
The Lack of STING Impairs the MHC-I Dependent Antigen Presentation and JAK/STAT Signaling in Murine Macrophages

 ,
, 
 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
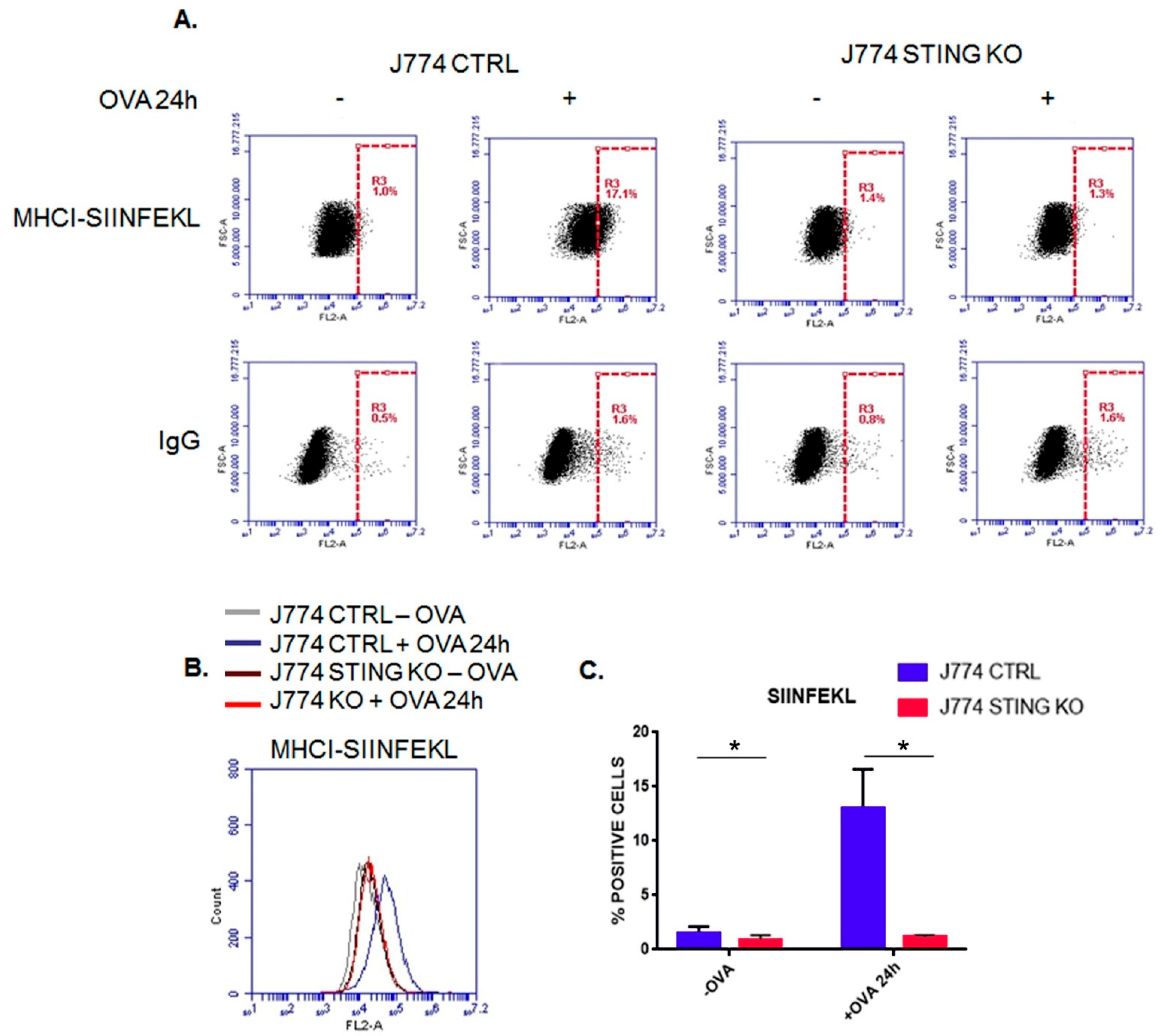
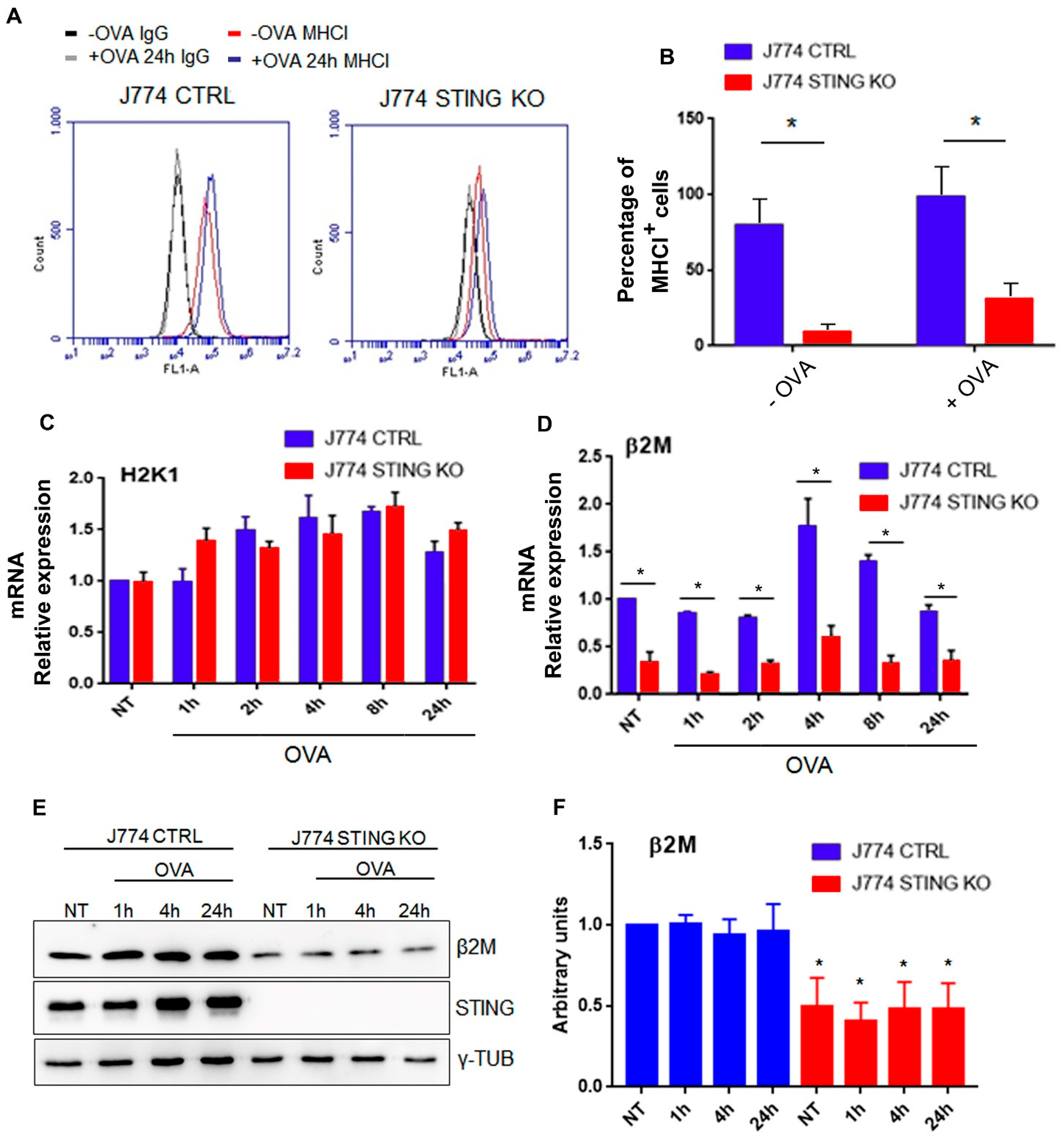
2.1. The Lack of STING Impairs the MHC-I-Dependent Antigen Presentation
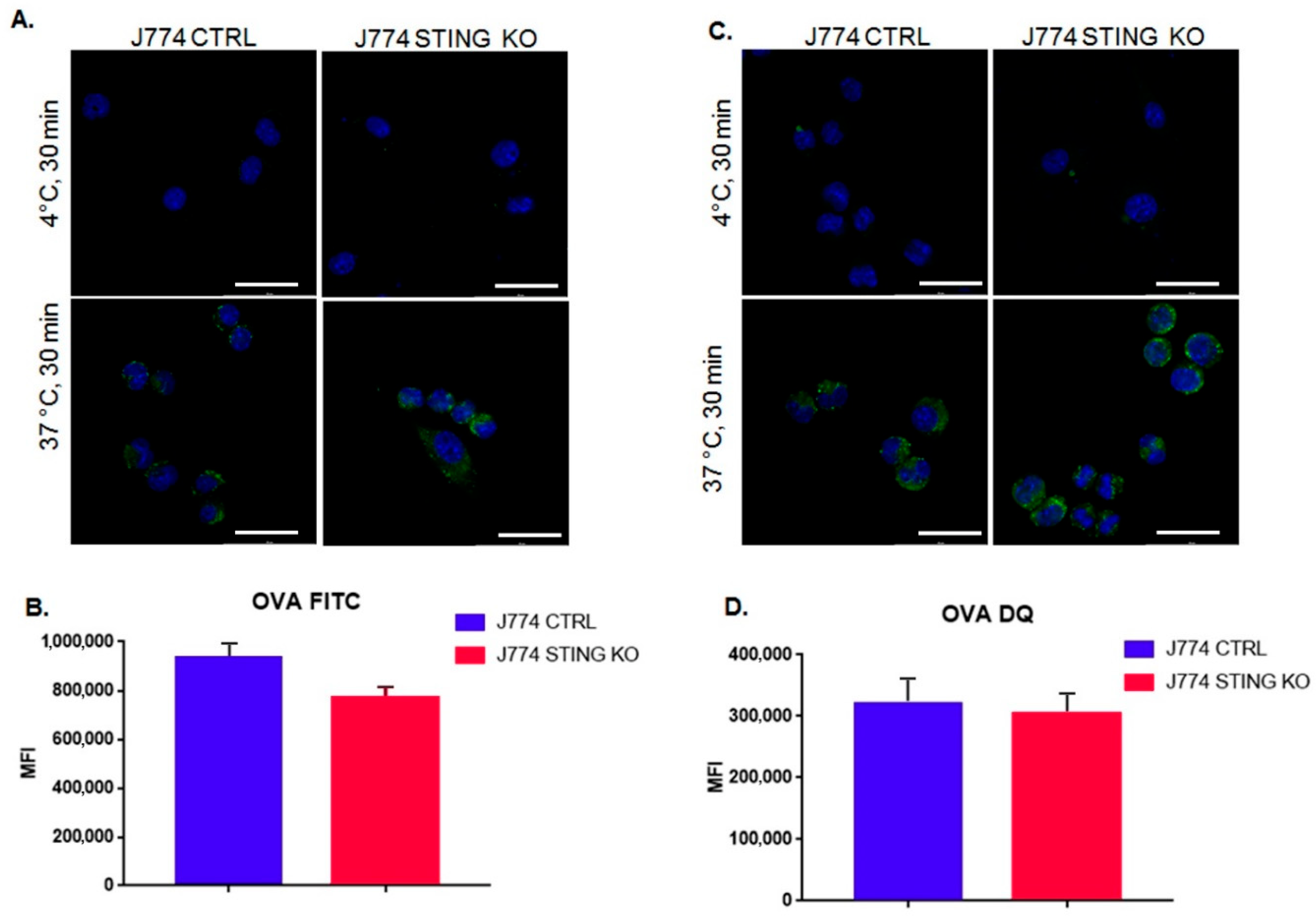
2.2. The Lack of STING Does Not Affect the Entry and Proteolysis of OVA Exogenous Protein and the Peptide-Loading Complex
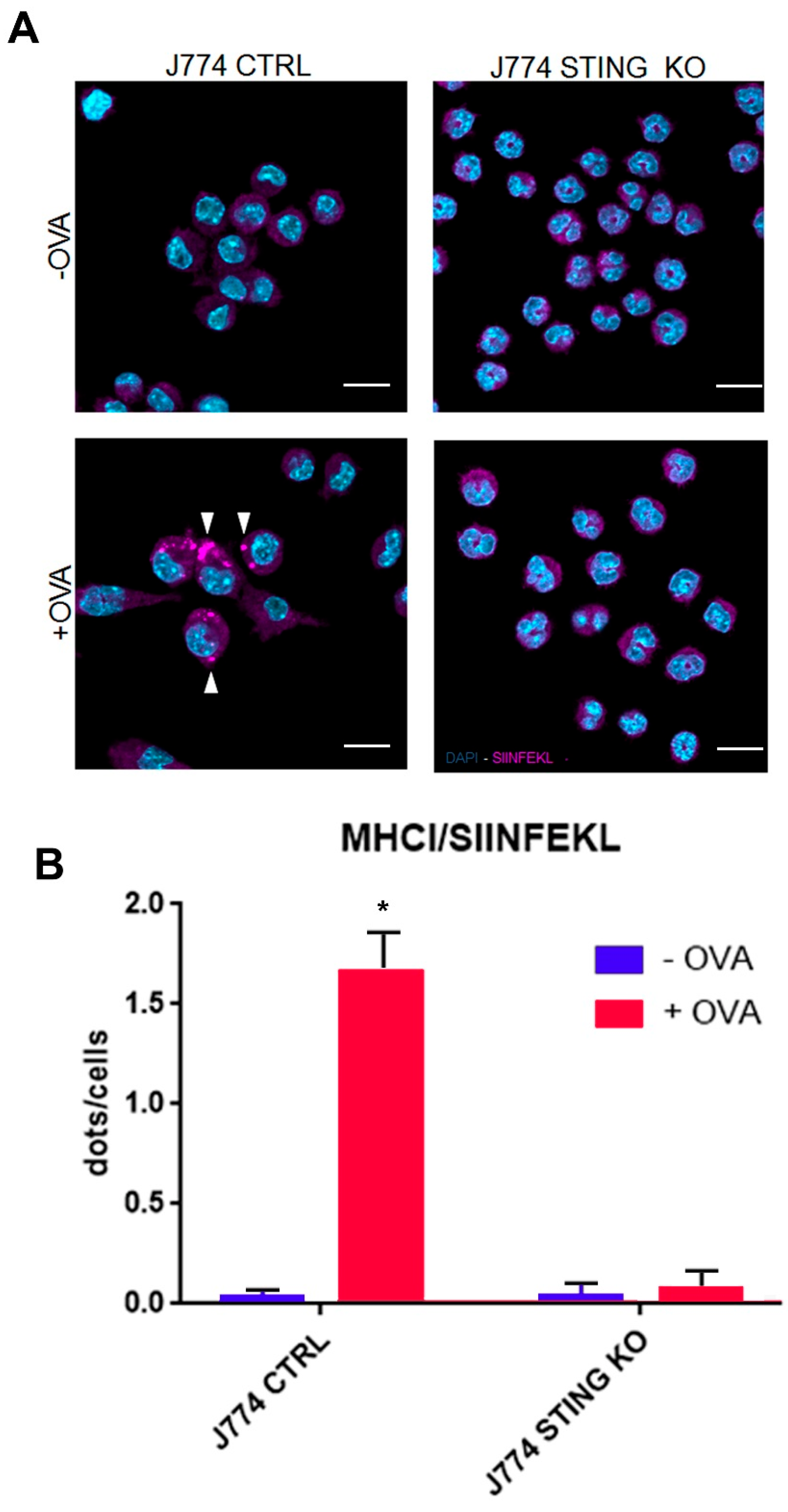
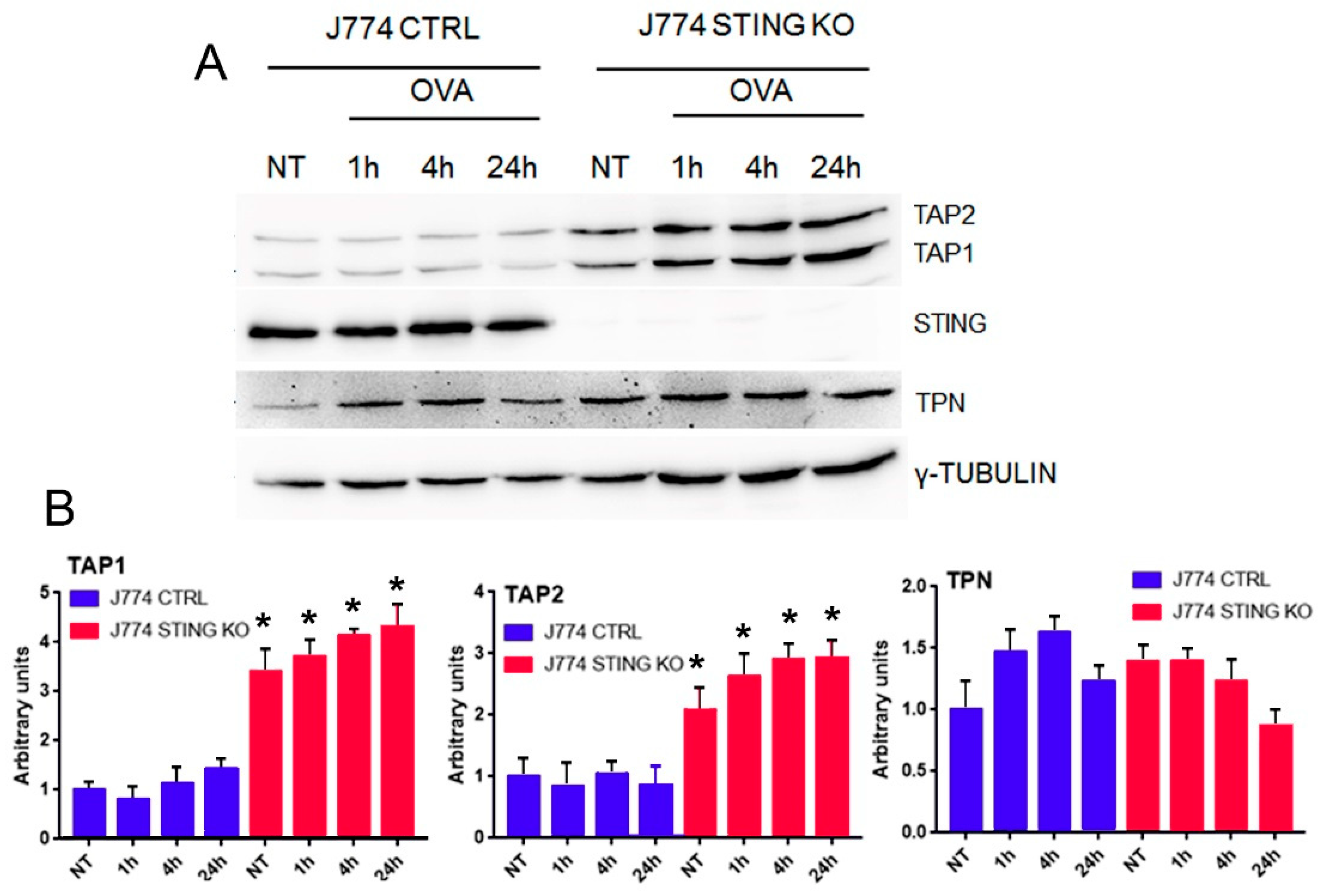
2.3. STING Is Required for an Efficient Expression of β2-Microglobulin and for the Formation of the MHC-I Complex
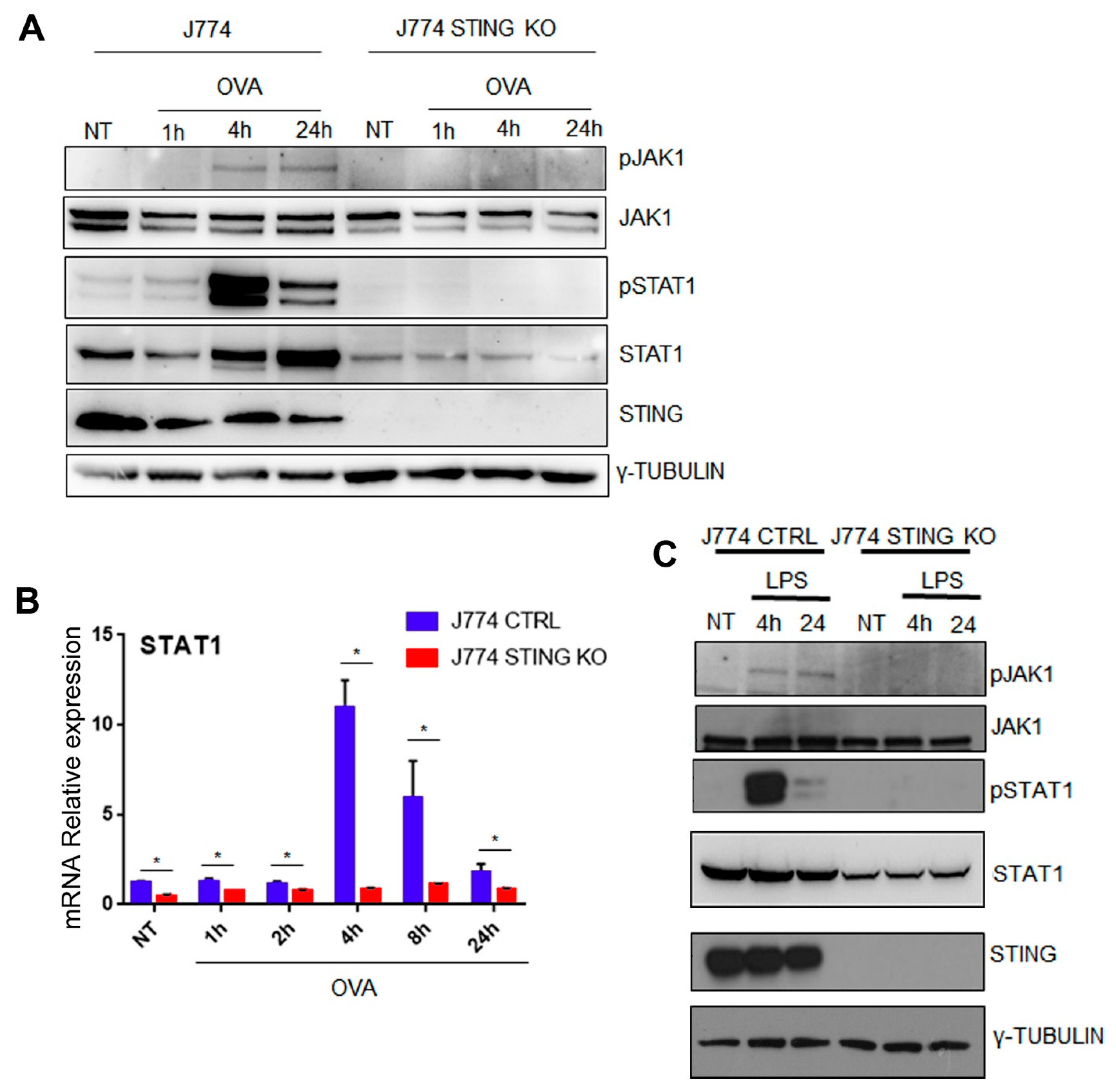
2.4. The Lack of STING Impairs the JAK/STAT Signaling
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Antibodies
4.3. Plasmids
4.4. Cell Culture, Treatments, and Viral Transductions
4.5. Western Blot Analysis
4.6. Uptake and Proteolysis Assay
4.7. Fluorescence Microscopy
4.8. Flow Cytometry
4.9. Quantitative Real-Time PCR (RT-qPCR)
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huang, Y.H.; Liu, X.Y.; Du, X.X.; Jiang, Z.F.; Su, X.D. The structural basis for the sensing and binding of cyclic di-gmp by sting. Nat. Struct. Mol. Biol. 2012, 19, 728–730. [Google Scholar] [CrossRef] [PubMed]
- Srikanth, S.; Woo, J.S.; Wu, B.; El-Sherbiny, Y.M.; Leung, J.; Chupradit, K.; Rice, L.; Seo, G.J.; Calmettes, G.; Ramakrishna, C.; et al. The ca(2+) sensor stim1 regulates the type i interferon response by retaining the signaling adaptor sting at the endoplasmic reticulum. Nat. Immunol. 2019, 20, 152–162. [Google Scholar] [CrossRef] [Green Version]
- Motwani, M.; Pesiridis, S.; Fitzgerald, K.A. DNA sensing by the cgas-sting pathway in health and disease. Nat. Rev. Genet. 2019, 20, 657–674. [Google Scholar] [CrossRef]
- Liu, S.; Cai, X.; Wu, J.; Cong, Q.; Chen, X.; Li, T.; Du, F.; Ren, J.; Wu, Y.T.; Grishin, N.V.; et al. Phosphorylation of innate immune adaptor proteins mavs, sting, and trif induces irf3 activation. Science 2015, 347, aaa2630. [Google Scholar] [CrossRef] [Green Version]
- Abe, T.; Barber, G.N. Cytosolic-DNA-mediated, sting-dependent proinflammatory gene induction necessitates canonical nf-kappab activation through tbk1. J. Virol. 2014, 88, 5328–5341. [Google Scholar] [CrossRef] [Green Version]
- Fang, R.; Wang, C.; Jiang, Q.; Lv, M.; Gao, P.; Yu, X.; Mu, P.; Zhang, R.; Bi, S.; Feng, J.M.; et al. Nemo-ikkbeta are essential for irf3 and nf-kappab activation in the cgas-sting pathway. J. Immunol. 2017, 199, 3222–3233. [Google Scholar] [CrossRef] [Green Version]
- Blum, J.S.; Wearsch, P.A.; Cresswell, P. Pathways of antigen processing. Annu. Rev. Immunol. 2013, 31, 443–473. [Google Scholar] [CrossRef] [Green Version]
- Albano, F.; Vecchio, E.; Renna, M.; Iaccino, E.; Mimmi, S.; Caiazza, C.; Arcucci, A.; Avagliano, A.; Pagliara, V.; Donato, G.; et al. Insights into thymus development and viral thymic infections. Viruses 2019, 11, 836. [Google Scholar] [CrossRef] [Green Version]
- Adams, E.J.; Luoma, A.M. The adaptable major histocompatibility complex (mhc) fold: Structure and function of nonclassical and mhc class i-like molecules. Annu. Rev. Immunol. 2013, 31, 529–561. [Google Scholar] [CrossRef]
- Oancea, G.; O’Mara, M.L.; Bennett, W.F.; Tieleman, D.P.; Abele, R.; Tampe, R. Structural arrangement of the transmission interface in the antigen abc transport complex tap. Proc. Natl. Acad. Sci. USA 2009, 106, 5551–5556. [Google Scholar] [CrossRef] [PubMed]
- Ortmann, B.; Copeman, J.; Lehner, P.J.; Sadasivan, B.; Herberg, J.A.; Grandea, A.G.; Riddell, S.R.; Tampe, R.; Spies, T.; Trowsdale, J.; et al. A critical role for tapasin in the assembly and function of multimeric mhc class i-tap complexes. Science 1997, 277, 1306–1309. [Google Scholar] [CrossRef] [Green Version]
- Hebert, D.N.; Garman, S.C.; Molinari, M. The glycan code of the endoplasmic reticulum: Asparagine-linked carbohydrates as protein maturation and quality-control tags. Trends Cell Biol. 2005, 15, 364–370. [Google Scholar] [CrossRef]
- Saveanu, L.; Carroll, O.; Lindo, V.; Del Val, M.; Lopez, D.; Lepelletier, Y.; Greer, F.; Schomburg, L.; Fruci, D.; Niedermann, G.; et al. Concerted peptide trimming by human erap1 and erap2 aminopeptidase complexes in the endoplasmic reticulum. Nat. Immunol. 2005, 6, 689–697. [Google Scholar] [CrossRef] [PubMed]
- Dersh, D.; Yewdell, J.W.; Wei, J. A siinfekl-based system to measure mhc class i antigen presentation efficiency and kinetics. Methods Mol. Biol. 2019, 1988, 109–122. [Google Scholar]
- Elsayed, S.; Hammer, A.S.; Kalvenes, M.B.; Florvaag, E.; Apold, J.; Vik, H. Antigenic and allergenic determinants of ovalbumin. I. Peptide mapping, cleavage at the methionyl peptide bonds and enzymic hydrolysis of native and carboxymethyl oa. Int. Arch. Allergy Appl. Immunol. 1986, 79, 101–107. [Google Scholar] [CrossRef]
- Elsayed, S.; Holen, E.; Haugstad, M.B. Antigenic and allergenic determinants of ovalbumin. II. The reactivity of the nh2 terminal decapeptide. Scand. J. Immunol. 1988, 27, 587–591. [Google Scholar] [CrossRef]
- Mant, A.; Chinnery, F.; Elliott, T.; Williams, A.P. The pathway of cross-presentation is influenced by the particle size of phagocytosed antigen. Immunology 2012, 136, 163–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muno, D.; Kominami, E.; Mizuochi, T. Generation of both mhc class i- and class ii-restricted antigenic peptides from exogenously added ovalbumin in murine phagosomes. FEBS Lett. 2000, 478, 178–182. [Google Scholar] [CrossRef] [Green Version]
- Chandra, J.; Miao, Y.; Romoff, N.; Frazer, I.H. Epithelium expressing the e7 oncoprotein of hpv16 attracts immune-modulatory dendritic cells to the skin and suppresses their antigen-processing capacity. PLoS ONE 2016, 11, e0152886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oura, J.; Tamura, Y.; Kamiguchi, K.; Kutomi, G.; Sahara, H.; Torigoe, T.; Himi, T.; Sato, N. Extracellular heat shock protein 90 plays a role in translocating chaperoned antigen from endosome to proteasome for generating antigenic peptide to be cross-presented by dendritic cells. Int. Immunol. 2011, 23, 223–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shastri, N.; Gonzalez, F. Endogenous generation and presentation of the ovalbumin peptide/kb complex to t cells. J. Immunol. 1993, 150, 2724–2736. [Google Scholar]
- Drew, P.D.; Franzoso, G.; Becker, K.G.; Bours, V.; Carlson, L.M.; Siebenlist, U.; Ozato, K. Nf kappa b and interferon regulatory factor 1 physically interact and synergistically induce major histocompatibility class i gene expression. J. Interferon Cytokine Res. 1995, 15, 1037–1045. [Google Scholar] [CrossRef] [PubMed]
- Gobin, S.J.; Biesta, P.; Van den Elsen, P.J. Regulation of human beta 2-microglobulin transactivation in hematopoietic cells. Blood 2003, 101, 3058–3064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rein, T.; Muller, M.; Zorbas, H. In vivo footprinting of the irf-1 promoter: Inducible occupation of a gas element next to a persistent structural alteration of the DNA. Nucleic Acids Res. 1994, 22, 3033–3037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Froechlich, G.; Caiazza, C.; Gentile, C.; D’Alise, A.M.; De Lucia, M.; Langone, F.; Leoni, G.; Cotugno, G.; Scisciola, V.; Nicosia, A.; et al. Integrity of the antiviral sting-mediated DNA sensing in tumor cells is required to sustain the immunotherapeutic efficacy of herpes simplex oncolytic virus. Cancers 2020, 12, 3407. [Google Scholar] [CrossRef]
- Guan, J.; Lu, C.; Jin, Q.; Lu, H.; Chen, X.; Tian, L.; Zhang, Y.; Ortega, J.; Zhang, J.; Siteni, S.; et al. Mlh1 deficiency-triggered DNA hyperexcision by exonuclease 1 activates the cgas-sting pathway. Cancer Cell 2021, 39, 109–121.e105. [Google Scholar] [CrossRef] [PubMed]
- Barnowski, C.; Ciupka, G.; Tao, R.; Jin, L.; Busch, D.H.; Tao, S.; Drexler, I. Efficient induction of cytotoxic t cells by viral vector vaccination requires sting-dependent dc functions. Front. Immunol. 2020, 11, 1458. [Google Scholar] [CrossRef] [PubMed]
- Woznica, A.; Kumar, A.; Sturge, C.R.; Xing, C.; King, N.; Pfeiffer, J.K. Sting mediates immune responses in the closest living relatives of animals. Elife 2021, 10, e70436. [Google Scholar] [CrossRef]
- Hogquist, K.A.; Jameson, S.C.; Heath, W.R.; Howard, J.L.; Bevan, M.J.; Carbone, F.R. T cell receptor antagonist peptides induce positive selection. Cell 1994, 76, 17–27. [Google Scholar] [CrossRef]
- Shields, M.J.; Assefi, N.; Hodgson, W.; Kim, E.J.; Ribaudo, R.K. Characterization of the interactions between mhc class i subunits: A systematic approach for the engineering of higher affinity variants of beta 2-microglobulin. J. Immunol. 1998, 160, 2297–2307. [Google Scholar] [PubMed]
- Tatake, R.J.; Ferrone, S.; Zeff, R.A. The role of beta-2 microglobulin in temperature-sensitive and interferon-gamma-induced exocytosis of hla class i molecules. Transplantation 1992, 54, 395–403. [Google Scholar] [CrossRef]
- Frank, D.A.; Mahajan, S.; Ritz, J. Fludarabine-induced immunosuppression is associated with inhibition of stat1 signaling. Nat. Med. 1999, 5, 444–447. [Google Scholar] [CrossRef]
- Lam, K.C.; Araya, R.E.; Huang, A.; Chen, Q.; Di Modica, M.; Rodrigues, R.R.; Lopes, A.; Johnson, S.B.; Schwarz, B.; Bohrnsen, E.; et al. Microbiota triggers sting-type i ifn-dependent monocyte reprogramming of the tumor microenvironment. Cell 2021, 184, 5338–5356.e21. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, Y.; Hong, W.; Wang, B.; Chen, Y.; Yang, P.; Zhou, J.; Fan, J.; Zeng, Z.; Du, S. Gut microbiota modulate radiotherapy-associated antitumor immune responses against hepatocellular carcinoma via sting signaling. Gut Microbes 2022, 14, 2119055. [Google Scholar] [CrossRef]
- Vitagliano, L.; Fiume, G.; Scognamiglio, P.L.; Doti, N.; Cannavo, R.; Puca, A.; Pedone, C.; Scala, G.; Quinto, I.; Marasco, D. Structural and functional insights into ikappab-alpha/hiv-1 tat interaction. Biochimie 2011, 93, 1592–1600. [Google Scholar] [CrossRef] [PubMed]
- Caiazza, C.; D’Agostino, M.; Passaro, F.; Faicchia, D.; Mallardo, M.; Paladino, S.; Pierantoni, G.M.; Tramontano, D. Effects of long-term citrate treatment in the pc3 prostate cancer cell line. Int. J. Mol. Sci. 2019, 20, 2613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vecchio, E.; Caiazza, C.; Mimmi, S.; Avagliano, A.; Iaccino, E.; Brusco, T.; Nistico, N.; Maisano, D.; Aloisio, A.; Quinto, I.; et al. Metabolites profiling of melanoma interstitial fluids reveals uridine diphosphate as potent immune modulator capable of limiting tumor growth. Front. Cell Dev. Biol. 2021, 9, 730726. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative pcr and the 2(-delta delta c(t)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Albano, F.; Chiurazzi, F.; Mimmi, S.; Vecchio, E.; Pastore, A.; Cimmino, C.; Frieri, C.; Iaccino, E.; Pisano, A.; Golino, G.; et al. The expression of inhibitor of bruton’s tyrosine kinase gene is progressively up regulated in the clinical course of chronic lymphocytic leukaemia conferring resistance to apoptosis. Cell Death Dis. 2018, 9, 13. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caiazza, C.; Brusco, T.; D’Alessio, F.; D’Agostino, M.; Avagliano, A.; Arcucci, A.; Ambrosino, C.; Fiume, G.; Mallardo, M. The Lack of STING Impairs the MHC-I Dependent Antigen Presentation and JAK/STAT Signaling in Murine Macrophages. Int. J. Mol. Sci. 2022, 23, 14232. https://doi.org/10.3390/ijms232214232
Caiazza C, Brusco T, D’Alessio F, D’Agostino M, Avagliano A, Arcucci A, Ambrosino C, Fiume G, Mallardo M. The Lack of STING Impairs the MHC-I Dependent Antigen Presentation and JAK/STAT Signaling in Murine Macrophages. International Journal of Molecular Sciences. 2022; 23(22):14232. https://doi.org/10.3390/ijms232214232
Chicago/Turabian StyleCaiazza, Carmen, Teresa Brusco, Federica D’Alessio, Massimo D’Agostino, Angelica Avagliano, Alessandro Arcucci, Concetta Ambrosino, Giuseppe Fiume, and Massimo Mallardo. 2022. "The Lack of STING Impairs the MHC-I Dependent Antigen Presentation and JAK/STAT Signaling in Murine Macrophages" International Journal of Molecular Sciences 23, no. 22: 14232. https://doi.org/10.3390/ijms232214232
APA StyleCaiazza, C., Brusco, T., D’Alessio, F., D’Agostino, M., Avagliano, A., Arcucci, A., Ambrosino, C., Fiume, G., & Mallardo, M. (2022). The Lack of STING Impairs the MHC-I Dependent Antigen Presentation and JAK/STAT Signaling in Murine Macrophages. International Journal of Molecular Sciences, 23(22), 14232. https://doi.org/10.3390/ijms232214232