
The Crystal Structure of Mouse Ces2c, a Potential Ortholog of Human CES2, Shows Structural Similarities in Substrate Regulation and Product Release to Human CES1


 , and
, and
Abstract
:1. Introduction
2. Results
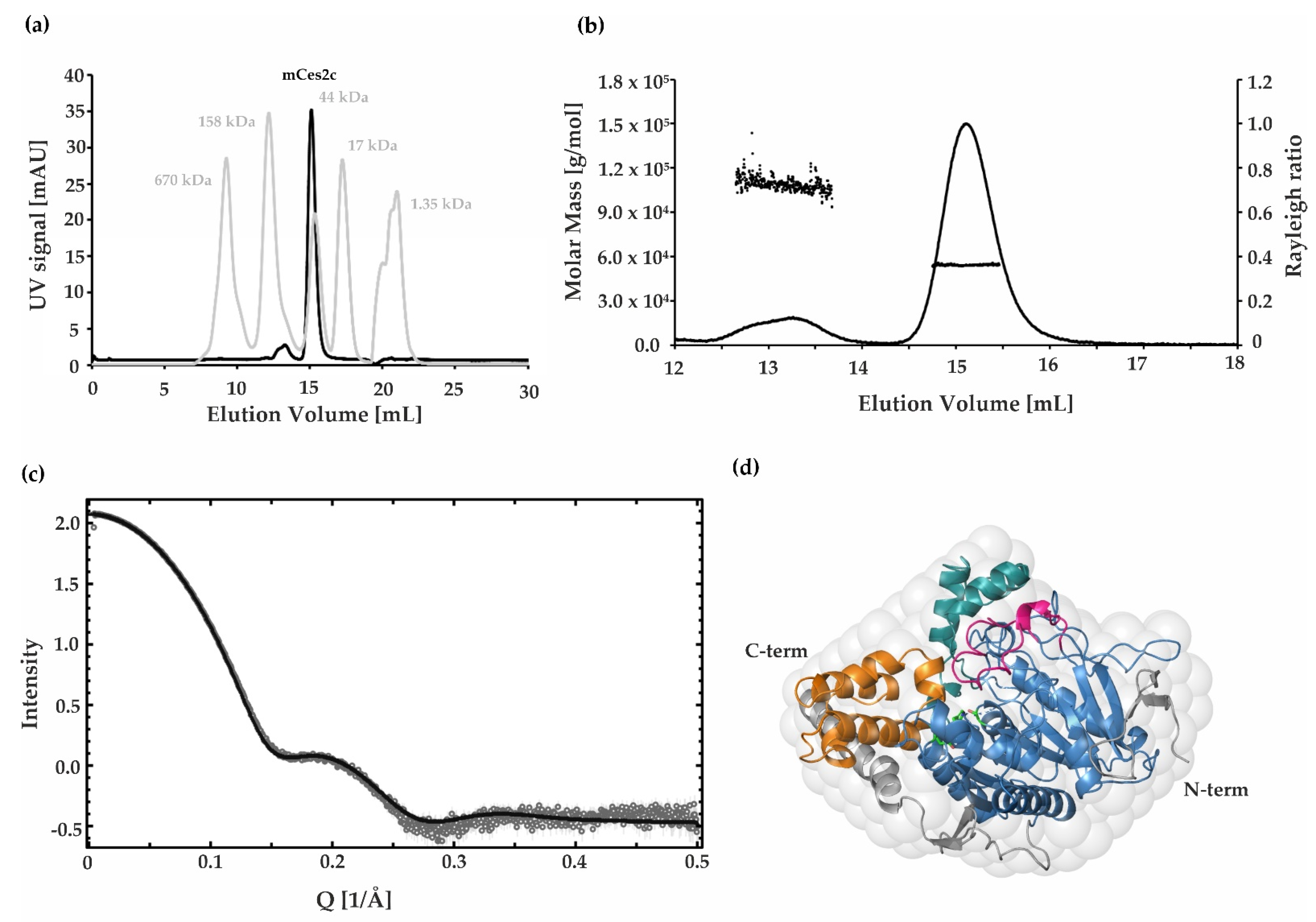
2.1. Purified mCes2c Is Active and Unfolds Irreversibly at a TM of 60 °C
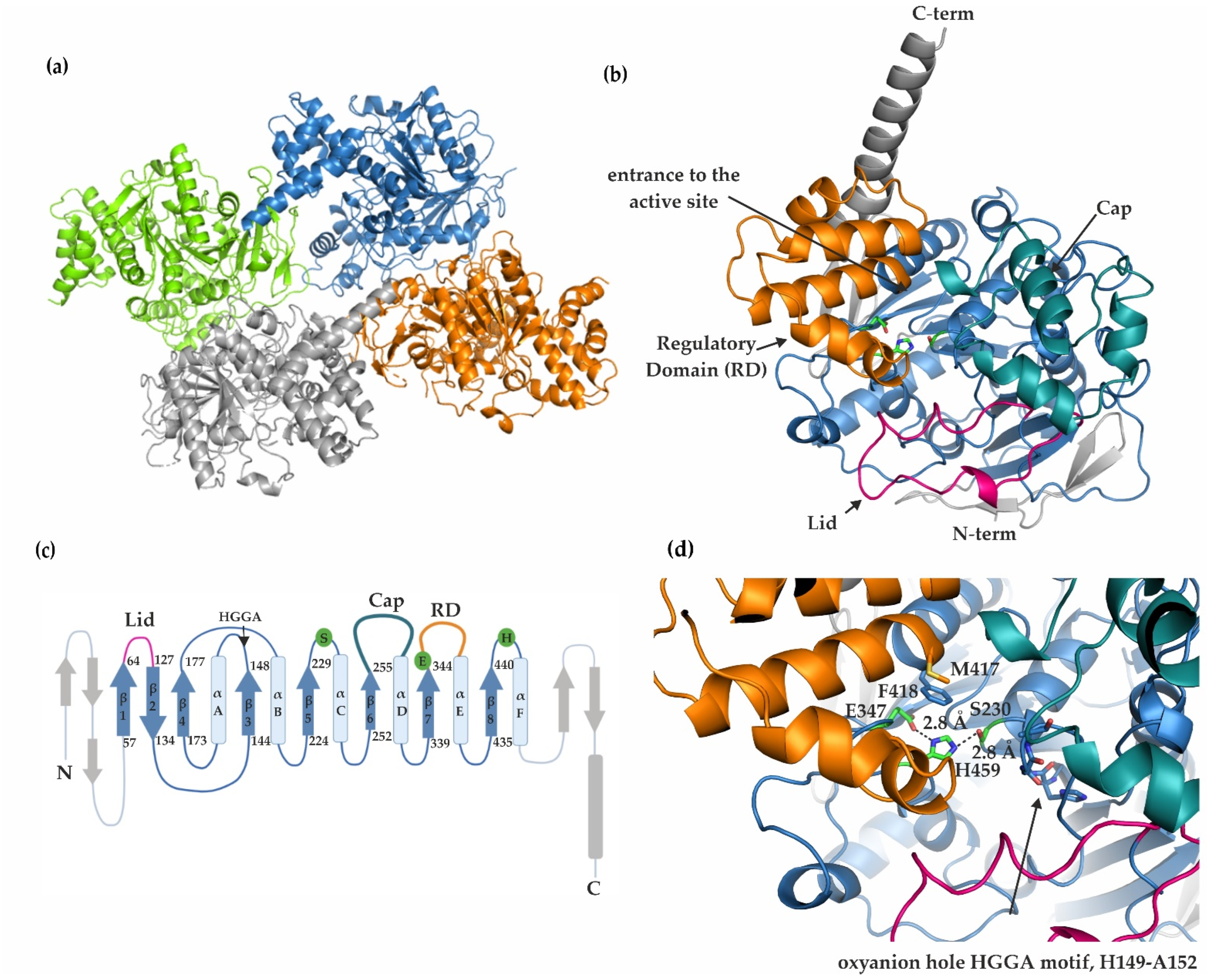
2.2. mCes2c Adopts an α/β-Hydrolase Fold with Residues S230, E347, and H459 Forming a Catalytic Triad
2.3. The Cap, the Regulatory Domain, and a Flexible Lid Region Restrict Access to the Active Site of mCes2c
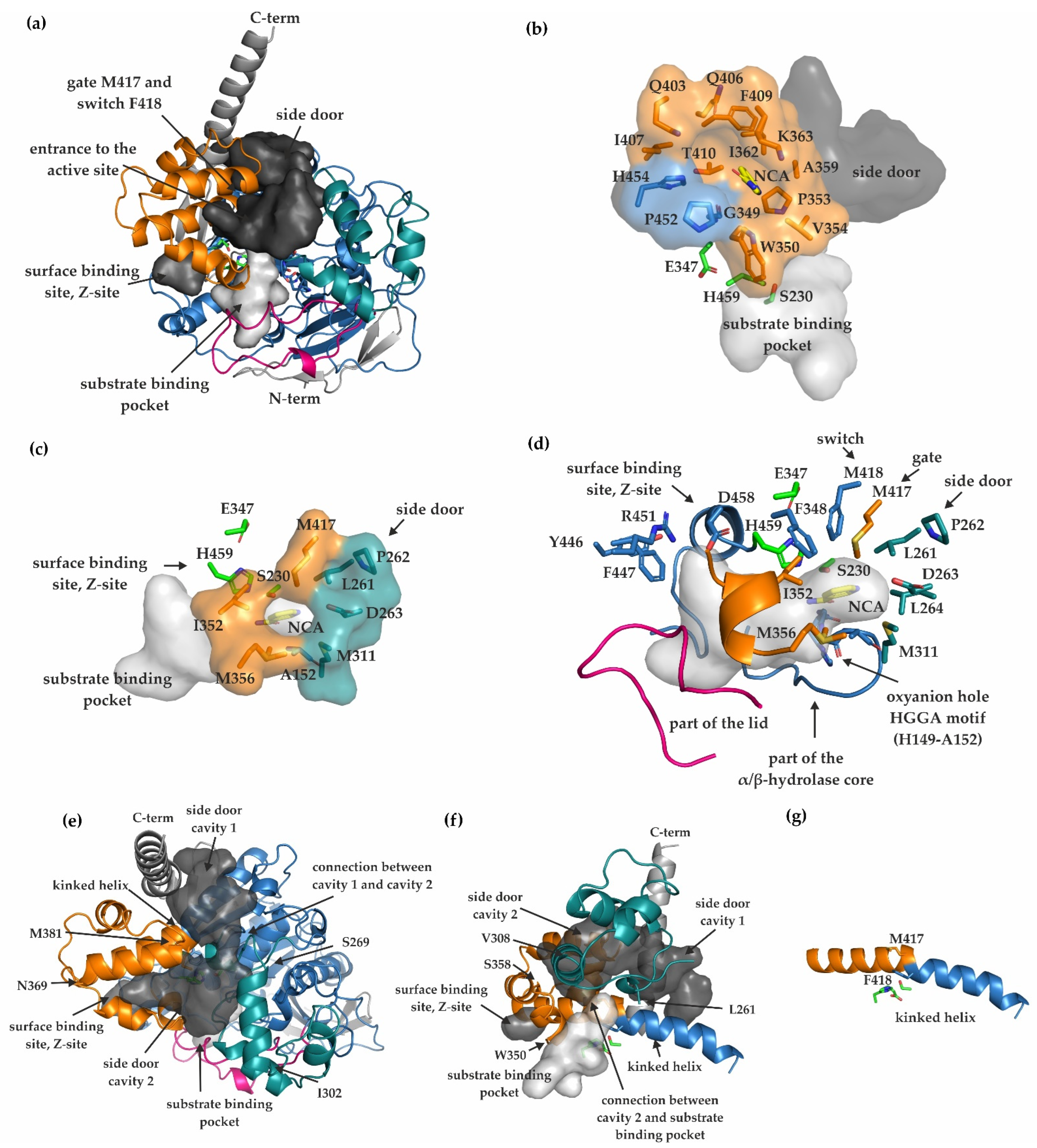
2.4. Substrate Binding Pocket, Active Site, Product Release, and Surface Binding Site of mCes2c
2.5. SAXS, SEC, and SEC MALS Demonstrate That mCes2c Is Monomeric in Solution
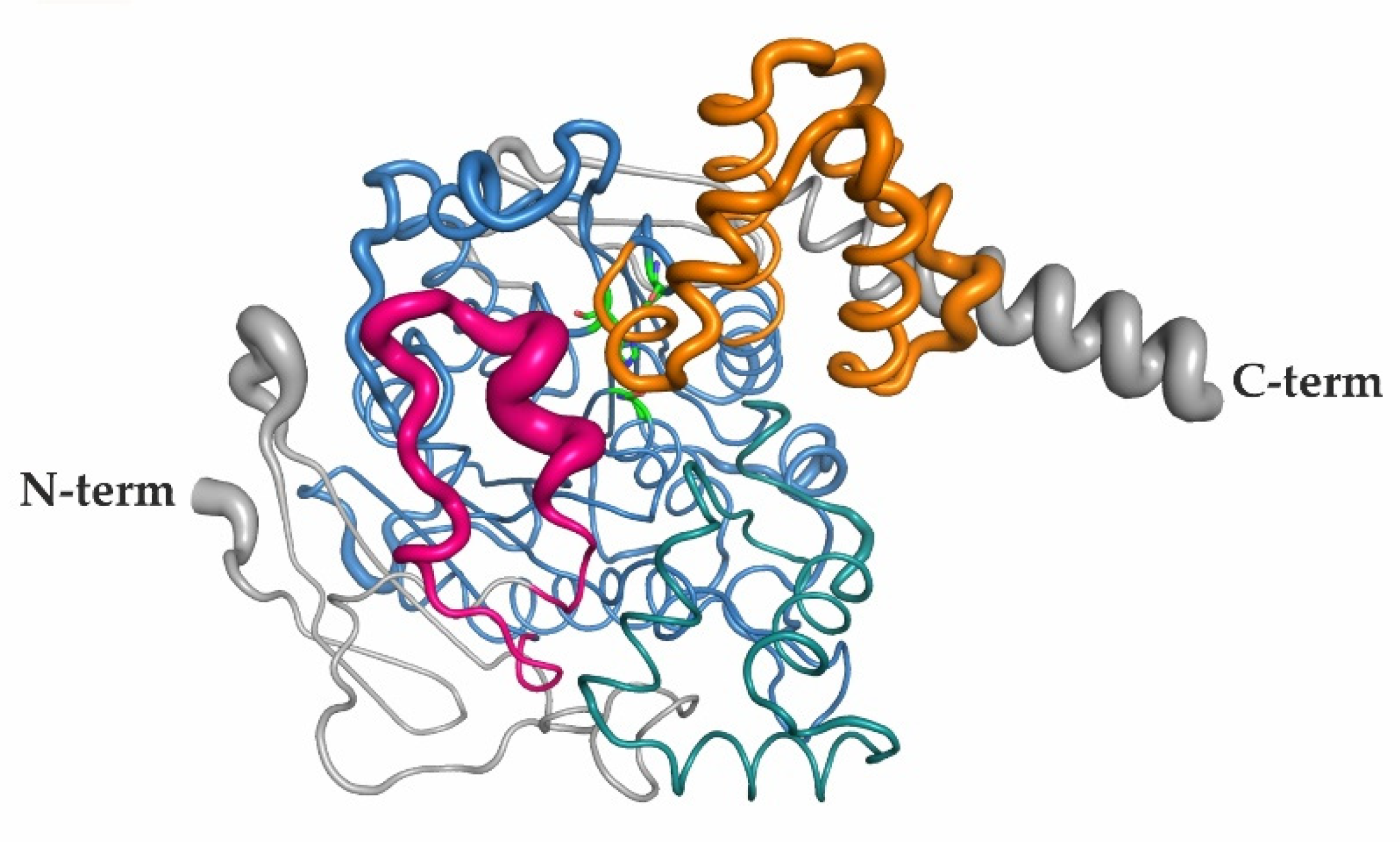
2.6. Elevated B-Factors Indicate Higher Flexibility and Thermal Motion of the RD and the Lid Region
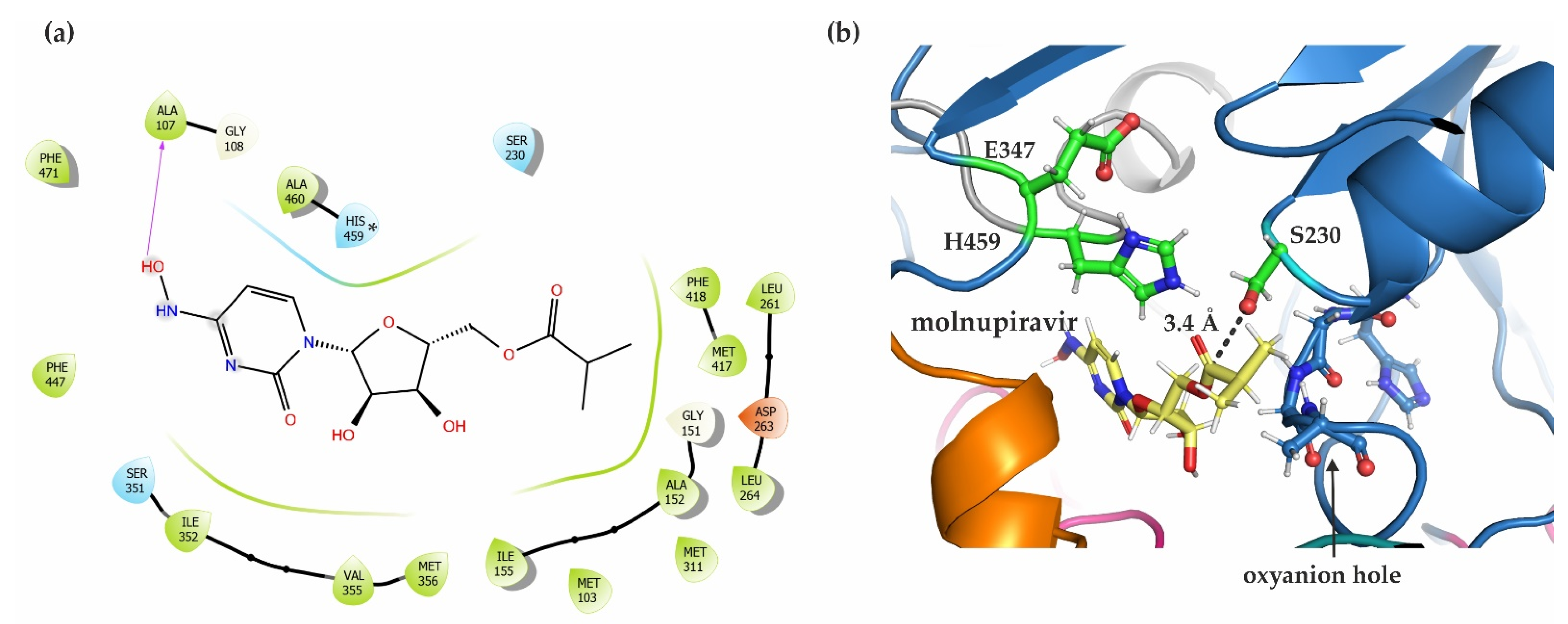
2.7. In Silico Docking Studies Show a Possible Binding Pose of the COVID-19 Prodrug Molnupiravir in the Active Site of mCes2c
3. Discussion
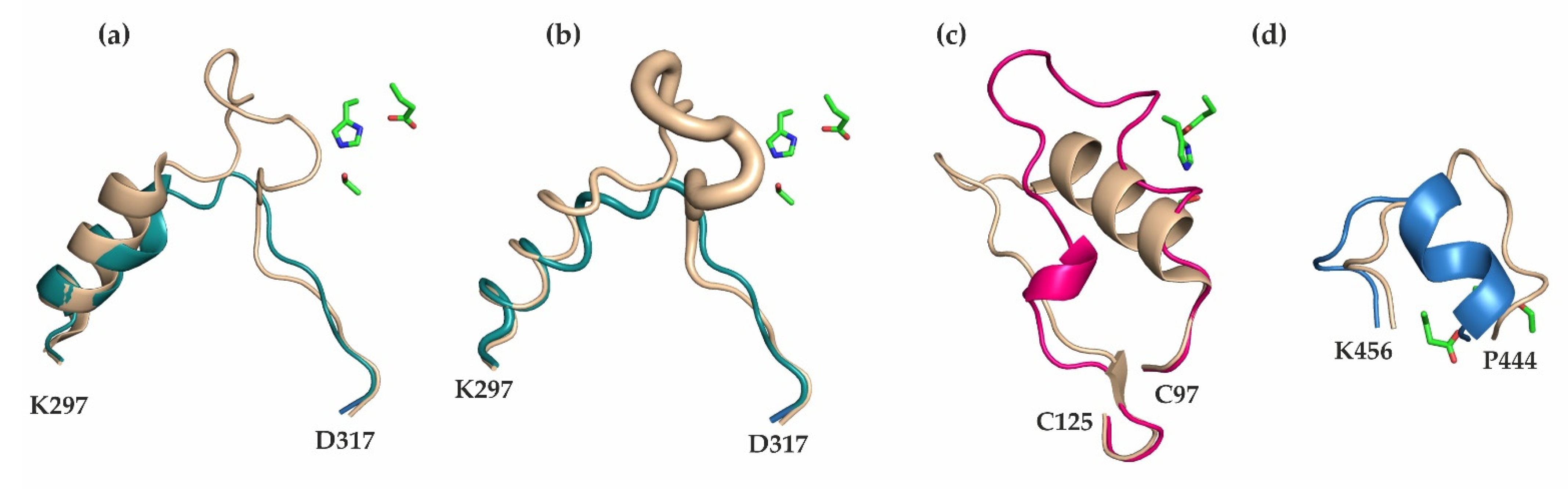
3.1. Differences in the Entrance to the Substrate Binding Pocket of mCes2c Compared to hCES1
3.2. Similarities in the Product Release between mCes2c and hCES1
3.3. The Oligomerization State of mCes2c Is Different to hCES1
3.4. hCES2/mCes2c and Their Ability to Activate the Antiviral Prodrug Molnupiravir
4. Materials and Methods
4.1. Nomenclature
4.2. Materials
4.3. Cloning, Expression and Purification of mCes2c
4.4. Biophysical and Biochemical Characterization of mCes2c
4.4.1. Activity Assay
4.4.2. Protein Characterization Using Size Exclusion Chromatography (SEC) and Multi-Angle Light Scattering (MALS)
4.4.3. Thermostability—Differential Scanning Fluorimetry
4.4.4. Thermostability—Circular Dichroism Spectroscopy
4.4.5. Mass-Spectrometry
4.5. Structural Characterization
4.5.1. Small-Angle X-ray Scattering (SAXS) Measurements
4.5.2. Crystallization, Data Processing, and Refinement
4.6. In Silico Docking Studies with the Novel COVID-19 Prodrug Molnupiravir
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gibson-Smith, D.; Bot, M.; Paans, N.P.G.; Visser, M.; Brouwer, I.; Penninx, B.W.J.H. The Role of Obesity Measures in the Development and Persistence of Major Depressive Disorder. J. Affect. Disord. 2016, 198, 222–229. [Google Scholar] [CrossRef]
- Alberca, R.W.; Oliveira, L.d.M.; Branco, A.C.C.C.; Pereira, N.Z.; Sato, M.N. Obesity as a Risk Factor for COVID-19: An Overview. Crit. Rev. Food Sci. Nutr. 2021, 61, 2262–2276. [Google Scholar] [CrossRef]
- Kim, H.; Lee, D.S.; An, T.H.; Park, H.J.; Kim, W.K.; Bae, K.H.; Oh, K.J. Metabolic Spectrum of Liver Failure in Type 2 Diabetes and Obesity: From Nafld to Nash to Hcc. Int. J. Mol. Sci. 2021, 22, 4495. [Google Scholar] [CrossRef] [PubMed]
- Hofer, P.; Taschler, U.; Schreiber, R.; Kotzbeck, P.; Schoiswohl, G. The Lipolysome—A Highly Complex and Dynamic Protein Network Orchestrating Cytoplasmic Triacylglycerol Degradation. Metabolites 2020, 10, 147. [Google Scholar] [CrossRef]
- Kulminskaya, N.; Oberer, M. Protein-Protein Interactions Regulate the Activity of Adipose Triglyceride Lipase in Intracellular Lipolysis. Biochimie 2020, 169, 62–68. [Google Scholar] [CrossRef]
- Ruby, M.A.; Massart, J.; Hunerdosse, D.M.; Schönke, M.; Correia, J.C.; Louie, S.M.; Ruas, J.L.; Näslund, E.; Nomura, D.K.; Zierath, J.R. Human Carboxylesterase 2 Reverses Obesity-Induced Diacylglycerol Accumulation and Glucose Intolerance. Cell Rep. 2017, 18, 636–646. [Google Scholar] [CrossRef] [Green Version]
- Lian, J.; Nelson, R.; Lehner, R. Carboxylesterases in Lipid Metabolism: From Mouse to Human. Protein Cell 2018, 9, 178–195. [Google Scholar] [CrossRef]
- Chalhoub, G.; Kolleritsch, S.; Maresch, L.K.; Taschler, U.; Pajed, L.; Tilp, A.; Eisner, H.; Rosina, P.; Kien, B.; Radner, F.P.W.; et al. Carboxylesterase 2 Proteins Are Efficient Diglyceride and Monoglyceride Lipases Possibly Implicated in Metabolic Disease. J. Lipid Res. 2021, 62, 100075. [Google Scholar] [CrossRef] [PubMed]
- Tardelli, M.; Bruschi, F.V.; Trauner, M. The Role of Metabolic Lipases in the Pathogenesis and Management of Liver Disease. Hepatology 2020, 72, 1117–1126. [Google Scholar] [CrossRef] [Green Version]
- Quiroga, A.D.; Lehner, R. Pharmacological Intervention of Liver Triacylglycerol Lipolysis: The Good, the Bad and the Ugly. Biochem. Pharmacol. 2018, 155, 233–241. [Google Scholar] [CrossRef]
- Maresch, L.K.; Benedikt, P.; Feiler, U.; Eder, S.; Zierler, K.A.; Taschler, U.; Kolleritsch, S.; Eichmann, T.O.; Schoiswohl, G.; Leopold, C.; et al. Intestine-Specific Overexpression of Carboxylesterase 2c Protects Mice from Diet-Induced Liver Steatosis and Obesity. Hepatol. Commun. 2019, 3, 227–245. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Pan, X.; Hu, S.; Zhu, Y.; Bawa, F.C.; Li, Y.; Yin, L.; Zhang, Y. Hepatocyte-Specific Expression of Human Carboxylesterase 2 Attenuates Nonalcoholic Steatohepatitis in Mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2021, 320, G166–G174. [Google Scholar] [CrossRef]
- Song, Y.Q.; Guan, X.Q.; Weng, Z.M.; Wang, Y.Q.; Chen, J.; Jin, Q.; Fang, S.Q.; Fan, B.; Cao, Y.F.; Hou, J.; et al. Discovery of a Highly Specific and Efficacious Inhibitor of Human Carboxylesterase 2 by Large-Scale Screening. Int. J. Biol. Macromol. 2019, 137, 261–269. [Google Scholar] [CrossRef]
- Satoh, T.; Hosokawa, M. The Mammalian Carboxylesterases: From Molecules to Functions. Annu. Rev. Pharmacol. Toxicol. 1998, 38, 257–288. [Google Scholar] [CrossRef]
- Hosokawa, M.; Furihata, T.; Yaginuma, Y.; Yamamoto, N.; Koyano, N.; Fujii, A.; Nagahara, Y.; Satoh, T.; Chiba, K. Genomic Structure and Transcriptional Regulation of the Rat, Mouse, and Human Carboxylesterase Genes. Drug Metab. Rev. 2007, 39, 1–15. [Google Scholar] [CrossRef]
- Holmes, R.S.; Wright, M.W.; Laulederkind, S.J.F.; Cox, L.A.; Hosokawa, M.; Imai, T.; Ishibashi, S.; Lehner, R.; Miyazaki, M.; Perkins, E.J.; et al. Recommended Nomenclature for Five Mammalian Carboxylesterase Gene Families: Human, Mouse, and Rat Genes and Proteins. Mamm. Genome 2010, 21, 427–441. [Google Scholar] [CrossRef] [Green Version]
- Fleming, C.D.; Bencharit, S.; Edwards, C.C.; Hyatt, J.L.; Tsurkan, L.; Bai, F.; Fraga, C.; Morton, C.L.; Howard-Williams, E.L.; Potter, P.M.; et al. Structural Insights into Drug Processing by Human Carboxylesterase 1: Tamoxifen, Mevastatin, and Inhibition by Benzil. J. Mol. Biol. 2005, 352, 165–177. [Google Scholar] [CrossRef]
- De Souza, V.A.; Scott, D.J.; Nettleship, J.E.; Rahman, N.; Charlton, M.H.; Walsh, M.A.; Owens, R.J. Comparison of the Structure and Activity of Glycosylated and Aglycosylated Human Carboxylesterase 1. PLoS ONE 2015, 10, e0143919. [Google Scholar] [CrossRef] [PubMed]
- Hemmert, A.C.; Otto, T.C.; Wierdl, M.; Edwards, C.C.; Fleming, C.D.; MacDonald, M.; Cashman, J.R.; Potter, P.M.; Cerasoli, D.M.; Redinbo, M.R. Human Carboxylesterase 1 Stereoselectively Binds the Nerve Agent Cyclosarin and Spontaneously Hydrolyzes the Nerve Agent Sarin. Mol. Pharmacol. 2010, 77, 508–516. [Google Scholar] [CrossRef] [Green Version]
- Bencharit, S.; Morton, C.L.; Xue, Y.; Potter, P.M.; Redinbo, M.R. Structural Basis of Heroin and Cocaine Metabolism by a Promiscuous Human Drug-Processing Enzyme. Nat. Struct. Biol. 2003, 10, 349–356. [Google Scholar] [CrossRef]
- Bencharit, S.; Edwards, C.C.; Morton, C.L.; Howard-Williams, E.L.; Kuhn, P.; Potter, P.M.; Redinbo, M.R. Multisite Promiscuity in the Processing of Endogenous Substrates by Human Carboxylesterase 1. J. Mol. Biol. 2006, 363, 201–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleming, C.D.; Edwards, C.C.; Kirby, S.D.; Maxwell, D.M.; Potter, P.M.; Cerasoli, D.M.; Redinbo, M.R. Crystal Structures of Human Carboxylesterase 1 in Covalent Complexes with the Chemical Warfare Agents Soman and Tabun. Biochemistry 2007, 46, 5063–5071. [Google Scholar] [CrossRef] [Green Version]
- Bencharit, S.; Morton, C.L.; Hyatt, J.L.; Kuhn, P.; Danks, M.K.; Potter, P.M.; Redinbo, M.R. Crystal Structure of Human Carboxylesterase 1 Complexed with the Alzheimer’s Drug Tacrine. Chem. Biol. 2003, 10, 341–349. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Zou, L.; Jin, Q.; Hou, J.; Ge, G.; Yang, L. Human Carboxylesterases: A Comprehensive Review. Acta Pharm. Sin. B 2018, 8, 699–712. [Google Scholar] [CrossRef] [PubMed]
- Sanghani, S.P.; Quinney, S.K.; Fredenburg, T.B.; Sun, Z.; Davis, W.I.; Murry, D.J.; Cummings, O.W.; Seitz, D.E.; Bosron, W.F. Carboxylesterases Expressed in Human Colon Tumor Tissue and Their Role in CPT-11 Hydrolysis. Clin. Cancer Res. 2003, 9, 4983–4991. [Google Scholar] [PubMed]
- Shen, Y.; Eades, W.; Liu, W.; Yan, B. The COVID-19 Oral Drug Molnupiravir Is a CES2 Substrate: Potential Drug-Drug Interactions and Impact of CES2 Genetic Polymorphism In Vitro. Drug Metab. Dispos. 2022, 50, 1151–1160. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.D.; Taylor, A.M.; Tong, E.Y.; Repa, J.J. Carboxylesterases Are Uniquely Expressed among Tissues and Regulated by Nuclear Hormone Receptors in the Mouse. Drug Metab. Dispos. 2013, 41, 40–49. [Google Scholar] [CrossRef] [Green Version]
- Furihata, T.; Hosokawa, M.; Nakata, F.; Satoh, T.; Chiba, K. Purification, Molecular Cloning, and Functional Expression of Inducible Liver Acylcarnitine Hydrolase in C57BL/6 Mouse, Belonging to the Carboxylesterase Multigene Family. Arch. Biochem. Biophys. 2003, 416, 101–109. [Google Scholar] [CrossRef]
- Li, Y.; Zalzala, M.; Jadhav, K.; Xu, Y.; Kasumov, T.; Yin, L.; Zhang, Y. Carboxylesterase 2 Prevents Liver Steatosis by Modulating Lipolysis, Endoplasmic Reticulum Stress, and Lipogenesis and Is Regulated by Hepatocyte Nuclear Factor 4 Alpha in Mice. Hepatology 2016, 63, 1860–1874. [Google Scholar] [CrossRef] [Green Version]
- Bauer, T.L.; Buchholz, P.C.F.; Pleiss, J. The Modular Structure of α/β-Hydrolases. FEBS J. 2020, 287, 1035–1053. [Google Scholar] [CrossRef]
- Bencharit, S.; Morton, C.L.; Howard-Williams, E.L.; Danks, M.K.; Potter, P.M.; Redinbo, M.R. Structural Insights into Cpt-11 Activation by Mammalian Carboxylesterases. Nat. Struct. Biol. 2002, 9, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Lee, Y.J.; Kanayama, T.; Kondo, Y.; Nishio, K.; Mukai, K.; Haba, M.; Hosokawa, M. Design, Synthesis and Biological Evaluation of Water-Soluble Phenytoin Prodrugs Considering the Substrate Recognition Ability of Human Carboxylesterase 1. Eur. J. Pharm. Sci. 2020, 152, 105455. [Google Scholar] [CrossRef] [PubMed]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-Based Map of the Human Proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.J.; Toots, M.; Lee, S.; Lee, M.E.; Ludeke, B.; Luczo, J.M.; Ganti, K.; Cox, R.M.; Sticher, Z.M.; Edpuganti, V.; et al. Orally Efficacious Broad-Spectrum Ribonucleoside Analog Inhibitor of Influenza and Respiratory Syncytial Viruses. Antimicrob. Agents Chemother. 2018, 62, e00766-18. [Google Scholar] [CrossRef] [Green Version]
- Painter, W.P.; Holman, W.; Bush, J.A.; Almazedi, F.; Malik, H.; Eraut, N.C.J.E.; Morin, M.J.; Szewczyk, L.J.; Painter, R. Human safety, tolerability, and pharmacokinetics of Molnupiravir, a Novel Broad-Spectrum Oral Antiviral Agent with Activity against SARS-CoV-2. Antimicrob. Agents Chemother. 2021, 65, e02428-20. [Google Scholar] [CrossRef]
- Kabinger, F.; Stiller, C.; Schmitzová, J.; Dienemann, C.; Kokic, G.; Hillen, H.S.; Höbartner, C.; Cramer, P. Mechanism of Molnupiravir-Induced SARS-CoV-2 Mutagenesis. Nat. Struct. Mol. Biol. 2021, 28, 740–746. [Google Scholar] [CrossRef]
- Fischer, W.A.; Eron, J.J.; Holman, W.; Cohen, M.S.; Fang, L.; Szewczyk, L.J.; Sheahan, T.P.; Baric, R.; Mollan, K.R.; Wolfe, C.R.; et al. A Phase 2a Clinical Trial of Molnupiravir in Patients with COVID-19 Shows Accelerated SARS-CoV-2 RNA Clearance and Elimination of Infectious Virus. Sci. Transl. Med. 2022, 14, eabl7430. [Google Scholar] [CrossRef]
- Babusis, D.; Yue, Q.; Ma, B.; Murray, B.P.; Subramanian, R.; Xie, X.; Zou, J.; Bilello, J.P.; Li, L. Key Metabolic Enzymes Involved in Remdesivir Activation In human lung cells. Antimicrob. Agents Chemother. 2021, 65, e00602-21. [Google Scholar]
- Manalastas-Cantos, K.; Konarev, P.V.; Hajizadeh, N.R.; Kikhney, A.G.; Petoukhov, M.V.; Molodenskiy, D.S.; Panjkovich, A.; Mertens, H.D.T.; Gruzinov, A.; Borges, C.; et al. ATSAS 3.0: Expanded Functionality and New Tools for Small-Angle Scattering Data Analysis. J. Appl. Crystallogr. 2021, 54, 343–355. [Google Scholar] [CrossRef]
- Kabsch, W. XDS. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [Green Version]
- McNicholas, S.; Potterton, E.; Wilson, K.S.; Noble, M.E.M. Presenting Your Structures: The CCP4mg Molecular-Graphics Software. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Krissinel, E.; Uski, V.; Lebedev, A.; Winn, M.; Ballard, C. Distributed Computing for Macromolecular Crystallography. Acta Crystallogr. Sect. D Struct. Biol. 2018, 74, 143–151. [Google Scholar] [CrossRef] [Green Version]
- Emsley, P.; Cowtan, K. Coot: Model-Building Tools for Molecular Graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [Green Version]
- Croll, T.I. ISOLDE: A Physically Realistic Environment for Model Building into Low-Resolution Electron-Density Maps. Acta Crystallogr. Sect. D Struct. Biol. 2018, 74, 519–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Echols, N.; Headd, J.J.; Hung, L.W.; Jain, S.; Kapral, G.J.; Grosse Kunstleve, R.W.; et al. The Phenix Software for Automated Determination of Macromolecular Structures. Methods 2011, 55, 94–106. [Google Scholar] [CrossRef] [Green Version]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B.; et al. MolProbity: More and Better Reference Data for Improved All-Atom Structure Validation. Protein Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New Data Content and Improved Web Interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein-Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Contents | mCes2c |
|---|---|
| PDB code | 8AXC |
| Data collection | |
| Data collection facility | Synchrotron, PETRA III, DESY BEAMLINE P11 Hamburg |
| Detector | DECTRIS EIGER2 X 16M |
| Wavelength (Å) | 1.0 |
| Resolution range (low–high Å) | 49–2.12 (2.19–2.12) |
| Space group | P212121 |
| No. of molecules in the asymmetric unit | 4 |
| Unit cell (Å, °) | a = 98.05 b = 143.59 c = 183.75 α = β = γ = 90 |
| Total reflections | 1,006,260 (89,693) |
| Unique reflections | 145,761 (14,158) |
| Multiplicity | 6.9 (6.3) |
| Completeness (%) | 98.99 (97.40) |
| Mean I/σ | 16.58 (3.67) |
| CC1/2 | 0.99 (0.87) |
| Refinement statistics | |
| R-merge | 0.090 (0.54) |
| Reflections used in refinement | 145,744 (14,158) |
| Reflections used for R-free | 7342 (711) |
| R-work | 0.166 (0.213) |
| R-free | 0.195 (0.242) |
| Number of non-hydrogen atoms | 18,010 |
| Macromolecules | 16,430 |
| Ligands | 63 |
| Water atoms | 1517 |
| No. of protein residues | 2069 |
| Model geometry | |
| RMSD bonds (Å) | 0.014 |
| RMSD angles (°) | 1.75 |
| Ramachandran distribution | |
| Most favored (%) | 97.27 |
| Additionally allowed (%) | 2.73 |
| Outliers (%) | 0 |
| Average B-factor (A2) | 35.23 |
| Macromolecules | 34.92 |
| Ligands | 37.41 |
| Solvent | 38.43 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eisner, H.; Riegler-Berket, L.; Gamez, C.F.R.; Sagmeister, T.; Chalhoub, G.; Darnhofer, B.; Jazleena, P.J.; Birner-Gruenberger, R.; Pavkov-Keller, T.; Haemmerle, G.; et al. The Crystal Structure of Mouse Ces2c, a Potential Ortholog of Human CES2, Shows Structural Similarities in Substrate Regulation and Product Release to Human CES1. Int. J. Mol. Sci. 2022, 23, 13101. https://doi.org/10.3390/ijms232113101
Eisner H, Riegler-Berket L, Gamez CFR, Sagmeister T, Chalhoub G, Darnhofer B, Jazleena PJ, Birner-Gruenberger R, Pavkov-Keller T, Haemmerle G, et al. The Crystal Structure of Mouse Ces2c, a Potential Ortholog of Human CES2, Shows Structural Similarities in Substrate Regulation and Product Release to Human CES1. International Journal of Molecular Sciences. 2022; 23(21):13101. https://doi.org/10.3390/ijms232113101
Chicago/Turabian StyleEisner, Helgit, Lina Riegler-Berket, Carlos Francisco Rodriguez Gamez, Theo Sagmeister, Gabriel Chalhoub, Barbara Darnhofer, P J Jazleena, Ruth Birner-Gruenberger, Tea Pavkov-Keller, Guenter Haemmerle, and et al. 2022. "The Crystal Structure of Mouse Ces2c, a Potential Ortholog of Human CES2, Shows Structural Similarities in Substrate Regulation and Product Release to Human CES1" International Journal of Molecular Sciences 23, no. 21: 13101. https://doi.org/10.3390/ijms232113101
APA StyleEisner, H., Riegler-Berket, L., Gamez, C. F. R., Sagmeister, T., Chalhoub, G., Darnhofer, B., Jazleena, P. J., Birner-Gruenberger, R., Pavkov-Keller, T., Haemmerle, G., Schoiswohl, G., & Oberer, M. (2022). The Crystal Structure of Mouse Ces2c, a Potential Ortholog of Human CES2, Shows Structural Similarities in Substrate Regulation and Product Release to Human CES1. International Journal of Molecular Sciences, 23(21), 13101. https://doi.org/10.3390/ijms232113101