
A Straightforward Method for the Isolation and Cultivation of Galleria mellonella Hemocytes


Abstract
:1. Introduction
2. Results and Discussion
2.1. Optimization of a G. mellonella Hemocyte Primary Culture
2.2. Quantification of Hemocyte Viability
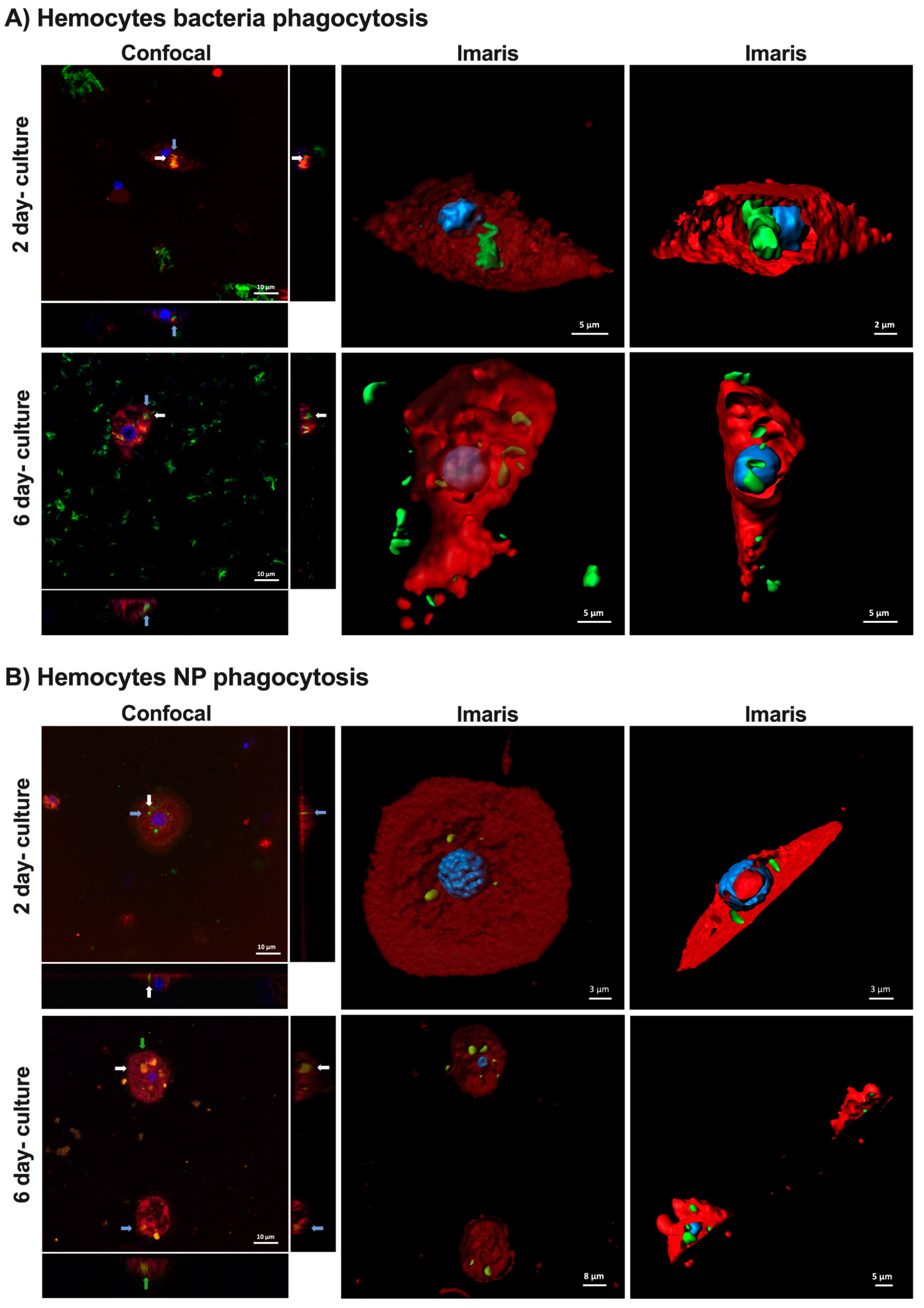
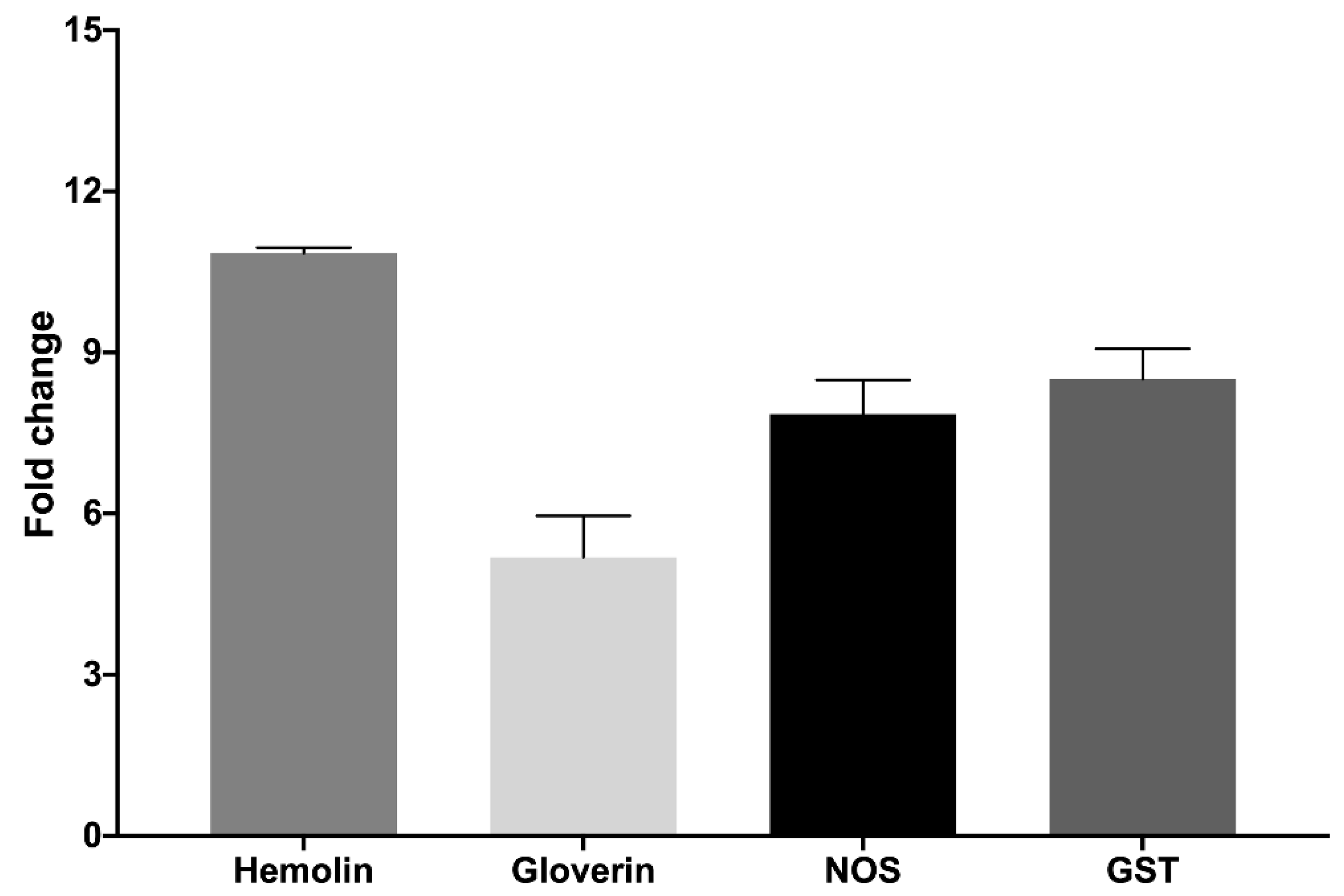
2.3. Hemocyte Functionality and Activity
3. Materials and Methods
3.1. Galleria mellonella Maintenance and Hemolymph Extraction
3.2. Maintenance of a Hemocyte Primary Cell Culture
3.3. Quantification of Hemocyte Viability in Culture
3.3.1. PrestoBlue Cell Viability Assay
3.3.2. Live/Dead Cell Viability Assay
3.4. Hemocyte Imaging by Confocal Microscopy
3.5. RNA Extraction, Reverse Transcription, and Real-Time PCR
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pereira, M.; Rossi, C.; da Silva, G.; Rosa, J.; Bazzolli, D. Galleria mellonella as an Infection Model: An in-Depth Look at Why It Works and Practical Considerations for Successful Application. Pathog. Dis. 2020, 78, ftaa056. [Google Scholar] [CrossRef] [PubMed]
- Ménard, G.; Rouillon, A.; Cattoir, V.; Donnio, P.Y. Galleria mellonella as a Suitable Model of Bacterial Infection: Past, Present and Future. Front. Cell. Infect. Microbiol. 2021, 11, 782733. [Google Scholar] [CrossRef] [PubMed]
- Strand, M.R. The Insect Cellular Immune Response. Insect. Sci. 2008, 15, 1–14. [Google Scholar] [CrossRef]
- Wojda, I. Immunity of the Greater Wax Moth Galleria mellonella. Insect. Sci. 2017, 24, 342–357. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, C.; Brehélin, M. Insect Haemocytes: What Type of Cell Is That? J. Insect. Physiol. 2006, 52, 417–429. [Google Scholar] [CrossRef] [PubMed]
- Singkum, P.; Suwanmanee, S.; Pumeesat, P.; Luplertlop, N. A Powerful in Vivo Alternative Model in Scientific Research: Galleria mellonella. Acta Microbiol. Immunol. Hung. 2019, 66, 31–55. [Google Scholar] [CrossRef] [PubMed]
- Stączek, S.; Zdybicka-Barabas, A.; Wiater, A.; Pleszczyńska, M.; Cytryńska, M. Activation of Cellular Immune Response in Insect Model Host Galleria mellonella by Fungal α-1,3-Glucan. Pathog. Dis. 2020, 78, 62. [Google Scholar] [CrossRef]
- Potts, R.W.A.; Gutierrez, A.P.; Cortés-Araya, Y.; Houston, R.D.; Bean, T.P. Developments in Marine Invertebrate Primary Culture Reveal Novel Cell Morphologies in the Model Bivalve Crassostrea Gigas. PeerJ 2020, 2020, e9180. [Google Scholar] [CrossRef]
- Arunkarthick, S.; Asokan, R.; Aravintharaj, R.; Niveditha, M.; Kumar, N.K.K. A Review of Insect Cell Culture: Establishment, Maintenance and Applications in Entomological Research. J. Entomol. Sci. 2017, 52, 261–273. [Google Scholar] [CrossRef]
- Lynn, D.E. Methods for the Development of Cell Lines from Insects. J. Tissue Cult. Methods 1989, 12, 23–29. [Google Scholar] [CrossRef]
- Cao, A.; Mercado, L.; Ramos-Martinez, J.I.; Barcia, R. Primary Cultures of Hemocytes from Mytilus Galloprovincialis Lmk.: Expression of IL-2Rα Subunit. Aquaculture 2003, 216, 1–8. [Google Scholar] [CrossRef]
- Abdel-Mageed, H.M.; AbuelEzz, N.Z.; Radwan, R.A.; Mohamed, S.A. Nanoparticles in Nanomedicine: A Comprehensive Updated Review on Current Status, Challenges and Emerging Opportunities. J. Microencapsul. 2021, 38, 414–436. [Google Scholar] [CrossRef] [PubMed]
- Joris, F.; Manshian, B.B.; Peynshaert, K.; de Smedt, S.C.; Braeckmans, K.; Soenen, S.J. Assessing Nanoparticle Toxicity in Cell-Based Assays: Influence of Cell Culture Parameters and Optimized Models for Bridging the in Vitro–in Vivo Gap. Chem. Soc. Rev. 2013, 42, 8339–8359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senior, N.J.; Titball, R.W. Isolation and Primary Culture of Galleria mellonella Hemocytes for Infection Studies. F1000Res 2020, 9, 1392. [Google Scholar] [CrossRef]
- Oers, M.M.; Lynn, D.E. Insect Cell Culture. Encycl. Life Sci. 2010, 1–5. [Google Scholar] [CrossRef]
- Savas, I. A New Approach for Classification of Major Larval Hemocytes (Prohemocytes, Plasmatocytes and Granulocytes) in the Greater Wax Moth, Galleria mellonella L. (Lepidoptera: Pyralidae) by Acridine Orange Staining. Turk. J. Entomol. 2012, 36, 163–168. [Google Scholar]
- Boguś, M.I.; Wieloch, W.; Ligeȩza-Zuber, M. Coronatin-2 from the Entomopathogenic Fungus Conidiobolus Coronatus Kills Galleria mellonella Larvae and Incapacitates Hemocytes. Bull. Entomol. Res. 2017, 107, 66–76. [Google Scholar] [CrossRef]
- Duressa, T.F.; Huybrechts, R. Development of Primary Cell Cultures Using Hemocytes and Phagocytic Tissue Cells of Locusta Migratoria: An Application for Locust Immunity Studies. In Vitro Cell. Dev. Biol.-Anim. 2016, 52, 100–106. [Google Scholar] [CrossRef]
- Thansa, K.; Kruangkum, T.; Pudgerd, A.; Chaichandee, L.; Amparyup, P.; Suebsing, R.; Chotwiwatthanakun, C.; Vanichviriyakit, R.; Sritunyalucksana, K. Establishment of Hematopoietic Tissue Primary Cell Cultures from the Giant Freshwater Prawn Macrobrachium Rosenbergii. Cytotechnology 2021, 73, 141. [Google Scholar] [CrossRef]
- Dantas-Lima, J.J.; Corteel, M.; Oanh, D.T.H.; Bossier, P.; Sorgeloos, P.; Nauwynck, H.J. Development of Two Haemocyte Culture Systems (in Attachment and in Suspension) for Shrimp Immunity Studies. Aquaculture 2012, 366–367, 17–26. [Google Scholar] [CrossRef]
- Ratcliffe, N.A.; Rowley, A.F. Cellular Defense Reactions of Insect Hemocytes In Vitro: Phagocytosis in a New Suspension Culture System. J. Invertebr. Pathol. 1975, 26, 225–233. [Google Scholar] [CrossRef]
- Li, F.; Zheng, Z.; Li, H.; Fu, R.; Xu, L.; Yang, F. Crayfish Hemocytes Develop along the Granular Cell Lineage. Sci. Rep. 2021, 11, 13099. [Google Scholar] [CrossRef] [PubMed]
- Nga, N.T.H.; Ngoc, T.T.B.; Trinh, N.T.M.; Thuoc, T.L.; Thao, D.T.P. Optimization and Application of MTT Assay in Determining Density of Suspension Cells. Anal. Biochem. 2020, 610, 113937. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; McCanna, D.J.; Sivak, J.G. Use of the Viability Reagent PrestoBlue in Comparison with AlamarBlue and MTT to Assess the Viability of Human Corneal Epithelial Cells. J. Pharmacol. Toxicol. Methods 2015, 71, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Boulos, L.; Prévost, M.; Barbeau, B.; Coallier, J.; Desjardins, R. LIVE/DEAD BacLight: Application of a New Rapid Staining Method for Direct Enumeration of Viable and Total Bacteria in Drinking Water. J. Microbiol. Methods 1999, 37, 77–86. [Google Scholar] [CrossRef]
- Ratcliffe, N.A.; Mead, G.P.; Renwrantz, L.R. Insect Haemocyte Separation—An Essential Prerequisite to Progress in Understanding Insect Cellular Immunity. In Immunity in Invertebrates; Springer: Berlin/Heidelberg, Germany, 1986; pp. 3–11. [Google Scholar]
- Vargas-Albores, F.; Guzmin, M.A.; Ochoa, J.L. An Anticoagulant Solution for Haemolymph Collection and Prophenoloxidase Studies of Penaeid Shrimp (Penaeus Californiensis). Comp. Biochem. Physiol. 1993, 106, 299–303. [Google Scholar] [CrossRef]
- Söderhäll, K.; Smith, V.J. Separation of the Haemocyte Populations of Carcinus Maenas and Other Marine Decapods, and Prophenoloxidase Distribution. Dev. Comp. Immunol. 1983, 7, 229–239. [Google Scholar] [CrossRef]
- Blanco-Cabra, N.; Movellan, J.; Marradi, M.; Gracia, R.; Salvador, C.; Dupin, D.; Loinaz, I.; Torrents, E. Neutralization of Ionic Interactions by Dextran-Based Single-Chain Nanoparticles Improves Tobramycin Diffusion into a Mature Biofilm. Npj Biofilms Microbiomes 2022, 8, 52. [Google Scholar] [CrossRef]
- Trevijano-Contador, N.; Zaragoza, O. Immune Response of Galleria mellonella against Human Fungal Pathogens. J. Fungi 2019, 5, 3. [Google Scholar] [CrossRef] [Green Version]
- Jung, J.; Sajjadian, S.M.; Kim, Y. Hemolin, an Immunoglobulin-like Peptide, Opsonizes Nonself Targets for Phagocytosis and Encapsulation in Spodoptera Exigua, a Lepidopteran Insect. J. Asia Pac. Entomol. 2019, 22, 947–956. [Google Scholar] [CrossRef]
- Eleftherianos, I.; Gökçen, F.; Felföldi, G.; Millichap, P.J.; Trenczek, T.E.; Ffrench-constant, R.H.; Reynolds, S.E. The Immunoglobulin Family Protein Hemolin Mediates Cellular Immune Responses to Bacteria in the Insect Manduca Sexta. Cell. Microbiol 2007, 9, 1137–1147. [Google Scholar] [CrossRef] [PubMed]
- Asai, M.; Sheehan, G.; Li, Y.; Robertson, B.D.; Kavanagh, K.; Langford, P.R.; Newton, S.M. Innate Immune Responses of Galleria mellonella to Mycobacterium Bovis BCG Challenge Identified Using Proteomic and Molecular Approaches. Front. Cell. Infect. Microbiol. 2021, 11, 7. [Google Scholar] [CrossRef] [PubMed]
- de Barros, P.P.; Rossoni, R.D.; Ribeiro, F.D.C.; Silva, M.P.; de Souza, C.M.; Jorge, A.O.C.; Junqueira, J.C. Two Sporulated Bacillus Enhance Immunity in Galleria mellonella Protecting against Candida Albicans. Microb. Pathog. 2019, 132, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.X.; Zhong, X.; Yi, H.Y.; Yu, X.Q. Manduca Sexta Gloverin Binds Microbial Components and Is Active against Bacteria and Fungi. Dev. Comp. Immunol. 2012, 38, 275. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Sun, Z.; Ma, S.; Liu, X.; Xia, H.; Chen, K. Molecular Mechanism and Potential Application of Bacterial Infection in the Silkworm, Bombyx Mori. Dev. Comp. Immunol. 2022, 131, 104381. [Google Scholar] [CrossRef] [PubMed]
- Eleftherianos, I.; Felföldi, G.; Ffrench-Constant, R.H.; Reynolds, S.E. Induced Nitric Oxide Synthesis in the Gut of Manduca Sexta Protects against Oral Infection by the Bacterial Pathogen Photorhabdus luminescens. Insect. Mol. Biol. 2009, 18, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Vogel, H.; Altincicek, B.; Glöckner, G.; Vilcinskas, A. A Comprehensive Transcriptome and Immune-Gene Repertoire of the Lepidopteran Model Host Galleria mellonella. BMC Genom. 2011, 12, 308. [Google Scholar] [CrossRef] [Green Version]
- Tripathy, A.; Mohanty, A.K.; Kar, S.K. Role of Glutathione S-Transferase (GST) in Mosquito Physiology and Anti-Plasmodial Activity. Int. J. Sci. Res. Publ. 2016, 6, 593. [Google Scholar]
- Dubovskiy, I.M.; Martemyanov, V.V.; Vorontsova, Y.L.; Rantala, M.J.; Gryzanova, E.V.; Glupov, V.V. Effect of Bacterial Infection on Antioxidant Activity and Lipid Peroxidation in the Midgut of Galleria mellonella L. Larvae (Lepidoptera, Pyralidae). Comp. Biochem. Physiol.-C Toxicol. Pharmacol. 2008, 148, 1–5. [Google Scholar] [CrossRef]
- Kaczmarek, A.; Wrońska, A.K.; Kazek, M.; Boguś, M.I. Octanoic Acid-An Insecticidal Metabolite of Conidiobolus Coronatus (Entomopthorales) That Affects Two Majors Antifungal Protection Systems in Galleria mellonella (Lepidoptera): Cuticular Lipids and Hemocytes. Int. J. Mol. Sci. 2022, 23, 5204. [Google Scholar] [CrossRef]
- Campo-Pérez, V.; Cendra, M.D.M.; Julián, E.; Torrents, E. Easily Applicable Modifications to Electroporation Conditions Improve the Transformation Efficiency Rates for Rough Morphotypes of Fast-Growing Mycobacteria. New Biotechnol. 2021, 63, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Crespo, A.; Blanco-Cabra, N.; Torrents, E. Aerobic Vitamin B12 Biosynthesis Is Essential for Pseudomonas Aeruginosa Class II Ribonucleotide Reductase Activity during Planktonic and Biofilm Growth. Front. Microbiol. 2018, 9, 986. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Medium | Fetal Bovine Serum (FBS) | Temperature | CO2 | Day 1 Cell Viability | |
|---|---|---|---|---|---|
| 1 | DMEM | 10% (v/v) | 37 °C | Yes | 0% |
| 2 | DMEM | 10% (v/v) | 25 °C | No | <10% |
| 3 | Grace | 10% (v/v) | 37 °C | Yes | <5% |
| 4 | Grace | 15% (v/v) | 37 °C | Yes | <5% |
| 5 | Grace | 20% (v/v) | 37 °C | Yes | <5% |
| 6 | Grace | 10% (v/v) | 37 °C | No | <5% |
| 7 | Grace | 10% (v/v) | 25 °C | No | ≈15% |
| 8 | Grace | 15% (v/v) | 25 °C | No | ≈15% |
| 9 | Grace | 20% (v/v) | 25 °C | No | ≈15% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Admella, J.; Torrents, E. A Straightforward Method for the Isolation and Cultivation of Galleria mellonella Hemocytes. Int. J. Mol. Sci. 2022, 23, 13483. https://doi.org/10.3390/ijms232113483
Admella J, Torrents E. A Straightforward Method for the Isolation and Cultivation of Galleria mellonella Hemocytes. International Journal of Molecular Sciences. 2022; 23(21):13483. https://doi.org/10.3390/ijms232113483
Chicago/Turabian StyleAdmella, Joana, and Eduard Torrents. 2022. "A Straightforward Method for the Isolation and Cultivation of Galleria mellonella Hemocytes" International Journal of Molecular Sciences 23, no. 21: 13483. https://doi.org/10.3390/ijms232113483
APA StyleAdmella, J., & Torrents, E. (2022). A Straightforward Method for the Isolation and Cultivation of Galleria mellonella Hemocytes. International Journal of Molecular Sciences, 23(21), 13483. https://doi.org/10.3390/ijms232113483