
Exploration of Tools for the Interpretation of Human Non-Coding Variants

 , , , ,
, , , , 
 , and
, and
Abstract
:1. Introduction
2. Methods
2.1. Inclusion Criteria
2.2. The Controlled Scenario: Knowledge-Based and Computational Performance
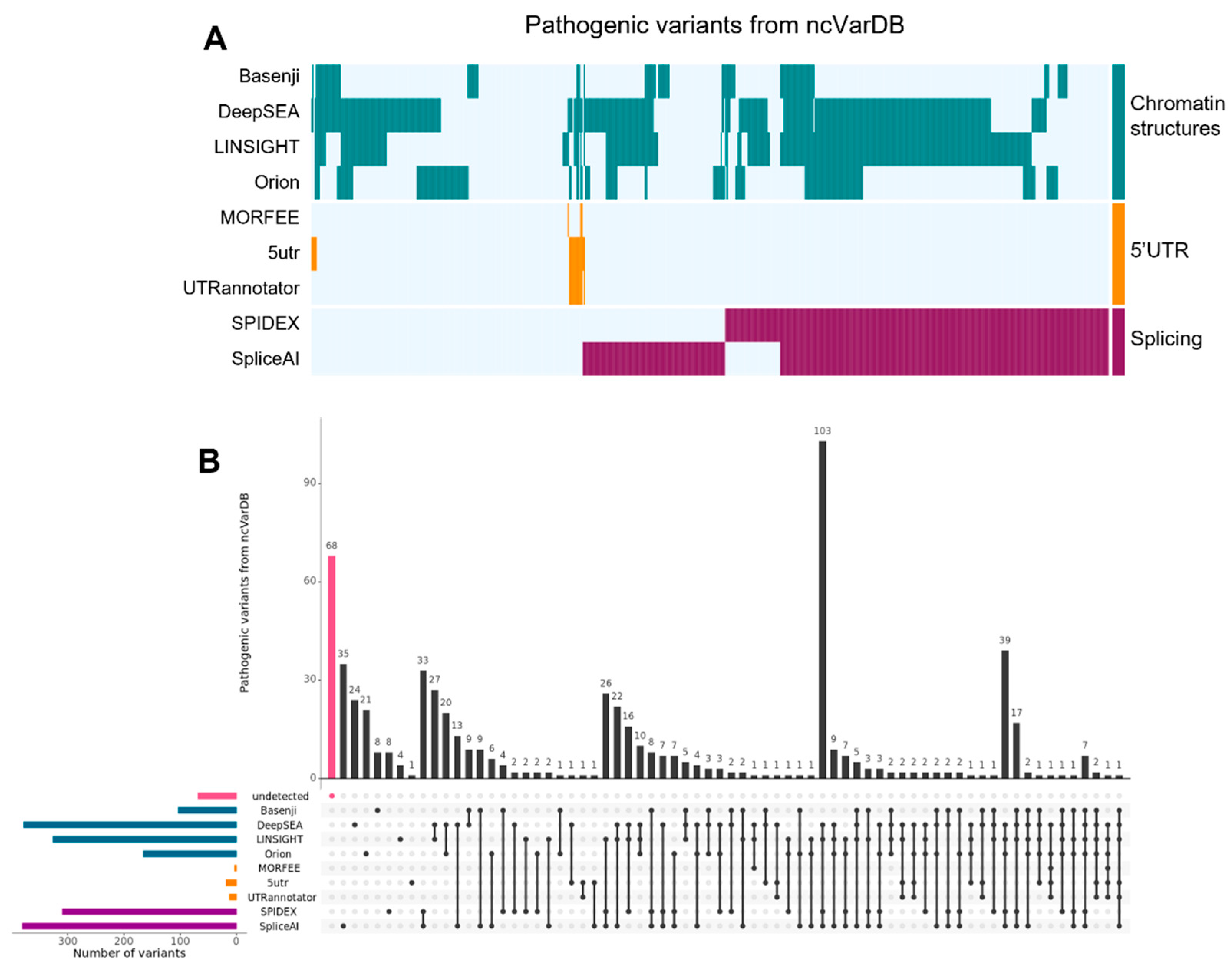
3. Results
3.1. Method Selection
3.2. Tools for Identifying NCV Affecting UTRs
3.3. Tools for Identifying NCV Affecting Splicing Sites
3.4. Tools for Identifying NCV Affecting Genome Accessibility and Mutation Intolerance
3.5. Phenotype-Based Functional Prediction Tool
4. Discussion
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Rands, C.M.; Meader, S.; Ponting, C.P.; Lunter, G. 8.2% of the Human Genome Is Constrained: Variation in Rates of Turnover across Functional Element Classes in the Human Lineage. PLoS Genet. 2014, 10, e1004525. [Google Scholar] [CrossRef] [Green Version]
- Kellis, M.; Wold, B.; Snyder, M.P.; Bernstein, B.E.; Kundaje, A.; Marinov, G.K.; Ward, L.D.; Birney, E.; Crawford, G.E.; Dekker, J.; et al. Defining functional DNA elements in the human genome. Proc. Natl. Acad. Sci. USA 2014, 111, 6131–6138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, M.; Yang, X.; Ren, X.; Maliskova, L.; Li, B.; Jones, I.R.; Wang, C.; Jacob, F.; Wu, K.; Traglia, M.; et al. Mapping cis -regulatory chromatin contacts in neural cells links neuropsychiatric disorder risk variants to target genes. Nat. Genet. 2019, 51, 1252–1262. [Google Scholar] [CrossRef]
- Rhind, N.; Gilbert, D.M. DNA replication timing. Cold Spring Harb. Perspect. Biol. 2013, 5, a010132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koren, A.; Handsaker, R.E.; Kamitaki, N.; Karlić, R.; Ghosh, S.; Polak, P.; Eggan, K.; McCarroll, S.A. Genetic variation in human DNA replication timing. Cell 2014, 159, 1015–1026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McInerney-Leo, A.M.; Duncan, E.L. Massively Parallel Sequencing for Rare Genetic Disorders: Potential and Pitfalls. Front. Endocrinol. 2020, 11, 628946. [Google Scholar] [CrossRef]
- Ellingford, J.M.; Ahn, J.W.; Bagnall, R.D.; Baralle, D.; Barton, S.; Campbell, C.; Downes, K.; Ellard, S.; Duff-Farrier, C.; FitzPatrick, D.R.; et al. Recommendations for clinical interpretation of variants found in non-coding regions of the genome. Genome Med. 2022, 14, 73. [Google Scholar] [CrossRef]
- Wang, Z.; Zhao, G.; Li, B.; Fang, Z.; Chen, Q.; Wang, X.; Luo, T.; Wang, Y.; Zhou, Q.; Li, K.; et al. Performance comparison of computational methods for the prediction of the function and pathogenicity of non-coding variants. Genom. Proteom. Bioinform. 2022; in press. [Google Scholar] [CrossRef]
- Rojano, E.; Seoane, P.; Ranea, J.A.G.; Perkins, J.R. Regulatory variants: From detection to predicting impact. Brief. Bioinform. 2019, 20, 1639–1654. [Google Scholar] [CrossRef] [Green Version]
- Pabinger, S.; Dander, A.; Fischer, M.; Snajder, R.; Sperk, M.; Efremova, M.; Krabichler, B.; Speicher, M.R.; Zschocke, J.; Trajanoski, Z. A survey of tools for variant analysis of next-generation genome sequencing data. Brief. Bioinform. 2014, 15, 256–278. [Google Scholar] [CrossRef]
- Gloss, B.S.; Dinger, M.E. Realizing the significance of noncoding functionality in clinical genomics. Exp. Mol. Med. 2018, 50, 1–8. [Google Scholar] [CrossRef] [Green Version]
- French, J.D.; Edwards, S.L. The Role of Noncoding Variants in Heritable Disease. Trends Genet. 2020, 36, 880–891. [Google Scholar] [CrossRef] [PubMed]
- Biggs, H.; Parthasarathy, P.; Gavryushkina, A.; Gardner, P.P. ncVarDB: A manually curated database for pathogenic non-coding variants and benign controls. Database 2020, 2020, baaa105. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, R.M.; Haussler, D.; Kent, W.J. The UCSC genome browser and associated tools. Brief. Bioinform. 2013, 14, 144–161. [Google Scholar] [CrossRef] [Green Version]
- Zook, J.M.; McDaniel, J.; Olson, N.D.; Wagner, J.; Parikh, H.; Heaton, H.; Irvine, S.A.; Trigg, L.; Truty, R.; McLean, C.Y.; et al. An open resource for accurately benchmarking small variant and reference calls. Nat. Biotechnol. 2019, 37, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Genome in a Bottle, NIST. 2012. Available online: https://www.nist.gov/programs-projects/genome-bottle (accessed on 7 June 2021).
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Uzun, Y.; Gao, P.; He, B.; Ma, X.; Wang, J.; Han, S.; Tan, K. Identifying noncoding risk variants using disease-relevant gene regulatory networks. Nat. Commun. 2018, 9, 702. [Google Scholar] [CrossRef] [Green Version]
- Kelley, D.R.; Reshef, Y.A.; Bileschi, M.; Belanger, D.; McLean, C.Y.; Snoek, J. Sequential regulatory activity prediction across chromosomes with convolutional neural networks. Genome Res. 2018, 28, 739–750. [Google Scholar] [CrossRef] [Green Version]
- Alipanahi, B.; Delong, A.; Weirauch, M.T.; Frey, B.J. Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat. Biotechnol. 2015, 33, 831–838. [Google Scholar] [CrossRef]
- Zhou, J.; Troyanskaya, O.G. Predicting effects of noncoding variants with deep learning-based sequence model. Nat. Methods 2015, 12, 931–934. [Google Scholar] [CrossRef]
- Lee, D.; Gorkin, D.U.; Baker, M.; Strober, B.J.; Asoni, A.L.; McCallion, A.S.; Beer, M.A. A method to predict the impact of regulatory variants from DNA sequence. Nat. Genet. 2015, 47, 955–961. [Google Scholar] [CrossRef] [Green Version]
- Backenroth, D.; He, Z.; Kiryluk, K.; Boeva, V.; Pethukova, L.; Khurana, E.; Christiano, A.; Buxbaum, J.D.; Ionita-Laza, I. FUN-LDA: A Latent Dirichlet Allocation Model for Predicting Tissue-Specific Functional Effects of Noncoding Variation: Methods and Applications. Am. J. Hum. Genet. 2018, 102, 920–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahary, D.; Golan, Y.; Mazor, Y.; Zelig, O.; Barshir, R.; Twik, M.; Stein, T.I.; Rosner, G.; Kariv, R.; Chen, F.; et al. Genome analysis and knowledge-driven variant interpretation with TGex. BMC Med. Genom. 2019, 12, 200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Q.; Hu, Y.; Sun, J.; Cheng, Y.; Cheung, K.-H.; Zhao, H. A Statistical Framework to Predict Functional Non-Coding Regions in the Human Genome Through Integrated Analysis of Annotation Data. Sci. Rep. 2015, 5, 10576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smedley, D.; Schubach, M.; Jacobsen, J.O.B.; Köhler, S.; Zemojtel, T.; Spielmann, M.; Jäger, M.; Hochheiser, H.; Washington, N.L.; McMurry, J.A.; et al. A Whole-Genome Analysis Framework for Effective Identification of Pathogenic Regulatory Variants in Mendelian Disease. Am. J. Hum. Genet. 2016, 99, 595–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desmet, F.-O.; Hamroun, D.; Lalande, M.; Collod-Béroud, G.; Claustres, M.; Béroud, C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009, 37, e67. [Google Scholar] [CrossRef] [Green Version]
- Amlie-Wolf, A.; Tang, M.; Mlynarski, E.E.; Kuksa, P.P.; Valladares, O.; Katanic, Z.; Tsuang, D.; Brown, C.D.; Schellenberg, G.D.; Wang, L.-S. INFERNO: Inferring the molecular mechanisms of noncoding genetic variants. Nucleic Acids Res. 2018, 46, 8740–8753. [Google Scholar] [CrossRef] [Green Version]
- Vitsios, D.; Dhindsa, R.S.; Middleton, L.; Gussow, A.B.; Petrovski, S. Prioritizing non-coding regions based on human genomic constraint and sequence context with deep learning. Nat. Commun. 2021, 12, 1504. [Google Scholar] [CrossRef]
- Huang, Y.-F.; Gulko, B.; Siepel, A. Fast, scalable prediction of deleterious noncoding variants from functional and population genomic data. Nat. Genet. 2017, 49, 618–624. [Google Scholar] [CrossRef] [Green Version]
- Aïssi, D.; Soukarieh, O.; Proust, C.; Jaspard-Vinassa, B.; Fautrad, P.; Ibrahim-Kosta, M.; Leal-Valentim, F.; Roux, M.; Bacq-Daian, D.; Olaso, R.; et al. MORFEE: A new tool for detecting and annotating single nucleotide variants creating premature ATG codons from VCF files. bioRxiv 2020. [Google Scholar] [CrossRef]
- Gussow, A.B.; Copeland, B.R.; Dhindsa, R.S.; Wang, Q.; Petrovski, S.; Majoros, W.H.; Allen, A.S.; Goldstein, D.B. Orion: Detecting regions of the human non-coding genome that are intolerant to variation using population genetics. PLoS ONE 2017, 12, e0181604. [Google Scholar] [CrossRef] [Green Version]
- Boyle, A.P.; Hong, E.L.; Hariharan, M.; Cheng, Y.; Schaub, M.A.; Kasowski, M.; Karczewski, K.J.; Park, J.; Hitz, B.C.; Weng, S.; et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012, 22, 1790–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, H.Y.; Alipanahi, B.; Lee, L.J.; Bretschneider, H.; Merico, D.; Yuen, R.K.C.; Hua, Y.; Gueroussov, S.; Najafabadi, H.S.; Hughes, T.R.; et al. The human splicing code reveals new insights into the genetic determinants of disease. Science 2015, 347, 1254806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaganathan, K.; Panagiotopoulou, S.K.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019, 176, 535–548.e24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Wakeling, M.; Ware, J.; Whiffin, N. Annotating high-impact 5′untranslated region variants with the UTRannotator. Bioinformatics 2021, 37, 1171–1173. [Google Scholar] [CrossRef] [PubMed]
- Stelzer, G.; Plaschkes, I.; Oz-Levi, D.; Alkelai, A.; Olender, T.; Zimmerman, S.; Twik, M.; Belinky, F.; Fishilevich, S.; Nudel, R.; et al. VarElect: The phenotype-based variation prioritizer of the GeneCards Suite. BMC Genom. 2016, 17, 444. [Google Scholar] [CrossRef] [Green Version]
- Fishilevich, S.; Nudel, R.; Rappaport, N.; Hadar, R.; Plaschkes, I.; Stein, T.I.; Rosen, N.; Kohn, A.; Twik, M.; Safran, M.; et al. GeneHancer: Genome-wide integration of enhancers and target genes in GeneCards. Database 2017, 2017, bax028. [Google Scholar] [CrossRef] [Green Version]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [Green Version]
- Quang, D.; Chen, Y.; Xie, X. DANN: A deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics 2015, 31, 761–763. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Chen, R.; Wang, Q.; Wei, Q.; Ji, Y.; Zheng, G.; Zhong, X.; Cox, N.J.; Li, B. De novo pattern discovery enables robust assessment of functional consequences of non-coding variants. Bioinformatics 2019, 35, 1453–1460. [Google Scholar] [CrossRef]
- Ionita-Laza, I.; McCallum, K.; Xu, B.; Buxbaum, J.D. A spectral approach integrating functional genomic annotations for coding and noncoding variants. Nat. Genet. 2016, 48, 214–220. [Google Scholar] [CrossRef] [Green Version]
- Rogers, M.F.; Shihab, H.A.; Mort, M.; Cooper, D.N.; Gaunt, T.R.; Campbell, C. FATHMM-XF: Accurate prediction of pathogenic point mutations via extended features. Bioinformatics 2018, 34, 511–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, G.R.S.; Dunham, I.; Zeggini, E.; Flicek, P. Functional annotation of non-coding sequence variants. Nat. Methods 2014, 11, 294–296. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ullah, A.Z.D.; Chelala, C. IW-Scoring: An Integrative Weighted Scoring framework for annotating and prioritizing genetic variations in the noncoding genome. Nucleic Acids Res. 2018, 46, e47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Zhao, F. Prioritization and functional assessment of noncoding variants associated with complex diseases. Genome Med. 2018, 10, 53. [Google Scholar] [CrossRef] [Green Version]
- Bodea, C.A.; Mitchell, A.A.; Bloemendal, A.; Day-Williams, A.G.; Runz, H.; Sunyaev, S.R. PINES: Phenotype-informed tissue weighting improves prediction of pathogenic noncoding variants. Genome Biol. 2018, 19, 173. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Hombach, D.; Köhler, S.; Cooper, D.N.; Schuelke, M.; Seelow, D. Regulation Spotter: Annotation and interpretation of extratranscriptic DNA variants. Nucleic Acids Res. 2019, 47, W106–W113. [Google Scholar] [CrossRef]
- Sophia Genetics, Sophia Genetics. (n.d.). Available online: https://www.interactive-biosoftware.com/ (accessed on 1 July 2021).
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Katainen, R.; Donner, I.; Cajuso, T.; Kaasinen, E.; Palin, K.; Mäkinen, V.; Aaltonen, L.A.; Pitkänen, E. Discovery of potential causative mutations in human coding and noncoding genome with the interactive software BasePlayer. Nat. Protoc. 2018, 13, 2580–2600. [Google Scholar] [CrossRef]
- Zhang, S.; He, Y.; Liu, H.; Zhai, H.; Huang, D.; Yi, X.; Dong, X.; Wang, Z.; Zhao, K.; Zhou, Y.; et al. regBase: Whole genome base-wise aggregation and functional prediction for human non-coding regulatory variants. Nucleic Acids Res. 2019, 47, e134. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [Green Version]
- SNPnexus: A Web Server for Functional Annotation of Human Genome Sequence Variation (2020 Update)|Nucleic Acids Research|Oxford Academic, (n.d.). Available online: https://academic.oup.com/nar/article/48/W1/W185/5851388 (accessed on 1 July 2021).
- Desvignes, J.-P.; Bartoli, M.; Delague, V.; Krahn, M.; Miltgen, M.; Béroud, C.; Salgado, D. VarAFT: A variant annotation and filtration system for human next generation sequencing data. Nucleic Acids Res. 2018, 46, W545–W553. [Google Scholar] [CrossRef] [PubMed]
- Hindorff, L.A.; Sethupathy, P.; Junkins, H.A.; Ramos, E.M.; Mehta, J.P.; Collins, F.S.; Manolio, T.A. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl. Acad. Sci. USA 2009, 106, 9362–9367. [Google Scholar] [CrossRef] [Green Version]
- MacArthur, J.; Bowler, E.; Cerezo, M.; Gil, L.; Hall, P.; Hastings, E.; Junkins, H.; McMahon, A.; Milano, A.; Morales, J.; et al. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res. 2017, 45, D896–D901. [Google Scholar] [CrossRef]
- Condit, C.M.; Achter, P.J.; Lauer, I.; Sefcovic, E. The changing meanings of “mutation”: A contextualized study of public discourse. Hum. Mutation 2002, 19, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Mignone, F.; Gissi, C.; Liuni, S.; Pesole, G. Untranslated regions of mRNAs. Genome Biol. 2002, 3, REVIEWS0004. [Google Scholar] [CrossRef] [PubMed]
- Whiffin, N.; Karczewski, K.J.; Zhang, X.; Chothani, S.; Smith, M.J.; Evans, D.G.; Roberts, A.M.; Quaife, N.M.; Schafer, S.; Rackham, O.; et al. Characterising the loss-of-function impact of 5′ untranslated region variants in 15,708 individuals. Nat. Commun. 2020, 11, 2523. [Google Scholar] [CrossRef] [PubMed]
- Kozak, M. The scanning model for translation: An update. J. Cell Biol. 1989, 108, 229–241. [Google Scholar] [CrossRef]
- ImperialCardioGenetics/UTRannotator, ImperialCardioGenetics. 2021. Available online: https://github.com/ImperialCardioGenetics/UTRannotator/blob/40e30db003d72435eb3744ee747029294abbcf3a/Supplementary_Information.pdf (accessed on 11 June 2021).
- Leklab/5utr, Leklab. 2020. Available online: https://github.com/leklab/5utr (accessed on 21 January 2021).
- Human 5′ UTR Design and Variant Effect Prediction from a Massively Parallel Translation Assay|Nature Biotechnology, (n.d.). Available online: https://www.nature.com/articles/s41587-019-0164-5 (accessed on 11 June 2021).
- Murat, P.; Marsico, G.; Herdy, B.; Ghanbarian, A.; Portella, G.; Balasubramanian, S. RNA G-quadruplexes at upstream open reading frames cause DHX36- and DHX9-dependent translation of human mRNAs. Genome Biol. 2018, 19, 229. [Google Scholar] [CrossRef]
- Anna, A.; Monika, G. Splicing mutations in human genetic disorders: Examples, detection, and confirmation. J. Appl. Genet. 2018, 59, 253–268. [Google Scholar] [CrossRef]
- Lord, J.; Gallone, G.; Short, P.J.; McRae, J.F.; Ironfield, H.; Wynn, E.H.; Gerety, S.S.; He, L.; Kerr, B.; Johnson, D.S.; et al. Deciphering Developmental Disorders study, Pathogenicity and selective constraint on variation near splice sites. Genome Res. 2019, 29, 159–170. [Google Scholar] [CrossRef] [Green Version]
- Soemedi, R.; Cygan, K.J.; Rhine, C.L.; Wang, J.; Bulacan, C.; Yang, J.; Bayrak-Toydemir, P.; McDonald, J.; Fairbrother, W.G. Pathogenic variants that alter protein code often disrupt splicing. Nat. Genet. 2017, 49, 848–855. [Google Scholar] [CrossRef] [PubMed]
- Tazi, J.; Bakkour, N.; Stamm, S. Alternative splicing and disease. Biochim. Biophys. Acta 2009, 1792, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.K.; Cooper, T.A. Pre-mRNA splicing in disease and therapeutics. Trends Mol. Med. 2012, 18, 472–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaz-Drago, R.; Custódio, N.; Carmo-Fonseca, M. Deep intronic mutations and human disease. Hum. Genet. 2017, 136, 1093–1111. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Burge, C.B. Splicing regulation: From a parts list of regulatory elements to an integrated splicing code. RNA 2008, 14, 802–813. [Google Scholar] [CrossRef] [Green Version]
- Butkiewicz, M.; Blue, E.E.; Leung, Y.Y.; Jian, X.; Marcora, E.; Renton, A.E.; Kuzma, A.; Wang, L.-S.; Koboldt, D.C.; Haines, J.L.; et al. Functional annotation of genomic variants in studies of late-onset Alzheimer’s disease. Bioinformatics 2018, 34, 2724–2731. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Cheng, T.; Jin, S.; Guo, Y.; Wu, Y.; Liu, D.; Xu, X.; Sun, Y.; Li, Z.; He, H.; et al. Genome-wide open chromatin regions and their effects on the regulation of silk protein genes in Bombyx mori. Sci. Rep. 2017, 7, 12919. [Google Scholar] [CrossRef] [Green Version]
- Gronau, I.; Arbiza, L.; Mohammed, J.; Siepel, A. Inference of natural selection from interspersed genomic elements based on polymorphism and divergence. Mol. Biol. Evol. 2013, 30, 1159–1171. [Google Scholar] [CrossRef] [Green Version]
- Arbiza, L.; Gronau, I.; Aksoy, B.A.; Hubisz, M.J.; Gulko, B.; Keinan, A.; Siepel, A. Genome-wide inference of natural selection on human transcription factor binding sites. Nat. Genet. 2013, 45, 723–729. [Google Scholar] [CrossRef]
- Symmons, O.; Uslu, V.V.; Tsujimura, T.; Ruf, S.; Nassari, S.; Schwarzer, W.; Ettwiller, L.; Spitz, F. Functional and topological characteristics of mammalian regulatory domains. Genome Res. 2014, 24, 390–400. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, B.S.; Layer, R.M.; Quinlan, A.R. Vcfanno: Fast, flexible annotation of genetic variants. Genome Biol. 2016, 17, 118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulko, B.; Hubisz, M.J.; Gronau, I.; Siepel, A. A method for calculating probabilities of fitness consequences for point mutations across the human genome. Nat. Genet. 2015, 47, 276–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, G.; Hu, R.; Dai, Y.; Manuel, A.M.; Zhao, Z.; Jia, P. Predicting regulatory variants using a dense epigenomic mapped CNN model elucidated the molecular basis of trait-tissue associations. Nucleic Acids Res. 2020, 49, 53–66. [Google Scholar] [CrossRef] [PubMed]
- Dey, K.K.; van de Geijn, B.; Kim, S.S.; Hormozdiari, F.; Kelley, D.R.; Price, A.L. Evaluating the informativeness of deep learning annotations for human complex diseases. Nat. Commun. 2020, 11, 4703. [Google Scholar] [CrossRef] [PubMed]
- Peng, P.-C.; Khoueiry, P.; Girardot, C.; Reddington, J.P.; Garfield, D.A.; Furlong, E.E.M.; Sinha, S. The Role of Chromatin Accessibility in cis-Regulatory Evolution. Genome Biol. Evol. 2019, 11, 1813–1828. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Wu, K.; Liu, Z.; Yao, X.; Yuan, S.; Tao, W.; Yi, L.; Yu, G.; Hou, Z.; Fan, D.; et al. Chromatin Accessibility Landscape in Human Early Embryos and Its Association with Evolution. Cell 2018, 173, 248–259.e15. [Google Scholar] [CrossRef] [Green Version]
- Cipriani, V.; Pontikos, N.; Arno, G.; Sergouniotis, P.I.; Lenassi, E.; Thawong, P.; Danis, D.; Michaelides, M.; Webster, A.R.; Moore, A.T.; et al. An Improved Phenotype-Driven Tool for Rare Mendelian Variant Prioritization: Benchmarking Exomiser on Real Patient Whole-Exome Data. Genes 2020, 11, 460. [Google Scholar] [CrossRef]
- Köhler, S.; Gargano, M.; Matentzoglu, N.; Carmody, L.C.; Lewis-Smith, D.; Vasilevsky, N.A.; Danis, D.; Balagura, G.; Baynam, G.; Brower, A.M.; et al. The Human Phenotype Ontology in 2021. Nucleic Acids Res. 2021, 49, D1207–D1217. [Google Scholar] [CrossRef]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef]
- Schubach, M. Remm Score. 2016. Available online: https://zenodo.org/record/1197579 (accessed on 1 July 2021).
- Smedley, D.; Jacobsen, J.O.B.; Jäger, M.; Köhler, S.; Holtgrewe, M.; Schubach, M.; Siragusa, E.; Zemojtel, T.; Buske, O.J.; Washington, N.L.; et al. Next-generation diagnostics and disease-gene discovery with the Exomiser. Nat. Protoc. 2015, 10, 2004–2015. [Google Scholar] [CrossRef] [Green Version]
- Cech, T.R.; Steitz, J.A. The noncoding RNA revolution-trashing old rules to forge new ones. Cell 2014, 157, 77–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peschansky, V.J.; Wahlestedt, C. Non-coding RNAs as direct and indirect modulators of epigenetic regulation. Epigenetics 2014, 9, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Wu, W.; Chen, Q.; Chen, M. Non-Coding RNAs and their Integrated Networks. J. Integr. Bioinform. 2019, 16, 20190027. [Google Scholar] [CrossRef] [PubMed]
- Klemm, S.L.; Shipony, Z.; Greenleaf, W.J. Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet. 2019, 20, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Calo, E.; Wysocka, J. Modification of enhancer chromatin: What, how and why? Mol. Cell 2013, 49, 825–837. [Google Scholar] [CrossRef] [Green Version]
- Whyte, W.A.; Orlando, D.A.; Hnisz, D.; Abraham, B.J.; Lin, C.Y.; Kagey, M.H.; Rahl, P.B.; Lee, T.I.; Young, R.A. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 2013, 153, 307–319. [Google Scholar] [CrossRef] [Green Version]
- Parker, S.C.J.; Stitzel, M.L.; Taylor, D.L.; Orozco, J.M.; Erdos, M.R.; Akiyama, J.A.; van Bueren, K.L.; Chines, P.S.; Narisu, N.; Black, B.L.; et al. Chromatin stretch enhancer states drive cell-specific gene regulation and harbor human disease risk variants. Proc. Natl. Acad. Sci. USA 2013, 110, 17921–17926. [Google Scholar] [CrossRef] [Green Version]
- Hnisz, D.; Abraham, B.J.; Lee, T.I.; Lau, A.; Saint-André, V.; Sigova, A.A.; Hoke, H.A.; Young, R.A. Super-enhancers in the control of cell identity and disease. Cell 2013, 155, 934–947. [Google Scholar] [CrossRef] [Green Version]
- Deaton, A.M.; Bird, A. CpG islands and the regulation of transcription. Genes Dev. 2011, 25, 1010–1022. [Google Scholar] [CrossRef]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [Green Version]
- Landrum, M.J.; Chitipiralla, S.; Brown, G.R.; Chen, C.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; Kaur, K.; Liu, C.; et al. ClinVar: Improvements to accessing data. Nucleic Acids Res. 2020, 48, D835–D844. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Sanderford, M.D.; Patel, R.; Chandrashekar, P.; Gibson, G.; Kumar, S. Biological relevance of computationally predicted pathogenicity of noncoding variants. Nat. Commun. 2019, 10, 330. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Category | Pathogenic Variants | Benign Variants |
|---|---|---|
| 5′UTR | 64 | 639 |
| intergenic | 10 | 100 |
| non-coding RNA | 72 | 738 |
| intronic | 539 | 5389 |
| 3′UTR | 36 | 359 |
| Scheme | Notes | Aim | Target | Var Type | Ref./ Year | License | Input | Output | Allowed Ref. Genome | Repository/Homepage |
|---|---|---|---|---|---|---|---|---|---|---|
| INVESTIGATORS | ||||||||||
| ARVIN | 1 | Predicting causal non-coding variants. It infers non-coding risk variants starting from known causal variants in gene promoters and enhancers for a number of diseases | SNPs on enhancers | SNVs/ Indels | [18] | free | SNPs in bed format + Enhancer-promoter interaction file + GWAVA features + FunSeq features + Differential expression p-values file | Prediction score for a list of snps for being risk or non-risk snps | Hg19 | https://github.com/gaolong/arvin |
| Basenji | Tested | Annotating every mutation in the genome with its influence on present chromatin accessibility and latent potential for accessibility | Epigenetic regions | SNVs | [19] | free | VCF | SNP activity difference (SAD) score—difference between the predicted accessibility of the alternative and the reference alleles | Hg19 | https://github.com/calico/basenji |
| DeepBind | 1 | Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning | Binding sites | SNVs | [20] | free | (1) a list of model IDs, and (2) a list of DNA/RNA sequences | Weighted ensemble of position weight matrices or a ‘mutation map’ that indicates how variations affect binding within a specific sequence | Hg19 | https://github.com/jisraeli/DeepBind.git |
| DeepSEA | Tested | Predicting genomic variant effects on a wide range of chromatin features at the variant position: transcription factors binding, DNase I hypersensitive sites, and histone marks in multiple human cell types | General purpose | SNVs/Indels | [21] | free | VCF/FASTA/BED (single file) | Multiple files analyzing chromatin feature probabilities and a functional significance score | Hg19 | http://deepsea.princeton.edu/help/ |
| DeltaSVM | 1 | Quantifying the effect of variants in regulatory non-coding regions | Regulatory non-coding regions | SNVs | [22] | free | Reference FASTA file (19 bp sequences centered at the SNPs with reference alleles) + Alternate FASTA file (19 bp sequences centered at the SNPs with alternate alleles) + SVM weight file (available for download from the webpage) | DeltaSVM scores, allowing the prediction of risk-conferring SNPs | Hg19, Hg38 | http://www.beerlab.org/deltasvm/ |
| Epossum2 | 1 | Predicting the impact of DNA variants on transcription factor binding | Binding sites | SNVs/Indels | - | free | Single/few variants (in VCF-like format), but does not accept a VCF file directly + one or more TFs | For each variant, a blue/red box indicates the likelihood of increased/reduced TF binding | Hg19 | https://www.genecascade.org/ePOSSUM2/ |
| FUN-LDA | 1 | Predicting Tissue-Specific Functional Effects of Non-coding Variation | General purpose | SNVs/Indels | [23] | free | .csv, .txt or .gz file without header (chr, hg19 coordinate or rs number), <100,000 var | FUN-LDA scores for each Roadmap tissue + Eigen and Eigen-PC raw and phred scores) | Hg19 | https://github.com/cran/FUNLDA |
| Geneyx (former Tgex’) | 2 | Working with GeneHancer and VarElect, it translates the finding of a variant in a non-coding region into a variant-to-gene-to-phenotype annotation | SNVs in enhancers, promoters, and ncRNA genes | SV/SNVs | [24] | license | VCF | Report containing prioritized variants, together with their annotation and analysis | Hg19 | https://geneyx.com/geneyxanalysis/ |
| GenoCanyon | 1 | Inferring the functional potential of each position in the human genome | Conserved regions | SNV | [25] | free | Chromosomal region/prediction score for the region; pre-computed scores are available | GenoCanyon score stating whether a genomic locus is functional or non-functional | Hg19 | http://zhaocenter.org/GenoCanyon_FAQ.html |
| Genomiser | Tested | Scoring the relevance of variation in the non-coding genome, and also associating regulatory variants to specific Mendelian diseases | General purpose | SNVs/Indels | [26] | free | VCF, PED-file (only for multiple samples in one VCF), your patient’s HPO terms (use the HPO-Browser to find terms), the inheritance model if known, the output prefix for your output files | Annotates, filters, and prioritizes likely causative variants, formulating a score | Hg19 | https://github.com/exomiser/Exomiser |
| Human Splicing Finder | 2 | Identifying all splicing signals, including acceptor and donor splice sites, branch points, and auxiliary splicing signals (ESE and ESS) | Splicing | SNVs/Indels | [27] | license | Single mutations/VCF, submitted through the website by directly submitting a VCF file through an API | Pathogenicity prediction for any mutation potentially affecting splicing | Hg19 | https://www.genomnis.com/the-system-1 |
| INFERNO | 1 | Inferring the molecular mechanisms of causal non-coding variants | General purpose | SNVs/Indels | [28] | free | GWAS/TSV (chromosome\t rsID \t region name\t position) maximum of 8 Mb/rsIDs | Several files containing relevant tissue contexts, target genes, and downstream biological processes affected by functional variants | Hg19 | https://bitbucket.org/wanglab-upenn/inferno/src/master/ |
| JARVIS | 3 | Prioritizing non-coding variants in whole genomes, using human-lineage purifying selection features and primary sequence context | Conserved regions | SNVs | [29] | free | VCF and pre-calculated JARVIS scores gwRVIS—genome-wide intolerance to variation score | gwRVIS—genome-wide intolerance to variation score | Hg19, Hg38 | https://github.com/astrazeneca-cgr-publications/JARVIS |
| LINSIGHT | Tested | Improving the prediction of non-coding nucleotide sites at which mutations are likely to have deleterious fitness consequences | Conserved regions | SNVs | [30] | free | VCF and Precomputed LINSIGHT scores | LINSIGHT score, which measures the probability of negative selection on non-coding sites | Hg19 | https://github.com/CshlSiepelLab/LINSIGHT |
| MORFEE | Tested | Detecting variants creating new uORF (new uAUG) | 5′UTR | SNVs | [31] | free | VCF (ANNOVAR-annotated) | MORFEE annotation reporting the effect of the variant on 5′UTR | Hg19 | https://github.com/daissi/MORFEE |
| Orion | Tested | Detecting regions of the non-coding genome that are depleted of variation, suggesting that the regions are intolerant to mutations and subject to purifying selection in the human lineage | Conserved regions | SNVs | [32] | free | One or more gVCF files, a summary by position for all samples of either read depth or genotype quality (GQ), and a file containing mutation rates; pre-computed scores exist | Orion score, with higher values corresponding to a higher degree of intolerance | Hg19 | https://github.com/igm-team/orion-public |
| RegulomeDB | 1 | Annotating SNPs with known and predicted regulatory elements in the intergenic regions | Regions of DNase hypersensitivity, binding sites of TF, promoter regions | SNVs | [33] | free | dbSNP IDs/0-based coordinates in batch | Graphic output + table containing experiments (often localized according to tissues) regarding various aspects (chromatin state, accessibility, motifs, chip data, qtl) | Hg19 | https://regulomedb.org/regulome-search/ |
| SPIDEX | Tested | Assessing whether a variant causes dysregulation of a splicing event | Splicing | SNVs | [34] | free | SPANR: maximum of 40 SNV at a time; SPIDEX: VCF of SNV | Score evaluating whether a certain splicing isoform is more enriched under the presence/absence of a given variant | Hg19 | http://download.openbioinformatics.org/spidex_download_form.php |
| SpliceAI | Tested | Identifying variants impacting splice sites | Splicing | SNVs/Indels | [35] | free | VCF | Delta score, highlighting the probability of the variant being splice-altering | Hg19, Hg38 | https://github.com/Illumina/SpliceAI |
| 5utr | Tested | Providing different annotations relevant to 5′UTR (untranslated region) variants | 5′UTR | SNVs | - | free | VCF | Annotation reporting the effect of the variant on 5′UTR | Hg19, Hg38 | https://github.com/leklab/5utr |
| UTRannotator | Tested | Annotating high-impact five prime UTR variants either creating new upstream ORFs or disrupting existing upstream ORFs | 5′UTR | SNVs/Indels | [36] | free | VCF | Annotation reporting the effect of the variant on 5′UTR | Hg19, Hg38 | https://github.com/ImperialCardioGenetics/UTRannotator |
| VarElect GeneHancer | 1, 2 | Inferring direct and indirect links between genes (or enhancers and promoters included in GeneHancer) and phenotypes—GeneHancer is a database of human regulatory elements (enhancers and promoters) and their inferred target genes | Enhancers | N.A. | [37,38] | license | Gene symbols and phenotypes | List of input genes with a score that tells how much each gene is associated with the requested phenotype | Hg19, Hg38 | https://github.com/ucscGenomeBrowser/kent/blob/master/src/hg/makeDb/doc/geneHancer.txt |
| INTEGRATORS | ||||||||||
| CADD | Scoring the deleteriousness of single nucleotide variants as well as insertion/deletions variants | General purpose | SNVs/Indels | [39] | Free * | VCF or .tsv.gz | CADD score, measuring the deleteriousness of SNVs and indels | Hg19, Hg38 | https://github.com/kircherlab/CADD-scripts | |
| DANN | Predict pathogenicity of SNVs and indels using deep neural network | General purpose | SNVs/Indels | [40] | free pyTorch implementation | VCF or .tsv.gz | DANN score, measuring the pathogenicity of SNVs | N.A. | https://cbcl.ics.uci.edu/public_data/DANN/ | |
| DVAR | Genome-wide functional scores | General purpose | SNVs/Indels | [41] | free | .tsv (chromosome, position, the ref nucleotides, the obs nucleotides, and the rs number) | Produced functional cluster labels and scores the importance of each variant | Hg19 | https://www.vumc.org/cgg/dvar | |
| Eigen v1.0 | Eigen uses a variety of functional annotations in both coding and non-coding regions and combines them into one single measure of functional importance | General purpose | SNVs | [42] | free | VCF | Eigen score, measuring how functional the variant is | Hg19, Hg38 | http://www.columbia.edu/~ii2135/eigen.html | |
| FATHMM-XF (old -MKL) | Functional predictor for SNVs | SNVs | SNVs | [43] | free | VCF or csv (chr, pos, ref n, mut n) | Score highlighting the variant pathogenicity | Hg19, Hg38 | https://github.com/HAShihab/fathmm-MKL | |
| GWAVA | Predicting the functional impact of non-coding genetic variants based on a wide range of annotations of non-coding elements, along with genome-wide properties such as evolutionary conservation and GC-content | General purpose | SNVs/Indels | [44] | free | Multiple variant identifiers (in BED format) or chromosomal regions | GWAVA score, with higher scores indicating variants predicted as more likely to be functional | Hg19 | https://www.sanger.ac.uk/tool/gwava/ | |
| IW-Scoring | Scoring (integrates in a weighted way the outputs of other software, chosen by the user from a list) | Non-coding variations | SNVs/Indels | [45] | free | In batches up to 100 K SNPs/InDels (also VCF format, but not exclusively) | Two separate linear weighted functional scoring schemas for known and novel variations, respectively, which differentiate functionally significant variations from others | Hg19 | https://snp-nexus.org/IW-Scoring/index.html | |
| PAFA | Genome-wide functional scores | General purpose | SNVs/Indels | [46] | free | VCF (<100.000 var) max 2 Mb | Prioritization of variants + functional score | Hg19, Hg38 | http://159.226.67.237:8080/pafa/ | |
| PINES | Predicting the functional impact of non-coding variants by integrating epigenetic annotations in a phenotype-dependent manner | General purpose | SNVs | [47] | free | List of intronic or intergenic variants one rs per line | Identification and prioritization of functional non-coding SNPs | Hg19 | https://github.com/PINES-scoring/PINES | |
| RegulationSpotter | Integrating data from various sources to show whether a variant lies within a regulatory region and has the potential to impair gene expression | Intolerant regions | SNVs/Indels | [48] | free | Single variant or VCF format | Summary table with a graphical matrix depicting key aspects of all analyzed variants | Hg19 | https://www.regulationspotter.org/ | |
| AGGREGATORS | ||||||||||
| Alamut (Batch) | Annotator | General purpose | SNVs/Indels | [49] | license | VCF, tab-delimited files | Annotated variants | Hg19, Hg38 | https://www.interactive-biosoftware.com/alamut-batch/ | |
| ANNOVAR | Annotator | General purpose | SNVs/Indels | [50] | free | VCF | Annotated variants | Hg19, Hg38 | https://github.com/WGLab/doc-ANNOVAR | |
| BasePlayer | Large-scale discovery tool for genomic variants allowing for complex comparative variant analyses | General purpose | SNVs/Indels | [51] | free | BAM or VCF (+BED) | Graphical user interface for variants visualization (built-in genome browser, interactive variant analysis, and data integration tracks) | Hg38 | https://github.com/rkataine/BasePlayer | |
| RegBase | Integrating non-coding regulatory prediction scores and composite prediction models from existing tools | Non-coding regulatory variants | SNVs | [52] | free * | Chromosome coordinates/query_file.bed | Variants prioritization | Hg19 | https://github.com/mulinlab/regBase | |
| SnpEff | Annotating and predicting the effects of genetic variants | General purpose | SNVs/Indels | [53] | free | VCF | Annotated variants | Hg19, Hg38 | https://github.com/pcingola/SnpEff | |
| SNPNexus | Variants annotation tool designed to simplify and assist in the selection and prioritization of known and novel genomic alterations | General purpose | SNVs/Indels | [54] | free * | Single and batch | Annotated variants | Hg19, Hg38 | https://www.snp-nexus.org/v4/ | |
| VarAFT | Provides experiments’ quality, annotates, and allows the filtration of VCF files; annotates and pinpoints human disease-causing mutations through access to multiple layers of information | General purpose | SNVs/Indels | [55] | free | VCF or ANN 4.1 | Graphical user interface that allows the simultaneous annotation, filtration, and breadth and depth of coverage analysis | Hg19, Hg38 | https://varaft.eu/ | |
| VEP Predictor | Annotator (but specific modules for non-coding variants exist) | General purpose | SNVs/Indels | [17] | free | VCF, rsID or HGVS notations | Annotated variants | Hg19, Hg38 | https://github.com/Ensembl/ensembl-vep | |
| Tool | Number of Annotated Variants from ncVarDB | Computational Performance | Parallel Paradigm | TP/FP/TN/FN | Specificity | Sensitivity | Precision | Accuracy |
|---|---|---|---|---|---|---|---|---|
| UTRs | ||||||||
| UTRannotator | 478 | 1 m–10 m | yes | 13/24/396/45 | 0.943 | 0.224 | 0.351 | 0.856 |
| 5utr | 478 | 1 m–10 m | yes | 20/78/342/38 | 0.814 | 0.345 | 0.204 | 0.757 |
| MORFEE | 433 | 10 m–30 m | no | 3/14/368/48 | 0.963 | 0.059 | 0.176 | 0.857 |
| Splicing sites | ||||||||
| SpliceAI | 4854 | 30 m–1 h | yes | 389/1/4249/n.a. | 0.9998 | --- | 0.997 | --- |
| SPIDEX | 756 | <1 m | Yes 1 | 314/19/273/n.a. | 0.935 | --- | 0.943 | --- |
| Genome accessibility and mutation intolerance | ||||||||
| DeepSEA | 7879 | >1 d | no | 388/243/6980/n.a. | 0.966 | --- | 0.615 | --- |
| Orion | 4373 | 1 m–10 m | Yes 2 | 169/311/3482/n.a. | 0.918 | --- | 0.352 | --- |
| LINSIGHT | 1240 | <1 m | Yes 2 | 334/59/676/n.a. | 0.9197 | --- | 0.8499 | --- |
| Basenji | 7946 | >1 d (on CPU) 5 h–24 h (on GPU) 3 | yes | 117/1263/5962/n.a. | 0.825 | --- | 0.085 | --- |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tabarini, N.; Biagi, E.; Uva, P.; Iovino, E.; Pippucci, T.; Seri, M.; Cavalli, A.; Ceccherini, I.; Rusmini, M.; Viti, F. Exploration of Tools for the Interpretation of Human Non-Coding Variants. Int. J. Mol. Sci. 2022, 23, 12977. https://doi.org/10.3390/ijms232112977
Tabarini N, Biagi E, Uva P, Iovino E, Pippucci T, Seri M, Cavalli A, Ceccherini I, Rusmini M, Viti F. Exploration of Tools for the Interpretation of Human Non-Coding Variants. International Journal of Molecular Sciences. 2022; 23(21):12977. https://doi.org/10.3390/ijms232112977
Chicago/Turabian StyleTabarini, Nicole, Elena Biagi, Paolo Uva, Emanuela Iovino, Tommaso Pippucci, Marco Seri, Andrea Cavalli, Isabella Ceccherini, Marta Rusmini, and Federica Viti. 2022. "Exploration of Tools for the Interpretation of Human Non-Coding Variants" International Journal of Molecular Sciences 23, no. 21: 12977. https://doi.org/10.3390/ijms232112977
APA StyleTabarini, N., Biagi, E., Uva, P., Iovino, E., Pippucci, T., Seri, M., Cavalli, A., Ceccherini, I., Rusmini, M., & Viti, F. (2022). Exploration of Tools for the Interpretation of Human Non-Coding Variants. International Journal of Molecular Sciences, 23(21), 12977. https://doi.org/10.3390/ijms232112977