

Intrinsically Disordered Proteins: An Overview


{kind=link}
{kind=link}
Abstract
:1. Introduction
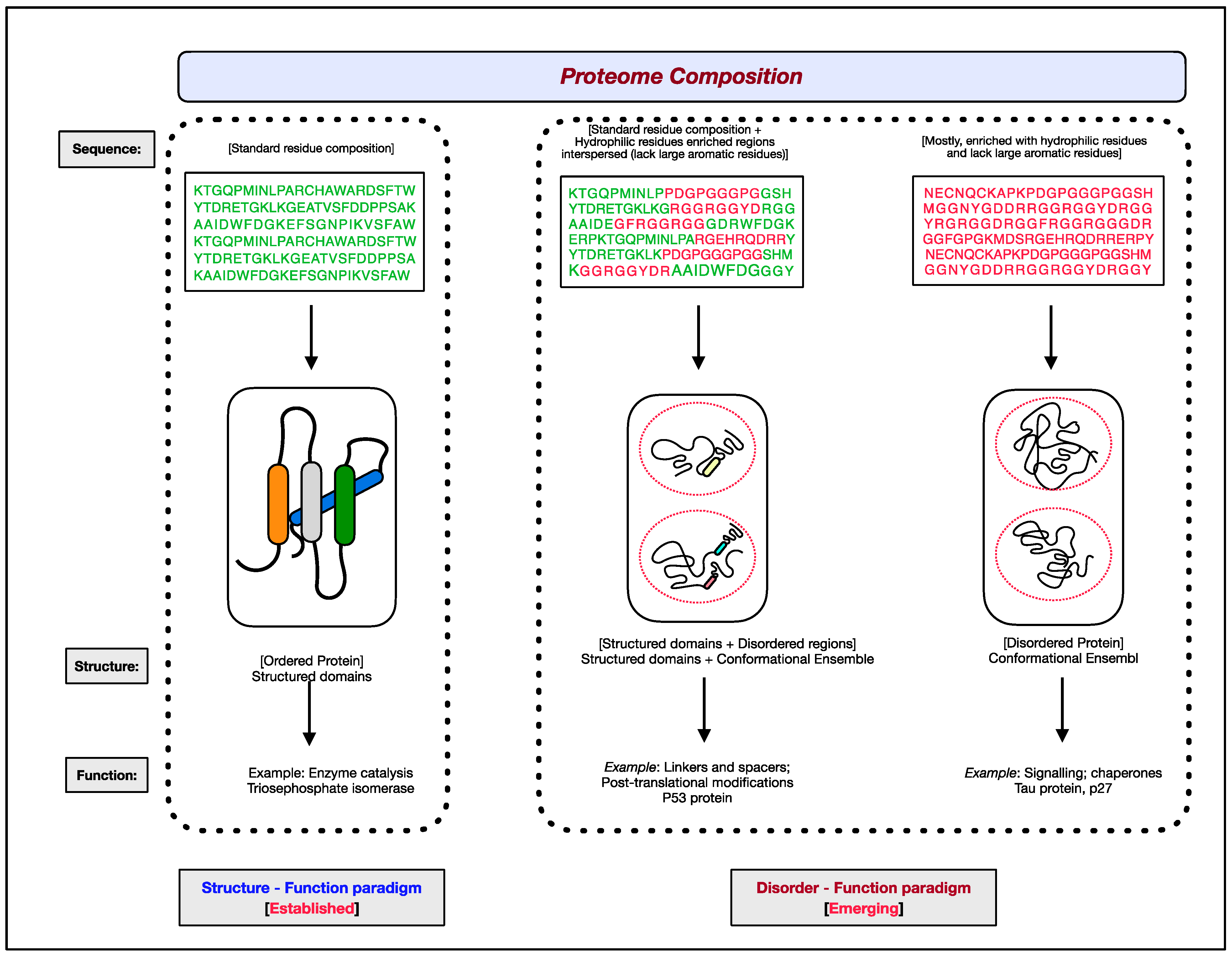
1.1. Structure–Function Paradigm
1.2. Disorder–Function Paradigm
2. Intrinsic Protein Disorder
2.1. Natural Abundance of Intrinsically Disordered Proteins
2.2. Sequence Characteristics of Intrinsically Disordered Proteins
2.3. Structural Aspects of Intrinsically Disordered Proteins
2.4. Functional Classification of Intrinsically Disordered Proteins
2.4.1. Entropy Chains
2.4.2. Modification Sites
2.4.3. Disordered Chaperones
2.4.4. Molecular Recognition Effectors
2.4.5. Molecular Assemblers
2.4.6. Molecular Recognition Scavengers
2.4.7. Metal Sponges
2.5. Functional Elements of Intrinsically Disordered Proteins
2.5.1. Short Linear Motifs
2.5.2. Molecular Recognition Features
2.5.3. Intrinsically Disordered Domains
3. Experimental Approaches for Assessing Intrinsic Protein Disorder
3.1. Indirect Methods
3.2. Direct Methods
3.2.1. X-ray Crystallography
3.2.2. Circular Dichroism (CD)
3.2.3. Nuclear Magnetic Resonance (NMR)
3.2.4. Small-Angle X-ray Scattering (SAXS)
3.2.5. Cryo-Electron Microscopy (Cryo-EM)
4. Computational Tools for Disorder Prediction
4.1. Propensity-Based Predictors
4.2. Machine Learning Algorithms (MLAs) Based Predictors
4.3. Inter-Residue Contact-Based Predictors
5. Evolution of IDPs/IDRs
6. IDPs/IDRs in Diseases
7. IDPs/IDRs as Drug Targets
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Babu, M.M.; Kriwacki, R.W.; Pappu, R.V. Structural biology. Versatility from protein disorder. Science 2012, 337, 1460–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.; et al. Classification of intrinsically disordered regions and proteins. Chem. Rev. 2014, 114, 6589–6631. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Ye, S.; Li, X.; Lu, W. Emerging Structure-Function Paradigm of Endocrine FGFs in Metabolic Diseases. Trends Pharmacol. Sci. 2019, 40, 142–153. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.H.; Hoelz, A. The Structure of the Nuclear Pore Complex (An Update). Annu. Rev. Biochem. 2019, 88, 725–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uversky, V.N. Protein intrinsic disorder and structure-function continuum. Prog. Mol. Biol. Transl. Sci. 2019, 166, 1–17. [Google Scholar] [PubMed]
- Mak, H.; Thurston, T.L.M. Interesting Biochemistries in the Structure and Function of Bacterial Effectors. Front. Cell Infect. Microbiol. 2021, 11, 608860. [Google Scholar] [CrossRef]
- Pinet, L.; Assrir, N.; van Heijenoort, C. Expanding the Disorder-Function Paradigm in the C-Terminal Tails of Erbbs. Biomolecules 2021, 11, 1690. [Google Scholar] [CrossRef]
- Ghag, G.; Holler, C.J.; Taylor, G.; Kukar, T.L.; Uversky, V.N.; Rangachari, V. Disulfide bonds and disorder in granulin-3: An unusual handshake between structural stability and plasticity. Protein Sci. 2017, 9, 1759–1772. [Google Scholar] [CrossRef] [Green Version]
- Davey, N.E. The functional importance of structure in unstructured protein regions. Curr. Opin. Struct. Biol. 2019, 56, 155–163. [Google Scholar] [CrossRef]
- Bondos, S.E.; Dunker, A.K.; Uversky, V.N. On the roles of intrinsically disordered proteins and regions in cell communication and signaling. Cell Commun. Signal. 2021, 19, 88. [Google Scholar] [CrossRef]
- Dunker, A.K.; Babu, M.M.; Barbar, E.; Blackledge, M.; Bondos, S.E.; Dosztányi, Z.; Dyson, H.J.; Forman-Kay, J.; Fuxreiter, M.; Gsponer, J.; et al. What’s in a name? Why these proteins are intrinsically disordered. Intrinsically Disord. Proteins 2013, 1, e24157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bickers, S.C.; Sayewich, J.S.; Kanelis, V. Intrinsically disordered regions regulate the activities of ATP binding cassette transporters. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183202. [Google Scholar] [CrossRef] [PubMed]
- Brodsky, S.; Jana, T.; Mittelman, K.; Chapal, M.; Kumar, D.K.; Carmi, M.; Barkai, N. Intrinsically Disordered Regions Direct Transcription Factor In Vivo Binding Specificity. Mol. Cell 2020, 79, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Jamecna, D.; Antonny, B. Intrinsically disordered protein regions at membrane contact sites. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2021, 1866, 159020. [Google Scholar] [CrossRef]
- Ahrens, J.B.; Nunez-Castilla, J.; Siltberg-Liberles, J. Evolution of intrinsic disorder in eukaryotic proteins. Cell. Mol. Life Sci. 2017, 74, 3163–3174. [Google Scholar] [CrossRef]
- Protter, D.S.W.; Rao, B.S.; Van Treeck, B.; Lin, Y.; Mizoue, L.; Rosen, M.K.; Parker, R. Intrinsically Disordered Regions Can Contribute Promiscuous Interactions to RNP Granule Assembly. Cell Rep. 2018, 22, 1401–1412. [Google Scholar] [CrossRef] [Green Version]
- Macossay-Castillo, M.; Marvelli, G.; Guharoy, M.; Jain, A.; Kihara, D.; Tompa, P.; Wodak, S.J. The Balancing Act of Intrinsically Disordered Proteins: Enabling Functional Diversity while Minimizing Promiscuity. J. Mol. Biol. 2019, 431, 1650–1670. [Google Scholar] [CrossRef]
- Chakrabarti, P.; Chakravarty, D. Intrinsically disordered proteins/regions and insight into their biomolecular interactions. Biophysical Chemistry 2022, 283, 106769. [Google Scholar] [CrossRef]
- Huang, Y.H.; Rose, P.W.; Hsu, C.N. Citing a Data Repository: A Case Study of the Protein Data Bank. PLoS ONE 2015, 10, e0136631. [Google Scholar] [CrossRef]
- Fischer, E. Einfiuss der Configurationa Uf die Wirkung der Enzyme. Ber. Dtsch. Chem. Ges. 1894, 11, 3479–3483. [Google Scholar] [CrossRef]
- Borgia, A.; Borgia, M.; Bugge, K.; Kissling, V.M.; Heidarsson, P.O.; Fernandes, C.B.; Sottini, A.; Soranno, A.; Buholzer, K.J.; Nettels, D.; et al. Extreme disorder in an ultrahigh-affinity protein complex. Nature 2018, 555, 61–66. [Google Scholar] [CrossRef] [Green Version]
- Koshland, D. Application of a theory of enzyme specificity to protein synthesis. Proc. Natl. Acad. Sci. USA 1958, 44, 98–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirzaie, M. Identification of native protein structures captured by principal interactions. BMC Bioinform. 2019, 20, 604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef] [PubMed]
- Lermyte, F. Roles, Characteristics, and Analysis of Intrinsically Disordered Proteins: A Minireview. Life 2020, 10, 320. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zeng, Y.; Liu, Y.; Gao, M.; Liu, S.; Su, Z.; Huang, Y. Electrostatic interactions in molecular recognition of intrinsically disordered proteins. J. Biomol. Struct. Dyn. 2020, 38, 4883–4894. [Google Scholar] [CrossRef]
- Wright, P.E.; Dyson, H.J. Intrinsically unstructured proteins: Re-assessing the protein structure-function paradigm. J. Mol. Biol. 1999, 293, 321–331. [Google Scholar] [CrossRef] [Green Version]
- Dunker, A.K.; Lawson, J.D.; Brown, C.J.; Williams, R.M.; Romero, P.; Oh, J.S.; Oldfield, C.J.; Campen, A.M.; Ratliff, C.M.; Hipps, K.W.; et al. Intrinsically disordered protein. J. Mol. Graph. Model. 2001, 19, 26–59. [Google Scholar] [CrossRef] [Green Version]
- Tompa, P. Intrinsically unstructured proteins. Trends Biochem. Sci. 2002, 27, 527–533. [Google Scholar] [CrossRef]
- Uversky, V.; Gillespie, J.; Fink, A. Why Are “Natively Unfolded” Proteins Unstructured Under Physiologic Conditions? Proteins Struct. Funct. Genet. 2000, 41, 415–427. [Google Scholar] [CrossRef]
- Wallin, S. Intrinsically disordered proteins: Structural and functional dynamics. Res. Rep. Biol. 2017, 8, 7–16. [Google Scholar] [CrossRef] [Green Version]
- Dunker, A.K.; Garner, E.; Guilliot, S.; Romero, P.; Albrecht, K.; Hart, J.; Obradovic, Z.; Kissinger, G.; Villafranca, J.E. Protein disorder and the evolution of molecular recognition: Theory, predictions and observations. Pac. Symp. Biocomput. 1998, 3, 473–484. [Google Scholar]
- Uversky, V.N.; Dunker, A.K. Understanding protein non-folding. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2010, 1804, 1231–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uversky, V.N. Intrinsically disordered proteins from A to Z. Int. J. Biochem. Cell Biol. 2011, 43, 1090–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uversky VN The mysterious unfoldome: Structureless, under appreciated, yet vital part of any given proteome. J. Biomed. Biotechnol. 2010, 2010, 568068.
- Ward, J.J.; Sodhi, J.S.; McGuffin, L.J.; Buxton, B.F.; Jones, D.T. Prediction and Functional Analysis of Native Disorder in Proteins from the Three Kingdoms of Life. J. Mol. Biol. 2004, 337, 635–645. [Google Scholar] [CrossRef]
- Dunker, A.K.; Obradovic, Z.; Romero, P.; Garner, E.C. Intrinsic Protein Disorder in Complete Genomes. Genome Inform. 2000, 11, 161–171. [Google Scholar]
- Hu, G.; Wang, K.; Song, J.; Uversky, V.N.; Kurgan, L. Taxonomic Landscape of the Dark Proteomes: Whole-Proteome Scale Interplay Between Structural Darkness, Intrinsic Disorder, and Crystallization Propensity. Proteomics 2018, 18, e1800243. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Kaushik, R.; Tennakoon, C.; Uversky, V.N.; Longhi, S.; Zhang, K.Y.J.; Bhatia, S. Comprehensive Intrinsic Disorder Analysis of 6108 Viral Proteomes: From the Extent of Intrinsic Disorder Penetrance to Functional Annotation of Disordered Viral Proteins. J. Proteome Res. 2021, 20, 2704–2713. [Google Scholar] [CrossRef]
- Monzon, A.M.; Necci, M.; Quaglia, F.; Walsh, I.; Zanotti, G.; Piovesan, D.; Tosatto, S.C.E. Experimentally Determined Long Intrinsically Disordered Protein Regions Are Now Abundant in the Protein Data Bank. Int. J. Mol. Sci. 2020, 21, 4496. [Google Scholar] [CrossRef]
- Tompa, P.; Dosztanyi, Z.; Simon, I. Prevalent Structural Disorder in E. coli and S. cerevisiae Proteomes. J. Proteome Res. 2006, 5, 1996–2000. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Brown, C.J.; Lawson, J.D.; Iakoucheva, L.M.; Obradovic, Z. Intrinsic Disorder and Protein Function. Biochemistry 2002, 41, 6573–6582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunker, A.K.; Cortese, M.S.; Romero, P.; Iakoucheva, L.M.; Uversky, V.N. Flexible nets. The roles of intrinsic disorder in protein interaction networks. FEBS J. 2005, 272, 5129–5148. [Google Scholar] [CrossRef] [PubMed]
- Dyson, H.J.; Wright, P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005, 6, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V. What does it mean to be natively unfolded? Eur. J. Biochem. 2002, 269, 2–12. [Google Scholar] [CrossRef]
- Uversky, V.N. Natively unfolded proteins: A point where biology waits for physics. Protein Sci. 2002, 11, 739–756. [Google Scholar] [CrossRef] [Green Version]
- Dyson, H.J.; Wright, P.E. Coupling of folding and binding for unstructured proteins. Curr. Opin. Struct. Biol. 2002, 12, 54–60. [Google Scholar] [CrossRef]
- Radivojac, P.; Iakoucheva, L.M.; Oldfield, C.J.; Obradovic, Z.; Uversky, V.N.; Dunker, A.K. Intrinsic Disorder and Functional Proteomics. Biophys. J. 2007, 92, 1439–1456. [Google Scholar] [CrossRef] [Green Version]
- Romero, P.; Obradovic, Z.; Li, X.; Garner, E.; Brown, C.; Dunker, A.K. Sequence complexity of disordered protein. Proteins Struct. Funct. Genet. 2001, 42, 38–48. [Google Scholar] [CrossRef]
- Williams, R.; Obradovi, Z.; Mathura, V.; Braun, W.; Garner, E.; Young, J.; Takayama, S.; Brown, C.J.; Dunker, A.K. The protein non-folding problem: Amino acid determinants of intrinsic order and disorder. Pac. Symp. Biocomput. 2001, 6, 89–100. [Google Scholar]
- Holt, C.; Raynes, J.K.; Carver, J.A. Sequence characteristics responsible for protein-protein interactions in the intrinsically disordered regions of caseins, amelogenins, and small heat-shock proteins. Biopolymers 2019, 110, e23319. [Google Scholar] [CrossRef] [PubMed]
- Kastano, K.; Mier, P.; Dosztanyi, Z.; Promponas, V.J.; Andrade-Navarro, M.A. Functional Tuning of Intrinsically Disordered Regions in Human Proteins by Composition Bias. Biomolecules 2022, 12, 1486. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, R.; Nagarajaram, H.A. Amino acid substitution scoring matrices specific to intrinsically disordered regions in proteins. Sci Rep. 2019, 9, 16380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trivedi, R.; Nagarajaram, H.A. Substitution scoring matrices for proteins—An overview. Protein Sci. 2020, 29, 2150–2163. [Google Scholar] [CrossRef]
- Jarnot, P.; Ziemska-Legiecka, J.; Grynberg, M.; Gruca, A. Insights from analyses of low complexity regions with canonical methods for protein sequence comparison. Brief Bioinform. 2022, 23, bbac299. [Google Scholar] [CrossRef]
- McFadden, W.M.; Yanowitz, J.L. idpr: A package for profiling and analyzing Intrinsically Disordered Proteins in R. PLoS ONE 2022, 17, e0266929. [Google Scholar] [CrossRef]
- Tompa, P. Structure and Function of Intrinsically Disordered Proteins; CRC Press: Boca Raton, FL, USA; Taylor and Francis Group: Abingdon, UK, 2010. [Google Scholar]
- Uversky, V.N.; Oldfield, C.J.; Dunker, A.K. Showing your ID: Intrinsic disorder as an ID for recognition, regulation and cell signaling. J. Mol. Recognit. 2005, 18, 343–384. [Google Scholar] [CrossRef]
- Uversky, V. Intrinsic Disorder-based Protein Interactions and their Modulators. Curr. Pharm. Des. 2013, 19, 4191–4213. [Google Scholar] [CrossRef]
- Uversky, V.N. The most important thing is the tail: Multitudinous functionalities of intrinsically disordered protein termini. FEBS Lett. 2013, 587, 1891–1901. [Google Scholar] [CrossRef] [Green Version]
- Uversky, V.N. Unusual biophysics of intrinsically disordered proteins. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2013, 1834, 932–951. [Google Scholar] [CrossRef]
- Morffy, N.; Strader, L.C. Structural Aspects of Auxin Signaling. Cold Spring Harb. Perspect. Biol. 2022, 14, a039883. [Google Scholar] [CrossRef] [PubMed]
- Crick, S.L.; Jayaraman, M.; Frieden, C.; Wetzel, R.; Pappu, R.V. Fluorescence correlation spectroscopy shows that monomeric poly- glutamine molecules form collapsed structures in aqueous solutions. Proc. Natl. Acad. Sci. USA 2006, 103, 16764–16769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walters, R.H.; Murphy, R.M. Examining Polyglutamine Peptide Length: A Connection between Collapsed Conformations and Increased Aggregation. J. Mol. Biol. 2009, 393, 978–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukhopadhyay, S.; Krishnan, R.; Lemke, E.A.; Lindquist, S.; Deniz, A.A. A natively unfolded yeast prion monomer adopts an ensemble of collapsed and rapidly fluctuating structures. Proc. Natl. Acad. Sci. USA 2007, 104, 2649–2654. [Google Scholar] [CrossRef]
- Wang, X.; Vitalis, A.; Wyczalkowski, M.A.; Pappu, R.V. Characterizing the conformational ensemble of monomeric polyglutamine. Proteins Struct. Funct. Bioinform. 2006, 63, 297–311. [Google Scholar] [CrossRef]
- Mukhopadhyay, S. The Dynamism of Intrinsically Disordered Proteins: Binding-Induced Folding, Amyloid Formation, and Phase Separation. J. Phys. Chem. B 2020, 124, 11541–11560. [Google Scholar] [CrossRef]
- Dunker, A.K.; Uversky, V.N. Drugs for ‘protein clouds’: Targeting intrinsically disordered transcription factors. Curr. Opin. Pharmacol. 2010, 10, 782–788. [Google Scholar] [CrossRef]
- Choy, W.Y.; Forman-Kay, J.D. Calculation of ensembles of structures representing the unfolded state of an SH3 domain. J. Mol. Biol. 2001, 308, 1011–1032. [Google Scholar] [CrossRef]
- Strodel, B. Energy Landscapes of Protein Aggregation and Conformation Switching in Intrinsically Disordered Proteins. J. Mol. Biol. 2021, 433, 167182. [Google Scholar] [CrossRef]
- Dunker, A.K.; Obradovic, Z. The protein trinity—Linking function and disorder. Nat. Biotechnol. 2001, 19, 805–806. [Google Scholar] [CrossRef]
- Dunker, A.K.; Brown, C.J.; Obradovic, Z. Identification and Functions of Usefully Disordered Proteins. Adv. Protein Chem. 2002, 62, 25–49. [Google Scholar] [PubMed]
- Toto, A.; Malagrino, F.; Visconti, L.; Troilo, F.; Pagano, L.; Brunori, M.; Jemth, P.; Gianni, S. Templated folding of intrinsically disordered proteins. J. Biol. Chem. 2020, 295, 6586–6593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malagrino, F.; Visconti, L.; Pagano, L.; Toto, A.; Troilo, F.; Gianni, S. Understanding the Binding Induced Folding of Intrinsically Disordered Proteins by Protein Engineering: Caveats and Pitfalls. Int. J. Mol. Sci. 2020, 21, 3484. [Google Scholar] [CrossRef] [PubMed]
- Malagrino, F.; Diop, A.; Pagano, L.; Nardella, C.; Toto, A.; Gianni, S. Unveiling induced folding of intrinsically disordered proteins—Protein engineering, frustration and emerging themes. Curr. Opin. Struct. Biol. 2022, 72, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Kriwacki, R.W.; Hengst, L.; Tennant, L.; Reed, S.I.; Wright, P.E. Structural studies of p2lWa1CiPl/Sdil in the free and Cdk2-bound state: Conformational disorder mediates binding diversity. Proc. Natl. Acad. Sci. USA 1996, 93, 11504–11509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uversky, V.N. Analyzing IDPs in Interactomes. Methods Mol Biol. 2020, 2141, 895–945. [Google Scholar]
- Fuxreiter, M.; Simon, I.; Friedrich, P.; Tompa, P. Preformed Structural Elements Feature in Partner Recognition by Intrinsically Unstructured Proteins. J. Mol. Biol. 2004, 338, 1015–1026. [Google Scholar] [CrossRef]
- Choi, U.B.; Sanabria, H.; Smirnova, T.; Bowen, M.E.; Weninger, K.R. Spontaneous Switching among Conformational Ensembles in Intrinsically Disordered Proteins. Biomolecules 2019, 9, 114. [Google Scholar] [CrossRef] [Green Version]
- Follis, A.V.; Llambi, F.; Kalkavan, H.; Yao, Y.; Phillips, A.H.; Park, C.G.; Marassi, F.M.; Green, D.R.; Kriwacki, R.W. Regulation of apoptosis by an intrinsically disordered region of Bcl-xL. Nat. Chem. Biol. 2018, 14, 458–465. [Google Scholar] [CrossRef]
- Miao, Y.; Tipakornsaowapak, T.; Zheng, L.; Mu, Y.; Lewellyn, E. Phospho-regulation of intrinsically disordered proteins for actin assembly and endocytosis. FEBS J. 2018, 285, 2762–2784. [Google Scholar] [CrossRef]
- Csizmok, V.; Forman-Kay, J.D. Complex regulatory mechanisms mediated by the interplay of multiple post-translational modifications. Curr. Opin. Struct. Biol. 2018, 48, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Bludau, I.; Willems, S.; Zeng, W.F.; Strauss, M.T.; Hansen, F.M.; Tanzer, M.C.; Karayel, O.; Schulman, B.A.; Mann, M. The structural context of posttranslational modifications at a proteome-wide scale. PLoS Biol. 2022, 20, e3001636. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Han, Y.; Li, P.; Su, Z.; Huang, Y. The Role of Post-Translational Modifications on the Structure and Function of Tau Protein. J. Mol. Neurosci. 2022, 72, 1557–1571. [Google Scholar] [CrossRef] [PubMed]
- Jeffery, C.J. Moonlighting proteins. Trends Biochem. Sci. 1999, 24, 8–11. [Google Scholar] [CrossRef]
- Jeffery, C.J. Molecular mechanisms for multitasking: Recent crystal structures of moonlighting proteins. Curr. Opin. Struct. Biol. 2004, 14, 663–668. [Google Scholar] [CrossRef]
- Meng, F.; Kurgan, L. High-throughput prediction of disordered moonlighting regions in protein sequences. Proteins 2018, 86, 1097–1110. [Google Scholar] [CrossRef]
- Balcerak, A.; Trebinska-Stryjewska, A.; Konopinski, R.; Wakula, M.; Grzybowska, E.A. RNA-protein interactions: Disorder, moonlighting and junk contribute to eukaryotic complexity. Open Biol. 2019, 9, 190096. [Google Scholar] [CrossRef] [Green Version]
- Rafiee, M.R.; Zagalak, J.A.; Sidorov, S.; Steinhauser, S.; Davey, K.; Ule, J.; Luscombe, N.M. Chromatin-contact atlas reveals disorder-mediated protein interactions and moonlighting chromatin-associated RBPs. Nucleic Acids Res. 2021, 49, 13092–13107. [Google Scholar] [CrossRef]
- Gsponer, J.; Babu, M.M. The rules of disorder or why disorder rules. Prog. Biophys. Mol. Biol. 2009, 99, 94–103. [Google Scholar] [CrossRef]
- Tompa, P. The interplay between structure and function in intrinsically unstructured proteins. FEBS Lett. 2005, 579, 3346–3354. [Google Scholar] [CrossRef] [Green Version]
- Sickmeier, M.; Hamilton, J.A.; LeGall, T.; Vacic, V.; Cortese, M.S.; Tantos, A.; Szabo, B.; Tompa, P.; Chen, J.; Uversky, V.N.; et al. DisProt: The Database of Disordered Proteins. Nucleic Acids Res. 2007, 35, D786–D793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piovesan, D.; Tosatto, S.C.E. Mobi 2.0: An improved method to define intrinsic disorder, mobility and linear binding regions in protein structures. Bioinformatics 2018, 34, 122–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vymetal, J.; Vondrasek, J.; Hlouchova, K. Sequence Versus Composition: What Prescribes IDP Biophysical Properties? Entropy 2019, 21, 654. [Google Scholar] [CrossRef] [Green Version]
- Thomasen, F.E.; Lindorff-Larsen, K. Conformational ensembles of intrinsically disordered proteins and flexible multidomain proteins. Biochem. Soc. Trans. 2022, 50, 541–554. [Google Scholar] [CrossRef]
- Weber, B.; Hora, M.; Kazman, P.; Gobl, C.; Camilloni, C.; Reif, B.; Buchner, J. The antibody light-chain linker regulates domain orientation and amyloidogenicity. J. Mol. Biol. 2018, 430, 4925–4940. [Google Scholar] [CrossRef] [PubMed]
- Murrali, M.G.; Felli, I.C.; Pierattelli, R. Adenoviral E1A Exploits Flexibility and Disorder to Target Cellular Proteins. Biomolecules 2020, 10, 1541. [Google Scholar] [CrossRef] [PubMed]
- Daughdrill, G.W.; Narayanaswami, P.; Gilmore, S.H.; Belczyk, A.; Brown, C.J. Dynamic behavior of an intrinsically unstructured linker domain is conserved in the face of negligible amino acid sequence conservation. J. Mol. Evol. 2007, 65, 277–288. [Google Scholar] [CrossRef]
- Cunningham, C.C.; Leclerc, N.; Flanagan, L.A.; Lu, M.; Janmey, P.A.; Kosik, K.S. Microtubule-associated protein 2c reorganizes both microtubules and microfilaments into distinct cytological structures in an actin-binding protein-280-deficient melanoma cell line. J. Cell Biol. 1997, 136, 845–857. [Google Scholar] [CrossRef] [Green Version]
- Fahmi, M.; Ito, M. Evolutionary Approach of Intrinsically Disordered CIP/KIP Proteins. Sci. Rep. 2019, 9, 1575. [Google Scholar] [CrossRef] [Green Version]
- Szabo, B.; Horvath, T.; Schad, E.; Murvai, N.; Tantos, A.; Kalmar, L.; Chemes, L.B.; Han, K.H.; Tompa, P. Intrinsically Disordered Linkers Impart Processivity on Enzymes by Spatial Confinement of Binding Domains. Int. J. Mol. Sci. 2019, 20, 2119. [Google Scholar] [CrossRef] [Green Version]
- Cornish, J.; Chamberlain, S.G.; Owen, D.; Mott, H.R. Intrinsically disordered proteins and membranes: A marriage of convenience for cell signalling? Biochem. Soc. Trans. 2020, 48, 2669–2689. [Google Scholar] [CrossRef] [PubMed]
- Fuglebakk, E.; Reuter, N. A model for hydrophobic protrusions on peripheral membrane proteins. PLoS Comput. Biol. 2018, 14, e1006325. [Google Scholar] [CrossRef] [PubMed]
- Deryusheva, E.; Nemashkalova, E.; Galloux, M.; Richard, C.A.; Eleouet, J.F.; Kovacs, D.; Van Belle, K.; Tompa, P.; Uversky, V.; Permyakov, S. Does Intrinsic Disorder in Proteins Favor Their Interaction with Lipids? Proteomics 2019, 19, e1800098. [Google Scholar] [CrossRef] [PubMed]
- Diella, F.; Haslam, N.; Chica, C.; Budd, A.; Michael, S.; Brown, N.P.; Trave, G.; Gibson, T.J. Understanding eukaryotic linear motifs and their role in cell signaling and regulation. Front. Biosci. 2008, 13, 6580–6603. [Google Scholar] [CrossRef] [Green Version]
- Galea, C.A.; Wang, Y.; Sivakolundu, S.G.; Kriwacki, R.W. Regulation of Cell Division by Intrinsically Unstructured Proteins: Intrinsic Flexibility, Modularity, and Signaling Conduits. Biochemistry 2008, 47, 7598–7609. [Google Scholar] [CrossRef]
- Hoermann, B.; Kohn, M. Evolutionary crossroads of cell signaling: PP1 and PP2A substrate sites in intrinsically disordered regions. Biochem. Soc. Trans. 2021, 49, 1065–1074. [Google Scholar] [CrossRef]
- Galea, C.A.; Pagala, V.R.; Obenauer, J.C.; Park, C.G.; Slaughter, C.A.; Kriwacki, R.W. Proteomic studies of the intrinsically unstructured mammalian proteome. J Proteome Res. 2006, 5, 2839–2948. [Google Scholar] [CrossRef]
- Bode, A.M.; Dong, Z. Post-translational modification of p53 in tumorigenesis. Nat. Rev. Cancer 2004, 4, 793–805. [Google Scholar] [CrossRef]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [Green Version]
- Jin, F.; Grater, F. How multisite phosphorylation impacts the conformations of intrinsically disordered proteins. PLoS Comput. Biol. 2021, 17, e1008939. [Google Scholar] [CrossRef]
- Kumar, R.; Thompson, E.B. Role of Phosphorylation in the Modulation of the Glucocorticoid Receptor’s Intrinsically Disordered Domain. Biomolecules 2019, 9, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorton, B.M.; Shechter, D. Cellular consequences of arginine methylation. Cell. Mol. Life Sci. 2019, 76, 2933–2956. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, R.; Barta, A.; Semrad, K. Strategies for RNA folding and assembly. Nat. Rev. Mol. Cell Biol. 2004, 5, 908–919. [Google Scholar] [CrossRef] [PubMed]
- Young, J.C.; Agashe, V.R.; Siegers, K.; Hartl, F.U. Pathways of chaperone-mediated protein folding in the cytosol. Nat. Rev. Mol. Cell Biol. 2004, 5, 781–791. [Google Scholar] [CrossRef]
- Kovacs, D.; Tompa, P. Diverse functional manifestations of intrinsic structural disorder in molecular chaperones. Biochem. Soc. Trans. 2012, 40, 963–968. [Google Scholar] [CrossRef]
- Rogers, J.M.; Steward, A.; Clarke, J. Folding and Binding of an Intrinsically Disordered Protein: Fast, but Not ‘Diffusion-Limited’. J. Am. Chem. Soc. 2013, 135, 1415–1422. [Google Scholar] [CrossRef]
- Tompa, P.; Csermely, P. The role of structural disorder in the function of RNA and protein chaperones. FASEB J. 2004, 18, 1169–1175. [Google Scholar] [CrossRef] [Green Version]
- Lindner, R.A.; Kapur, A.; Mariani, M.; Titmuss, S.J.; Carver, J.A. Structural alterations of alpha-crystallin during its chaperone action. Eur. J. Biochem. 1998, 258, 170–183. [Google Scholar] [CrossRef]
- Treweek, T.M.; Rekas, A.; Walker, M.J.; Carver, J.A. A quantitative NMR spectroscopic examination of the flexibility of the C-terminal extensions of the molecular chaperones, αA- and αB-crystallin. Exp Eye Res. 2010, 91, 691–699. [Google Scholar] [CrossRef]
- Holmstrom, E.D.; Liu, Z.; Nettels, D.; Best, R.B.; Schuler, B. Disordered RNA chaperones can enhance nucleic acid folding via local charge screening. Nat. Commun. 2019, 10, 2453. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.M.; Kwon, S.B.; Son, A.; Kim, D.H.; Kim, K.H.; Lim, J.; Kwon, Y.G.; Kang, J.S.; Lee, B.K.; Byun, Y.H.; et al. Stabilization of Intrinsically Disordered DKK2 Protein by Fusion to RNA-Binding Domain. Int. J. Mol. Sci. 2019, 20, 2847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yakubu, U.M.; Morano, K.A. Suppression of aggregate and amyloid formation by a novel intrinsically disordered region in metazoan Hsp110 chaperones. J. Biol. Chem. 2021, 296, 100567. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.Y.; Minetti, C.A.; Remeta, D.P.; Breslauer, K.J.; Chen, K.Y. HSF1, Aging, and Neurodegeneration. Adv. Exp. Med. Biol. 2022, in press. [Google Scholar]
- Murvai, N.; Kalmar, L.; Szabo, B.; Schad, E.; Micsonai, A.; Kardos, J.; Buday, L.; Han, K.H.; Tompa, P.; Tantos, A. Cellular Chaperone Function of Intrinsically Disordered Dehydrin ERD14. Int. J. Mol. Sci. 2021, 22, 6190. [Google Scholar] [CrossRef] [PubMed]
- Mitrea, D.M.; Kriwacki, R.W. Regulated unfolding of proteins in signaling. FEBS Lett. 2013, 587, 1081–1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bardwell, J.C.; Jakob, U. Conditional disorder in chaperone action. Trends Biochem. Sci. 2012, 37, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Tempra, C.; Scollo, F.; Pannuzzo, M.; Lolicato, F.; La Rosa, C. A unifying framework for amyloid-mediated membrane damage: The lipid-chaperone hypothesis. Biochim. Biophys. Acta Proteins Proteom. 2022, 1870, 140767. [Google Scholar] [CrossRef]
- Sugase, K.; Dyson, H.J.; Wright, P.E. Mechanism of coupled folding and binding of an intrinsically disordered protein. Nature 2007, 447, 1021–1025. [Google Scholar] [CrossRef]
- Wright, P.E.; Dyson, H.J. Linking folding and binding. Curr. Opin. Struct. Biol. 2009, 19, 31–38. [Google Scholar] [CrossRef] [Green Version]
- Gianni, S.; Jemth, P. Affinity versus specificity in coupled binding and folding reactions. Protein Eng. Des. Sel. 2019, 32, 355–357. [Google Scholar] [CrossRef]
- Zou, R.; Zhou, Y.; Wang, Y.; Kuang, G.; Ågren, H.; Wu, J.; Tu, Y. Free Energy Profile and Kinetics of Coupled Folding and Binding of the Intrinsically Disordered Protein p53 with MDM2. J. Chem. Inf. Model. 2020, 60, 1551–1558. [Google Scholar] [CrossRef] [PubMed]
- Morris, O.M.; Torpey, J.H.; Isaacson, R.L. Intrinsically disordered proteins: Modes of binding with emphasis on disordered domains. Open Biol. 2021, 11, 210222. [Google Scholar] [CrossRef] [PubMed]
- Schuler, B.; Borgia, A.; Borgia, M.B.; Heidarsson, P.O.; Holmstrom, E.D.; Nettels, D.; Sottini, A. Binding without folding—The biomolecular function of disordered polyelectrolyte complexes. Curr. Opin. Struct. Biol. 2020, 60, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Dreier, J.E.; Prestel, A.; Martins, J.M.; Brøndum, S.S.; Nielsen, O.; Garbers, A.E.; Suga, H.; Boomsma, W.; Rogers, J.M.; Hartmann-Petersen, R.; et al. A context-dependent and disordered ubiquitin-binding motif. Cell. Mol. Life Sci. 2022, 79, 484. [Google Scholar] [CrossRef] [PubMed]
- Robustelli, P.; Piana, S.; Shaw, D.E. Mechanism of Coupled Folding-upon-Binding of an Intrinsically Disordered Protein. J Am Chem Soc. 2020, 142, 11092–11101. [Google Scholar] [CrossRef]
- Kim, A.; Kakalis, L.; Abdul-Manan, N.; Liu, G.; Rosen, M. Autoinhibition and activation mechanisms of the Wiskott-Aldrich syndrome protein. Nature 2000, 404, 151–158. [Google Scholar] [CrossRef]
- Berlow, R.B.; Dyson, H.J.; Wright, P.E. Multivalency enables unidirectional switch-like competition between intrinsically disordered proteins. Proc. Natl. Acad. Sci. USA 2022, 119, e2117338119. [Google Scholar] [CrossRef]
- Ferreon, A.C.; Ferreon, J.C.; Wright, P.E.; Deniz, A.A. Modulation of allostery by protein intrinsic disorder. Nature 2013, 498, 390–394. [Google Scholar] [CrossRef] [Green Version]
- Radhakrishnan, I.; Perez-Alvarado, G.C.; Parker, D.; Dyson, H.J.; Montminy, M.R.; Wright, P.E. Solution structure of the KIX domain of CBP bound to the transactivation domain of CREB: A model for activator: Coactivator interactions. Cell 1997, 91, 741–752. [Google Scholar] [CrossRef] [Green Version]
- De Guzman, R.N.; Goto, N.K.; Dyson, H.J.; Wright, P.E. Structural basis for cooperative transcription factor binding to the CBP coactivator. J. Mol. Biol. 2006, 355, 1005–1013. [Google Scholar] [CrossRef]
- Berlow, R.B.; Dyson, H.J.; Wright, P.E. Expanding the Paradigm: Intrinsically Disordered Proteins and Allosteric Regulation. J Mol. Biol. 2018, 430, 2309–2320. [Google Scholar] [CrossRef] [PubMed]
- Hilser, V.J.; Thompson, E.B. Intrinsic disorder as a mechanism to optimize allosteric coupling in proteins. Proc. Natl. Acad. Sci. USA 2007, 104, 8311–8315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flock, T.; Weatheritt, R.J.; Latysheva, N.S.; Babu, M.M. Controlling entropy to tune the functions of intrinsically disordered regions. Curr. Opin. Struct. Biol. 2014, 26, 62–72. [Google Scholar] [CrossRef]
- Fuxreiter, M.; Tompa, P.; Simon, I.; Uversky, V.N.; Hansen, J.C.; Asturias, F.J. Malleable machines take shape in eukaryotic transcriptional regulation. Nat. Chem. Biol. 2008, 4, 728–737. [Google Scholar] [CrossRef] [Green Version]
- Peng, Z.; Oldfield, C.J.; Xue, B.; Mizianty, M.J.; Dunker, A.K.; Kurgan, L.; Uversky, V.N. A creature with a hundred waggly tails: Intrinsically disordered proteins in the ribosome. Cell. Mol. Life Sci. 2013, 71, 1477–1504. [Google Scholar] [CrossRef] [PubMed]
- Hegyi, H.; Schad, E.; Tompa, P. Structural disorder promotes assembly of protein complexes. BMC Struct. Biol. 2007, 7, 65. [Google Scholar] [CrossRef]
- Gunasekaran, K.; Tsai, C.J.; Kumar, S.; Zanuy, D.; Nussinov, R. Extended disordered proteins: Targeting function with less scaffold. Trends Biochem. Sci. 2003, 28, 81–85. [Google Scholar] [CrossRef]
- Cortese, M.S.; Uversky, V.N.; Dunker, A.K. Intrinsic disorder in scaffold proteins: Getting more from less. Prog. Biophys. Mol. Biol. 2008, 98, 85–106. [Google Scholar] [CrossRef] [Green Version]
- Clerc, I.; Sagar, A.; Barducci, A.; Sibille, N.; Bernado, P.; Cortes, J. The diversity of molecular interactions involving intrinsically disor dered proteins: A molecular modeling perspective. Comput. Struct. Biotechnol. J. 2021, 19, 3817–3828. [Google Scholar] [CrossRef]
- Shajani, Z.; Sykes, M.T.; Williamson, J.R. Assembly of bacterial ribosomes. Annu. Rev. Biochem. 2011, 80, 501–526. [Google Scholar] [CrossRef] [Green Version]
- Bondos, S.E.; Dunker, A.K.; Uversky, V.N. Intrinsically disordered proteins play diverse roles in cell signaling. Cell Commun. Signal. 2022, 20, 20. [Google Scholar] [CrossRef] [PubMed]
- Fung, H.Y.J.; Birol, M.; Rhoades, E. IDPs in macromolecular complexes: The roles of multivalent interactions in diverse assemblies. Curr Opin Struct Biol. 2018, 49, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Holt, C. Unfolded phosphopolypeptides enable soft and hard tissues to coexist in the same organism with relative ease. Curr. Opin. Struct. Biol. 2013, 23, 420–425. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Hernandez, M.; Romero, I.; Sanchez-Ballesta, M.T.; Merodio, C.; Escribano, M.I. Functional characterization of VviDHN2 and VviDHN4 dehydrin isoforms from Vitis vinifera (L.): An in silico and in vitro approach. Plant Physiol. Biochem. 2021, 158, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Elko, E.A.; Manuel, A.M.; White, S.; Zito, E.; van der Vliet, A.; Anathy, V.; Janssen-Heininger, Y.M.W. Oxidation of peroxiredoxin-4 induces oligomerization and promotes interaction with proteins governing protein folding and endoplasmic reticulum stress. J. Biol. Chem. 2021, 296, 100665. [Google Scholar] [CrossRef]
- Bidwell LM, McManus ME, Gaedigk A, Kakuta Y, Negishi M, Pedersen L, Martin JL Crystal structure of human catecholamine sulfotransferase. J. Mol. Biol. 1999, 293, 521–530.
- Barney, B.M.; LoBrutto, R.; Francisco, W.A. Characterization of a small metal binding protein from Nitrosomonas europaea. Biochemistry 2004, 43, 11206–11213. [Google Scholar] [CrossRef]
- Jobby, M.K.; Sharma, Y. Calcium-binding crystallins from Yersinia pestis. Characterization of two single betagamma-crystallin domains of a putative exported protein. J. Biol. Chem. 2005, 280, 1209–1216. [Google Scholar] [CrossRef] [Green Version]
- Davey, N.E.; Van Roey, K.; Weatheritt, R.J.; Toedt, G.; Uyar, B.; Altenberg, B.; Budd, A.; Diella, F.; Dinkel, H.; Gibson, T.J. Attributes of short linear motifs. Mol. BioSyst. 2012, 8, 268–281. [Google Scholar] [CrossRef]
- Fuxreiter, M.; Tompa, P.; Simon, I. Local structural disorder imparts plasticity on linear motifs. Bioinformatics 2007, 23, 950–956. [Google Scholar] [CrossRef]
- Lee, S.; Kim, D.; Han, J.; Cha, E.; Lim, J.; Cho, Y.; Lee, C.; Han, K. Understanding Pre-Structured Motifs (PreSMos) in Intrinsically Unfolded Proteins. Curr. Protein Pept. Sci. 2012, 13, 34–54. [Google Scholar] [CrossRef] [PubMed]
- Mohan, A.; Oldfield, C.J.; Radivojac, P.; Vacic, V.; Cortese, M.S.; Dunker, A.K.; Uversky, V.N. Analysis of Molecular Recognition Features (MoRFs). J. Mol. Biol. 2006, 362, 1043–1059. [Google Scholar] [CrossRef] [PubMed]
- Oldfield, C.; Cheng, Y.; Cortese, M.; Romero, P.; Uversky, V.; Dunker, A.K. Coupled Folding and Binding with R-Helix-Forming Molecular Recognition. Biochemistry 2005, 44, 12454–12470. [Google Scholar] [CrossRef] [PubMed]
- Hinds, M.G.; Smits, C.; Fredericks-Short, R.; Risk, J.M.; Bailey, M.; Huang, D.C.; Day, C.L. Bim, Bad and Bmf: Intrinsically unstructured BH3-only proteins that undergo a localized conformational change upon binding to prosurvival Bcl-2 targets. Cell Death Differ. 2007, 14, 128–136. [Google Scholar] [CrossRef] [Green Version]
- Dinkel, H.; Michael, S.; Weatheritt, R.J.; Davey, N.E.; Van Roey, K.; Altenberg, B.; Toedt, G.; Uyar, B.; Seiler, M.; Budd, A.; et al. ELM—The database of eukaryotic linear motifs. Nucleic Acids Res. 2012, 40, D242–D251. [Google Scholar] [CrossRef] [Green Version]
- Van Roey, K.; Uyar, B.; Weatheritt, R.J.; Dinkel, H.; Seiler, M.; Budd, A.; Gibson, T.J.; Davey, N.E. Short Linear Motifs: Ubiquitous and Functionally Diverse Protein Interaction Modules Directing Cell Regulation. Chem. Rev. 2014, 114, 6733–6778. [Google Scholar] [CrossRef]
- Dinkel, H.; Van Roey, K.; Michael, S.; Davey, N.E.; Weatheritt, R.J.; Born, D.; Speck, T.; Kruger, D.; Grebnev, G.; Kuban, M.; et al. The eukaryotic linear motif resource ELM: 10 years and counting. Nucleic Acids Res. 2014, 42, D259–D266. [Google Scholar] [CrossRef]
- Dinkel, H.; Van Roey, K.; Michael, S.; Kumar, M.; Uyar, B.; Altenberg, B.; Milchevskaya, V.; Schneider, M.; Kühn, H.; Behrendt, A.; et al. ELM 2016—Data update and new functionality of the eukaryotic linear motif resource. Nucleic Acids Res. 2016, 44, D294–D300. [Google Scholar] [CrossRef]
- Tompa, P.; Davey, N.E.; Gibson, T.J.; Babu, M.M. A million peptide motifs for the molecular biologist. Mol. Cell 2014, 55, 161–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gouw, M.; Michael, S.; Sámano-Sánchez, H.; Kumar, M.; Zeke, A.; Lang, B.; Bely, B.; Chemes, L.B.; Davey, N.E.; Deng, Z.; et al. The eukaryotic linear motif resource—2018 update. Nucleic Acids Res. 2018, 46, D428–D434. [Google Scholar] [CrossRef]
- Zhou, X.; Kops, O.; Werner, A.; Lu, P.; Shen, M.; Stoller, G.; Kullertz, G.; Stark, M.; Fischer, G.; Lu, K. Pin1-Dependent Prolyl Isomerization Regulates Dephosphorylation of Cdc25C and Tau Proteins. Mol. Cell 2000, 6, 873–883. [Google Scholar] [CrossRef]
- Pop, C.; Salvesen, G.S. Human Caspases: Activation, Specificity, and Regulation. J. Biol. Chem. 2009, 284, 21777–21781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalderon, D.; Roberts, B.L.; Richardson, W.D.; Smith, A.E. A Short Amino Acid Sequence Able to Specify Nuclear Location. Cell 1984, 39, 499–509. [Google Scholar] [CrossRef]
- Schmid, E.M.; McMahon, H.T. Integrating molecular and network biology to decode endocytosis. Nature 2007, 448, 883–888. [Google Scholar] [CrossRef]
- Li, J.; McQuade, T.; Siemer, A.B.; Napetschnig, J.; Moriwaki, K.; Hsiao, Y.S.; Damko, E.; Moquin, D.; Walz, T.; McDermott, A.; et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 2012, 150, 339–350. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Banjade, S.; Cheng, H.; Kim, S.; Chen, B.; Guo, L.; Llaguno, M.; Hollingsworth, J.; King, D.; Banani, S.; et al. Phase transitions in the assembly of multivalent signalling proteins. Nature 2012, 483, 336–340. [Google Scholar] [CrossRef] [Green Version]
- Pfleger, C.; Kirschner, M. The KEN box: An APC recognition signal distinct from the D box targeted by Cdh1. Genes Dev. 2000, 14, 655–665. [Google Scholar] [CrossRef]
- He, J.; Chao, W.C.; Zhang, Z.; Yang, J.; Cronin, N.; Barford, D. Insights into Degron Recognition by APC/C Coactivators from the Structure of an Acm1-Cdh1 Complex. Mol. Cell 2013, 50, 649–660. [Google Scholar] [CrossRef] [Green Version]
- Gagliardi, P.A.; Primo, L. Irreversible Activation of Rho-activated Kinases Resulted from Evolution of Proteolytic Sites within Disordered Regions in Coiled-coil Domain. Mol. Biol. Evol. 2019, 36, 376–392. [Google Scholar] [CrossRef]
- Brautigan, D.; Shenolikar, S. Protein Serine/Threonine Phosphatases: Keys to Unlocking Regulators and Substrates. Annu. Rev. Biochem. 2018, 87, 921–964. [Google Scholar] [CrossRef] [Green Version]
- Hossain, M.; Bhalla, K.; Stillman, B. Multiple, short protein binding motifs in ORC1 and CDC6 control the initiation of DNA replication. Mol. Cell 2021, 81, 1951–1969. [Google Scholar] [CrossRef] [PubMed]
- Faustova, I.; Loog, M. SLiMs in intrinsically disordered protein regions regulate the cell cycle dynamics of ORC1-CDC6 interaction and pre-replicative complex assembly. Mol. Cell 2021, 81, 1861–1862. [Google Scholar] [CrossRef] [PubMed]
- Gibson, T.J. Cell regulation: Determined to signal discrete cooperation. Trends Biochem. Sci. 2009, 34, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Van Roey, K.; Dinkel, H.; Weatheritt, R.J.; Gibson, T.J.; Davey, N.E. The switches.ELM resource: A compendium of conditional regulatory interaction interfaces. Sci. Signal. 2013, 6, rs7. [Google Scholar] [CrossRef] [PubMed]
- Zanzoni, A.; Ribeiro, D.M.; Brun, C. Understanding protein multifunctionality: From short linear motifs to cellular functions. Cell Mol Life Sci. 2019, 76, 4407–4412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunker, A.K.; Oldfield, C.J.; Meng, J.; Romero, P.; Yang, J.Y.; Chen, J.; Vacic, V.; Obradovic, Z.; Uversky, V.N. The unfoldomics decade: An update on intrinsically disordered proteins. BMC Genom. 2008, 9, S1. [Google Scholar] [CrossRef] [Green Version]
- Espinoza-Fonseca, L.M. Reconciling binding mechanisms of intrinsically disordered proteins. Biochem. Biophys. Res. Commun. 2009, 382, 479–482. [Google Scholar] [CrossRef]
- Das, R.K.; Crick, S.L.; Pappu, R.V. N-Terminal Segments Modulate the α-Helical Propensities of the Intrinsically Disordered Basic Regions of bZIP Proteins. J. Mol. Biol. 2012, 416, 287–299. [Google Scholar] [CrossRef]
- Sharma, R.; Sharma, A.; Patil, A.; Tsunoda, T. Discovering MoRFs by trisecting intrinsically disordered protein sequence into terminals and middle regions. BMC Bioinform. 2019, 19, 378. [Google Scholar] [CrossRef]
- Yan, J.; Dunker, A.K.; Uversky, V.N.; Kurgan, L. Molecular recognition features (MoRFs) in three domains of life. Mol. Biosyst. 2016, 12, 697–710. [Google Scholar] [CrossRef] [Green Version]
- Chi, S.W.; Lee, S.H.; Kim, D.H.; Ahn, M.J.; Kim, J.S.; Woo, J.Y.; Torizawa, T.; Kainosho, M.; Han, K.H. Structural Details on mdm2-p53 Interaction. J. Biol. Chem. 2005, 280, 38795–38802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borcherds, W.; Kashtanov, S.; Wu, H.; Daughdrill, G.W. Structural divergence is more extensive than sequence divergence for a family of intrinsically disordered proteins. Proteins Struct. Funct. Bioinform. 2013, 81, 1686–1698. [Google Scholar] [CrossRef] [PubMed]
- Katuwawala, A.; Peng, Z.; Yang, J.; Kurgan, L. Computational Prediction of MoRFs, Short Disorder-to-order Transitioning Protein Binding Regions. Comput. Struct. Biotechnol. J. 2019, 17, 454–462. [Google Scholar] [CrossRef]
- Sun, X.; Malhis, N.; Zhao, B.; Xue, B.; Gsponer, J.; Rikkerink, E.H.A. Computational Disorder Analysis in Ethylene Response Factors Uncovers Binding Motifs Critical to Their Diverse Functions. Int. J. Mol. Sci. 2019, 21, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tompa, P.; Fuxreiter, M.; Oldfield, C.J.; Simon, I.; Dunker, A.K.; Uversky, V.N. Close encounters of the third kind: Disordered domains and the interactions of proteins. BioEssays 2009, 31, 328–335. [Google Scholar] [CrossRef]
- Chen, J.W.; Romero, P.; Uversky, V.N.; Dunker, A.K. Conservation of intrinsic disorder in protein domains and families: I. A database of conserved predicted disordered regions. J. Proteome Res. 2006, 5, 879–887. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.W.; Romero, P.; Uversky, V.N.; Dunker, A.K. Conservation of Intrinsic Disorder in Protein Domains and Families: II. Functions of Conserved Disorder. J. Proteome Res. 2006, 5, 888–898. [Google Scholar] [CrossRef]
- Zhou, J.; Oldfield, C.J.; Yan, W.; Shen, B.; Dunker, A.K. Intrinsically disordered domains: Sequence ➔ disorder ➔ function relationships. Protein Sci. 2019, 28, 1652–1663. [Google Scholar] [CrossRef]
- Buljan, M.; Frankish, A.; Bateman, A. Quantifying the mechanisms of domain gain in animal proteins. Genome Biol. 2010, 11, R74. [Google Scholar] [CrossRef] [Green Version]
- Russo, A.A.; Jeffrey, P.D.; Patten, A.K.; Massague, J.; Pavletich, N.P. Crystal structure of the p27Kip1 cyclin-dependent-kinase inhibitor bound to the cyclin A-Cdk2 complex. Nature 1996, 382, 325–331. [Google Scholar] [CrossRef]
- Galea, C.A.; Nourse, A.; Wang, Y.; Sivakolundu, S.G.; Heller, W.T.; Kriwacki, R.W. Role of intrinsic flexibility in signal transduction mediated by the cell cycle regulator, p27 Kip1. J. Mol. Biol. 2008, 376, 827–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pentony, M.M.; Jones, D.T. Modularity of intrinsic disorder in the human proteome. Proteins Struct. Funct. Bioinform. 2010, 78, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Teraguchi, S.; Patil, A.; Standley, D. Intrinsically disordered domains deviate significantly from random sequences in mammalian proteins. BMC Bioinform. 2010, 11, S7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manica, G.; Ghenea, S.; Munteanu, C.V.A.; Martin, E.C.; Butnaru, C.; Surleac, M.; Chiritoiu, G.N.; Alexandru, P.R.; Petrescu, A.J.; Petrescu, S.M. EDEM3 Domains Cooperate to Perform Its Overall Cell Functioning. Int. J. Mol. Sci. 2021, 22, 2172. [Google Scholar] [CrossRef]
- Rautureau, G.J.; Day, C.L.; Hinds, M.G. Intrinsically disordered proteins in bcl-2 regulated apoptosis. Int. J. Mol. Sci. 2010, 11, 1808–1824. [Google Scholar] [CrossRef] [Green Version]
- Kai, M. Roles of RNA-Binding Proteins in DNA Damage Response. Int. J. Mol. Sci. 2016, 17, 310. [Google Scholar] [CrossRef] [Green Version]
- Blocquel, D.; Habchi, J.; Gruet, A.; Blangy, S.; Longhi, S. Compaction and binding properties of the intrinsically disordered C-terminal domain of Henipavirus nucleoprotein as unveiled by deletion studies. Mol. BioSyst. 2012, 8, 392–410. [Google Scholar] [CrossRef]
- Etoh, Y.; Simon, M.; Green, H. Involucrin acts as transglutaminase substrate at multiple sites. Biochem. Biophys. Res. Commun. 1986, 136, 51–60. [Google Scholar] [CrossRef]
- Lynch, W.; Riseman, V.; Bretscher, A. Smooth Muscle CaldesmonIs an Extended Flexible Monomeric Protein in Solution That Can Readily Undergo Reversible Intra- and Intermolecular Sulfhydryl Cross-linking. J. Biol. Chem. 1987, 262, 7429–7437. [Google Scholar] [CrossRef]
- Weinreb, P.; Zhen, W.; Poon, A.; Conway, K.; Lansbury, P.J. NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry 1996, 35, 13709–13715. [Google Scholar] [CrossRef]
- Huber, R. Conformational flexibility in protein molecules. Nature 1979, 280, 538–539. [Google Scholar] [CrossRef] [PubMed]
- Ohgushi, M.; Wada, A. ‘Molten-globule state’: A compact form of globular proteins with mobile side chains. FEBS Lett. 1983, 164, 21–24. [Google Scholar] [CrossRef] [Green Version]
- Kuwajima, K. A Folding Model of a-Lactalbumin Deduced from the Three-state Denaturation Mechanism. J. Mol. Biol. 1977, 114, 241–258. [Google Scholar] [CrossRef]
- Fasman, G.D. Circular Dichroism and the Conformational Analysis of Biomolecules; Plenum Press: New York, NY, USA, 1996. [Google Scholar]
- Dyson, H.J.; Wright, P.E. Insights into the structure and dynamics of unfolded proteins from nuclear magnetic resonance. Adv. Protein Chem. 2002, 62, 311–340. [Google Scholar]
- Dyson, H.J.; Wright, P.E. Unfolded proteins and protein folding studied by NMR. Chem. Rev. 2004, 104, 3607–3622. [Google Scholar] [CrossRef]
- Poulsen, F.M. A Brief Introduction to NMR Spectroscopy of Proteins. 2002. Available online: https://users.cs.duke.edu/~brd/Teaching/Bio/asmb/Papers/Intro-reviews/flemming.pdf (accessed on 7 November 2022).
- Cavalli, A.; Salvatella, X.; Dobson, C.M.; Vendruscolo, M. Protein structure determination from NMR chemical shifts. Proc. Natl. Acad. Sci. USA 2007, 104, 9615–9620. [Google Scholar] [CrossRef] [Green Version]
- Kikhney, A.G.; Svergun, D.I. A practical guide to small angle X-ray scattering (SAXS) of flexible and intrinsically disordered proteins. FEBS Lett. 2015, 589, 2570–2577. [Google Scholar] [CrossRef] [Green Version]
- Korasick, D.A.; Tanner, J.J. Determination of protein oligomeric structure from small-angle X-ray scattering. Protein Sci. 2018, 27, 814–824. [Google Scholar] [CrossRef]
- Da Vela, S.; Svergun, D.I. Methods, development and applications of small-angle X-ray scattering to characterize biological macromolecules in solution. Curr. Res. Struct. Biol. 2020, 27, 164–170. [Google Scholar] [CrossRef]
- Grawert, T.W.; Svergun, D.I. Structural Modeling Using Solution Small-Angle X-ray Scattering (SAXS). J. Mol. Biol. 2020, 432, 3078–3092. [Google Scholar] [CrossRef]
- Jacques, D.A.; Trewhella, J. Small-angle scattering for structural biology--Expanding the frontier while avoiding the pitfalls. Protein Sci. 2010, 19, 642–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czaplewski, C.; Gong, Z.; Lubecka, E.A.; Xue, K.; Tang, C.; Liwo, A. Recent Developments in Data-Assisted Modeling of Flexible Proteins. Front. Mol. Biosci. 2021, 8, 765562. [Google Scholar] [CrossRef] [PubMed]
- Perez, J.; Vachette, P.; Russo, D.; Desmadril, M.; Durand, D. Heat-induced unfolding of neocarzinostatin, a small all-β protein investigated by small-angle X-ray scattering. J. Mol. Biol. 2001, 308, 721–743. [Google Scholar] [CrossRef] [PubMed]
- Bernado, P.; Blackledge, M. A Self-Consistent Description of the Conformational Behavior of Chemically Denatured Proteins from NMR and Small Angle Scattering. Biophys. J. 2009, 97, 2839–2845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernado, P.; Svergun, D.I. Structural analysis of intrinsically disordered proteins by small-angle X-ray scattering. Mol. Biosyst. 2012, 8, 151–167. [Google Scholar] [CrossRef]
- Jeffries, C.M.; Svergun, D.I. High-Throughput Studies of Protein Shapes and Interactions by Synchrotron Small-Angle X-ray Scattering. In Structural Proteomics; Methods in Molecular Biology; Owens, R., Ed.; Humana: New York, NY, USA, 2015; Volume 1261, pp. 277–301. [Google Scholar]
- Graewert, M.A.; Jeffries, C.M. Sample and Buffer Preparation for SAXS. Adv. Exp. Med. Biol. 2017, 1009, 11–30. [Google Scholar]
- David, G.; Perez, J. Combined sampler robot and high-performance liquid chromatography: A fully automated system for biological small-angle X-ray scattering experiments at the Synchrotron SOLEIL SWING beamline. J. Appl. Crystallogr. 2009, 42, 892–900. [Google Scholar] [CrossRef]
- Schulte, T.; Lofling, J.; Mikaelsson, C.; Kikhney, A.; Hentrich, K.; Diamante, A.; Ebel, C.; Normark, S.; Svergun, D.; Birgitta, H.N.; et al. The basic keratin 10-binding domain of the virulence-associated pneumococcal serine-rich protein PsrP adopts a novel MSCRAMM fold. Open Biol. 2014, 41, 30090. [Google Scholar] [CrossRef]
- Rodriguez, Z.P. Conjugation of NMR and SAXS for flexible and multidomain protein structure determination: From sample preparation to model refinement. Prog. Biophys. Mol. Biol. 2020, 150, 140–144. [Google Scholar] [CrossRef]
- Gast, K.; Damaschun, H.; Eckert, K.; Schulze-Forster, K.; Maurer, H.R.; Muller-Frohne, M.; Zirwer, D.; Czarnecki, J.; Damaschun, G. Prothymosin alpha: A biologically active protein with random coil conformation. Biochemistry 1995, 34, 13211–13218. [Google Scholar] [CrossRef]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petoukhov, M.V.; Franke, D.; Shkumatov, A.V.; Tria, G.; Kikhney, A.G.; Gajda, M.; Gorba, C.; Mertens, H.D.T.; Konarev, P.V.; Svergun, D.I. New developments in the ATSAS program package for small-angle scattering data analysis. J. Appl. Crystallogr. 2012, 45, 342–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gijsbers, A.; Sánchez-Puig, N.; Gao, Y.; Peters, P.J.; Ravelli, R.B.G.; Siliqi, D. Structural Analysis of the Partially Disordered Protein EspK from Mycobacterium Tuberculosis. Crystals 2021, 11, 18. [Google Scholar] [CrossRef]
- Nogales, E. The development of cryo-EM into a mainstream structural biology technique. Nat. Methods 2016, 13, 24–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoemaker, S.C.; Ando, N. X-rays in the cryo-electron microscopy era: Structural biology’s dynamic future. Bichemistry 2018, 57, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Hanske, J.; Sadian, Y.; Muller, C.W. The cryo-em resolution revolution and transcription complexes. Curr. Opin. Struct. Biol. 2018, 52, 8–15. [Google Scholar] [CrossRef]
- Nwanochie, E.; Uversky, V.N. Structure Determination by Single-Particle Cryo-Electron Microscopy: Only the Sky (and Intrinsic Disorder) is the Limit. Int. J. Mol. Sci. 2019, 20, 4186. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y. Single-particle cryo-EM-How did it get here and where will it go. Science 2018, 361, 876–880. [Google Scholar] [CrossRef] [Green Version]
- Musselman, C.A.; Kutateladze, T.G. Characterization of functional disordered regions within chromatin-associated proteins. IScience 2021, 24, 102070. [Google Scholar] [CrossRef]
- Bharat, T.A.M.; Russo, C.J.; Lowe, J.; Passmore, L.A.; Scheres, S.H.W. Advances in single-particle electron cryo microscopy structure determination applied to sub-tomogram averaging. Structure 2015, 23, 1743–1753. [Google Scholar] [CrossRef] [Green Version]
- Yan, Z.; Bai, X.; Yan, C.; Wu, J.; Li, Z.; Xie, T.; Peng, W.; Yin, C.; Li, X.; Scheres, S.H.W.; et al. Structure of the rabbit ryanodine receptor ryr1 at near-atomic resolution. Nature 2015, 517, 50–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibbs, E.B.; Cook, E.C.; Showalter, S.A. Application of NMR to studies of intrinsically disordered proteins. Arch. Biochem. Biophys. 2017, 628, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Geraets, J.A.; Pothula, K.R.; Schroder, G.F. Integrating cryo-EM and NMR data. Curr. Opin. Struc. Biol. 2020, 61, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Prilusky, J.; Felder, C.E.; Zeev-Ben-Mordehai, T.; Rydberg, E.H.; Man, O.; Beckmann, J.S.; Silman, I.; Sussman, J.L. FoldIndex(C): A simple tool to predict whether a given protein sequence is intrinsically unfolded. Bioinformatics 2005, 21, 3435–3438. [Google Scholar] [CrossRef]
- Liu, J.; Rost, B. NORSp: Predictions of long regions without regular secondary structure. Nucleic Acids Res. 2003, 31, 3833–3835. [Google Scholar] [CrossRef] [Green Version]
- Linding, R.; Russell, R.; Neduva, V.; Gibson, T. GlobPlot: Exploring protein sequences for globularity and disorder. Nucleic Acids Res. 2003, 31, 3701–3708. [Google Scholar] [CrossRef] [Green Version]
- Coeytaux, K.; Poupon, A. Prediction of unfolded segments in a protein sequence based on amino acid composition. Bioinformatics 2005, 21, 1891–1900. [Google Scholar] [CrossRef] [Green Version]
- Di Domenico, T.; Walsh, I.; Martin, A.J.; Tosatto, S.C. MobiDB: A comprehensive database of intrinsic protein disorder annotations. Bioinformatics 2012, 28, 2080–2081. [Google Scholar] [CrossRef] [Green Version]
- Fukuchi, S.; Sakamoto, S.; Nobe, Y.; Murakami, S.D.; Amemiya, T.; Hosoda, K.; Koike, R.; Hiroaki, H.; Ota, M. IDEAL: Intrinsically Disordered proteins with Extensive Annotations and Literature. Nucleic Acids Res. 2012, 40, D507–D511. [Google Scholar] [CrossRef]
- Romero, P.; Obradovic, Z.; Dunker, A.K. Folding minimal sequences: The lower bound for sequence complexity of globular proteins. FEBS Lett. 1999, 462, 363–367. [Google Scholar] [CrossRef] [Green Version]
- Vullo, A.; Bortolami, O.; Pollastri, G.; Tosatto, S.C. Spritz: A server for the prediction of intrinsically disordered regions in protein sequences using kernel machines. Nucleic Acids Res. 2006, 34, W164–W168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linding, R.; Jensen, L.; Diella, F.; Bork, P.; Gibson, T.; Russell, R. Protein Disorder Prediction: Implications for Structural Proteomics. Structure 2003, 11, 1453–1459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.R.; Thomson, R.; McNeil, P.; Esnouf, R.M. RONN: The bio-basis function neural network technique applied to the detection of natively disordered regions in proteins. Bioinformatics 2005, 21, 3369–3376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, J.J.; McGuffin, L.J.; Bryson, K.; Buxton, B.F.; Jones, D.T. The DISOPRED server for the prediction of protein disorder. Bioinformatics 2004, 20, 2138–2139. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.T.; Cozzetto, D. DISOPRED3: Precise disordered region predictions with annotated protein-binding activity. Bioinformatics 2015, 31, 857–863. [Google Scholar] [CrossRef] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Zweckstetter, M. NMR hawk-eyed view of AlphaFold2 structures. Protein Sci. 2021, 30, 2333–2337. [Google Scholar] [CrossRef]
- AlQuraishi, M. Protein-structure prediction revolutionized. Nature 2021, 596, 487–488. [Google Scholar] [CrossRef]
- He, X.H.; You, C.Z.; Jiang, H.L.; Jiang, Y.; Xu, H.E.; Cheng, X. AlphaFold2 versus experimental structures: Evaluation on G protein-coupled receptors. Acta Pharmacol. Sin. 2022. [Google Scholar] [CrossRef]
- Ruff, K.M.; Pappu, R.V. AlphaFold and Implications for Intrinsically Disordered Proteins. J. Mol. Biol. 2021, 433, 167208. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef] [PubMed]
- Tunyasuvunakool, K.; Adler, J.; Wu, Z.; Green, T.; Zielinski, M.; Zidek, A.; Bridgland, A.; Cowie, A.; Meyer, C.; Laydon, A.; et al. Highly accurate protein structure prediction for the human proteome. Nature 2021, 596, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Dosztanyi, Z.; Csizmok, V.; Tompa, P.; Simon, I. IUPred: Web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics 2005, 21, 3433–3434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dosztanyi, Z.; Csizmok VTompa, P.; Simon, I. The Pairwise Energy Content Estimated from Amino Acid Composition Discriminates between Folded and Intrinsically Unstructured Proteins. J. Mol. Biol. 2005, 347, 827–839. [Google Scholar] [CrossRef] [PubMed]
- Garbuzynskiy, S.O.; Lobanov, M.Y.; Galzitskaya, O.V. To be folded or to be unfolded? Protein Sci. 2008, 13, 2871–2877. [Google Scholar] [CrossRef] [PubMed]
- Schlessinger, A.; Punta, M.; Rost, B. Natively unstructured regions in proteins identified from contact predictions. Bioinformatics 2007, 23, 2376–2384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue BDunbrack, R.L.; Williams, R.W.; Dunker, A.K.; Uversky, V.N. PONDR-FIT: A meta-predictor of intrinsically disordered amino acids. Biochim. Biophys. Acta 2010, 1804, 996–1010. [Google Scholar]
- Lieutaud, P.; Canard, B.; Longhi, S. MeDor: A metaserver for predicting protein disorder. BMC Genom. 2008, 9, S25. [Google Scholar] [CrossRef] [Green Version]
- Ishida, T.; Kinoshita, K. Prediction of disordered regions in proteins based on the meta-approach. Bioinformatics 2008, 24, 1344–1348. [Google Scholar] [CrossRef] [Green Version]
- Meng, F.; Uversky, V.N.; Kurgan, L. Comprehensive review of methods for prediction of intrinsic disorder and its molecular functions. Cell. Mol. Life Sci. 2017, 74, 3069–3090. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, X.; Liu, B. A comprehensive review and comparison of existing computational methods for intrinsically disordered protein and region prediction. Brief Bioinform. 2019, 20, 330–346. [Google Scholar] [CrossRef] [PubMed]
- Kurgan, L. Resources for computational prediction of intrinsic disorder in proteins. Methods 2022, 204, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Jordan, I.; Kondrashov, F.; Adzhubei, I.A.; Wolf, Y.; Koonin, E.; Kondrashov, A.; Sunyaev, S. A universal trend of amino acid gain and loss in protein evolution. Nature 2005, 433, 633–638. [Google Scholar] [CrossRef] [PubMed]
- Brooks, D.J.; Fresco, J.R. Increased Frequency of Cysteine, Tyrosine, and Phenylalanine Residues Since the Last Universal Ancestor. Mol. Cell. Proteom. 2002, 1, 125–131. [Google Scholar] [CrossRef] [Green Version]
- Cristofari, G.; Darlix, J.L. The ubiquitous nature of RNA chaperone proteins. Prog. Nucl. Acid Res. Mol. Biol. 2002, 72, 223–268. [Google Scholar]
- Doolittle, W.F. Uprooting the tree of life. Sci. Am. 2000, 282, 90–95. [Google Scholar] [CrossRef]
- Theobald, D.L. A formal test of the theory of universal common ancestry. Nature 2010, 465, 219–222. [Google Scholar] [CrossRef]
- Van Oss, S.B.; Carvunis, A.R. De novo gene birth. PLoS Genet. 2019, 15, e1008160. [Google Scholar] [CrossRef] [Green Version]
- Tompa, P. Intrinsically unstructured proteins evolve by repeat expansion. BioEssays 2003, 25, 847–855. [Google Scholar] [CrossRef]
- Lise, S.; Jones, D. Sequence Patterns Associated with Disordered Regions in Proteins. Proteins Struct. Funct. Genet. 2005, 58, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.J.; Johnson, A.K.; Dunker, A.K.; Daughdrill, G.W. Evolution and disorder. Curr. Opin. Struct. Biol. 2011, 21, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Pancsa, R.; Tompa, P. Structural Disorder in Eukaryotes. PLoS ONE 2012, 7, e34687. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.; Kirchner, M.; Uyar, B.; Cheng, J.Y.; Russo, G.; Hernandez-Miranda, L.R.; Szymborska, A.; Zauber, H.; Rudolph, I.M.; Willnow, T.E.; et al. Mutations in Disordered Regions Can Cause Disease by Creating Dileucine Motifs. Cell 2018, 175, 239–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pajkos, M.; Zeke, A.; Dosztányi, Z. Ancient Evolutionary Origin of Intrinsically Disordered Cancer Risk Regions. Biomolecules 2020, 10, 1115. [Google Scholar] [CrossRef] [PubMed]
- Meszaros, B.; Hajdu-Soltesz, B.; Zeke, A.; Dosztanyi, Z. Mutations of Intrinsically Disordered Protein Regions Can Drive Cancer but Lack Therapeutic Strategies. Biomolecules 2021, 11, 381. [Google Scholar] [CrossRef]
- Seera, S.; Nagarajaram, H.A. Effect of Disease Causing Missense Mutations on Intrinsically Disordered Regions in Proteins. Protein Pept. Lett. 2022, 29, 254–267. [Google Scholar] [CrossRef]
- Peng, Z.; Uversky, V.; Kurgan, L. Genes encoding intrinsic disorder in Eukaryota have high GC content. Intrinsically Disord. Proteins 2016, 4, e1262225. [Google Scholar] [CrossRef]
- Basile, W.; Sachenkova, O.; Light, S.; Elofsson, A. High GC content causes orphan proteins to be intrinsically disordered. PLoS Comput. Biol. 2017, 13, e1005375. [Google Scholar] [CrossRef] [Green Version]
- Brown, C.J.; Takayama, S.; Campen, A.M.; Vise, P.; Marshall, T.W.; Oldfield, C.J.; Williams, C.J.; Dunker, A.K. Evolutionary Rate Heterogeneity in Proteins with Long Disordered Regions. J. Mol. Evol. 2002, 55, 104–110. [Google Scholar] [CrossRef]
- Homma, K.; Anbo, H.; Noguchi, T.; Fukuchi, S. Both Intrinsically Disordered Regions and Structural Domains Evolve Rapidly in Immune-Related Mammalian Proteins. Int. J. Mol. Sci. 2018, 19, 3860. [Google Scholar] [CrossRef] [Green Version]
- Pancsa, R.; Zsolyomi, F.; Tompa, P. Co-Evolution of Intrinsically Disordered Proteins with Folded Partners Witnessed by Evolutionary Couplings. Int. J. Mol. Sci. 2018, 19, 3315. [Google Scholar] [CrossRef] [PubMed]
- Zarin, T.; Strome, B.; Nguyen Ba, A.N.; Alberti, S.; Forman-Kay, J.D.; Moses, A.M. Proteome-wide signatures of function in highly diverged intrinsically disordered regions. eLife 2019, 8, e46883. [Google Scholar] [CrossRef] [PubMed]
- Bellay, J.; Han, S.; Michaut, M.; Kim, T.; Costanzo, M.; Andrews, B.J.; Boone, C.; Bader, G.D.; Myers, C.L.; Kim, P.M. Bringing order to protein disorder through comparative genomics and genetic interactions. Genome Biol. 2011, 12, R14. [Google Scholar] [CrossRef] [PubMed]
- Covaceuszach, S.; Peche, L.Y.; Konarev, P.V.; Lamba, D. A combined evolutionary and structural approach to disclose the primary structural determinants essential for proneurotrophins biological functions. Comput. Struct. Biotechnol. J. 2021, 19, 2891–2904. [Google Scholar] [CrossRef]
- Millard, P.S.; Bugge, K.; Marabini, R.; Boomsma, W.; Burow, M.; Kragelund, B.B. IDDomainSpotter: Compositional bias reveals domains in long disordered protein regions-Insights from transcription factors. Protein Sci. 2020, 29, 169–183. [Google Scholar] [CrossRef] [Green Version]
- Cohan, M.C.; Shinn, M.K.; Lalmansingh, J.M.; Pappu, R.V. Uncovering Non-random Binary Patterns Within Sequences of Intrinsically Disordered Proteins. J. Mol. Biol. 2022, 434, 167373. [Google Scholar] [CrossRef]
- Lu, A.X.; Lu, A.X.; Pritisanac, I.; Zarin, T.; Forman-Kay, J.D.; Moses, A.M. Discovering molecular features of intrinsically disordered regions by using evolution for contrastive learning. PLoS Comput. Biol. 2022, 18, e1010238. [Google Scholar] [CrossRef]
- Gsponer, J.; Futschik, M.E.; Teichmann, S.A.; Babu, M.M. Tight Regulation of Unstructured Proteins: From Transcript Synthesis to Protein Degradation. Science 2008, 322, 1365–1368. [Google Scholar] [CrossRef] [Green Version]
- Uversky, V.N.; Oldfield, C.J.; Dunker, A.K. Intrinsically disordered proteins in human diseases: Introducing the D2 concept. Annu. Rev. Biophys. 2008, 37, 215–246. [Google Scholar] [CrossRef]
- Langella, E.; Buonanno, M.; De Simone, G.; Monti, S.M. Intrinsically disordered features of carbonic anhydrase IX proteoglycan-like domain. Cell. Mol. Life Sci. 2021, 78, 2059–2067. [Google Scholar] [CrossRef]
- Chang, B.S.; Minn, A.J.; Muchmore, S.W.; Fesik, S.W.; Thompson, C.B. Identification of a novel regulatory domain in Bcl-X(L) and Bcl-2. EMBO J. 1997, 16, 968–977. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Mok, K.H.; Muhandiram, R.; Park, K.; Suk, J.; Kim, D.; Chang, J.; Sung, Y.C.; Choi, K.Y.; Han, K. Local Structural Elements in the Mostly Unstructured Transcriptional Activation Domain of Human p53. J. Biol. Chem. 2000, 275, 29426–29432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garg, N.; Kumar, P.; Gadhave, K.; Giri, R. The dark proteome of cancer: Intrinsic disorderedness and functionality of HIF-1α along with its interacting proteins. Prog. Mol. Biol. Transl. Sci. 2019, 166, 371–403. [Google Scholar] [PubMed]
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease and Down’s syndrome: Sharing of a unique cerebrovascular amyloid fibril protein. Biochem. Biophys. Res. Commun. 1984, 122, 1131–1135. [Google Scholar] [CrossRef]
- Lee, V.; Balin, B.; Otvos, L.J.; Trojanowski, J. A68: A major subunit of paired helical filaments and derivatized forms of normal Tau. Science 1991, 251, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Ueda, K.; Fukushima, H.; Masliah, E.; Xia, Y.; Iwai, A.; Yoshimoto, M.; Otero, D.; Kondo, J.; Ihara, Y.; Saitoh, T. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 11282–11286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prusiner, S. Shattuck lecture: Neurodegenerative diseases and prions. N. Engl. J. Med. 2001, 344, 1516–1526. [Google Scholar] [CrossRef]
- Eftekharzadeh BHyman, B.T.; Wegmann, S. Structural studies on the mechanism of protein aggregation in age related neurodegenerative diseases. Mech. Ageing Dev. 2016, 156, 1–13. [Google Scholar] [CrossRef]
- Kovacs, G.G. Concepts and classification of neurodegenerative diseases. Handb. Clin. Neurol. 2017, 145, 301–307. [Google Scholar]
- Martinelli, A.H.S.; Lopes, F.C.; John, E.B.O.; Carlini, C.R.; Ligabue-Braun, R. Modulation of Disordered Proteins with a Focus on Neurodegenerative Diseases and Other Pathologies. Int. J. Mol. Sci. 2019, 20, 1322. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, P.; Uversky, V.N. Intrinsically Disordered Proteins in Chronic Diseases. Biomolecules 2019, 9, 147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monti, S.M.; De Simone, G.; Langella, E. The Amazing World of IDPs in Human Diseases. Biomolecules 2021, 11, 333. [Google Scholar] [CrossRef] [PubMed]
- Monti, S.M.; De Simone, G.; Langella, E. The Amazing World of IDPs in Human Diseases II. Biomolecules 2022, 12, 369. [Google Scholar] [CrossRef] [PubMed]
- Brundin, P.; Melki, R. Prying into the Prion Hypothesis for Parkinson’s Disease. J. Neurosci. 2017, 37, 9808–9818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weller, J.; Budson, A. Current understanding of Alzheimer’s disease diagnosis and treatment. F1000Research 2018, 7, 1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dos Santos Picanco, L.C.; Ozela, P.F.; de Fatima de Brito Brito, M.; Pinheiro, A.A.; Padilha, E.C.; Braga, F.S.; de Paula da Silva, C.H.T.; Dos Santos, C.B.R.; Rosa, J.M.C.; da Silva Hage-Melim, L.I. Alzheimer’s Disease: A Review from the Pathophysiology to Diagnosis, New Perspectives for Pharmacological Treatment. Curr. Med. Chem. 2018, 25, 3141–3159. [Google Scholar] [CrossRef] [PubMed]
- Ledeen, R.W.; Wu, G. Gangliosides, α-Synuclein, and Parkinson’s Disease. Prog. Mol. Biol. Transl. Sci. 2018, 156, 435–454. [Google Scholar]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [Green Version]
- Coskuner, O.; Uversky, V.N. Intrinsically disordered proteins in various hypotheses on the pathogenesis of Alzheimer’s and Parkinson’s diseases. Prog. Mol. Biol. Transl. Sci. 2019, 166, 145–223. [Google Scholar]
- Foster, E.M.; Dangla-Valls, A.; Lovestone, S.; Ribe, E.M.; Buckley, N.J. Clusterin in Alzheimer’s Disease: Mechanisms, Genetics, and Lessons from Other Pathologies. Front. Neurosci. 2019, 13, 164. [Google Scholar] [CrossRef] [Green Version]
- Coskuner-Weber, O.; Mirzanli, O.; Uversky, V.N. Intrinsically disordered proteins and proteins with intrinsically disordered regions in neurodegenerative diseases. Biophys. Rev. 2022, 14, 679–707. [Google Scholar] [CrossRef] [PubMed]
- Akbayrak, I.Y.; Caglayan, S.I.; Ozcan, Z.; Uversky, V.N.; Coskuner-Weber, O. Current Challenges and Limitations in the Studies of Intrinsically Disordered Proteins in Neurodegenerative Diseases by Computer Simulations. Curr. Alzheimer Res. 2020, 17, 805–818. [Google Scholar] [CrossRef] [PubMed]
- Roterman, I.; Stapor, K.; Fabian, P.; Konieczny, L. In Silico Modeling of the Influence of Environment on Amyloid Folding Using FOD-M Model. Int. J. Mol. Sci. 2021, 19, 10587. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkov, P.; Myers, N.; Adler, J.; Shaul, Y. Degradation of Intrinsically Disordered Proteins by the NADH 26S Proteasome. Biomolecules 2020, 10, 1642. [Google Scholar] [CrossRef]
- Gadhave, K.; Kumar, P.; Kapuganti, S.K.; Uversky, V.N.; Giri, R. Unstructured Biology of Proteins from Ubiquitin-Proteasome System: Roles in Cancer and Neurodegenerative Diseases. Biomolecules 2020, 10, 796. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Alarcon, D.; Claveria-Gimeno, R.; Vega, S.; Jorge-Torres, O.C.; Esteller, M.; Abian, O.; Velazquez-Campoy, A. Molecular Context-Dependent Effects Induced by Rett Syndrome-Associated Mutations in MeCP2. Biomolecules 2020, 10, 1533. [Google Scholar] [CrossRef]
- Ortega-Alarcon, D.; Claveria-Gimeno, R.; Vega, S.; Jorge-Torres, O.C.; Esteller, M.; Abian, O.; Velazquez-Campoy, A. Stabilization Effect of Intrinsically Disordered Regions on Multidomain Proteins: The Case of the Methyl-CpG Protein 2, MeCP2. Biomolecules 2021, 11, 1216. [Google Scholar] [CrossRef]
- Neira, J.L.; Rizzuti, B.; Jimenez-Alesanco, A.; Palomino-Schätzlein, M.; Abian, O.; Velazquez-Campoy, A.; Iovanna, J.L. A Phosphorylation-Induced Switch in the Nuclear Localization Sequence of the Intrinsically Disordered NUPR1 Hampers Binding to Importin. Biomolecule 2020, 10, 1313. [Google Scholar] [CrossRef]
- Wong, E.T.C.; So, V.; Guron, M.; Kuechler, E.R.; Malhis, N.; Bui, J.M.; Gsponer, J. Protein-Protein Interactions Mediated by Intrinsically Disordered Protein Regions Are Enriched in Missense Mutations. Biomolecules 2020, 10, 1097. [Google Scholar] [CrossRef]
- Raut, K.K.; Ponniah, K.; Pascal, S.M. Structural Analysis of the cl-Par-4 Tumor Suppressor as a Function of Ionic Environment. Biomolecules 2021, 11, 3386. [Google Scholar] [CrossRef]
- Wang, J.; Cao, Z.; Zhao, L.; Li, S. Novel strategies for drug discovery based on Intrinsically Disordered Proteins (IDPs). Int. J. Mol. Sci. 2011, 12, 3205–3219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melo, A.M.; Coraor, J.; Alpha-Cobb, G.; Elbaum-Garfinkle, S.; Nath, A.; Rhoades, E. A functional role for intrinsic disorder in the tau-tubulin complex. Proc. Natl. Acad. Sci. USA 2016, 113, 14336–14341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, G.; Wu, Z.; Uversky, V.N.; Kurgan, L. Functional Analysis of Human Hub Proteins and Their Interactors Involved in the Intrinsic Disorder-Enriched Interactions. Int. J. Mol. Sci. 2017, 18, 2761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Necci, M.; Piovesan, D. CAID Predictors, DisProt Curators, Tosatto SCE Critical assessment of protein intrinsic disorder prediction. Nat. Methods 2021, 18, 472–481. [Google Scholar] [CrossRef] [PubMed]
- Necci, M.; Piovesan, D.; Tosatto, S.C. Large-scale analysis of intrinsic disorder flavors and associated functions in the protein sequence universe. Protein Sci. 2016, 25, 2164–2174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, X.; Li, X.; Lin, X. A Review on Applications of Computational Methods in Drug Screening and Design. Molecules 2020, 25, 1375. [Google Scholar] [CrossRef] [Green Version]
- Larsson, P.; Engqvist, H.; Biermann, J.; Werner Rönnerman, E.; Forssell-Aronsson, E.; Kovács, A.; Karlsson, P.; Helou, K.; Parris, T.Z. Optimization of cell viability assays to improve replicability and reproducibility of cancer drug sensitivity screens. Sci. Rep. 2020, 10, 5798. [Google Scholar] [CrossRef] [Green Version]
- Kitaeva, K.V.; Rutland, C.S.; Rizvanov, A.A.; Solovyeva, V.V. Cell Culture Based in vitro Test Systems for Anticancer Drug Screening. Front. Bioeng. Biotechnol. 2020, 8, 322. [Google Scholar] [CrossRef] [Green Version]
- Rothenaigner, I.; Hadian, K. Brief Guide: Experimental Strategies for High-Quality Hit Selection from Small-Molecule Screening Campaigns. SLAS Discov. 2021, 7, 851–854. [Google Scholar] [CrossRef]
- Tanoli, Z.; Aldahdooh, J.; Alam, F.; Wang, Y.; Seemab, U.; Fratelli, M.; Pavlis, P.; Hajduch, M.; Bietrix, F.; Gribbon, P.; et al. Minimal information for chemosensitivity assays (MICHA): A next-generation pipeline to enable the FAIRification of drug screening experiments. Brief Bioinform. 2022, 23, bbab350. [Google Scholar] [CrossRef]
- Rizzuti, B.; Lan, W.; Santofimia-Castaño, P.; Zhou, Z.; Velázquez-Campoy, A.; Abián, O.; Peng, L.; Neira, J.L.; Xia, Y.; Iovanna, J.L. Design of Inhibitors of the Intrinsically Disordered Protein NUPR1: Balance between Drug Affinity and Target Function. Biomolecules 2021, 11, 1453. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, S.; Lopus, M.; Hosur, R.V. Targeting disorders in unstructured and structured proteins in various diseases. Biophys. Chem. 2022, 281, 106742. [Google Scholar] [CrossRef] [PubMed]
- Metallo, S.J. Intrinsically disordered proteins are potential drug targets. Curr. Opin. Chem. Biol. 2010, 14, 481–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, D.; Sharma, N.; Giri, R. Therapeutic Interventions of Cancers Using Intrinsically Disordered Proteins as Drug Targets: C-Myc as Model System. Cancer Inform. 2017, 16, 1176935117699408. [Google Scholar] [CrossRef]
- Chen, H.; Liu, H.; Qing, G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct Target Ther. 2018, 3, 5. [Google Scholar] [CrossRef] [Green Version]
- Na, I.; Choi, S.; Son, S.H.; Uversky, V.N.; Kim, C.G. Drug Discovery Targeting the Disorder-To-Order Transition Regions through the Conformational Diversity Mimicking and Statistical Analysis. Int. J. Mol. Sci. 2020, 21, 5248. [Google Scholar] [CrossRef]
- Kim, M.Y.; Na, I.; Kim, J.S.; Son, S.H.; Choi, S.; Lee, S.E.; Kim, J.H.; Jang, K.; Alterovitz, G.; Chen, Y.; et al. Rational discovery of antimetastatic agents targeting the intrinsically disordered region of MBD2. Sci. Adv. 2019, 5, eaav9810. [Google Scholar] [CrossRef] [Green Version]
- Brabletz, T.; Kalluri, R.; Nieto, M.A.; Weinberg, R.A. EMT in cancer. Nat. Rev. Cancer 2018, 18, 128–134. [Google Scholar] [CrossRef]
- Cheng, Y.; LeGall, T.; Oldfield, C.J.; Mueller, J.P.; Van, Y.Y.; Romero, P.; Cortese, M.S.; Uversky, V.N.; Dunker, A.K. Rational drug design via intrinsically disordered protein. Trends Biotechnol. 2006, 24, 435–442. [Google Scholar] [CrossRef]
- Uversky, V.N. Intrinsically disordered proteins and novel strategies for drug discovery. Expert Opin. Drug Discov. 2012, 7, 475–488. [Google Scholar] [CrossRef]
- Uversky, V.N. Intrinsic Disorder, Protein-Protein Interactions, and Disease. Adv. Protein Chem. Struct. Biol. 2018, 110, 85–121. [Google Scholar] [PubMed]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In Vivo Activation of the p53 Pathway by Small-Molecule Antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neira, J.L.; Bintz, J.; Arruebo, M.; Rizzuti, B.; Bonacci, T.; Vega, S.; Lanas, A.; Velázquez-Campoy, A.; Iovanna, J.L.; Abián, O. Identification of a Drug Targeting an Intrinsically Disordered Protein Involved in Pancreatic Adenocarcinoma. Sci. Rep. 2017, 7, 39732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, P.; Choudhary, S.; Mukherjee, S.; Sengupta, D.; Sharma, S.; Hosur, R.V. Molten globule nature of Plasmodium falciparum P2 homo-tetramer. Biochem. Biophys. Rep. 2015, 1, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Santofimia-Castano, P.; Rizzuti, B.; Xia, Y.; Abian, O.; Peng, L.; Velazquez-Campoy, A.; Neira, J.L.; Iovanna, J. Targeting intrinsically disordered proteins involved in cancer. Cell Mol Life Sci. 2020, 77, 1695–1707. [Google Scholar] [CrossRef] [Green Version]
- Pietrobono, D.; Giacomelli, C.; Trincavelli, M.L.; Daniele, S.; Martini, C. Inhibitors of protein aggregates as novel drugs in neurodegenerative diseases. Glob. Drugs Ther. 2017, 2, 1–5. [Google Scholar]
- Save, S.S.; Rachineni, K.; Hosur, R.V.; Choudhary, S. Natural compound safranal driven inhibition and dis-aggregation of α-synuclein fibrils. Int. J. Biol. Macromol. 2019, 141, 585–595. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trivedi, R.; Nagarajaram, H.A. Intrinsically Disordered Proteins: An Overview. Int. J. Mol. Sci. 2022, 23, 14050. https://doi.org/10.3390/ijms232214050
Trivedi R, Nagarajaram HA. Intrinsically Disordered Proteins: An Overview. International Journal of Molecular Sciences. 2022; 23(22):14050. https://doi.org/10.3390/ijms232214050
Chicago/Turabian StyleTrivedi, Rakesh, and Hampapathalu Adimurthy Nagarajaram. 2022. "Intrinsically Disordered Proteins: An Overview" International Journal of Molecular Sciences 23, no. 22: 14050. https://doi.org/10.3390/ijms232214050
APA StyleTrivedi, R., & Nagarajaram, H. A. (2022). Intrinsically Disordered Proteins: An Overview. International Journal of Molecular Sciences, 23(22), 14050. https://doi.org/10.3390/ijms232214050