
Neurodegenerative Disorder Risk in Krabbe Disease Carriers

 , , , and
, , , and
Abstract
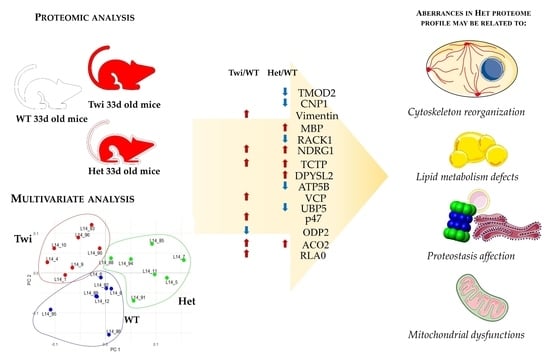
:
1. Introduction
2. Results and Discussion
2.1. Cytoskeleton (Re)Organization and Its Possible Roles in Myelination Dynamics of KD Carriers
2.2. Lipid Metabolism Defects in Krabbe Disease
2.3. Proteostasis Affection in Het Mice
2.4. Mitochondrial and Energy Supply Dysfunctions
3. Materials and Methods
3.1. Animal Model
3.2. Whole Brain Preparation
3.3. Proteomic Analyses
3.4. Statistical Analysis
3.5. Protein Identification by MALDI TOF-TOF Mass Spectrometry
3.6. Western Blot Analysis
3.7. Enrichment Analysis by MetaCore™ Software
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
Abbreviations
| %Vol | Percentage relative volume |
| 2DE | Two-dimensional electrophoresis |
| 2D | Two-dimensional |
| ACN | Acetonitrile |
| ACO2 | Mitochondrial aconitase |
| AD | Alzheimer disease |
| ALS | Amyotrophic lateral sclerosis |
| AP-1 | Activator protein 1 |
| APP | Amyloid-beta precursor protein |
| ATPB | ATP synthase subunit beta, mitochondrial |
| BMP | Bone morphogenetic protein |
| CHAPS | 3-[(3-cholamidopropyl) dimethylammonio]-1-propanesulfonate |
| CHCA | α-cyano-4-hydroxycinnamic acid |
| CNP | 2′,3′-cyclic-nucleotide 3′-phosphodiesterase |
| CNS | Central nervous system |
| DTE | 1,4-Dithioerythritol |
| DPYSL2 | Dihydropyrimidinase-Related Protein 2 |
| DUB | Deubiquitinase |
| ER | Endoplasmic reticulum |
| ERAD | Endoplasmic reticulum-associated degradation |
| FC | Fold change |
| GALC | Galactosylceramidase |
| GalCer | Galactosylceramide |
| GFAP | Glial fibrillary acidic protein |
| GWAS | Genome-wide association studies |
| HIF-1A | Hypoxia-inducible factor 1-alpha |
| Het | Heterozygous mice (galc +/−) |
| HRM-PCR | High resolution melting polymerase chain reaction |
| IBMPFD | Inclusion body myopathy with early-onset Paget disease and frontotemporal dementia 1 |
| IEF | Isoelectrofocusing |
| IF | Intermediate filament |
| KD | Krabbe disease |
| LacCer | Lactosylceramide |
| LB | Lewy body |
| LSD | Lysosomal storage disease |
| MALDI | Matrix assisted laser desorption/ionization |
| MAP | Microtubule-associated-protein |
| MBP | Myelin basic protein |
| NDRG1 | N-myc downstream regulated 1 |
| NS | Nervous system |
| OS | Oxidative stress |
| p47 | NSFL1 cofactor p47 |
| PBS | Phosphate buffer saline |
| PC1 | First principal component |
| PC2 | Second principal component |
| PC3 | Third principal component |
| PCA | Principal component analysis |
| PCs | Principal components |
| PD | Parkinson disease |
| PDC | Pyruvate dehydrogenase complex |
| PHS | Public health service |
| PKC | Protein kinase C |
| PFE | Plasma fibrinogen extravasation |
| PMF | Peptide mass fingerprinting |
| PN | Proteostasis network |
| PNS | Peripheral nervous system |
| PP2A | Protein phosphatase 2A |
| Psy | Psychosine |
| PwMS | People with multiple sclerosis |
| RACK1 | Receptor of activated protein c kinase 1 |
| SDS | Sodium dodecyl sulfate |
| SPN | Shortest-path-network |
| STAT1 | Activator of transcription-1 |
| STAT3 | Activator of transcription-3 |
| TCA | Tricarboxylic acid |
| TCTP | Translationally-controlled tumor protein |
| TOF | Time of flight |
| TW | Homozygous twitcher mice (galc −/−) |
| UCHL5 | Ubiquitin carboxyl-terminal hydrolase 5 |
| UPS | Ubiquitination proteasome system |
| VCP | Transitional endoplasmic reticulum ATPase; alternative name: valosin-containing protein |
| VIME | Vimentin |
| WB | Western blot |
| WT | Homozygous wild-type mice (galc +/+) |
References
- Komatsuzaki, S.; Zielonka, M.; Mountford, W.K.; Kölker, S.; Hoffmann, G.F.; Garbade, S.F.; Ries, M. Clinical Characteristics of 248 Patients with Krabbe Disease: Quantitative Natural History Modeling Based on Published Cases. Genet. Med. 2019, 21, 2208–2215. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K. Globoid Cell Leukodystrophy (Krabbe’s Disease): Update. J. Child Neurol. 2003, 18, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Ryckman, A.E.; Brockhausen, I.; Walia, J.S. Metabolism of Glycosphingolipids and Their Role in the Pathophysiology of Lysosomal Storage Disorders. Int. J. Mol. Sci. 2020, 21, 6881. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, S.; Castelvetri, L.C.; Simons, M. Metabolism and Functions of Lipids in Myelin. Biochim. Biophys. Acta 2015, 1851, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Giussani, P.; Prinetti, A.; Tringali, C. The Role of Sphingolipids in Myelination and Myelin Stability and Their Involvement in Childhood and Adult Demyelinating Disorders. J. Neurochem. 2021, 156, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Reza, S.; Ugorski, M.; Suchański, J. Glucosylceramide and Galactosylceramide, Small Glycosphingolipids with Significant Impact on Health and Disease. Glycobiology 2021, 31, 1416–1434. [Google Scholar] [CrossRef] [PubMed]
- Potter, G.B.; Petryniak, M.A. Neuroimmune Mechanisms in Krabbe’s Disease. J. Neurosci. Res. 2016, 94, 1341–1348. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Xu, Y.; Benitez, B.A.; Nagree, M.S.; Dearborn, J.T.; Jiang, X.; Guzman, M.A.; Woloszynek, J.C.; Giaramita, A.; Yip, B.K.; et al. Genetic Ablation of Acid Ceramidase in Krabbe Disease Confirms the Psychosine Hypothesis and Identifies a New Therapeutic Target. Proc. Natl. Acad. Sci. USA 2019, 116, 20097–20103. [Google Scholar] [CrossRef] [Green Version]
- Papini, N.; Giallanza, C.; Brioschi, L.; Ranieri, F.R.; Giussani, P.; Mauri, L.; Ciampa, M.G.; Viani, P.; Tringali, C. Galactocerebrosidase Deficiency Induces an Increase in Lactosylceramide Content: A New Hallmark of Krabbe Disease? Int. J. Biochem. Cell Biol. 2022, 145, 106184. [Google Scholar] [CrossRef]
- Lim, S.M.; Choi, B.-O.; Oh, S.-I.; Choi, W.J.; Oh, K.-W.; Nahm, M.; Xue, Y.; Choi, J.H.; Choi, J.Y.; Kim, Y.-E.; et al. Patient Fibroblasts-Derived Induced Neurons Demonstrate Autonomous Neuronal Defects in Adult-Onset Krabbe Disease. Oncotarget 2016, 7, 74496–74509. [Google Scholar] [CrossRef]
- Kreher, C.; Favret, J.; Weinstock, N.I.; Maulik, M.; Hong, X.; Gelb, M.H.; Wrabetz, L.; Feltri, M.L.; Shin, D. Neuron-Specific Ablation of the Krabbe Disease Gene Galactosylceramidase in Mice Results in Neurodegeneration. PLoS Biol. 2022, 20, e3001661. [Google Scholar] [CrossRef] [PubMed]
- Weinstock, N.I.; Kreher, C.; Favret, J.; Nguyen, D.; Bongarzone, E.R.; Wrabetz, L.; Feltri, M.L.; Shin, D. Brainstem Development Requires Galactosylceramidase and Is Critical for Pathogenesis in a Model of Krabbe Disease. Nat. Commun. 2020, 11, 5356. [Google Scholar] [CrossRef] [PubMed]
- Sahai, I.; Baris, H.; Kimonis, V.; Levy, H.L. Krabbe Disease: Severe Neonatal Presentation with a Family History of Multiple Sclerosis. J. Child Neurol. 2005, 20, 826–828. [Google Scholar] [CrossRef] [PubMed]
- Sawcer, S.; Hellenthal, G.; Pirinen, M.; Spencer, C.C.A.; Patsopoulos, N.A.; Moutsianas, L.; Dilthey, A.; Su, Z.; Freeman, C.; Hunt, S.E.; et al. Genetic Risk and a Primary Role for Cell-Mediated Immune Mechanisms in Multiple Sclerosis. Nature 2011, 476, 214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Hou, X.; Liang, Y.; Xu, F.; Zhang, X.; Cui, P.; Xing, G.; Wang, X.; Jiang, W. Gene-Based Tests of a Genome-Wide Association Study Dataset Highlight Novel Multiple Sclerosis Risk Genes. Front. Neurosci. 2021, 15, 614528. [Google Scholar] [CrossRef] [PubMed]
- Scott-Hewitt, N.J.; Folts, C.J.; Noble, M.D. Heterozygous Carriers of Galactocerebrosidase Mutations That Cause Krabbe Disease Have Impaired Microglial Function and Defective Repair of Myelin Damage. Neural. Regen. Res. 2018, 13, 393–401. [Google Scholar] [CrossRef]
- Abdelkarim, H.; Marshall, M.S.; Scesa, G.; Smith, R.A.; Rue, E.; Marshall, J.; Elackattu, V.; Stoskute, M.; Issa, Y.; Santos, M.; et al. α-Synuclein Interacts Directly but Reversibly with Psychosine: Implications for α-Synucleinopathies. Sci. Rep. 2018, 8, 12462. [Google Scholar] [CrossRef] [Green Version]
- Smith, B.R.; Santos, M.B.; Marshall, M.S.; Cantuti-Castelvetri, L.; Lopez-Rosas, A.; Li, G.; van Breemen, R.; Claycomb, K.I.; Gallea, J.I.; Celej, M.S.; et al. Neuronal Inclusions of α-Synuclein Contribute to the Pathogenesis of Krabbe Disease. J. Pathol. 2014, 232, 509–521. [Google Scholar] [CrossRef] [Green Version]
- Nasir, G.; Chopra, R.; Elwood, F.; Ahmed, S.S. Krabbe Disease: Prospects of Finding a Cure Using AAV Gene Therapy. Front. Med. 2021, 8, 760236. [Google Scholar] [CrossRef]
- Hult, M.; Darin, N.; von Döbeln, U.; Månsson, J.-E. Epidemiology of Lysosomal Storage Diseases in Sweden. Acta Paediatr. 2014, 103, 1258–1263. [Google Scholar] [CrossRef]
- Laquerrière, A.; Bekri, S.; Suzuki, K.; Harding, B.N. Sphingolipidoses and Related Disorders. In Developmental Neuropathology; Wiley Online Library: Hoboken, NJ, USA, 2018; pp. 313–367. ISBN 978-1-119-01311-2. [Google Scholar]
- Liu, Y.; Gibson, J.; Wheeler, J.; Kwee, L.C.; Santiago-Turla, C.M.; Akafo, S.K.; Lichter, P.R.; Gaasterland, D.E.; Moroi, S.E.; Challa, P.; et al. GALC Deletions Increase the Risk of Primary Open-Angle Glaucoma: The Role of Mendelian Variants in Complex Disease. PLoS ONE 2011, 6, e27134. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Cho, M.H.; Hersh, C.P.; McDonald, M.-L.N.; Wells, J.M.; Dransfield, M.T.; Bowler, R.P.; Lynch, D.A.; Lomas, D.A.; Crapo, J.D.; et al. IREB2 and GALC Are Associated with Pulmonary Artery Enlargement in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Cell Mol. Biol. 2015, 52, 365–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heller, G.J.; Marshall, M.S.; Issa, Y.; Marshall, J.N.; Nguyen, D.; Rue, E.; Pathmasiri, K.C.; Domowicz, M.S.; van Breemen, R.B.; Tai, L.M.; et al. Waning Efficacy in a Long-Term AAV-Mediated Gene Therapy Study in the Murine Model of Krabbe Disease. Mol. Ther. 2021, 29, 1883–1902. [Google Scholar] [CrossRef] [PubMed]
- Signorini, C.; Cardile, V.; Pannuzzo, G.; Graziano, A.C.E.; Durand, T.; Galano, J.-M.; Oger, C.; Leoncini, S.; Cortelazzo, A.; Lee, J.C.-Y.; et al. Increased Isoprostanoid Levels in Brain from Murine Model of Krabbe Disease-Relevance of Isoprostanes, Dihomo-Isoprostanes and Neuroprostanes to Disease Severity. Free Radic. Biol. Med. 2019, 139, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Taniike, M. Murine Model of Genetic Demyelinating Disease: The Twitcher Mouse. Microsc. Res. Tech. 1995, 32, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Kodama, S.; Igisu, H.; Siegel, D.A.; Suzuki, K. Glycosylceramide Synthesis in the Developing Spinal Cord and Kidney of the Twitcher Mouse, an Enzymatically Authentic Model of Human Krabbe Disease. J. Neurochem. 1982, 39, 1314–1318. [Google Scholar] [CrossRef]
- Marshall, M.S.; Bongarzone, E.R. Beyond Krabbe’s Disease: The Potential Contribution of Galactosylceramidase Deficiency to Neuronal Vulnerability in Late-Onset Synucleinopathies. J. Neurosci. Res. 2016, 94, 1328–1332. [Google Scholar] [CrossRef] [Green Version]
- Chiu, F.C.; Sacchi, R.S.; Claudio, L.; Kobayashi, S.; Suzuki, K. Coexpression of Glial Fibrillary Acidic Protein and Vimentin in the Central and Peripheral Nervous Systems of the Twitcher Mutant. Glia 1988, 1, 105–112. [Google Scholar] [CrossRef]
- Rama Rao, K.V.; Kielian, T. Astrocytes and Lysosomal Storage Diseases. Neuroscience 2016, 323, 195–206. [Google Scholar] [CrossRef] [Green Version]
- Hol, E.M.; Pekny, M. Glial Fibrillary Acidic Protein (GFAP) and the Astrocyte Intermediate Filament System in Diseases of the Central Nervous System. Curr. Opin. Cell Biol. 2015, 32, 121–130. [Google Scholar] [CrossRef]
- Landi, C.; Luddi, A.; Bianchi, L.; Pannuzzo, G.; Pavone, V.; Piomboni, P.; Bini, L. Proteostasis Network Alteration in Lysosomal Storage Disorders: Insights from the Mouse Model of Krabbe Disease. J. Neurosci. Res. 2020, 98, 718–733. [Google Scholar] [CrossRef] [PubMed]
- Saffari, A.; Kölker, S.; Hoffmann, G.F.; Ebrahimi-Fakhari, D. Linking Mitochondrial Dysfunction to Neurodegeneration in Lysosomal Storage Diseases. J. Inherit. Metab. Dis. 2017, 40, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Benitez-King, G.; Ramírez-Rodríguez, G.; Ortíz, L.; Meza, I. The Neuronal Cytoskeleton as a Potential Therapeutical Target in Neurodegenerative Diseases and Schizophrenia. Curr. Drug. Targets CNS Neurol. Disord. 2004, 3, 515–533. [Google Scholar] [CrossRef] [PubMed]
- Kounakis, K.; Tavernarakis, N. The Cytoskeleton as a Modulator of Aging and Neurodegeneration. Adv. Exp. Med. Biol. 2019, 1178, 227–245. [Google Scholar] [CrossRef]
- Peter, F.; Rost, S.; Rolfs, A.; Frech, M.J. Activation of PKC Triggers Rescue of NPC1 Patient Specific IPSC Derived Glial Cells from Gliosis. Orphanet. J. Rare Dis. 2017, 12, 145. [Google Scholar] [CrossRef]
- Bauer, P.O.; Hudec, R.; Goswami, A.; Kurosawa, M.; Matsumoto, G.; Mikoshiba, K.; Nukina, N. ROCK-Phosphorylated Vimentin Modifies Mutant Huntingtin Aggregation via Sequestration of IRBIT. Mol. Neurodegener. 2012, 7, 43. [Google Scholar] [CrossRef] [Green Version]
- Jang, B.; Kim, M.J.; Lee, Y.J.; Ishigami, A.; Kim, Y.S.; Choi, E.K. Vimentin Citrullination Probed by a Novel Monoclonal Antibody Serves as a Specific Indicator for Reactive Astrocytes in Neurodegeneration. Neuropathol. Appl. Neurobiol. 2020, 46, 751–769. [Google Scholar] [CrossRef]
- Feuillette, S.; Deramecourt, V.; Laquerriere, A.; Duyckaerts, C.; Delisle, M.-B.; Maurage, C.-A.; Blum, D.; Buée, L.; Frébourg, T.; Campion, D.; et al. Filamin-A and Myosin VI Colocalize with Fibrillary Tau Protein in Alzheimer’s Disease and FTDP-17 Brains. Brain Res. 2010, 1345, 182–189. [Google Scholar] [CrossRef]
- Tsujikawa, K.; Hamanaka, K.; Riku, Y.; Hattori, Y.; Hara, N.; Iguchi, Y.; Ishigaki, S.; Hashizume, A.; Miyatake, S.; Mitsuhashi, S.; et al. Actin-Binding Protein Filamin-A Drives Tau Aggregation and Contributes to Progressive Supranuclear Palsy Pathology. Sci. Adv. 2022, 8, eabm5029. [Google Scholar] [CrossRef]
- Zaka, M.; Rafi, M.A.; Rao, H.Z.; Luzi, P.; Wenger, D.A. Insulin-like Growth Factor-1 Provides Protection against Psychosine-Induced Apoptosis in Cultured Mouse Oligodendrocyte Progenitor Cells Using Primarily the PI3K/Akt Pathway. Mol. Cell. Neurosci. 2005, 30, 398–407. [Google Scholar] [CrossRef]
- Wan, H.-L.; Hong, X.-Y.; Zhao, Z.-H.; Li, T.; Zhang, B.-G.; Liu, Q.; Wang, Q.; Zhao, S.; Wang, J.-Z.; Shen, X.-F.; et al. STAT3 Ameliorates Cognitive Deficits via Regulation of NMDAR Expression in an Alzheimer’s Disease Animal Model. Theranostics 2021, 11, 5511–5524. [Google Scholar] [CrossRef] [PubMed]
- Czubowicz, K.; Jęśko, H.; Wencel, P.; Lukiw, W.J.; Strosznajder, R.P. The Role of Ceramide and Sphingosine-1-Phosphate in Alzheimer’s Disease and Other Neurodegenerative Disorders. Mol. Neurobiol. 2019, 56, 5436–5455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herdegen, T.; Waetzig, V. AP-1 Proteins in the Adult Brain: Facts and Fiction about Effectors of Neuroprotection and Neurodegeneration. Oncogene 2001, 20, 2424–2437. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Yan, J.; Chang, Y.; Yan, S.S.; Shi, H. Hypoxia Inducible Factor-1 as a Target for Neurodegenerative Diseases. Curr. Med. Chem. 2011, 18, 4335–4343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Yu, L.; Xu, Y.; Tang, X.; Wang, X. Substantia Nigra Smad3 Signaling Deficiency: Relevance to Aging and Parkinson’s Disease and Roles of Microglia, Proinflammatory Factors, and MAPK. J. Neuroinflamm. 2020, 17, 342. [Google Scholar] [CrossRef]
- Kaleli, H.N.; Ozer, E.; Kaya, V.O.; Kutlu, O. Protein Kinase C Isozymes and Autophagy during Neurodegenerative Disease Progression. Cells 2020, 9, 553. [Google Scholar] [CrossRef] [Green Version]
- Rai, S.N.; Dilnashin, H.; Birla, H.; Singh, S.S.; Zahra, W.; Rathore, A.S.; Singh, B.K.; Singh, S.P. The Role of PI3K/Akt and ERK in Neurodegenerative Disorders. Neurotox. Res. 2019, 35, 775–795. [Google Scholar] [CrossRef]
- Torrent, L.; Ferrer, I. PP2A and Alzheimer Disease. Curr. Alzheimer Res. 2012, 9, 248–256. [Google Scholar] [CrossRef]
- Ma, J.; Wu, R.; Zhang, Q.; Wu, J.; Lou, J.; Zheng, Z.; Ding, J.; Yuan, Z. DJ-1 Interacts with RACK1 and Protects Neurons from Oxidative-Stress-Induced Apoptosis. Biochem. J. 2014, 462, 489–497. [Google Scholar] [CrossRef]
- Sklan, E.H.; Podoly, E.; Soreq, H. RACK1 Has the Nerve to Act: Structure Meets Function in the Nervous System. Prog Neurobiol 2006, 78, 117–134. [Google Scholar] [CrossRef]
- Osmanagic-Myers, S.; Wiche, G. Plectin-RACK1 (Receptor for Activated C Kinase 1) Scaffolding: A Novel Mechanism to Regulate Protein Kinase C Activity. J. Biol. Chem. 2004, 279, 18701–18710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCahill, A.; Warwicker, J.; Bolger, G.B.; Houslay, M.D.; Yarwood, S.J. The RACK1 Scaffold Protein: A Dynamic Cog in Cell Response Mechanisms. Mol. Pharmacol. 2002, 62, 1261–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Battaini, F.; Pascale, A.; Paoletti, R.; Govoni, S. The Role of Anchoring Protein RACK1 in PKC Activation in the Ageing Rat Brain. Trends Neurosci. 1997, 20, 410–415. [Google Scholar] [CrossRef]
- Kishimoto, A.; Nishiyama, K.; Nakanishi, H.; Uratsuji, Y.; Nomura, H.; Takeyama, Y.; Nishizuka, Y. Studies on the Phosphorylation of Myelin Basic Protein by Protein Kinase C and Adenosine 3′:5′-Monophosphate-Dependent Protein Kinase. J. Biol. Chem. 1985, 260, 12492–12499. [Google Scholar] [CrossRef]
- Whitaker, J.N. Myelin Basic Protein and Multiple Sclerosis. Ital. J. Neurol. Sci. 1983, 4, 153–157. [Google Scholar] [CrossRef]
- Gaboreanu, A.-M.; Hrstka, R.; Xu, W.; Shy, M.; Kamholz, J.; Lilien, J.; Balsamo, J. Myelin Protein Zero/P0 Phosphorylation and Function Require an Adaptor Protein Linking It to RACK1 and PKC Alpha. J. Cell Biol. 2007, 177, 707–716. [Google Scholar] [CrossRef] [Green Version]
- Tomás, J.; Durães, J.; Lacerda, L.; Macário, M.C. Adolescent-Onset Krabbe Disease with an Initial Diagnosis of Multiple Sclerosis and a Novel Mutation. BMJ Case Rep. 2015, 2015, bcr2015210625. [Google Scholar] [CrossRef] [Green Version]
- Vassall, K.A.; Bamm, V.V.; Harauz, G. MyelStones: The Executive Roles of Myelin Basic Protein in Myelin Assembly and Destabilization in Multiple Sclerosis. Biochem. J. 2015, 472, 17–32. [Google Scholar] [CrossRef]
- Smirnova, E.V.; Rakitina, T.V.; Ziganshin, R.H.; Arapidi, G.P.; Saratov, G.A.; Kudriaeva, A.A.; Belogurov, A.A. Comprehensive Atlas of the Myelin Basic Protein Interaction Landscape. Biomolecules 2021, 11, 1628. [Google Scholar] [CrossRef]
- Harauz, G.; Ladizhansky, V.; Boggs, J.M. Structural Polymorphism and Multifunctionality of Myelin Basic Protein. Biochemistry 2009, 48, 8094–8104. [Google Scholar] [CrossRef]
- GS Chirivi, R. Citrullination: A Target for Disease Intervention in Multiple Sclerosis and Other Inflammatory Diseases? J. Clin. Cell Immunol. 2013, 4, 146. [Google Scholar] [CrossRef]
- Moscarello, M.A.; Mastronardi, F.G.; Wood, D.D. The Role of Citrullinated Proteins Suggests a Novel Mechanism in the Pathogenesis of Multiple Sclerosis. Neurochem. Res. 2007, 32, 251–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harauz, G.; Musse, A.A. A Tale of Two Citrullines—Structural and Functional Aspects of Myelin Basic Protein Deimination in Health and Disease. Neurochem. Res. 2007, 32, 137–158. [Google Scholar] [CrossRef] [PubMed]
- Traina, G.; Federighi, G.; Macchi, M.; Bernardi, R.; Durante, M.; Brunelli, M. Modulation of Myelin Basic Protein Gene Expression by Acetyl-L-Carnitine. Mol. Neurobiol. 2011, 44, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Al-Abdi, L.; Al Murshedi, F.; Elmanzalawy, A.; Al Habsi, A.; Helaby, R.; Ganesh, A.; Ibrahim, N.; Patel, N.; Alkuraya, F.S. CNP Deficiency Causes Severe Hypomyelinating Leukodystrophy in Humans. Hum. Genet. 2020, 139, 615–622. [Google Scholar] [CrossRef]
- Mustapha, O.; Oke, B.; Offen, N.; Sirén, A.-L.; Olopade, J. Neurobehavioral and Cytotoxic Effects of Vanadium during Oligodendrocyte Maturation: A Protective Role for Erythropoietin. Environ. Toxicol. Pharmacol. 2014, 38, 98–111. [Google Scholar] [CrossRef]
- Disruption of Cnp1 Uncouples Oligodendroglial Functions in Axonal Support and Myelination|Nature Genetics. Available online: https://www.nature.com/articles/ng1095z (accessed on 15 June 2022).
- Jones, R.L.; Wong, E.K.; Ibsen, K.H.; Leopold, I.H. Erythrocyte Membrane 2′, 3′-Cyclic Nucleotide 3′-Phosphodiesterase Activity in Multiple Sclerosis. Metab. Pediatr. Syst. Ophthalmol. 1983, 7, 25–30. [Google Scholar]
- Muraro, P.A.; Kalbus, M.; Afshar, G.; McFarland, H.F.; Martin, R. T Cell Response to 2′,3′-Cyclic Nucleotide 3′-Phosphodiesterase (CNPase) in Multiple Sclerosis Patients. J. Neuroimmunol. 2002, 130, 233–242. [Google Scholar] [CrossRef]
- Mori, K.; Muto, Y.; Kokuzawa, J.; Yoshioka, T.; Yoshimura, S.; Iwama, T.; Okano, Y.; Sakai, N. Neuronal Protein NP25 Interacts with F-Actin. Neurosci. Res. 2004, 48, 439–446. [Google Scholar] [CrossRef]
- Ratié, L.; Ware, M.; Jagline, H.; David, V.; Dupé, V. Dynamic Expression of Notch-Dependent Neurogenic Markers in the Chick Embryonic Nervous System. Front. Neuroanat. 2014, 8, 158. [Google Scholar] [CrossRef]
- Arnaud, L.; Benech, P.; Greetham, L.; Stephan, D.; Jimenez, A.; Jullien, N.; García-González, L.; Tsvetkov, P.O.; Devred, F.; Sancho-Martinez, I.; et al. The Alzheimer’s Disease Risk Factor APOE4 Drives pro-Inflammation in Human Astrocytes via HDAC-Dependent Repression of TAGLN3. bioRxiv 2021. [Google Scholar] [CrossRef]
- Krugmann, B.; Radulescu, A.; Appavou, M.-S.; Koutsioubas, A.; Stingaciu, L.R.; Dulle, M.; Förster, S.; Stadler, A.M. Membrane Stiffness and Myelin Basic Protein Binding Strength as Molecular Origin of Multiple Sclerosis. Sci. Rep. 2020, 10, 16691. [Google Scholar] [CrossRef] [PubMed]
- Ellen, T.P.; Ke, Q.; Zhang, P.; Costa, M. NDRG1, a Growth and Cancer Related Gene: Regulation of Gene Expression and Function in Normal and Disease States. Carcinogenesis 2008, 29, 2–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.-T.; Ongusaha, P.P.; Hong, Y.-K.; Kurdistani, S.K.; Nakamura, M.; Lu, K.P.; Lee, S.W. Function of Drg1/Rit42 in P53-Dependent Mitotic Spindle Checkpoint. J. Biol. Chem. 2004, 279, 38597–38602. [Google Scholar] [CrossRef] [Green Version]
- Jeon, H.-J.; You, S.Y.; Park, Y.S.; Chang, J.W.; Kim, J.-S.; Oh, J.S. TCTP Regulates Spindle Microtubule Dynamics by Stabilizing Polar Microtubules during Mouse Oocyte Meiosis. Biochim. Et Biophys. Acta BBA-Mol. Cell Res. 2016, 1863, 630–637. [Google Scholar] [CrossRef]
- Schonkeren, S.L.; Massen, M.; van der Horst, R.; Koch, A.; Vaes, N.; Melotte, V. Nervous NDRGs: The N-Myc Downstream-Regulated Gene Family in the Central and Peripheral Nervous System. Neurogenetics 2019, 20, 173–186. [Google Scholar] [CrossRef] [Green Version]
- Okuda, T.; Higashi, Y.; Kokame, K.; Tanaka, C.; Kondoh, H.; Miyata, T. Ndrg1-Deficient Mice Exhibit a Progressive Demyelinating Disorder of Peripheral Nerves. Mol. Cell. Biol. 2004, 24, 3949–3956. [Google Scholar] [CrossRef] [Green Version]
- Marechal, D.; Dansu, D.K.; Castro, K.; Patzig, J.; Magri, L.; Inbar, B.; Gacias, M.; Moyon, S.; Casaccia, P. N-Myc Downstream Regulated Family Member 1 (NDRG1) Is Enriched in Myelinating Oligodendrocytes and Impacts Myelin Degradation in Response to Demyelination. Glia 2022, 70, 321–336. [Google Scholar] [CrossRef]
- Haq, E.; Giri, S.; Singh, I.; Singh, A.K. Molecular Mechanism of Psychosine-Induced Cell Death in Human Oligodendrocyte Cell Line. J. Neurochem. 2003, 86, 1428–1440. [Google Scholar] [CrossRef]
- March-Diaz, R.; Lara-Ureña, N.; Romero-Molina, C.; Heras-Garvin, A.; Ortega-de San Luis, C.; Alvarez-Vergara, M.I.; Sanchez-Garcia, M.A.; Sanchez-Mejias, E.; Davila, J.C.; Rosales-Nieves, A.E.; et al. Hypoxia Compromises the Mitochondrial Metabolism of Alzheimer’s Disease Microglia via HIF1. Nat. Aging. 2021, 1, 385–399. [Google Scholar] [CrossRef]
- Graziano, A.C.E.; Parenti, R.; Avola, R.; Cardile, V. Krabbe Disease: Involvement of Connexin43 in the Apoptotic Effects of Sphingolipid Psychosine on Mouse Oligodendrocyte Precursors. Apoptosis 2016, 21, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Hatami, M.; Salmani, T.; Arsang-Jang, S.; Davood Omrani, M.; Mazdeh, M.; Ghafouri-Fard, S.; Sayad, A.; Taheri, M. STAT5a and STAT6 Gene Expression Levels in Multiple Sclerosis Patients. Cytokine 2018, 106, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Giri, S.; Jatana, M.; Rattan, R.; Won, J.-S.; Singh, I.; Singh, A.K. Galactosylsphingosine (Psychosine)-Induced Expression of Cytokine-Mediated Inducible Nitric Oxide Synthases via AP-1 and C/EBP: Implications for Krabbe Disease. FASEB J. 2002, 16, 661–672. [Google Scholar] [CrossRef]
- Skedsmo, F.S.; Tranulis, M.A.; Espenes, A.; Prydz, K.; Matiasek, K.; Gunnes, G.; Hermansen, L.C.; Jäderlund, K.H. Cell and Context-Dependent Sorting of Neuropathy-Associated Protein NDRG1–Insights from Canine Tissues and Primary Schwann Cell Cultures. BMC Vet. Res. 2019, 15, 121. [Google Scholar] [CrossRef]
- MA, W.; NA, M.; TANG, C.; WANG, H.; LIN, Z. Overexpression of N-Myc Downstream-Regulated Gene 1 Inhibits Human Glioma Proliferation and Invasion via Phosphoinositide 3-Kinase/AKT Pathways. Mol. Med. Rep. 2015, 12, 1050–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, Z.; Purtell, K.; Lachance, V.; Wold, M.S.; Chen, S.; Yue, Z. Autophagy Receptors and Neurodegenerative Diseases. Trends Cell Biol. 2017, 27, 491–504. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.R.; Ghafouri, M.; Mukerjee, R.; Bagashev, A.; Chabrashvili, T.; Sawaya, B.E. Role of P53 in Neurodegenerative Diseases. Neurodegener. Dis. 2012, 9, 68–80. [Google Scholar] [CrossRef] [Green Version]
- Raivich, G.; Behrens, A. Role of the AP-1 Transcription Factor c-Jun in Developing, Adult and Injured Brain. Prog. Neurobiol. 2006, 78, 347–363. [Google Scholar] [CrossRef]
- Qin, L.; Shu, L.; Zhong, J.; Pan, H.; Guo, J.; Sun, Q.; Yan, X.; Tang, B.; Xu, Q. Association of HIF1A and Parkinson’s Disease in a Han Chinese Population Demonstrated by Molecular Inversion Probe Analysis. Neurol. Sci. 2019, 40, 1927–1931. [Google Scholar] [CrossRef]
- Luckhaus, C.; Sand, P.G. Estrogen Receptor 1 Gene (ESR1) Variants in Alzheimer’s Disease. Results of a Meta-Analysis. Aging Clin. Exp. Res. 2007, 19, 165–168. [Google Scholar] [CrossRef]
- Wetzel, M.K.; Naska, S.; Laliberté, C.L.; Rymar, V.V.; Fujitani, M.; Biernaskie, J.A.; Cole, C.J.; Lerch, J.P.; Spring, S.; Wang, S.-H.; et al. P73 Regulates Neurodegeneration and Phospho-Tau Accumulation during Aging and Alzheimer’s Disease. Neuron 2008, 59, 708–721. [Google Scholar] [CrossRef]
- Natarajan, C.; Sriram, S.; Muthian, G.; Bright, J.J. Signaling through JAK2-STAT5 Pathway Is Essential for IL-3-Induced Activation of Microglia. Glia 2004, 45, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Kokame, K.; Kato, H.; Miyata, T. Homocysteine-Respondent Genes in Vascular Endothelial Cells Identified by Differential Display Analysis. GRP78/BiP and Novel Genes. J. Biol. Chem. 1996, 271, 29659–29665. [Google Scholar] [CrossRef] [Green Version]
- Kurdistani, S.K.; Arizti, P.; Reimer, C.L.; Sugrue, M.M.; Aaronson, S.A.; Lee, S.W. Inhibition of Tumor Cell Growth by RTP/Rit42 and Its Responsiveness to P53 and DNA Damage. Cancer Res. 1998, 58, 4439–4444. [Google Scholar] [PubMed]
- Piquemal, D.; Joulia, D.; Balaguer, P.; Basset, A.; Marti, J.; Commes, T. Differential Expression of the RTP/Drg1/Ndr1 Gene Product in Proliferating and Growth Arrested Cells. Biochim. Biophys. Acta 1999, 1450, 364–373. [Google Scholar] [CrossRef] [Green Version]
- Hara, K.; Hasegawa, T.; Ooi, H.; Koya, T.; Tanabe, Y.; Tsukada, H.; Igarashi, K.; Suzuki, E.; Arakawa, M.; Gejyo, F. Inhibitory Role of Eosinophils on Cell Surface Plasmin Generation by Bronchial Epithelial Cells: Inhibitory Effects of Transforming Growth Factor β. Lung 2001, 179, 9–20. [Google Scholar] [CrossRef]
- van Belzen, N.; Dinjens, W.N.; Diesveld, M.P.; Groen, N.A.; van der Made, A.C.; Nozawa, Y.; Vlietstra, R.; Trapman, J.; Bosman, F.T. A Novel Gene Which Is Up-Regulated during Colon Epithelial Cell Differentiation and down-Regulated in Colorectal Neoplasms. Lab. Investig. 1997, 77, 85–92. [Google Scholar]
- King, R.H.M.; Chandler, D.; Lopaticki, S.; Huang, D.; Blake, J.; Muddle, J.R.; Kilpatrick, T.; Nourallah, M.; Miyata, T.; Okuda, T.; et al. Ndrg1 in Development and Maintenance of the Myelin Sheath. Neurobiol. Dis. 2011, 42, 368–380. [Google Scholar] [CrossRef]
- Mitchelmore, C.; Büchmann-Møller, S.; Rask, L.; West, M.J.; Troncoso, J.C.; Jensen, N.A. NDRG2: A Novel Alzheimer’s Disease Associated Protein. Neurobiol. Dis. 2004, 16, 48–58. [Google Scholar] [CrossRef]
- Merlot, A.M.; Porter, G.M.; Sahni, S.; Lim, E.G.; Peres, P.; Richardson, D.R. The Metastasis Suppressor, NDRG1, Differentially Modulates the Endoplasmic Reticulum Stress Response. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 2094–2110. [Google Scholar] [CrossRef]
- Hunter, M.; Angelicheva, D.; Tournev, I.; Ingley, E.; Chan, D.C.; Watts, G.F.; Kremensky, I.; Kalaydjieva, L. NDRG1 Interacts with APO A-I and A-II and Is a Functional Candidate for the HDL-C QTL on 8q24. Biochem. Biophys. Res. Commun 2005, 332, 982–992. [Google Scholar] [CrossRef] [PubMed]
- Chun, J.; Giovannoni, G.; Hunter, S.F. Sphingosine 1-Phosphate Receptor Modulator Therapy for Multiple Sclerosis: Differential Downstream Receptor Signalling and Clinical Profile Effects. Drugs 2021, 81, 207–231. [Google Scholar] [CrossRef] [PubMed]
- White, A.B.; Givogri, M.I.; Lopez-Rosas, A.; Cao, H.; van Breemen, R.; Thinakaran, G.; Bongarzone, E.R. Psychosine Accumulates in Membrane Microdomains in the Brain of Krabbe Patients, Disrupting the Raft Architecture. J. Neurosci. 2009, 29, 6068–6077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, K.; Webster, H. deF. Effects of Psychosine (Galactosylsphingosine) on the Survival and the Fine Structure of Cultured Schwann Cells. J. Neuropathol. Exp. Neurol. 1993, 52, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Wenger, D.A.; Luzi, P.; Rafi, M.A. Advances in the Diagnosis and Treatment of Krabbe Disease. Int. J. Neonatal. Screen 2021, 7, 57. [Google Scholar] [CrossRef]
- Martinez, L.O.; Jacquet, S.; Esteve, J.-P.; Rolland, C.; Cabezón, E.; Champagne, E.; Pineau, T.; Georgeaud, V.; Walker, J.E.; Tercé, F.; et al. Ectopic Beta-Chain of ATP Synthase Is an Apolipoprotein A-I Receptor in Hepatic HDL Endocytosis. Nature 2003, 421, 75–79. [Google Scholar] [CrossRef]
- Ebanks, B.; Chakrabarti, L. Mitochondrial ATP Synthase Is a Target of Oxidative Stress in Neurodegenerative Diseases. Front. Mol. Biosci. 2022, 9, 854321. [Google Scholar] [CrossRef]
- Cai, K.; El-Merahbi, R.; Loeffler, M.; Mayer, A.E.; Sumara, G. Ndrg1 Promotes Adipocyte Differentiation and Sustains Their Function. Sci. Rep. 2017, 7, 7191. [Google Scholar] [CrossRef] [Green Version]
- Bartz, F.; Kern, L.; Erz, D.; Zhu, M.; Gilbert, D.; Meinhof, T.; Wirkner, U.; Erfle, H.; Muckenthaler, M.; Pepperkok, R.; et al. Identification of Cholesterol-Regulating Genes by Targeted RNAi Screening. Cell Metabolism. 2009, 10, 63–75. [Google Scholar] [CrossRef] [Green Version]
- Pietiäinen, V.; Vassilev, B.; Blom, T.; Wang, W.; Nelson, J.; Bittman, R.; Bäck, N.; Zelcer, N.; Ikonen, E. NDRG1 Functions in LDL Receptor Trafficking by Regulating Endosomal Recycling and Degradation. J. Cell Sci. 2013, 126, 3961–3971. [Google Scholar] [CrossRef] [Green Version]
- Martins-de-Souza, D.; Cassoli, J.S.; Nascimento, J.M.; Hensley, K.; Guest, P.C.; Pinzon-Velasco, A.M.; Turck, C.W. The Protein Interactome of Collapsin Response Mediator Protein-2 (CRMP2/DPYSL2) Reveals Novel Partner Proteins in Brain Tissue. Proteom. Clin. Appl. 2015, 9, 817–831. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.-H.; Tsai, J.-N.; Chang, S.-W.; Hsu, W.-T.; Yang, C.-P.; Hsiao, C.-W.; Shiau, M.-Y. Regulation of Adipogenesis and Lipid Deposits by Collapsin Response Mediator Protein 2. Int. J. Mol. Sci. 2020, 21, 2172. [Google Scholar] [CrossRef] [PubMed]
- Hensley, K.; Gabbita, S.P.; Venkova, K.; Hristov, A.; Johnson, M.F.; Eslami, P.; Harris-White, M.E. A Derivative of the Brain Metabolite Lanthionine Ketimine Improves Cognition and Diminishes Pathology in the 3 × Tg-AD Mouse Model of Alzheimer Disease. J. Neuropathol. Exp. Neurol. 2013, 72, 955–969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayaraj, G.G.; Hipp, M.S.; Hartl, F.U. Functional Modules of the Proteostasis Network. Cold Spring Harb. Perspect. Biol. 2020, 12, a033951. [Google Scholar] [CrossRef] [Green Version]
- Martínez, G.; Khatiwada, S.; Costa-Mattioli, M.; Hetz, C. ER Proteostasis Control of Neuronal Physiology and Synaptic Function. Trends Neurosci. 2018, 41, 610–624. [Google Scholar] [CrossRef]
- Ross, C.A.; Poirier, M.A. Protein Aggregation and Neurodegenerative Disease. Nat. Med. 2004, 10, S10–S17. [Google Scholar] [CrossRef]
- Roussel, B.D.; Kruppa, A.J.; Miranda, E.; Crowther, D.C.; Lomas, D.A.; Marciniak, S.J. Endoplasmic Reticulum Dysfunction in Neurological Disease. Lancet Neurol. 2013, 12, 105–118. [Google Scholar] [CrossRef] [Green Version]
- Bingol, B.; Sheng, M. Deconstruction for Reconstruction: The Role of Proteolysis in Neural Plasticity and Disease. Neuron 2011, 69, 22–32. [Google Scholar] [CrossRef] [Green Version]
- Engelender, S.; Stefanis, L.; Oddo, S.; Bellucci, A. Can We Treat Neurodegenerative Proteinopathies by Enhancing Protein Degradation? Mov. Disord. 2022, 37, 1346–1359. [Google Scholar] [CrossRef]
- Ruan, J.; Schlüter, D.; Wang, X. Deubiquitinating Enzymes (DUBs): DoUBle-Edged Swords in CNS Autoimmunity. J. Neuroinflamm. 2020, 17, 102. [Google Scholar] [CrossRef] [Green Version]
- Gadhave, K.; Kumar, P.; Kapuganti, S.K.; Uversky, V.N.; Giri, R. Unstructured Biology of Proteins from Ubiquitin-Proteasome System: Roles in Cancer and Neurodegenerative Diseases. Biomolecules 2020, 10, 796. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Ambasta, R.K.; Kumar, P. Ubiquitin Biology in Neurodegenerative Disorders: From Impairment to Therapeutic Strategies. Ageing Res. Rev. 2020, 61, 101078. [Google Scholar] [CrossRef] [PubMed]
- Volpi, V.G.; Touvier, T.; D’Antonio, M. Endoplasmic Reticulum Protein Quality Control Failure in Myelin Disorders. Front. Mol. Neurosci. 2017, 9, 162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papadopoulos, C.; Kirchner, P.; Bug, M.; Grum, D.; Koerver, L.; Schulze, N.; Poehler, R.; Dressler, A.; Fengler, S.; Arhzaouy, K.; et al. VCP/P97 Cooperates with YOD1, UBXD1 and PLAA to Drive Clearance of Ruptured Lysosomes by Autophagy. EMBO J. 2017, 36, 135–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrari, V.; Cristofani, R.; Tedesco, B.; Crippa, V.; Chierichetti, M.; Casarotto, E.; Cozzi, M.; Mina, F.; Piccolella, M.; Galbiati, M.; et al. Valosin Containing Protein (VCP): A Multistep Regulator of Autophagy. Int. J. Mol. Sci. 2022, 23, 1939. [Google Scholar] [CrossRef]
- Xia, D.; Tang, W.K.; Ye, Y. Structure and Function of the AAA+ ATPase P97/Cdc48p. Gene 2016, 583, 64–77. [Google Scholar] [CrossRef] [Green Version]
- Buchan, J.R.; Kolaitis, R.-M.; Taylor, J.P.; Parker, R. Eukaryotic Stress Granules Are Cleared by Autophagy and Cdc48/VCP Function. Cell 2013, 153, 1461–1474. [Google Scholar] [CrossRef] [Green Version]
- Hillert, E.-K.; Brnjic, S.; Zhang, X.; Mazurkiewicz, M.; Saei, A.A.; Mofers, A.; Selvaraju, K.; Zubarev, R.; Linder, S.; D’Arcy, P. Proteasome Inhibitor B-AP15 Induces Enhanced Proteotoxicity by Inhibiting Cytoprotective Aggresome Formation. Cancer Lett. 2019, 448, 70–83. [Google Scholar] [CrossRef] [Green Version]
- Deol, K.K.; Crowe, S.O.; Du, J.; Bisbee, H.A.; Guenette, R.G.; Strieter, E.R. Proteasome-Bound UCH37/UCHL5 Debranches Ubiquitin Chains to Promote Degradation. Mol. Cell 2020, 80, 796–809.e9. [Google Scholar] [CrossRef]
- Bello, A.I.; Goswami, R.; Brown, S.L.; Costanzo, K.; Shores, T.; Allan, S.; Odah, R.; Mohan, R.D. Deubiquitinases in Neurodegeneration. Cells 2022, 11, 556. [Google Scholar] [CrossRef]
- Yao, T. A Timer to Coordinate Substrate Processing by the 26S Proteasome. Nat. Struct. Mol. Biol. 2015, 22, 652–653. [Google Scholar] [CrossRef] [PubMed]
- Al-Shami, A.; Jhaver, K.G.; Vogel, P.; Wilkins, C.; Humphries, J.; Davis, J.J.; Xu, N.; Potter, D.G.; Gerhardt, B.; Mullinax, R.; et al. Regulators of the Proteasome Pathway, Uch37 and Rpn13, Play Distinct Roles in Mouse Development. PLoS ONE 2010, 5, e13654. [Google Scholar] [CrossRef] [PubMed]
- Ristic, G.; Tsou, W.-L.; Todi, S.V. An Optimal Ubiquitin-Proteasome Pathway in the Nervous System: The Role of Deubiquitinating Enzymes. Front. Mol. Neurosci. 2014, 7, 72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazumdar, T.; Gorgun, F.M.; Sha, Y.; Tyryshkin, A.; Zeng, S.; Hartmann-Petersen, R.; Jørgensen, J.P.; Hendil, K.B.; Eissa, N.T. Regulation of NF-KappaB Activity and Inducible Nitric Oxide Synthase by Regulatory Particle Non-ATPase Subunit 13 (Rpn13). Proc. Natl. Acad. Sci. USA 2010, 107, 13854–13859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benameur, T.; Soleti, R.; Panaro, M.A.; La Torre, M.E.; Monda, V.; Messina, G.; Porro, C. Curcumin as Prospective Anti-Aging Natural Compound: Focus on Brain. Molecules 2021, 26, 4794. [Google Scholar] [CrossRef]
- Factory, If Else RJLM. Available online: https://www.rjlm.ro/ (accessed on 27 September 2022).
- Priyam, N.; Savoy, A. The Role of Astrocytes in the Cause of Alzheimer’s Disease. J. Stud. Res. 2021, 10, 3. [Google Scholar] [CrossRef]
- Fang, Y.; Shen, X. Ubiquitin Carboxyl-Terminal Hydrolases: Involvement in Cancer Progression and Clinical Implications. Cancer Metastasis Rev. 2017, 36, 669–682. [Google Scholar] [CrossRef]
- Kim, W.-Y.; Snider, W.D. Functions of GSK-3 Signaling in Development of the Nervous System. Front. Mol. Neurosci. 2011, 4, 44. [Google Scholar] [CrossRef] [Green Version]
- Fomenkov, A.; Zangen, R.; Huang, Y.-P.; Osada, M.; Guo, Z.; Fomenkov, T.; Trink, B.; Sidransky, D.; Ratovitski, E.A. RACK1 and Stratifin Target DeltaNp63alpha for a Proteasome Degradation in Head and Neck Squamous Cell Carcinoma Cells upon DNA Damage. Cell Cycle 2004, 3, 1285–1295. [Google Scholar] [CrossRef]
- Liu, Y.V.; Semenza, G.L. RACK1 vs. HSP90: Competition for HIF-1α Degradation vs. Stabilization. Cell Cycle 2007, 6, 656–659. [Google Scholar] [CrossRef]
- Lachat, P.; Shaw, P.; Gebhard, S.; van Belzen, N.; Chaubert, P.; Bosman, F.T. Expression of NDRG1, a Differentiation-Related Gene, in Human Tissues. Histochem. Cell Biol. 2002, 118, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Hawkins-Salsbury, J.A.; Qin, E.Y.; Reddy, A.S.; Vogler, C.A.; Sands, M.S. Oxidative Stress as a Therapeutic Target in Globoid Cell Leukodystrophy. Exp. Neurol. 2012, 237, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Del Grosso, A.; Angella, L.; Tonazzini, I.; Moscardini, A.; Giordano, N.; Caleo, M.; Rocchiccioli, S.; Cecchini, M. Dysregulated Autophagy as a New Aspect of the Molecular Pathogenesis of Krabbe Disease. Neurobiol. Dis. 2019, 129, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Erskine, D.; Koss, D.; Korolchuk, V.I.; Outeiro, T.F.; Attems, J.; McKeith, I. Lipids, Lysosomes and Mitochondria: Insights into Lewy Body Formation from Rare Monogenic Disorders. Acta Neuropathol. 2021, 141, 511–526. [Google Scholar] [CrossRef] [PubMed]
- Shahmoradian, S.H.; Lewis, A.J.; Genoud, C.; Hench, J.; Moors, T.E.; Navarro, P.P.; Castaño-Díez, D.; Schweighauser, G.; Graff-Meyer, A.; Goldie, K.N.; et al. Lewy Pathology in Parkinson’s Disease Consists of Crowded Organelles and Lipid Membranes. Nat. Neurosci. 2019, 22, 1099–1109. [Google Scholar] [CrossRef] [Green Version]
- Cheng, A.; Kawahata, I.; Fukunaga, K. Fatty Acid Binding Protein 5 Mediates Cell Death by Psychosine Exposure through Mitochondrial Macropores Formation in Oligodendrocytes. Biomedicines 2020, 8, 635. [Google Scholar] [CrossRef]
- Voccoli, V.; Tonazzini, I.; Signore, G.; Caleo, M.; Cecchini, M. Role of Extracellular Calcium and Mitochondrial Oxygen Species in Psychosine-Induced Oligodendrocyte Cell Death. Cell Death Dis. 2014, 5, e1529. [Google Scholar] [CrossRef] [Green Version]
- Head, R.A.; Brown, R.M.; Zolkipli, Z.; Shahdadpuri, R.; King, M.D.; Clayton, P.T.; Brown, G.K. Clinical and Genetic Spectrum of Pyruvate Dehydrogenase Deficiency: Dihydrolipoamide Acetyltransferase (E2) Deficiency. Ann. Neurol. 2005, 58, 234–241. [Google Scholar] [CrossRef]
- Singhi, P.; De Meirleir, L.; Lissens, W.; Singhi, S.; Saini, A.G. Pyruvate Dehydrogenase-E1α Deficiency Presenting as Recurrent Demyelination: An Unusual Presentation and a Novel Mutation. JIMD Rep. 2013, 10, 107–111. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Zhang, J.; Yang, L.; Chen, Q.; Hao, Q.; Zhang, L.; Sun, H. A Dual-Function Chemical Probe for Detecting Erasers of Lysine Lipoylation. Front. Chem. Sci. Eng. 2022, 16, 121–127. [Google Scholar] [CrossRef]
- Udow, S.; Bunge, M.; Ryner, L.; Mhanni, A.A.; Salman, M.S. Prolonged Survival and Serial Magnetic Resonance Imaging/Magnetic Resonance Spectroscopy Changes in Infantile Krabbe Disease. Pediatr. Neurol. 2012, 47, 299–302. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Melberg, A.; Weis, J.; Månsson, J.-E.; Raininko, R. The Earliest MR Imaging and Proton MR Spectroscopy Abnormalities in Adult-Onset Krabbe Disease. Acta Neurol. Scand. 2007, 116, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Kang, P.B.; Hunter, J.V.; Kaye, E.M. Lactic Acid Elevation in Extramitochondrial Childhood Neurodegenerative Diseases. J. Child Neurol. 2001, 16, 657–660. [Google Scholar] [CrossRef] [PubMed]
- Meisingset, T.W.; Ricca, A.; Neri, M.; Sonnewald, U.; Gritti, A. Region- and Age-Dependent Alterations of Glial-Neuronal Metabolic Interactions Correlate with CNS Pathology in a Mouse Model of Globoid Cell Leukodystrophy. J. Cereb. Blood Flow Metab. 2013, 33, 1127–1137. [Google Scholar] [CrossRef] [Green Version]
- Bahr, L.S.; Bock, M.; Liebscher, D.; Bellmann-Strobl, J.; Franz, L.; Prüß, A.; Schumann, D.; Piper, S.K.; Kessler, C.S.; Steckhan, N.; et al. Ketogenic Diet and Fasting Diet as Nutritional Approaches in Multiple Sclerosis (NAMS): Protocol of a Randomized Controlled Study. Trials 2020, 21, 3. [Google Scholar] [CrossRef] [Green Version]
- Iñarrea, P.; Alarcia, R.; Alava, M.A.; Capablo, J.L.; Casanova, A.; Iñiguez, C.; Iturralde, M.; Larrodé, P.; Martín, J.; Mostacero, E.; et al. Mitochondrial Complex Enzyme Activities and Cytochrome c Expression Changes in Multiple Sclerosis. Mol. Neurobiol. 2014, 49, 1–9. [Google Scholar] [CrossRef]
- Park, J.S.; Kim, M.J.; Kim, S.Y.; Lim, B.C.; Kim, K.J.; Seong, M.-W.; Lee, J.S.; Chae, J.-H. Novel Compound Heterozygous ACO2 Mutations in an Infant with Progressive Encephalopathy: A Newly Identified Neurometabolic Syndrome. Brain Dev. 2020, 42, 680–685. [Google Scholar] [CrossRef]
- Quan, W.; Li, J.; Jin, X.; Liu, L.; Zhang, Q.; Qin, Y.; Pei, X.; Chen, J. Identification of Potential Core Genes in Parkinson’s Disease Using Bioinformatics Analysis. Parkinsons Dis. 2021, 2021, 1690341. [Google Scholar] [CrossRef]
- Bogenhagen, D.F.; Rousseau, D.; Burke, S. The Layered Structure of Human Mitochondrial DNA Nucleoids. J. Biol. Chem. 2008, 283, 3665–3675. [Google Scholar] [CrossRef] [Green Version]
- Bradford, M.M. A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Oakley, B.R.; Kirsch, D.R.; Morris, N.R. A Simplified Ultrasensitive Silver Stain for Detecting Proteins in Polyacrylamide Gels. Anal. Biochem. 1980, 105, 361–363. [Google Scholar] [CrossRef]
- Gharahdaghi, F.; Weinberg, C.R.; Meagher, D.A.; Imai, B.S.; Mische, S.M. Mass Spectrometric Identification of Proteins from Silver-Stained Polyacrylamide Gel: A Method for the Removal of Silver Ions to Enhance Sensitivity. Electrophoresis 1999, 20, 601–605. [Google Scholar] [CrossRef]
- Ihaka, R.; Gentleman, R. R: A Language for Data Analysis and Graphics. J. Comput. Graph. Stat. 1996, 5, 299–314. Available online: https://www.tandfonline.com/doi/abs/10.1080/10618600.1996.10474713 (accessed on 15 June 2022).
- Bianchi, L.; Bruzzese, F.; Leone, A.; Gagliardi, A.; Puglia, M.; Di Gennaro, E.; Rocco, M.; Gimigliano, A.; Pucci, B.; Armini, A.; et al. Proteomic Analysis Identifies Differentially Expressed Proteins after HDAC Vorinostat and EGFR Inhibitor Gefitinib Treatments in Hep-2 Cancer Cells. Proteomics 2011, 11, 3725–3742. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, L.; Altera, A.; Barone, V.; Bonente, D.; Bacci, T.; De Benedetto, E.; Bini, L.; Tosi, G.M.; Galvagni, F.; Bertelli, E. Untangling the Extracellular Matrix of Idiopathic Epiretinal Membrane: A Path Winding among Structure, Interactomics and Translational Medicine. Cells 2022, 11, 2531. [Google Scholar] [CrossRef]
- Angeloni, C.; Turroni, S.; Bianchi, L.; Fabbri, D.; Motori, E.; Malaguti, M.; Leoncini, E.; Maraldi, T.; Bini, L.; Brigidi, P.; et al. Novel Targets of Sulforaphane in Primary Cardiomyocytes Identified by Proteomic Analysis. PLoS ONE 2013, 8, e83283. [Google Scholar] [CrossRef] [Green Version]
- Bini, L.; Schvartz, D.; Carnemolla, C.; Besio, R.; Garibaldi, N.; Sanchez, J.-C.; Forlino, A.; Bianchi, L. Intracellular and Extracellular Markers of Lethality in Osteogenesis Imperfecta: A Quantitative Proteomic Approach. Int. J. Mol. Sci. 2021, 22, 429. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| a Spot N. | b Protein Name | c UniProtKB Accession Number | d MetaCore Name | e Mascot Search Results | f Fold Change | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Score | Expected | Matched Peptides/ Detected Peptides | Sequence Coverage % | Twi/Het | Het/WT | Twi/WT | ||||
| 1 | Ubiquitin carboxyl-terminal hydrolase 5 | P56399 | USP5 | 144 | 6.8 × 10−11 | 14/18 | 15 | 4.05 ¥ | 0.22 ¥ | 0.86 NS |
| 2 | Transitional endoplasmic reticulum ATPase | Q01853 | VCP | 226 | 4.3 × 10−19 | 22/27 | 25 | 2.52 ¥ | 0.59 NS | 1.48 NS |
| 3 | Aconitate hydratase, mitochondrial | Q99KI0 | ACON | 188 | 2.7 × 10−15 | 13/14 | 25 | 0.70 NS | 2.89 # | 2.02 NS |
| 4 | Aconitate hydratase, mitochondrial | Q99KI0 | ACON | 100 | 1.7 × 10−6 | 9/15 | 16 | 2.32 ¥ | 1.33 NS | 3.07 # |
| 6 | Aconitate hydratase, mitochondrial | Q99KI0 | ACON | 258 | 2.7 × 10−22 | 20/22 | 31 | 2.58 ¥ | 0.72 NS | 1.84 NS |
| 7 | Albumin | P07724 | Albumin | 414 | 6.8 × 10−38 | 29/32 | 52 | 2.66 NS | 0.26 ¥ | 0.67 NS |
| 9 | Dihydrolipoyllysine-residue acetyltransferase component of pyruvate dehydrogenase complex, mitochondrial | Q8BMF4 | ODP2 | 119 | 2.2 × 10−8 | 10/14 | 20 | 1.34 NS | 1.50 NS | 2 ¥ |
| 10 | Dihydropyrimidinase-related protein 2 | O08553 | CRMP2 | 233 | 8.6 × 10−20 | 17/23 | 43 | 2.00 # | 0.73 NS | 1.45 ¥ |
| 11 | Lipoamide acyltransferase component of branched-chain alpha-keto acid dehydrogenase complex, mitochondrial | P53395 | BCKD-E2 | 127 | 3.4 × 10−9 | 12/18 | 17 | 0.32 # | 1.69 NS | 0.54 NS |
| 12 | 2′,3′-cyclic-nucleotide 3′-phosphodiesterase | P16330 | CNP1 | 210 | 1.7 × 10−17 | 16/25 | 41 | 2.47 # | 0.47 # | 1.14 NS |
| 13 | Vimentin | P20152 | Vimentin | 98 | 3.00 × 10−6 | 7/9 | 19 | 12.75 ¥ | 0.46 NS | 5.77 ¥ |
| 14 | ATP synthase subunit beta, mitochondrial | P56480 | ATP5B | 171 | 1.4 × 10−13 | 13/17 | 32 | 3.98 ¥ | 0.33 ¥ | 1.29 NS |
| 15 | Protein NDRG1 | Q62433 | NDRG1 | 101 | 1.4 × 10−6 | 8/9 | 24 | 0.91 NS | 2.61 # | 2.35 ¥ |
| 16 | NSFL1 cofactor p47 | Q9CZ44 | p47 | 118 | 2.7 × 10−8 | 8/12 | 29 | 3.74 ¥ | 0.63 NS | 2.33 ¥ |
| 17 | Tropomodulin-2 | Q9JKK7 | Tropomodulin-2 | 100 | 1.7 × 10−6 | 9/19 | 33 | 3.06 ¥ | 0.28 ¥ | 0.86 NS |
| 18 | 60S acidic ribosomal protein P0 | P14869 | RPLP0 | 194 | 6.8 × 10−16 | 12/15 | 47 | 0.52 NS | 2.01 ¥ | 1.03 NS |
| 19 | Beta-soluble NSF attachment protein | P28663 | NAPB | 227 | 3.4 × 10−19 | 18/22 | 60 | 1.67 NS | 1.60 NS | 2.67 ¥ |
| 20 | Creatine kinase B-type | Q04447 | KCRB | 127 | 3.4 × 10−9 | 7/7 | 21 | 2.72 ¥ | 0.67 NS | 1.82 NS |
| 22 | Receptor of activated protein C kinase 1 | P68040 | RACK1 | 189 | 2.2 × 10−15 | 13/23 | 59 | 3.14 # | 0.42 ¥ | 1.30 NS |
| 23 | Protein-L-isoaspartate(D-aspartate) O-methyltransferase | P23506 | PIMT | 173 | 8.6 × 10−14 | 12/15 | 42 | 0.39 ¥ | 1.71 NS | 0.66 NS |
| 24 | Translationally-controlled tumor protein | P63028 | TPT1 | 136 | 4.3 × 10−10 | 10/18 | 44 | 1.05 NS | 2.7 ¥ | 2.83 # |
| 25 | Transgelin-3 | Q9R1Q8 | Transgelin-3 | 159 | 2.2 × 10−12 | 16/30 | 56 | 0.53 ¥ | 2.24 ¥ | 1.19 NS |
| 27 | Myelin basic protein | P04370 | Myelin basic protein | 151 | 1.4 × 10−11 | 10/13 | 30 | 0.59 ¥ | 2.12 # | 1.25 NS |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vantaggiato, L.; Shaba, E.; Carleo, A.; Bezzini, D.; Pannuzzo, G.; Luddi, A.; Piomboni, P.; Bini, L.; Bianchi, L. Neurodegenerative Disorder Risk in Krabbe Disease Carriers. Int. J. Mol. Sci. 2022, 23, 13537. https://doi.org/10.3390/ijms232113537
Vantaggiato L, Shaba E, Carleo A, Bezzini D, Pannuzzo G, Luddi A, Piomboni P, Bini L, Bianchi L. Neurodegenerative Disorder Risk in Krabbe Disease Carriers. International Journal of Molecular Sciences. 2022; 23(21):13537. https://doi.org/10.3390/ijms232113537
Chicago/Turabian StyleVantaggiato, Lorenza, Enxhi Shaba, Alfonso Carleo, Daiana Bezzini, Giovanna Pannuzzo, Alice Luddi, Paola Piomboni, Luca Bini, and Laura Bianchi. 2022. "Neurodegenerative Disorder Risk in Krabbe Disease Carriers" International Journal of Molecular Sciences 23, no. 21: 13537. https://doi.org/10.3390/ijms232113537
APA StyleVantaggiato, L., Shaba, E., Carleo, A., Bezzini, D., Pannuzzo, G., Luddi, A., Piomboni, P., Bini, L., & Bianchi, L. (2022). Neurodegenerative Disorder Risk in Krabbe Disease Carriers. International Journal of Molecular Sciences, 23(21), 13537. https://doi.org/10.3390/ijms232113537