD.
The time required for aligning and assembling the results depends on the computing power and also on the number of the queries (a query sequence or a list of many queries) run against the reference genome, the length of each query and the size of the genome. The best performances are obtained when the reference genome is interrogated with queries of tens to a few hundred/thousands of nucleotides. We do not recommend the usage of query sequences longer than 15,000 nucleotides.
A key issue is setting the parameter named Nucleus size (Settings/Parameters/Nucleus size). For query sequences longer than a few hundreds of nucleotides, a higher value than 10 (the minimal effective value) should be set for the Nucleus size in order to obtain the alignment results in a reasonable time-span. As an example, when the Nucleus size is set to 15–20, the user might obtain in just a few minutes a list of alignments for the interrogation of a moderate size reference genome with a 10,000 nucleotides query. An indication that the Nucleus size is used at values that are too low (10–15) for the length of the aligned queries is that the aligning process freezes at the stage assembling results shown in the status bar. In this case, the software should be restarted and a higher value of the Nucleus size may be tested. A drawback of GA_v2 that we aim to fix is that only one session of GA_v2 can be run at once. When two sessions of GA_v2 are simultaneously open, some RAM issues would occur and the computer must be restarted.
Picking depth parameter is key when searching if more than one insertion of an NT occurs in a reference sequence, regardless of whether the reference is a long read, a contig, or an assembled chromosome. By default, the Picking depth parameter is set to the value of 1. Therefore, once a query sequence is covered by an alignment versus a subsequence of a particular reference sequence, no other similar alignment is reported, even if identical subsequences occur at different coordinates. If there are two instances of an NT insertion in a contig, only one of them will be reported when Picking depth is set to 1.
We further detail two recommended pipelines for mapping NTs, namely Workflow 1 (WF1) and Workflow 2 (WF2), designed to map insertions of hobo and P mobile elements harbored by the genome of D. melanogaster Horezu strain.
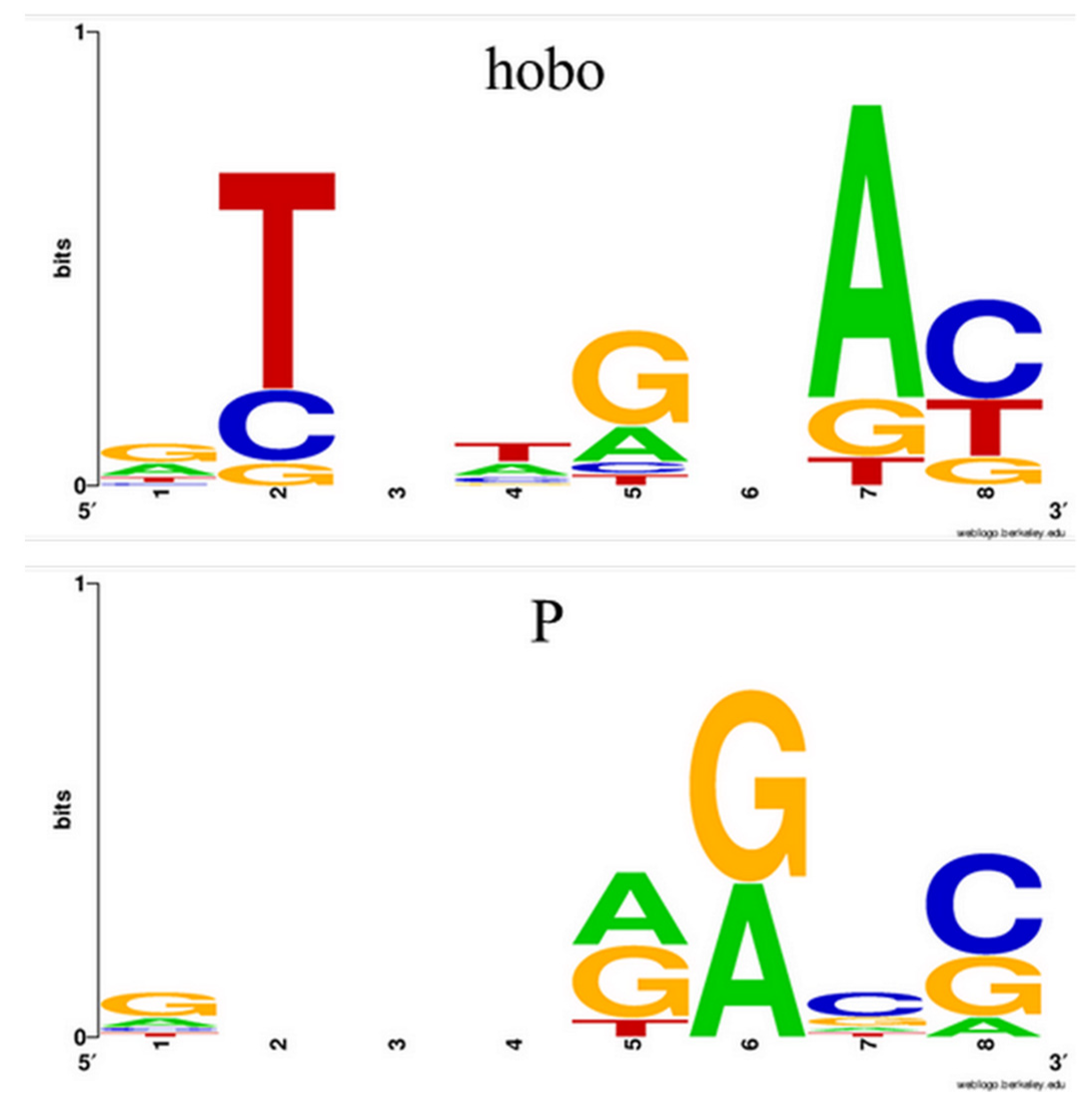
We chose to work with P-element and hobo NTs because they are both class II transposons and produce 8 bp length TSDs. Additionally, P elements spread relative recently in natural populations of
D. melanogaster [
12] but are completely absent in the reference genome. This aspect facilitates the identification of the P-element with GA_v2, as all the results obtained for a given ARG will be marked in blue-red and have a green TGN. Apart from the P-element, hobo is found in many genomic locations in the reference genome (~ 60 annotated copies). We included it in this study to exemplify the two types of output that GA_v2 offers when dealing with transposons existing in the reference: a blue alignment for those conserved and a blue-red output for those identified only in the analyzed ARG.
Each workflow makes use of specific GA_v2 packages and associated scripts. The value for the parameter Number of results impacts on results visualization. For WF1, we recommend a value around 1000 in order to find more positive results for NTs remnants. For WF2, the alignment score of a NT insertion with an almost intact TIR next to the green TGN is increased with 500 if the bonus option is activated. Therefore, a list of two results is usually enough to map an insertion of a DNA transposon. After finishing the WF1, the user must close the GA_v2 containing the ARG and only then start the WF2 with the GA_v2 package loaded with the CRG and the NTs reference sequences.
The WF1 package contains only the ARG in the GD and the user scans it with a query sequence represented by a selected subsequence of it, in order to find contigs with insertions of the NT. In this stage, there is no need to have the NT reference sequence loaded in the TD. Herein, we present examples for finding and mapping of hobo and P-element insertions in a collection of contigs generated with Flye tool [
13] starting from two sets of reads obtained by sequencing and re-sequencing of the Horezu strain. For each transposon, we used two query sequences, where the first one is represented by the first 60 nucleotides cropped from the 5’ end (Q5’) of the NT, and the second one contains the last 60 nucleotides from the 3’ (Q3’) end of the NT. The nucleotide length of these queries is an arbitrary one, but this interval of 60 nucleotides contains the TIRs of P-element (31 bp) or of hobo (12 bp), plus a subsequence that allows us to differentiate between the 5’ TIR and 3’ TIR of an NT when the insertions are in either I or II orientation.
The alignments that cover almost the entire query length and have a reasonable score (the user picks the threshold score; for a 60 nucleotides query, we recommend a score of at least 40) are visually checked to have the complete TIR. Then, they are exported as tabular data and are manipulated with specific scripts to export junction sequences required as queries in the WF2. A junction sequence is composed by either a Q5’ or a Q3’ plus flanking nucleotides extracted from the respective contig. The length of the flanking region is decided by the user and the size of the upstream flanking sequence of Q5’ may be different from the length of downstream flanking sequence of Q3’. Consecutive to our tests, we concluded that at least a few hundred or even thousands of nucleotides should be considered for extension if the NT is inserted in a repetitive sequence (such as in a different NT) or if the remnant is located quite close to an end of the ARG contig. The extraction script exports an ALERT string in the header of any contig sequence containing terminal remnants.
Frequently, GA_v2 detects positive alignment hits for both Q5’ and Q3’, but for some NT remnants it finds only a Q5’ or a Q3’, since one TIR has been lost during evolution. For each insertion having both TIRs, the script extracts an upstream flanking sequence for Q5’ and a downstream sequence for Q3’, regardless of whether the NT is inserted in the relative orientation plus (orientation 1) or minus (orientation 2). As a result, a collection of junction sequences (JQ5’s and JQ3’s) is constructed. A particular issue is found when an NT remnant has only one TIR; in this case, the extraction procedure is identical, as above, for (let us say) the present Q5’. However, to extract the paired JQ3’, a different procedure joins three consecutive subsequences: the Q5’ one, a downstream subsequence with the same length as the respective NT, and 2000 (adjustable) additional downstream nucleotides.
The recommended settings of the parameters for this step are Short, Nucleus size = 20–30 and Picking depth = 1.
In order to map hobo and P transposons in Horezu strain, we opted for collecting 3000 nucleotides long flanking sequences; therefore, the 5’ and 3’ JQ sequences had approximatively 3060 nucleotides. The JQ5’s and JQ3’s generated in WF1 were used as queries to map the insertions of the NT in the ARG, but versus CRG. The procedure requires a second GA_v2 package, containing only the CRG in the GD and the reference sequence of the NT in TD. We recommend activating the bonus option in this stage in order to prioritize the blue-red results in the list of exported alignments.
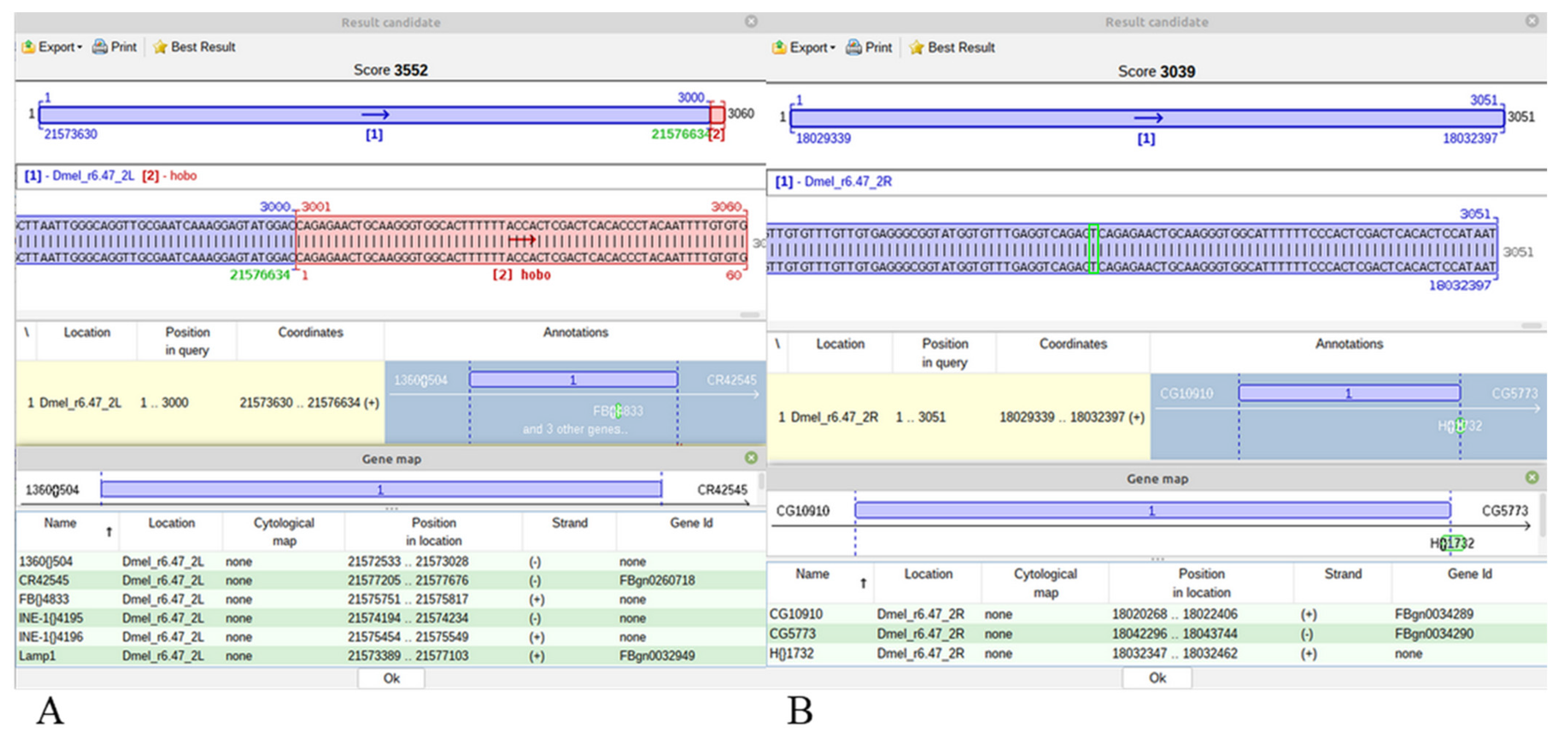
For a hobo insertion, a blue-red best scoring alignment is obtained only when it is specific for the Horezu strain, the ARG (
Figure 5A). Alternatively, a similar/identic insertion may exist in the CRG but it may be either annotated or not in FlyBase. In both cases, the best alignments of JQ5’ or JQ3’ are completely blue best scoring results (
Figure 5B). In these cases, no green TGN is to be highlighted, but the user can visualize the genomic region of insertion and may also easily compute the TGN coordinate, which is the first genomic nucleotide next to nucleotide 1 of Q5’ or to nucleotide 60 of Q3’.
The rationale of this output is that if a best blue scoring alignment covers the length of a JQ over a nucleotide interval of CRG, then GA_v2 does not compute any other shorter alignments in the same coordinate range of the query or reference sequences. Obviously, an alternative blue-red alignment with the same score is excluded in this case.
Using the aforementioned strategy, we have successfully mapped 25 hobo insertions (
Table 2). Fourteen hobo insertions are present only in Horezu genome and six genes are hit by insertions, namely
Lamp1,
Nlg4,
Mnr,
CG42541,
dnc and
bru3. The remaining 11 hobo insertions were found in both Horezu and
D. melanogaster r6.47 reference genotypes; 2 of them hit
Stlk,
fog and
CG42346 genes. Out of these 11 insertions, only 6 are currently annotated in the reference genome. The other five hobo insertions hit four genes (
l(3)80Fg,
Myo81F,
Pzl and
MFS17) and represent new annotated insertions mapped by GA_v2.
The mapping of the P-element is simpler because this NT is absent in the CRG of D. melanogaster; therefore, a JQ alignment is never a completely blue one. Since only the CRG fragment is covered by a blue sub-alignment of the JQ, the rest of the query is aligned versus the P-element reference sequence; therefore, a blue-red best scoring alignment is reported and the TGN is highlighted in green.
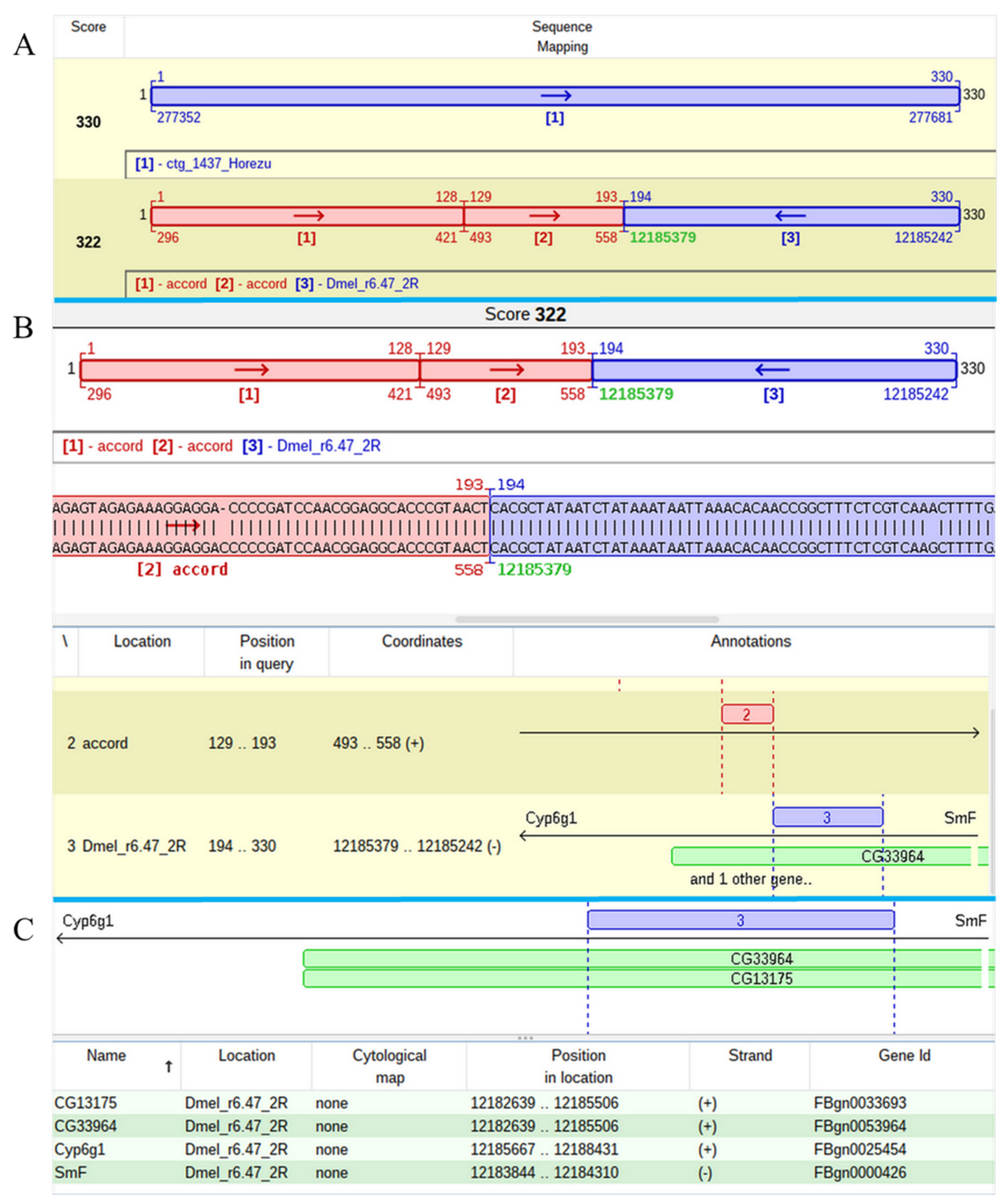
The mapping of one of two P-element insertions that was found in the contig 1437 of Horezu strain was a challenging task. The alignment of the respective JQ5 points to an insertion in the overlapped genes
CG13175 and
CG33964, but the result of the JQ3 analysis indicates that this insertion is located in
Cyp6g1, a gene located in the close proximity of
CG13175/
CG33964. The poor quality of the subalignments close to both TIRs of the P-element led us to manually annotate these regions and to find that they are actually fragments of the accord LTR retrotransposon. Basically, this P-element insertion hit an accord insertion located in its turn in the overlapped genes
CG13175/
CG33964 (
Figure 6).
Interestingly, an accord insertion in
Cyp6g1 is reported for various natural populations of
D. melanogaster and this insertional allele confers resistance to insecticides [
14,
15,
16]. We presume that this insertion of accord in
CG13175/
CG33964 is actually the insertional allele previously reported as being located at 291 bp upstream of the transcription start site of
Cyp6g1 [
14,
15,
16]. The increase in the mapping resolution obtained with GA_v2 is consistent with the fact that the nucleotide coordinate corresponding to 291 bp upstream of the transcription start site of
Cyp6g1 is actually very close to the TGN 12185379, pertaining to
CG13175/
CG33964 genes.
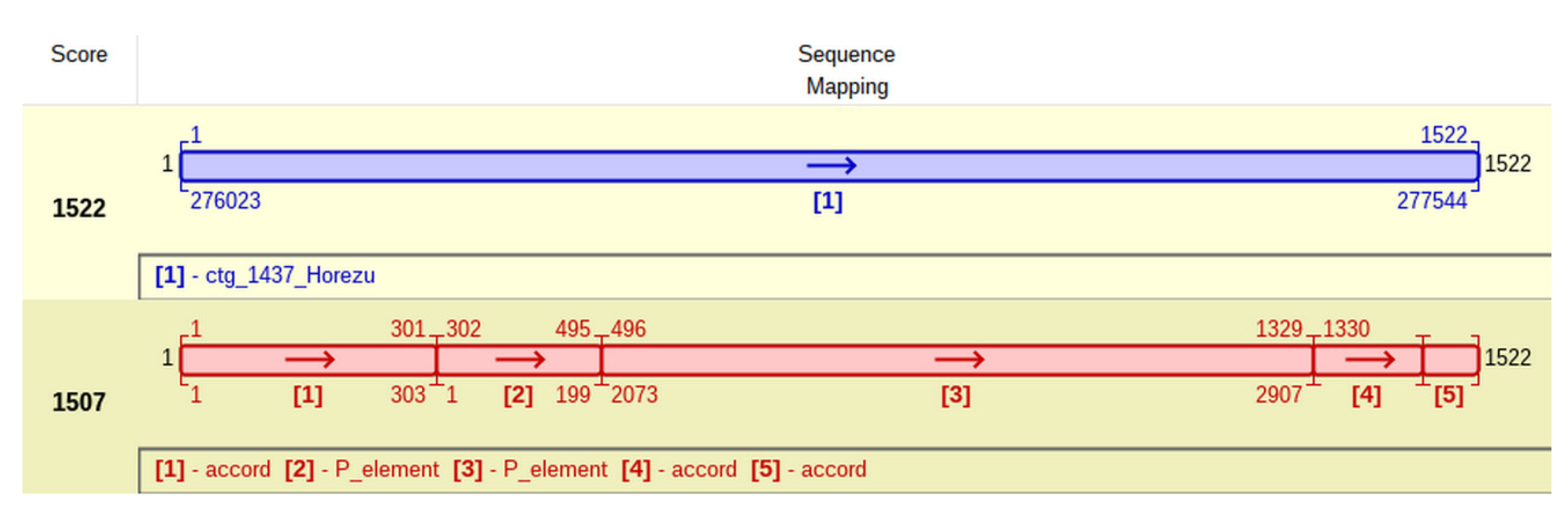
This particular insertion of P-element is located at coordinate 303 of accord (
Figure 7) and generates a TSD with the approximate sequence GTCTAGAC. It is tempting to ask if this peculiar P-element insertion, located in an accord insertional allele, commonly found in many
D. melanogaster natural populations, impedes on the adaptative resistance to insecticides of Horezu strain.
Regarding the P-element, all of its insertions are found, as expected, only in the Horezu strain (
Table 3) and affect four genes (
Ac13E,
spri,
shn and
NFAT).
In conclusion, this straightforward mapping strategy is true when the NT subsequence of a JQ is present in the ARG, but not in the CRG (as for the P-element), or when an NT is present in both the ARG and the CRG, but with instances of NT insertions present only in the ARG (the hobo case).
There is a distinction between mapping retrotransposons and DNA transposons, since DNA transposons are usually located in unique genomic regions or in low copy number sequences, such as genes, while retrotransposons are mainly located in repetitive sequences and often have repetitive sequence structure. Software, such as LoRTE (a tool also tested on
D. melanogaster in the describing paper) [
17] and RepeatMasker [
18], only report the chromosome and the coordinates of each resolvable insertion, a task readily performed by GA_v2, but these tools do not offer detailed local genomic annotations. Instead, GA_v2 is able to map insertions based on junction sequences with the highest available resolution [
5]; it reports detailed annotations in both GUI and in the tabular format; it works with any genome, regardless of its size; it works with any NTs sequence, regardless of its length; it may be run by using either fragments of an NT or with the whole reference sequence of an NT as a query; it is able to work both as a transposon tracker, performing the two workflows described in this paper, or as a standard aligner for nucleotide sequences, aligning certain sequences to one or more reference genomes (or the user can load a sequenced genome in the form of a contig list, in order to scan it with a query).
In contrast to the aforementioned mapping tools, GA_v2 is designed for the analysis of individual NTs of interest, not for a concurrent inquiry of the global NTs landscape of a genome. Although the user may load several genomes and transposons in GD and in TD, in both WF1 and WF2 a single transposon is processed, a mapping strategy, which, in our opinion, is less estimative and offers higher precision and more details about an NT of interest.
Due to the structural complexity of the LTR elements, the extraction of JQs in WF1 must be done carefully, which implies a step of manual curation by the user. In the following example we describe both the identification and mapping of the copia retrotransposon, as well as the potential issues encountered in WF1 or WF2. The LTRs of copia are 276 nucleotides long, and in order to discriminate between LTR5’ and LTR3’, we used the terminal 600 nucleotides query sequences from both ends of copia. Some alignments almost completely cover the query length, but others stand only for an LTR; therefore, a manual inquiry should be performed in order to avoid ambiguities between the identical LTR5’ and LTR3’.
In order to check over the results of the mapping strategy performed with Q5’ and Q3’ derived from the copia LTRs, we repeated the procedure starting from a single query represented by a subsequence of 100 nucleotides extracted from the middle of the copia reference sequence. The specific JQs derived from this subsequence allowed us to remap copia in Horezu genome and the two sets of results coincide. In WF2, we achieved better results when Give bonus to insertion candidates was not activated, because the number of concurrent blue-red alignments was reduced.
Thus, GA_v2 found 14 copia insertions harbored by 13 contigs. Out of them, four were unresolvable, while ten were mapped at least at chromosome level (
Table 4). Eight out of ten copia insertions are particular for the Horezu strain (blue-red alignments) and affect three genes (
Pzl,
Dscam4 and
Pkc53E). The remaining two insertions are present in both Horezu and the reference genome and map in Y chromosome (blue alignments). The apparent contradiction seen for contig 890, i.e., two distinct copia transposons mapping to different chromosomes, is most probably a consequence of genome missassembly. Comparatively, LoRTE only finds four copia insertions in the Horezu strain. This difference reveals a higher efficiency of GA_v2, at least for mapping this retrotransposon (
Table 5).
GA_v2 is also able to perform a high-throughput processing of a retrotransposon for further mapping and polymorphisms analysis. Nevertheless, the user should be warned that the mapping of overrepresented NTs involves various issues, which become more complex as the genome of interest is larger and the particular retrotransposon to be mapped occurs in very high number copies, such as SINEs and LINEs in humans. These problems are intrinsic for other mapping software, as they rely on specific flanking sequences, regardless of whether these sequences are only TSDs or extended sequences. To map either DNA transposons or retrotransposons with high accuracy, GA_v2 requires unique flanking DNA sequences which border the insertion. The key step of extracting specific flanking sequencing is performed in WF1, but care should be taken when interpreting the alignments of the respective JQs, since retrotransposons are often inserted in highly repetitive regions, such as similar or different NTs. Therefore, many flanking sequences bordering retrotransposons insertions may align almost perfectly in several places in the genome, preventing the user to map some insertions from an ARG at unique locations in the CRG. These cases may sometimes be solved by extending the length of the extracted flanking sequences and a manual curation of the GA_v2 output file. By default, some insertions cannot be mapped, either because of inherent sequencing and assembly issues, or due to their occurrence inside very long repetitive genomic landscapes.
A particular case is when the first few high-scoring alignments in the list of results for a particular JQ have very similar scores (differences of the magnitude order of tens) and map at various genomic nucleotides coordinates pertaining to the same chromosome of the CRG. Obviously, such insertions can be mapped at chromosome resolution, but not at genomic nucleotide coordinate or gene resolution.
To facilitate the interpretation of the colored results offered by GA_v2 in GUI, we further describe some technical aspects specific for the aligning and mapping strategy of GA_v2.
The two databases of GA_v2 are versatile and one may decide which database should be used to upload reference sequences. The same reference sequence may be uploaded in any of the databases or in both of them, either to have a positive control for a target alignment, or to get rid of the genomic restrictions. In order to have two (or more) blue subalignments coalesced in the same final alignment, there are two rules to be fulfilled. First of all, two blue subalignments are forbidden to share overlapped genomic coordinates (genomic distance ≥ 0). Then, only if the first condition is fulfilled, the maximal genomic distance between two subalignments should not be bigger than a customizable value (set in the source code and requiring the compilation of a new GA_v2 package when changed), which by default is 200. Therefore, whenever the query sequence is a gene/NT affected by a deletion > 200 nucleotides, GA_v2 would report two different final results for each of the two genic fragments, which border the deletion.
These two restrictions are not functional when GA_v2 composes blue-red or red-red alignments to allow the detection of NTs insertions, NTs self-insertions and large internal deletions affecting some transposon remnants. Care should be taken when contigs are loaded in the NT database, since they would be considered as transposons and there are no restriction rules. This may be advantageous when the user wants to check how many remnants of an NT are present in a long contig, as they would be composed in the same final alignment. On the other hand, if an NT remnant is affected by a big internal deletion, then the two partial alignments are presented as two discrete subalignments relative to the length of the NT reference sequence. It is always instructive to analyze the partial results used to compose the final results; in such a case, one may notice that the two transposon fragments are combined in the same alignment, regardless of whether they share overlapped nucleotide coordinates of the NT reference sequence.
Summarizing, GA_v2 is able to identify NTs insertions in long reads, i.e., generated with nanopore technologies, as well as in contigs generated by the assembly of reads obtained with previous generations of sequencing technologies. To scan for transposon insertions, the user must load the reads or the ARG contigs sequences in the GD and then use sets of Q5’ or the Q3’ sequences as queries. If a complete NT canonical sequence is used instead to scan for insertions, the WF1 should be adapted by the user in order to obtain equivalent JQs. Most of the existing tools that identify transposable elements in sequenced genomes involve dependencies that are usually of two types: databases (more or less available and updated to the latest genome version) and applications/packages (which can undergo changes or may become obsolete over time, leading to problems with the tool’s functionality).
An obvious advantage of GA_v2 is a friendly graphical user interface (GUI), which allows the user to visualize and to interpret the results of aligning and mapping, along with various genome annotation features. Most mapping programs do not have a graphical interface, hence raw alignment results are difficult to obtain and interpret. An example is LoRTE software, dedicated to the analysis of long read sequences and able to identify the presence/absence of NTs in a sequenced genome, comparative to a reference genome [
17]. LoRTE is written in Python 2.7 and depends on a recent version of the ncbi-blast+ package and BioPython. To run the program, five input files are needed: the reference genome, a .fasta file with reads, a list of annotated NTs (difficult to obtain for some species or for newer versions of genomes), a list of consensus NTs and a list of parameters that the user modifies according to their choice. The output does not include the actual alignments, but rather lists present/absent/new inserted NTs, with or without sequence polymorphisms. LoRTE indicates the chromosome and the coordinates where the NTs are located, but it does not show annotations of the local genomic landscape (the presence of genes or other NTs, TSDs, etc.). A drawback of LoRTE is that it cannot be run without the lists of consensus NTs or NTs annotations specific for the reference genome.
LoRTE does not use junction sequences, instead using only flanking sequences for the NTs and checking them on the CRG with MEGABLAST [
17]. On the contrast, GA_v2 uses JQs, which are a hallmark of our tool, which improves the resolution of mapping insertions with the highest resolution reported so far [
5], mainly when partial overlapping occurs between the IRs or LTRs and the proximal ends of the flanking sequences. Although GA_v2 is able to detect polymorphisms, such as new insertions/deletions of an NT in a population versus the reference genome, it is mainly focused on a comparative and very accurate mapping and annotation of each insertion of an NT of interest. As a result, GA_v2 offers mapping at chromosomal and/or genic level, and it does not compute the total NTs content in numbers and percentages.
Our TIR-dependent mapping strategy identified the same conserved hobo remnants in CRG r6.47 that were identified by LoRTE in an older reference genome (r5), for which the file with TE annotations is available. Apart from the hobo remnants that have one of the TIRs and which were identified with both GA and LoRTE, the latter tool also detected a number of remnants without TIRs, but which have internal sequences specific to the hobo transposon. The mapping strategy of GA_v2 described herein is customizable and may be adapted to search for NTs remnants, which have no TIRs. To initiate the WF1, various internal subsequences of the NT reference sequence can be selected and used as alternative queries to search for remnants that have been missed by TIR-dependent mapping strategy.
In summary, GA_v2 outperforms LoRTE when single transposons are mapped, regardless of their mechanism of transposition (
Table 5).
Another NTs mapping tool without a GUI is the transposon insertion finder (TIF), which was designed to identify insertions by using as queries the terminal sequences of NTs [
19]. A consistent limitation of this mapping tool is the exclusive requirement of TIF for short reads generated by Illumina, which are grouped by the presence of a specific TSD sequence.
When GA_v2 is compared with RepeatMasker, a program generally used to evaluate the content of transposons in sequenced genomes [
18], one can notice that the work strategies and the expected results are different. Unlike RepeatMasker, GA_v2 allows a detailed analysis of a specific NT of interest just by using an NT subsequence as a query to be run versus a database containing reads or contigs set as reference sequences. Both tools allow the user to find out the number of copies of an NT of interest, but RepeatMasker is able to mask the repetitive sequences in the analyzed genome and to calculate the proportion of total transposable elements in the genome. An advantage of RepeatMasker is the online version with GUI, but with a size limitation of 100 kb for the contigs scanned for insertions. To obtain accurate alignment and mapping results, GA_v2 relies on its own alignment heuristic described in detail elsewhere [
5]. Conversely, RepeatMasker can be run via an implementation of the Smith–Waterman algorithm or via BLAST aligner, an alternative that conducts to a decrease in the tool’s sensitivity. Even though RepeatMasker is one of the most used programs in the analysis of repetitive sequences, it also has some limitations concerning the accuracy of the identification of NTs residues [
20].
We verified if the EXTRACTION.sh script used in WF1 is functional on the genomes of different species downloaded from other database than FlyBase. For this purpose, we tested the script on the genome of
Caenorhabditis elegans from Ensembl release 107 [
21] and concluded that the collection of the JQs was generated as expected. Therefore, the mapping strategy of GA_v2 is not limited to
D. melanogaster or to the FlyBase format. Unfortunately, to our knowledge, there are no annotations for NTs available in Ensembl; therefore, GA_v2 does not show the NTs local landscape for the mapped insertions.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}