

Mitochondrial Fatty Acid β-Oxidation Disorders: From Disease to Lipidomic Studies—A Critical Review


 ,
,  , and
, and 
Abstract
:1. Introduction
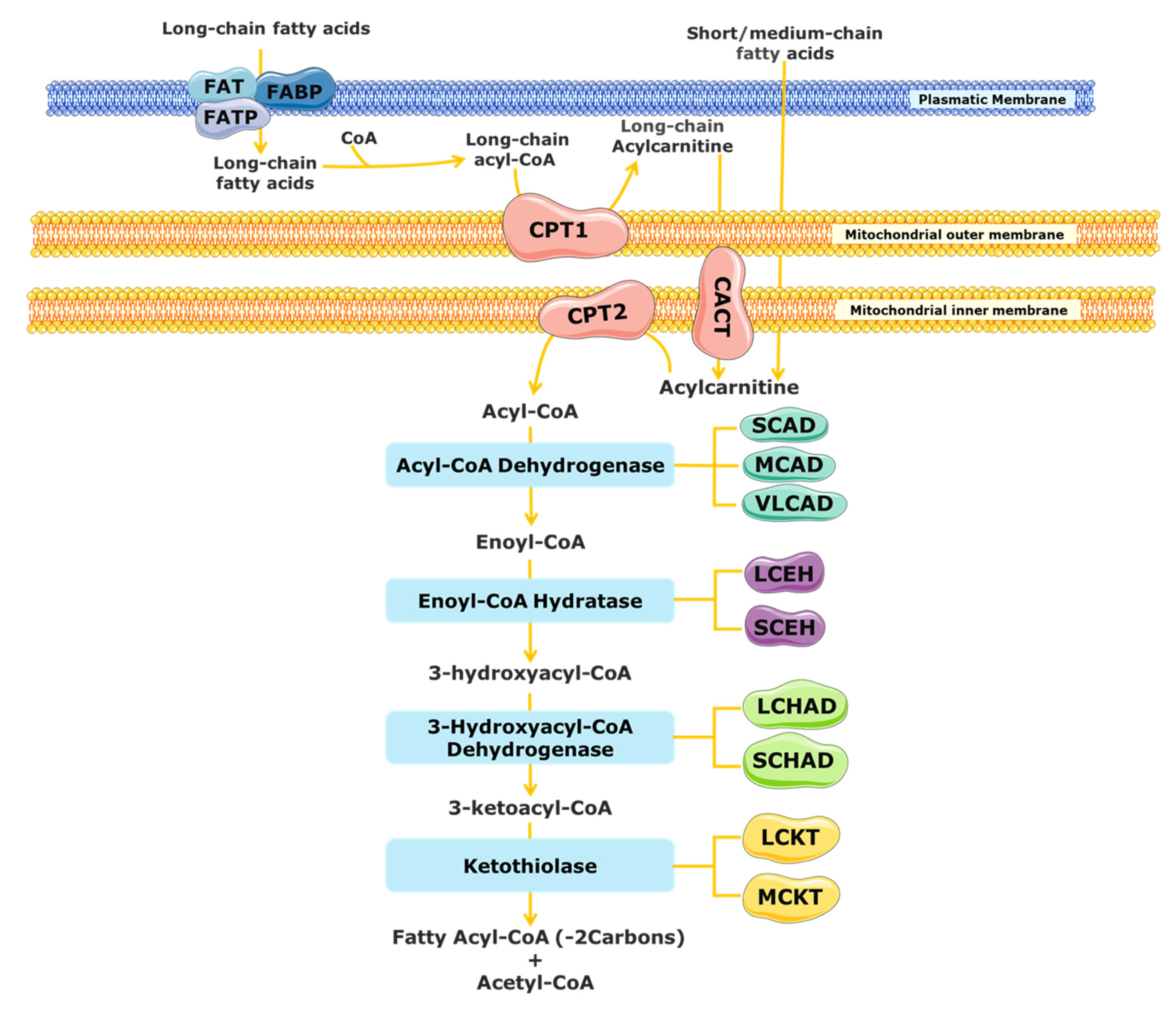
2. Mitochondrial Fatty Acid β-Oxidation
3. Mitochondrial Fatty Acid β-Oxidation Disorders (FAODs)

{kind=link}
{kind=link}
| Deficiency | Disorder Abbreviation | Worldwide Prevalence (Portugal in 2020) | Hypoketotic Hypoglycemia | Rhabdomyolysis | Cardiomyopathy | Skeletal Myopathy | Liver Dysfunction | Encephalopathy | Peripherical Neuropathy | Retinopathy | Acylcarnitine Marker (NBS) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Mitochondrial β-oxidation spiral | |||||||||||
| Medium-chain acyl-CoA dehydrogenase | MCADD | 1:4000 to 1:15,000 (1:7005) | X | - | - | - | X | X | - | - | CAR8:0, CAR8:0/CAR10:0 and CAR8:0/CAR2:0 |
| Very long chain acyl-CoA dehydrogenase | VLCADD | 1:30,000 to 1:100,000 (1:129,272) | X | X | X | X | X | - | - | - | CAR 14:1, CAR 14:2, CAR 14:1/CAR 2:0 and CAR 14:1/CAR 12:1 |
| Long-chain 3-hydroxyacyl-CoA dehydrogenase | LCHADD | 1:110,000 to 1:150,000 (1:94,800) * | X | X | X | X | X | - | X | X | CAR 16:0;O, CAR 18:1;O and CAR 18:0;O |
| Carnitine shuttle | |||||||||||
| Carnitine palmitoyl transferase deficiency type 1 | CPT1D | 1:500,000 (1:473,998) | X | - | - | - | X | - | - | - | Free carnitine/(CAR16:0 + CAR18:0) |
| Carnitine palmitoyl transferase deficiency type 2 | CPT2D | Rare (1:284,399) | X | X | X | X | X | - | - | - | (CAR16:0 + CAR18:0)/CAR2:0 |
| Carnitine–acylcarnitine translocase | CACTD | Rare (1:284,399) | X | X | X | X | - | - | - | (CAR16:0 + CAR18:0)/CAR2:0 | |
4. Oxidative Stress and Lipids in FAOD Pathogenesis
5. Deregulation of the Lipidome in FAODs
5.1. Changes in Fatty Acids, Acylcarnitines, and Complex Lipid Profiles in MCADD
| MCADD | |||
|---|---|---|---|
| Sample | Decreased | Increased | Reference |
| Plasma | - | FFAs (8:0, 10:0, 10:1 n-6) | [72] |
| - | FFAs (6:0, 8:0, 10:0, 10:1 n-6, 12:0, 12:1 14:0, 14:1 n-9, 14:2, 16:0, 16:1, 16:2, 18:0, 18:1, and 18:2); 3OH-FAs (6:0;O and 8:0;O) | [73] | |
| DBS | - | FFAs (8:0, 10:0, and 10:1 n-6) | [74] |
| - | CARs (6:0, 8:0, 10:0, and 10:1); PCs (16:0/9:0(COOH), 18:0/5:0(COOH), and 16:0/8:0(COOH)) | [44] | |
| Post-mortem liver tissue | - | TFAs (10:1 n-6, 12:1, 14:1, 14:2, and 16:2 n-6) | [75] |
5.2. Changes in Fatty Acids, Acylcarnitines, and Complex Lipid Profiles in LCHADD
| LCHADD | ||||
|---|---|---|---|---|
| Sample | Additional Information | Decreased | Increased | Reference |
| Plasma | - | FFAs (14:1 n-9, 14:2, and 16:1) | [72] | |
| - | 3OH-FAs (6:0;O, 8:0;O, 10:0;O, 12:0;O, 14:0;O, 14:1;O, 14:2;O, 16:0;O, 16:1;O, 16:2;O, 18:0;O, 18:1;O, and 18:2;O) | [73] | ||
| TFA 18:2 n-6 | - | [78] | ||
| - | CAR 12:0, CAR 14:0, CAR 14:1, CAR 14:2, CAR 16:0, CAR 18:1, CAR 18:2, CAR 14:0;O, CAR 14:1;O, CAR 16:0;O, and CAR18:1;O | [79] | ||
| LCHADD and CPT2D (considered as one study group) vs. healthy controls | HDL-C, PC 33:2, PC 34:0;O, PC 34:1;O, PC 34:3;O, PC 35:1, PC 35:2, PC 35:3, PC 36:2;O, PC(P-34:2), SM (d18:1/14:0), SM (d18:1/21:0), SM (d18:2/23:0), SM 33:1, SM 38:1, SM 40:2, SM 41:1, SM 43:1, SM 43:2, Cer (d18:1/23:0), Cer (40:1), Cer(42:1) and PE 36:3;O | TAG (14:0/14:0/14:0), TAG 44:1, TAG 46:2, TAG 46:3, TAG 56:6, TAG 58:9, and PC 40:5 | [80] | |
| Red blood cells | TFA 18:2 n-6 | [78] | ||
| DBS | CAR 14:0, CAR 14:1, CAR 14:2, CAR 16:0, CAR 18:1, CAR 14:0;O, CAR 14:1;O, CAR 16:0;O, and CAR 18:1;O | [79] | ||
| Human skin fibroblasts | Incubated with l-carnitine | Medium: CAR 5:0 | Cells: CAR 16:0, CAR 18:1, CAR 16:0;O, and CAR 18:1;O | [81] |
| Incubated with l-carnitine and palmitic acid | Medium: CAR 2:0, CAR 4:0, CAR 5:0, CAR 6:0, CAR 8:0 and CAR 10:0 | Medium and cells: CAR 16:0 and CAR 16:0;O | ||
| Incubated with l-carnitine and linoleic acid | Medium: CAR 2:0, CAR 4:0, CAR 5:0 and CAR 10:1 | Medium: CAR 14:2, CAR 18:2, and CAR 18:2;O Cells: CAR18:2;O | ||
| Investigate specific alterations (without detailed identification of lipid species) | PE, SM, PC and PE with C30 and C37 | PC; PC-O; CL; DAG; TAG; HexCer; PC/PE ratio; PC with C32 and C34; PC-O with C37; PE with C34, C36, and C38; LysoPL with C18; CL with C66 and C70; CL 64:3; and CL 64:4 | [82] | |
| Investigate effects of the incubation with MCTs (C7 and C8) | Upon C7: PC Upon C8: PC, CL, HexCer, PC/PE ratio | Upon C7: CL, DAG, TAG, and Cer Upon C8: PE, DAG, TAG, SM, Cer, and CL with C68 | [83] | |
5.3. Changes in Fatty Acids, Acylcarnitines, and Complex Lipid Profiles in VLCADD
| VLCADD | |||||
|---|---|---|---|---|---|
| Sample | Additional Information | Decreased | Increased | Reference | |
| Human | Plasma | - | FFAs (14:1 n-9, 14:2, and 16:1) | [72] | |
| - | FFAs (14:1 n-9, 14:2, and 16:2) 3OH-FAs (6:0;O and 8:0;O) | [73] | |||
| Patients with MCT (C7 or C8) supplementation vs. healthy controls | - | C8 or C7: CAR 17:0, CAR 20:0, CAR 22:0, and CAR 24:1 C7: LPE 17:0, LPC 15:0, PC 17:0/20:4, PC 17:0/22:6, PC 15:0/20:4, PC 15:0/22:6, PC 15:0/18:1, PC 17:0/16:1, PC 16:0/17:1, SM d18:2/23:1, SM d17:2/16:0, and SM d18:2/15:0 | [99] | ||
| Post-mortem liver and muscle tissue | - | TFAs (10:1 n-6, 12:1, 14:1 n-9, 14:2, and 16:2 n-6) | [75] | ||
| Mice | Liver tissue | MCT (C7 or C8) vs. LCT | C7 or C8: FAs (18:2 n-6, 18:3 n-3, 20:4 n-6, 20:5, 22:6 n-3 and PUFA) | C7 or C8: FAs (14:0, 16:1, 18:1 n-9, 18:2 n-6, 20:1, 20:2, 20:3 n-9, and SFA MUFA) and cholesterol C7: FAs (15:0, 17:1, 18:0, and 22:1) C8: FA 22:5 n-3 | [97] |
| Heart tissue | C7 or C8: FAs (18:2 n-6, 18:3 n-3 and PUFA) C8: 16:0 | C7 or C8: FAs (18:0, 20:1, 20:3 n-9, 22:4, SFA, and MUFA) C7: FAs (16:1, 17:1, and 22:4) C8: FAs (20:3 n-6 and 22:5 n-6) and cholesterol | |||
| Human | DBS | - | FFA 14:1 | [74] | |
| Mice | MCT-VLCAD-/- vs. LCT-VLCAD-/- | Male MCT-VLCAD-/-: CAR 4:0 and CAR 18:2 Female MCT-VLCAD-/-: CAR 3:0, CAR 18:2, and CAR 16:0;O | Male MCT-VLCAD-/-: CAR 16:0 and CAR 16:1 Female MCT-VLCAD-/-: CAR 4:0 | [98] | |
| MCT-VLCAD-/- vs. MCT-WT | - | Male MCT-VLCAD-/-: CAR 16:0, CAR 16:1, and CAR 18:0 Female MCT-VLCAD-/-: CAR 4:0 | |||
| LCT-VLCAD-/- vs. LCT-WT | - | Male LCT-VLCAD-/-: CAR 4:0 and CAR 16:0 Female LCT-VLCAD-/-: CAR 18:0 | |||
| Human | Fibroblasts | VLCADD vs. healthy controls | PE-O, PE-O with C36 and C38 | PC, PC-O, CL, TAG, LysoPL, and LysoPL with C14 | [82] |
| Investigate effects of the incubation with MCT (C7 or C8) | C7: PE C8: PC and PE | C7: SM, HexCer, DAG, TAG, and PC/PE ratio C8: SM, HexCer, DAG, TAG, and CL with C66 | [83] | ||
| WT vs. VLCAD | Male VLCAD: PE, PI, and SM Female VLCAD: PI | Male VLCAD: PC-O, PE-O, and HexCer Female VLCAD: PC, PE-O, Cer, HexCer, and SM | [101] | ||
| WT-C8 vs. VLCAD-C8 | Male VLCAD-C8: PC, PE, PI, and PSFemale VLCAD-C8: PE, PI, and PS | Male VLCAD-C8: PC-O, PE-O, and HexCer Female VLCAD-C8: PC, PG, Cer, HexCer, and SM | |||
| VLCAD vs. VLCAD-C8 | Male VLCAD-C8: PC and PE | Male VLCAD-C8: PC-O, and PE-OFemale VLCAD-C8: PC, LPC 16:0, LPC 16:1, LPC 18:0, and LPC 18:1 | |||
5.4. Changes in Fatty Acids, Acylcarnitines, and Complex Lipid Profiles in CPT2D
| CPT2D | |||
|---|---|---|---|
| Sample | Decreased | Increased | Reference |
| Plasma | - | FFAs (16:0 and 18:0) 3OH-FAs (6:0;O and 8:0;O) | [73] |
| Human skin fibroblasts | PC, PE-O, PC/PE ratio, PC and PE with C30 and C37, and PE-O with C36 and C38 | PE, DAG, TAG, LysoPL, PC with C32 and C34, PE with C34, C36 and C38, LysoPL with C18, and CL with C66 and C68 | [82] |
6. Concluding Remarks and Future of Lipidomics in FAODs
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Conflicts of Interest
References
- Moczulski, D.; Majak, I.; Mamczur, D. An Overview of Beta-Oxidation Disorders. Postep. Hig. Med. Dosw. 2009, 63, 266–277. [Google Scholar]
- Knottnerus, S.J.G.; Bleeker, J.C.; Wüst, R.C.I.; Ferdinandusse, S.; IJlst, L.; Wijburg, F.A.; Wanders, R.J.A.; Visser, G.; Houtkooper, R.H. Disorders of Mitochondrial Long-Chain Fatty Acid Oxidation and the Carnitine Shuttle. Rev. Endocr. Metab. Disord. 2018, 19, 93–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiekerkoetter, U.; Sun, B.; Zytkovicz, T.; Wanders, R.; Strauss, A.W.; Wendel, U. MS/MS-Based Newborn and Family Screening Detects Asymptomatic Patients with Very-Long-Chain Acyl-CoA Dehydrogenase Deficiency. J. Pediatr. 2003, 143, 335–342. [Google Scholar] [CrossRef] [Green Version]
- Ribas, G.S.; Vargas, C.R. Evidence That Oxidative Disbalance and Mitochondrial Dysfunction Are Involved in the Pathophysiology of Fatty Acid Oxidation Disorders. Cell. Mol. Neurobiol. 2020, 42, 521–532. [Google Scholar] [CrossRef]
- Ruiz-Sala, P.; Peña-Quintana, L. Biochemical Markers for the Diagnosis of Mitochondrial Fatty Acid Oxidation Diseases. J. Clin. Med. 2021, 10, 4855. [Google Scholar] [CrossRef]
- Hyötyläinen, T.; Orešič, M. Analytical Lipidomics in Metabolic and Clinical Research. Trends Endocrinol. Metab. 2015, 26, 671–673. [Google Scholar] [CrossRef]
- Guerra, I.M.S.; Ferreira, H.B.; Neves, B.; Melo, T.; Diogo, L.M.; Domingues, M.R.; Moreira, A.S.P. Lipids and Phenylketonuria: Current Evidences Pointed the Need for Lipidomics Studies. Arch. Biochem. Biophys. 2020, 688, 108431. [Google Scholar] [CrossRef]
- Zhao, Y.-Y.; Miao, H.; Cheng, X.-L.; Wei, F. Lipidomics: Novel Insight into the Biochemical Mechanism of Lipid Metabolism and Dysregulation-Associated Disease. Chem.-Biol. Interact. 2015, 240, 220–238. [Google Scholar] [CrossRef]
- Züllig, T.; Trötzmüller, M.; Köfeler, H.C. Lipidomics from Sample Preparation to Data Analysis: A Primer. Anal. Bioanal. Chem. 2020, 412, 2191–2209. [Google Scholar] [CrossRef] [Green Version]
- Herzog, K.; Pras-Raves, M.L.; Ferdinandusse, S.; Vervaart, M.A.T.; Luyf, A.C.M.; van Kampen, A.H.C.; Wanders, R.J.A.; Waterham, H.R.; Vaz, F.M. Plasma Lipidomics as a Diagnostic Tool for Peroxisomal Disorders. J. Inherit. Metab. Dis. 2018, 41, 489–498. [Google Scholar] [CrossRef] [Green Version]
- Ivanovová, E.; Piskláková, B.; Dobešová, D.; Kvasnička, A.; Friedecký, D. Novel LC-MS Tools for Diagnosing Inborn Errors of Metabolism. Microchem. J. 2021, 170, 106654. [Google Scholar] [CrossRef]
- Ismail, I.T.; Showalter, M.R.; Fiehn, O. Inborn Errors of Metabolism in the Era of Untargeted Metabolomics and Lipidomics. Metabolites 2019, 9, 242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houten, S.M.; Wanders, R.J.A. A General Introduction to the Biochemistry of Mitochondrial Fatty Acid β-Oxidation. J. Inherit. Metab. Dis. 2010, 33, 469–477. [Google Scholar] [CrossRef] [Green Version]
- Houten, S.M.; Violante, S.; Ventura, F.V.; Wanders, R.J.A. The Biochemistry and Physiology of Mitochondrial Fatty Acid β-Oxidation and Its Genetic Disorders. Annu. Rev. Physiol. 2016, 78, 23–44. [Google Scholar] [CrossRef] [Green Version]
- Sim, K.G.; Hammond, J.; Wilcken, B. Strategies for the Diagnosis of Mitochondrial Fatty Acid β-Oxidation Disorders. Clin. Chim. Acta 2002, 323, 37–58. [Google Scholar] [CrossRef]
- Kompare, M.; Rizzo, W.B. Mitochondrial Fatty-Acid Oxidation Disorders. Semin. Pediatr. Neurol. 2008, 15, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Cooper, D.E.; Young, P.A.; Klett, E.L.; Coleman, R.A. Physiological Consequences of Compartmentalized Acyl-CoA Metabolism. J. Biol. Chem. 2015, 290, 20023–20031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nsiah-Sefaa, A.; McKenzie, M. Combined defects in oxidative phosphorylation and fatty acid β-oxidation in mitochondrial disease. Biosci. Rep. 2016, 36, e00313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longo, N.; Frigeni, M.; Pasquali, M. Carnitine Transport and Fatty Acid Oxidation. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2016, 1863, 2422–2435. [Google Scholar] [CrossRef]
- Kerner, J.; Hoppel, C. Fatty Acid Import into Mitochondria. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2000, 1486, 1–17. [Google Scholar] [CrossRef]
- Wanders, R.J.A.; Ruiter, J.P.N.; IJlst, L.; Waterham, H.R.; Houten, S.M. The Enzymology of Mitochondrial Fatty Acid Beta-Oxidation and Its Application to Follow-up Analysis of Positive Neonatal Screening Results. J. Inherit. Metab. Dis. 2010, 33, 479–494. [Google Scholar] [CrossRef] [PubMed]
- Adeva-Andany, M.M.; Carneiro-Freire, N.; Seco-Filgueira, M.; Fernández-Fernández, C.; Mouriño-Bayolo, D. Mitochondrial β-Oxidation of Saturated Fatty Acids in Humans. Mitochondrion 2019, 46, 73–90. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, K.; Eaton, S. Mitochondrial β-Oxidation. Eur. J. Biochem. 2004, 271, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Lund, M.; Olsen, R.; Gregersen, N. A Short Introduction to Acyl-CoA Dehydrogenases; Deficiencies and Novel Treatment Strategies. Expert Opin. Orphan Drugs 2015, 3, 1375–1386. [Google Scholar] [CrossRef]
- Wanders, R.J.; Vreken, P.; den Boer, M.E.; Wijburg, F.A.; van Gennip, A.H.; IJlst, L. Disorders of Mitochondrial Fatty Acyl-CoA Beta-Oxidation. J. Inherit. Metab. Dis. 1999, 22, 442–487. [Google Scholar] [CrossRef]
- Rinaldo, P.; Matern, D.; Bennett, M.J. Fatty Acid Oxidation Disorders. Annu. Rev. Physiol. 2002, 64, 477–502. [Google Scholar] [CrossRef]
- Wajner, M.; Amaral, A.U. Mitochondrial Dysfunction in Fatty Acid Oxidation Disorders: Insights from Human and Animal Studies. Biosci. Rep. 2016, 36, e00281. [Google Scholar] [CrossRef] [Green Version]
- Olpin, S.E. Pathophysiology of Fatty Acid Oxidation Disorders and Resultant Phenotypic Variability. J. Inherit. Metab. Dis. 2013, 36, 645–658. [Google Scholar] [CrossRef]
- Merritt, J.L.; MacLeod, E.; Jurecka, A.; Hainline, B. Clinical Manifestations and Management of Fatty Acid Oxidation Disorders. Rev. Endocr. Metab. Disord. 2020, 21, 479–493. [Google Scholar] [CrossRef]
- Blau, N.; Duran, M.; Gibson, K.M.; Dionisi-Vici, C. Physician’s Guide to the Diagnosis, Treatment, and Follow-Up of Inherited Metabolic Diseases, 2nd ed.; Springer: Cham, Switzerland, 2014; ISBN 978-3-030-67727-5. [Google Scholar]
- Merritt, J.L.; Norris, M.; Kanungo, S. Fatty Acid Oxidation Disorders. Ann. Transl. Med. 2018, 6, 473. [Google Scholar] [CrossRef]
- Rovelli, V.; Manzoni, F.; Viau, K.; Pasquali, M.; Longo, N. Clinical and Biochemical Outcome of Patients with Very Long-Chain Acyl-CoA Dehydrogenase Deficiency. Mol. Genet. Metab. 2019, 127, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Loeber, J.G.; Platis, D.; Zetterström, R.H.; Almashanu, S.; Boemer, F.; Bonham, J.R.; Borde, P.; Brincat, I.; Cheillan, D.; Dekkers, E.; et al. Neonatal Screening in Europe Revisited: An ISNS Perspective on the Current State and Developments since 2010. Int. J. Neonatal Screen. 2021, 7, 15. [Google Scholar] [CrossRef]
- Lindner, M.; Hoffmann, G.F.; Matern, D. Newborn Screening for Disorders of Fatty-Acid Oxidation: Experience and Recommendations from an Expert Meeting. J. Inherit. Metab. Dis. 2010, 33, 521–526. [Google Scholar] [CrossRef] [PubMed]
- Vilarinho, L.; Rocha, H.; Sousa, C.; Marcão, A.; Fonseca, H.; Bogas, M.; Osório, R.V. Four Years of Expanded Newborn Screening in Portugal with Tandem Mass Spectrometry. J. Inherit. Metab. Dis. 2010, 33, S133–S138. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Taketani, T. Management and Diagnosis of Mitochondrial Fatty Acid Oxidation Disorders: Focus on Very-Long-Chain Acyl-CoA Dehydrogenase Deficiency. J. Hum. Genet. 2019, 64, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Wanders, R.J.A.; Visser, G.; Ferdinandusse, S.; Vaz, F.M.; Houtkooper, R.H. Mitochondrial Fatty Acid Oxidation Disorders: Laboratory Diagnosis, Pathogenesis, and the Complicated Route to Treatment. J. Lipid Atheroscler. 2020, 9, 313–333. [Google Scholar] [CrossRef] [PubMed]
- Morris, A.A.M.; Spiekerkoetter, U. Disorders of Mitochondrial Fatty Acid Oxidation & Riboflavin Metabolism. In Inborn Metabolic Diseases: Diagnosis and Treatment; Saudubray, J.-M., Baumgartner, M.R., Walter, J., Eds.; Springer: Berlin/Heidelberg, Germany, 2016; pp. 201–213. ISBN 978-3-662-49771-5. [Google Scholar]
- Spiekerkoetter, U.; Lindner, M.; Santer, R.; Grotzke, M.; Baumgartner, M.R.; Boehles, H.; Das, A.; Haase, C.; Hennermann, J.B.; Karall, D.; et al. Treatment Recommendations in Long-Chain Fatty Acid Oxidation Defects: Consensus from a Workshop. J. Inherit. Metab. Dis. 2009, 32, 498–505. [Google Scholar] [CrossRef]
- Calder, P.C. Fatty Acids and Inflammation: The Cutting Edge between Food and Pharma. Eur. J. Pharmacol. 2011, 668 (Suppl. 1), S50–S58. [Google Scholar] [CrossRef]
- Radzikowska, U.; Rinaldi, A.O.; Çelebi Sözener, Z.; Karaguzel, D.; Wojcik, M.; Cypryk, K.; Akdis, M.; Akdis, C.A.; Sokolowska, M. The Influence of Dietary Fatty Acids on Immune Responses. Nutrients 2019, 11, 2990. [Google Scholar] [CrossRef] [Green Version]
- El-Gharbawy, A.; Goldstein, A. Mitochondrial Fatty Acid Oxidation Disorders Associated with Cardiac Disease. Curr. Pathobiol. Rep. 2017, 5, 259–270. [Google Scholar] [CrossRef]
- Spiekerkoetter, U.; Wood, P.A. Mitochondrial Fatty Acid Oxidation Disorders: Pathophysiological Studies in Mouse Models. J. Inherit. Metab. Dis. 2010, 33, 539–546. [Google Scholar] [CrossRef] [PubMed]
- Najdekr, L.; Gardlo, A.; Mádrová, L.; Friedecký, D.; Janečková, H.; Correa, E.S.; Goodacre, R.; Adam, T. Oxidized Phosphatidylcholines Suggest Oxidative Stress in Patients with Medium-Chain Acyl-CoA Dehydrogenase Deficiency. Talanta 2015, 139, 62–66. [Google Scholar] [CrossRef] [PubMed]
- Schuck, P.F.; Ferreira, G.C.; Moura, A.P.; Busanello, E.N.B.; Tonin, A.M.; Dutra-Filho, C.S.; Wajner, M. Medium-Chain Fatty Acids Accumulating in MCAD Deficiency Elicit Lipid and Protein Oxidative Damage and Decrease Non-Enzymatic Antioxidant Defenses in Rat Brain. Neurochem. Int. 2009, 54, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Tonin, A.M.; Grings, M.; Knebel, L.A.; Zanatta, Â.; Moura, A.P.; Ribeiro, C.A.J.; Leipnitz, G.; Wajner, M. Disruption of Redox Homeostasis in Cerebral Cortex of Developing Rats by Acylcarnitines Accumulating in Medium-Chain Acyl-CoA Dehydrogenase Deficiency. Int. J. Dev. Neurosci. 2012, 30, 383–390. [Google Scholar] [CrossRef]
- Schuck, P.F.; Ceolato, P.C.; Ferreira, G.C.; Tonin, A.; Leipnitz, G.; Dutra-Filho, C.S.; Latini, A.; Wajner, M. Oxidative Stress Induction by Cis-4-Decenoic Acid: Relevance for MCAD Deficiency. Free Radic. Res. 2007, 41, 1261–1272. [Google Scholar] [CrossRef]
- Scaini, G.; Simon, K.R.; Tonin, A.M.; Busanello, E.N.B.; Moura, A.P.; Ferreira, G.C.; Wajner, M.; Streck, E.L.; Schuck, P.F. Toxicity of Octanoate and Decanoate in Rat Peripheral Tissues: Evidence of Bioenergetic Dysfunction and Oxidative Damage Induction in Liver and Skeletal Muscle. Mol. Cell. Biochem. 2012, 361, 329–335. [Google Scholar] [CrossRef]
- Amaral, A.U.; Cecatto, C.; da Silva, J.C.; Wajner, A.; dos Godoy, K.S.; Ribeiro, R.T.; Wajner, M. Cis-4-Decenoic and Decanoic Acids Impair Mitochondrial Energy, Redox and Ca2+ Homeostasis and Induce Mitochondrial Permeability Transition Pore Opening in Rat Brain and Liver: Possible Implications for the Pathogenesis of MCAD Deficiency. Biochim. Biophys. Acta (BBA)-Bioenerg. 2016, 1857, 1363–1372. [Google Scholar] [CrossRef]
- Reis de Assis, D.; Maria, R.D.C.; Borba Rosa, R.; Schuck, P.F.; Ribeiro, C.A.J.; da Costa Ferreira, G.; Dutra-Filho, C.S.; Terezinha de Souza Wyse, A.; Duval Wannmacher, C.M.; Santos Perry, M.L.; et al. Inhibition of Energy Metabolism in Cerebral Cortex of Young Rats by the Medium-Chain Fatty Acids Accumulating in MCAD Deficiency. Brain Res. 2004, 1030, 141–151. [Google Scholar] [CrossRef]
- Hickmann, F.H.; Cecatto, C.; Kleemann, D.; Monteiro, W.O.; Castilho, R.F.; Amaral, A.U.; Wajner, M. Uncoupling, Metabolic Inhibition and Induction of Mitochondrial Permeability Transition in Rat Liver Mitochondria Caused by the Major Long-Chain Hydroxyl Monocarboxylic Fatty Acids Accumulating in LCHAD Deficiency. Biochim. Biophys. Acta (BBA)-Bioenerg. 2015, 1847, 620–628. [Google Scholar] [CrossRef] [Green Version]
- Cecatto, C.; Godoy, K.D.S.; da Silva, J.C.; Amaral, A.U.; Wajner, M. Disturbance of Mitochondrial Functions Provoked by the Major Long-Chain 3-Hydroxylated Fatty Acids Accumulating in MTP and LCHAD Deficiencies in Skeletal Muscle. Toxicol. In Vitro 2016, 36, 1–9. [Google Scholar] [CrossRef]
- Tonin, A.M.; Ferreira, G.C.; Grings, M.; Viegas, C.M.; Busanello, E.N.; Amaral, A.U.; Zanatta, Â.; Schuck, P.F.; Wajner, M. Disturbance of Mitochondrial Energy Homeostasis Caused by the Metabolites Accumulating in LCHAD and MTP Deficiencies in Rat Brain. Life Sci. 2010, 86, 825–831. [Google Scholar] [CrossRef] [PubMed]
- Cecatto, C.; Hickmann, F.H.; Rodrigues, M.D.N.; Amaral, A.U.; Wajner, M. Deregulation of Mitochondrial Functions Provoked by Long-Chain Fatty Acid Accumulating in Long-Chain 3-Hydroxyacyl-CoA Dehydrogenase and Mitochondrial Permeability Transition Deficiencies in Rat Heart—Mitochondrial Permeability Transition Pore Opening as a Potential Contributing Pathomechanism of Cardiac Alterations in These Disorders. FEBS J. 2015, 282, 4714–4726. [Google Scholar] [CrossRef] [PubMed]
- Tonin, A.M.; Amaral, A.U.; Busanello, E.N.B.; Grings, M.; Castilho, R.F.; Wajner, M. Long-Chain 3-Hydroxy Fatty Acids Accumulating in Long-Chain 3-Hydroxyacyl-CoA Dehydrogenase and Mitochondrial Trifunctional Protein Deficiencies Uncouple Oxidative Phosphorylation in Heart Mitochondria. J. Bioenerg. Biomembr. 2013, 45, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Cecatto, C.; Wajner, A.; Vargas, C.R.; Wajner, S.M.; Amaral, A.U.; Wajner, M. High Vulnerability of the Heart and Liver to 3-Hydroxypalmitic Acid–Induced Disruption of Mitochondrial Functions in Intact Cell Systems. J. Cell. Biochem. 2018, 119, 7678–7686. [Google Scholar] [CrossRef]
- Tonin, A.M.; Amaral, A.U.; Busanello, E.N.; Gasparotto, J.; Gelain, D.P.; Gregersen, N.; Wajner, M. Mitochondrial Bioenergetics Deregulation Caused by Long-Chain 3-Hydroxy Fatty Acids Accumulating in LCHAD and MTP Deficiencies in Rat Brain: A Possible Role of MPTP Opening as a Pathomechanism in These Disorders? Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2014, 1842, 1658–1667. [Google Scholar] [CrossRef] [Green Version]
- Tonin, A.M.; Grings, M.; Busanello, E.N.B.; Moura, A.P.; Ferreira, G.C.; Viegas, C.M.; Fernandes, C.G.; Schuck, P.F.; Wajner, M. Long-Chain 3-Hydroxy Fatty Acids Accumulating in LCHAD and MTP Deficiencies Induce Oxidative Stress in Rat Brain. Neurochem. Int. 2010, 56, 930–936. [Google Scholar] [CrossRef]
- de Moraes, M.S.; Guerreiro, G.; Sitta, A.; de Moura Coelho, D.; Manfredini, V.; Wajner, M.; Vargas, C.R. Oxidative Damage in Mitochondrial Fatty Acids Oxidation Disorders Patients and the in Vitro Effect of L-Carnitine on DNA Damage Induced by the Accumulated Metabolites. Arch. Biochem. Biophys. 2020, 679, 108206. [Google Scholar] [CrossRef]
- Cecatto, C.; Amaral, A.U.; Wajner, A.; Wajner, S.M.; Castilho, R.F.; Wajner, M. Disturbance of Mitochondrial Functions Associated with Permeability Transition Pore Opening Induced by Cis-5-Tetradecenoic and Myristic Acids in Liver of Adolescent Rats. Mitochondrion 2020, 50, 1–13. [Google Scholar] [CrossRef]
- Cecatto, C.; Amaral, A.U.; da Silva, J.C.; Wajner, A.; Schimit, M.D.O.V.; da Silva, L.H.R.; Wajner, S.M.; Zanatta, Â.; Castilho, R.F.; Wajner, M. Metabolite Accumulation in VLCAD Deficiency Markedly Disrupts Mitochondrial Bioenergetics and Ca2+ Homeostasis in the Heart. FEBS J. 2018, 285, 1437–1455. [Google Scholar] [CrossRef] [Green Version]
- Cecatto, C.; Amaral, A.U.; Roginski, A.C.; Castilho, R.F.; Wajner, M. Impairment of Mitochondrial Bioenergetics and Permeability Transition Induction Caused by Major Long-Chain Fatty Acids Accumulating in VLCAD Deficiency in Skeletal Muscle as Potential Pathomechanisms of Myopathy. Toxicol. In Vitro 2020, 62, 104665. [Google Scholar] [CrossRef]
- Hoffmann, L.; Seibt, A.; Herebian, D.; Spiekerkoetter, U. Monounsaturated 14:1n-9 and 16:1n-9 Fatty Acids but Not 18:1n-9 Induce Apoptosis and Necrosis in Murine HL-1 Cardiomyocytes. Lipids 2014, 49, 25–37. [Google Scholar] [CrossRef] [PubMed]
- McCoin, C.S.; Knotts, T.A.; Adams, S.H. Acylcarnitines—Old Actors Auditioning for New Roles in Metabolic Physiology. Nat. Rev. Endocrinol. 2015, 11, 617–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seminotti, B.; Leipnitz, G.; Karunanidhi, A.; Kochersperger, C.; Roginskaya, V.Y.; Basu, S.; Wang, Y.; Wipf, P.; Van Houten, B.; Mohsen, A.-W.; et al. Mitochondrial Energetics Is Impaired in Very Long-Chain Acyl-CoA Dehydrogenase Deficiency and Can Be Rescued by Treatment with Mitochondria-Targeted Electron Scavengers. Hum. Mol. Genet. 2019, 28, 928–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagenbuchner, J.; Scholl-Buergi, S.; Karall, D.; Ausserlechner, M.J. Very Long-/ and Long Chain-3-Hydroxy Acyl CoA Dehydrogenase Deficiency Correlates with Deregulation of the Mitochondrial Fusion/Fission Machinery. Sci. Rep. 2018, 8, 3254. [Google Scholar] [CrossRef] [Green Version]
- Fahy, E.; Subramaniam, S.; Brown, H.A.; Glass, C.K.; Merrill, A.H.; Murphy, R.C.; Raetz, C.R.H.; Russell, D.W.; Seyama, Y.; Shaw, W.; et al. A Comprehensive Classification System for Lipids1. J. Lipid Res. 2005, 46, 839–861. [Google Scholar] [CrossRef] [Green Version]
- Fahy, E.; Subramaniam, S.; Murphy, R.C.; Nishijima, M.; Raetz, C.R.H.; Shimizu, T.; Spener, F.; van Meer, G.; Wakelam, M.J.O.; Dennis, E.A. Update of the LIPID MAPS Comprehensive Classification System for Lipids1. J. Lipid Res. 2009, 50, S9–S14. [Google Scholar] [CrossRef] [Green Version]
- Liebisch, G.; Fahy, E.; Aoki, J.; Dennis, E.A.; Durand, T.; Ejsing, C.S.; Fedorova, M.; Feussner, I.; Griffiths, W.J.; Köfeler, H.; et al. Update on LIPID MAPS Classification, Nomenclature, and Shorthand Notation for MS-Derived Lipid Structures. J. Lipid Res. 2020, 61, 1539–1555. [Google Scholar] [CrossRef]
- Alves, M.A.; Lamichhane, S.; Dickens, A.; McGlinchey, A.; Ribeiro, H.C.; Sen, P.; Wei, F.; Hyötyläinen, T.; Orešič, M. Systems Biology Approaches to Study Lipidomes in Health and Disease. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2021, 1866, 158857. [Google Scholar] [CrossRef]
- Wei, F.; Lamichhane, S.; Orešič, M.; Hyötyläinen, T. Lipidomes in Health and Disease: Analytical Strategies and Considerations. Trends Anal. Chem. 2019, 120, 115664. [Google Scholar] [CrossRef]
- Martínez, G.; Jiménez-Sánchez, G.; Divry, P.; Vianey-Saban, C.; Riudor, E.; Rodés, M.; Briones, P.; Ribes, A. Plasma Free Fatty Acids in Mitochondrial Fatty Acid Oxidation Defects. Clin. Chim. Acta 1997, 267, 143–154. [Google Scholar] [CrossRef]
- Costa, C.G.; Dorland, L.; Holwerda, U.; Tavares de Almeida, I.; Poll-The, B.-T.; Jakobs, C.; Duran, M. Simultaneous Analysis of Plasma Free Fatty Acids and Their 3-Hydroxy Analogs in Fatty Acid β-Oxidation Disorders. Clin. Chem. 1998, 44, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M.; Yoon, H.R.; Wasant, P.; Takahashi, Y.; Yamaguchi, S. A Sensitive and Simplified Method to Analyze Free Fatty Acids in Children with Mitochondrial Beta Oxidation Disorders Using Gas Chromatography/Mass Spectrometry and Dried Blood Spots. Clin. Chim. Acta 2002, 316, 117–121. [Google Scholar] [CrossRef]
- Onkenhout, W.; Venizelos, V.; Scholte, H.R.; de Klerk, J.B.C.; Poorthuis, B.J.H.M. Intermediates of Unsaturated Fatty Acid Oxidation Are Incorporated in Triglycerides but Not in Phospholipids in Tissues from Patients with Mitochondrial β-Oxidation Defects. J. Inherit. Metab. Dis. 2001, 24, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Zhong, S.; Li, L.; Shen, X.; Li, Q.; Xu, W.; Wang, X.; Tao, Y.; Yin, H. An Update on Lipid Oxidation and Inflammation in Cardiovascular Diseases. Free Radic. Biol. Med. 2019, 144, 266–278. [Google Scholar] [CrossRef] [PubMed]
- Freigang, S. The Regulation of Inflammation by Oxidized Phospholipids. Eur. J. Immunol. 2016, 46, 1818–1825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lund, A.M.; Dixon, M.A.; Vreken, P.; Leonard, J.V.; Morris, A.A.M. Plasma and Erythrocyte Fatty Acid Concentrations in Long-Chain 3-Hydroxyacyl-CoA Dehydrogenase Deficiency. J. Inherit. Metab. Dis. 2003, 26, 410–412. [Google Scholar] [CrossRef] [PubMed]
- Van Hove, J.L.K.; Kahler, S.G.; Feezor, M.D.; Ramakrishna, J.P.; Hart, P.; Treem, W.R.; Shen, J.-J.; Matern, D.; Millington, D.S. Acylcarnitines in Plasma and Blood Spots of Patients with Long-Chain 3-Hydroxyacyl-Coenzyme A Dehydrogenase Defiency. J. Inherit. Metab. Dis. 2000, 23, 571–582. [Google Scholar] [CrossRef]
- McCoin, C.S.; Piccolo, B.D.; Knotts, T.A.; Matern, D.; Vockley, J.; Gillingham, M.B.; Adams, S.H. Unique Plasma Metabolomic Signatures of Individuals with Inherited Disorders of Long-Chain Fatty Acid Oxidation. J. Inherit. Metab. Dis. 2016, 39, 399–408. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.J.; Matern, D.; Millington, D.S.; Hillman, S.; Feezor, M.D.; Bennett, M.J.; Qumsiyeh, M.; Kahler, S.G.; Chen, Y.-T.; Van Hove, J.L.K. Acylcarnitines in Fibroblasts of Patients with Long-Chain 3-Hydroxyacyl-CoA Dehydrogenase Deficiency and Other Fatty Acid Oxidation Disorders. J. Inherit. Metab. Dis. 2000, 23, 27–44. [Google Scholar] [CrossRef]
- Alatibi, K.I.; Hagenbuchner, J.; Wehbe, Z.; Karall, D.; Ausserlechner, M.J.; Vockley, J.; Spiekerkoetter, U.; Grünert, S.C.; Tucci, S. Different Lipid Signature in Fibroblasts of Long-Chain Fatty Acid Oxidation Disorders. Cells 2021, 10, 1239. [Google Scholar] [CrossRef]
- Alatibi, K.I.; Tholen, S.; Wehbe, Z.; Hagenbuchner, J.; Karall, D.; Ausserlechner, M.J.; Schilling, O.; Grünert, S.C.; Vockley, J.; Tucci, S. Lipidomic and Proteomic Alterations Induced by Even and Odd Medium-Chain Fatty Acids on Fibroblasts of Long-Chain Fatty Acid Oxidation Disorders. Int. J. Mol. Sci. 2021, 22, 10556. [Google Scholar] [CrossRef] [PubMed]
- van der Veen, J.N.; Kennelly, J.P.; Wan, S.; Vance, J.E.; Vance, D.E.; Jacobs, R.L. The Critical Role of Phosphatidylcholine and Phosphatidylethanolamine Metabolism in Health and Disease. Biochim. Biophys. Acta (BBA)-Biomembr. 2017, 1859, 1558–1572. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ge, M.; Ciani, L.; Kuriakose, G.; Westover, E.J.; Dura, M.; Covey, D.F.; Freed, J.H.; Maxfield, F.R.; Lytton, J.; et al. Enrichment of Endoplasmic Reticulum with Cholesterol Inhibits Sarcoplasmic-Endoplasmic Reticulum Calcium ATPase-2b Activity in Parallel with Increased Order of Membrane Lipids: Implications for Depletion of Endoplasmic Reticulum Calcium Stores and Apoptosis in Cholesterol-Loaded Macrophages. J. Biol. Chem. 2004, 279, 37030–37039. [Google Scholar] [CrossRef] [Green Version]
- Fu, S.; Yang, L.; Li, P.; Hofmann, O.; Dicker, L.; Hide, W.; Lin, X.; Watkins, S.M.; Ivanov, A.; Hotamisligil, G.S. Aberrant Lipid Metabolism Disrupts Calcium Homeostasis Causing Liver Endoplasmic Reticulum Stress in Obesity. Nature 2011, 473, 528–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braverman, N.E.; Moser, A.B. Functions of Plasmalogen Lipids in Health and Disease. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2012, 1822, 1442–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lessig, J.; Fuchs, B. Plasmalogens in Biological Systems: Their Role in Oxidative Processes in Biological Membranes, Their Contribution to Pathological Processes and Aging and Plasmalogen Analysis. Curr. Med. Chem. 2009, 16, 2021–2041. [Google Scholar] [CrossRef] [PubMed]
- Pujol-Lereis, L.M.; Liebisch, G.; Schick, T.; Lin, Y.; Grassmann, F.; Uchida, K.; Zipfel, P.F.; Fauser, S.; Skerka, C.; Weber, B.H.F. Evaluation of Serum Sphingolipids and the Influence of Genetic Risk Factors in Age-Related Macular Degeneration. PLoS ONE 2018, 13, e0200739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vidaurre, O.G.; Haines, J.D.; Katz Sand, I.; Adula, K.P.; Huynh, J.L.; McGraw, C.A.; Zhang, F.; Varghese, M.; Sotirchos, E.; Bhargava, P.; et al. Cerebrospinal Fluid Ceramides from Patients with Multiple Sclerosis Impair Neuronal Bioenergetics. Brain 2014, 137, 2271–2286. [Google Scholar] [CrossRef]
- Casares, D.; Escribá, P.V.; Rosselló, C.A. Membrane Lipid Composition: Effect on Membrane and Organelle Structure, Function and Compartmentalization and Therapeutic Avenues. Int. J. Mol. Sci. 2019, 20, 2167. [Google Scholar] [CrossRef] [Green Version]
- van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane Lipids: Where They Are and How They Behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Role of Cardiolipin in Mitochondrial Function and Dynamics in Health and Disease: Molecular and Pharmacological Aspects. Cells 2019, 8, 728. [Google Scholar] [CrossRef]
- Kreutzberger, A.J.B.; Ji, M.; Aaron, J.; Mihaljević, L.; Urban, S. Rhomboid Distorts Lipids to Break the Viscosity-Imposed Speed Limit of Membrane Diffusion. Science 2019, 363, eaao0076. [Google Scholar] [CrossRef] [PubMed]
- Sezgin, E.; Gutmann, T.; Buhl, T.; Dirkx, R.; Grzybek, M.; Coskun, Ü.; Solimena, M.; Simons, K.; Levental, I.; Schwille, P. Adaptive Lipid Packing and Bioactivity in Membrane Domains. PLoS ONE 2015, 10, e0123930. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.T.; Ramesh, T.; Toh, X.R.; Nguyen, L.N. Emerging Roles of Lysophospholipids in Health and Disease. Prog. Lipid Res. 2020, 80, 101068. [Google Scholar] [CrossRef] [PubMed]
- Tucci, S.; Behringer, S.; Spiekerkoetter, U. De Novo Fatty Acid Biosynthesis and Elongation in Very Long-Chain Acyl-CoA Dehydrogenase-Deficient Mice Supplemented with Odd or Even Medium-Chain Fatty Acids. FEBS J. 2015, 282, 4242–4253. [Google Scholar] [CrossRef] [Green Version]
- Tucci, S.; Flögel, U.; Spiekerkoetter, U. Sexual Dimorphism of Lipid Metabolism in Very Long-Chain Acyl-CoA Dehydrogenase Deficient (VLCAD−/−) Mice in Response to Medium-Chain Triglycerides (MCT). Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2015, 1852, 1442–1450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sklirou, E.; Alodaib, A.N.; Dobrowolski, S.F.; Mohsen, A.-W.A.; Vockley, J. Physiological Perspectives on the Use of Triheptanoin as Anaplerotic Therapy for Long Chain Fatty Acid Oxidation Disorders. Front. Genet. 2021, 11, 598760. [Google Scholar] [CrossRef]
- Tucci, S. Very Long-Chain Acyl-CoA Dehydrogenase (VLCAD-) Deficiency–Studies on Treatment Effects and Long-Term Outcomes in Mouse Models. J. Inherit. Metab. Dis. 2017, 40, 317–323. [Google Scholar] [CrossRef]
- Alatibi, K.I.; Wehbe, Z.; Spiekerkoetter, U.; Tucci, S. Sex-Specific Perturbation of Complex Lipids in Response to Medium-Chain Fatty Acids in Very Long-Chain Acyl-CoA Dehydrogenase Deficiency. FEBS J. 2020, 287, 3511–3525. [Google Scholar] [CrossRef]
- Ntambi, J.M. The Regulation of Stearoyl-CoA Desaturase (SCD). Prog. Lipid Res. 1995, 34, 139–150. [Google Scholar] [CrossRef]
- Opalka, J.R.; Gellerich, F.-N.; Zierz, S. Age and Sex Dependency of Carnitine Concentration in Human Serum and Skeletal Muscle. Clin. Chem. 2001, 47, 2150–2153. [Google Scholar] [CrossRef] [PubMed]
- Wehbe, Z.; Alatibi, K.; Jellusova, J.; Spiekerkoetter, U.; Tucci, S. The Fate of Medium-Chain Fatty Acids in Very Long-Chain Acyl-CoA Dehydrogenase Deficiency (VLCADD): A Matter of Sex? Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2019, 1864, 1591–1605. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, M.; Tsuchiya, S.; Aramaki, Y. Involvement of ERK, a MAP Kinase, in the Production of TGF-β by Macrophages Treated with Liposomes Composed of Phosphatidylserine. Biochem. Biophys. Res. Commun. 2004, 324, 1400–1405. [Google Scholar] [CrossRef] [PubMed]
- Fadok, V.A.; de Cathelineau, A.; Daleke, D.L.; Henson, P.M.; Bratton, D.L. Loss of Phospholipid Asymmetry and Surface Exposure of Phosphatidylserine Is Required for Phagocytosis of Apoptotic Cells by Macrophages and Fibroblasts. J. Biol. Chem. 2001, 276, 1071–1077. [Google Scholar] [CrossRef] [Green Version]
- Di Paolo, G.; De Camilli, P. Phosphoinositides in Cell Regulation and Membrane Dynamics. Nature 2006, 443, 651–657. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guerra, I.M.S.; Ferreira, H.B.; Melo, T.; Rocha, H.; Moreira, S.; Diogo, L.; Domingues, M.R.; Moreira, A.S.P. Mitochondrial Fatty Acid β-Oxidation Disorders: From Disease to Lipidomic Studies—A Critical Review. Int. J. Mol. Sci. 2022, 23, 13933. https://doi.org/10.3390/ijms232213933
Guerra IMS, Ferreira HB, Melo T, Rocha H, Moreira S, Diogo L, Domingues MR, Moreira ASP. Mitochondrial Fatty Acid β-Oxidation Disorders: From Disease to Lipidomic Studies—A Critical Review. International Journal of Molecular Sciences. 2022; 23(22):13933. https://doi.org/10.3390/ijms232213933
Chicago/Turabian StyleGuerra, Inês M. S., Helena B. Ferreira, Tânia Melo, Hugo Rocha, Sónia Moreira, Luísa Diogo, Maria Rosário Domingues, and Ana S. P. Moreira. 2022. "Mitochondrial Fatty Acid β-Oxidation Disorders: From Disease to Lipidomic Studies—A Critical Review" International Journal of Molecular Sciences 23, no. 22: 13933. https://doi.org/10.3390/ijms232213933
APA StyleGuerra, I. M. S., Ferreira, H. B., Melo, T., Rocha, H., Moreira, S., Diogo, L., Domingues, M. R., & Moreira, A. S. P. (2022). Mitochondrial Fatty Acid β-Oxidation Disorders: From Disease to Lipidomic Studies—A Critical Review. International Journal of Molecular Sciences, 23(22), 13933. https://doi.org/10.3390/ijms232213933