
The Four Homeostasis Knights: In Balance upon Post-Translational Modifications

 , , , and
, , , and
Abstract
:1. Introduction
2. Transcriptional and Translational Control of Protein Synthesis
2.1. Genetic and Epigenetic Control
2.2. Translational Control
3. Post-Translational Modifications
3.1. Types of PTMs
3.2. PTMs Crosstalk
3.3. PTMs in the Control of Oncogenes and TSGs Activity
- -
- RAS proteins as molecules integrating receptor signaling along pathways that control cellular growth;
- -
- c-MYC as a transcription factor acting as a key player in the development of many human cancers;
- -
- RB, as the first tumor suppressor gene identified;
- -
- p53, as the master gene in the preservation of genetic integrity, so far called “the guardian of the genome”.
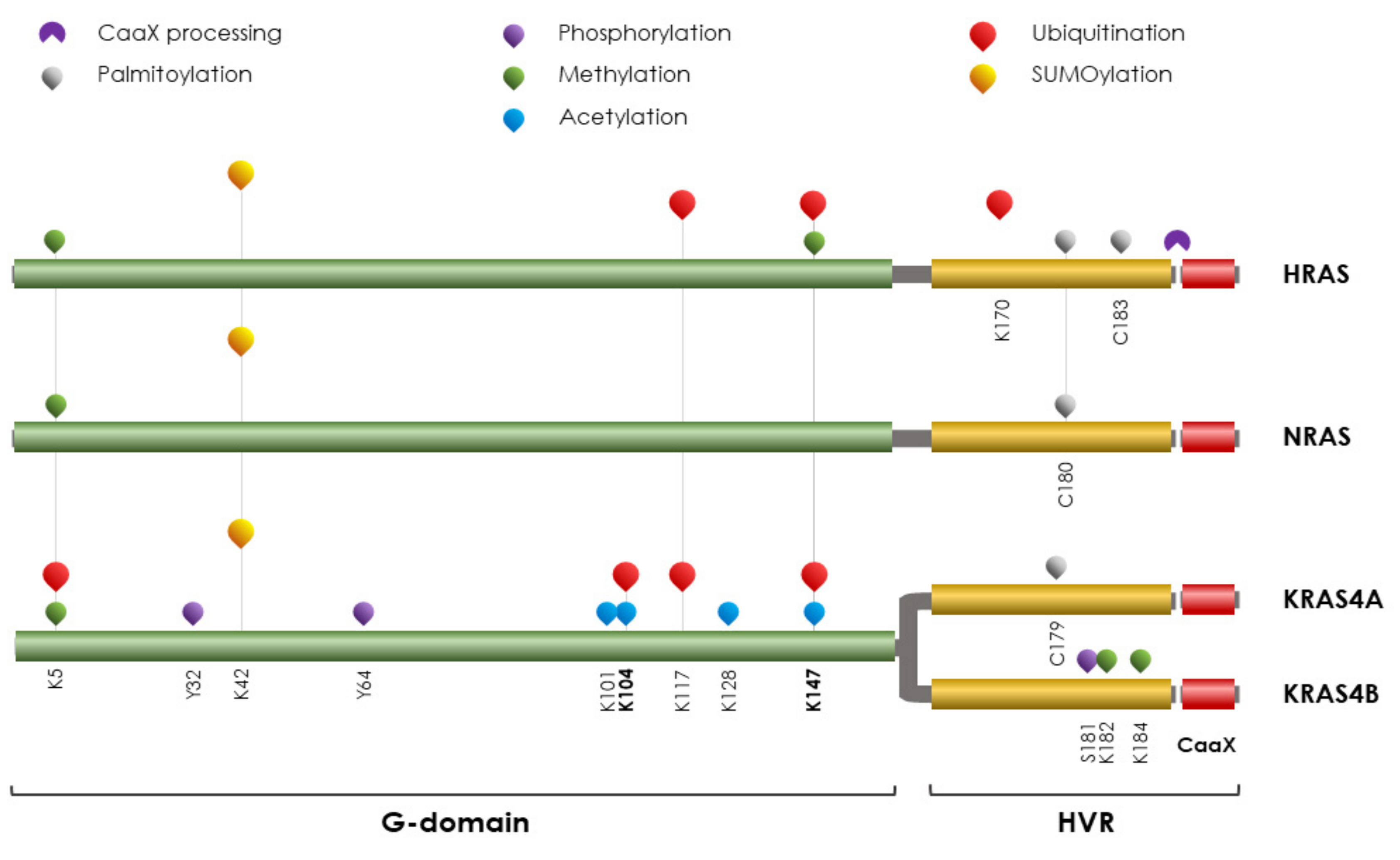
4. RAS Proteins

PTMs on RAS
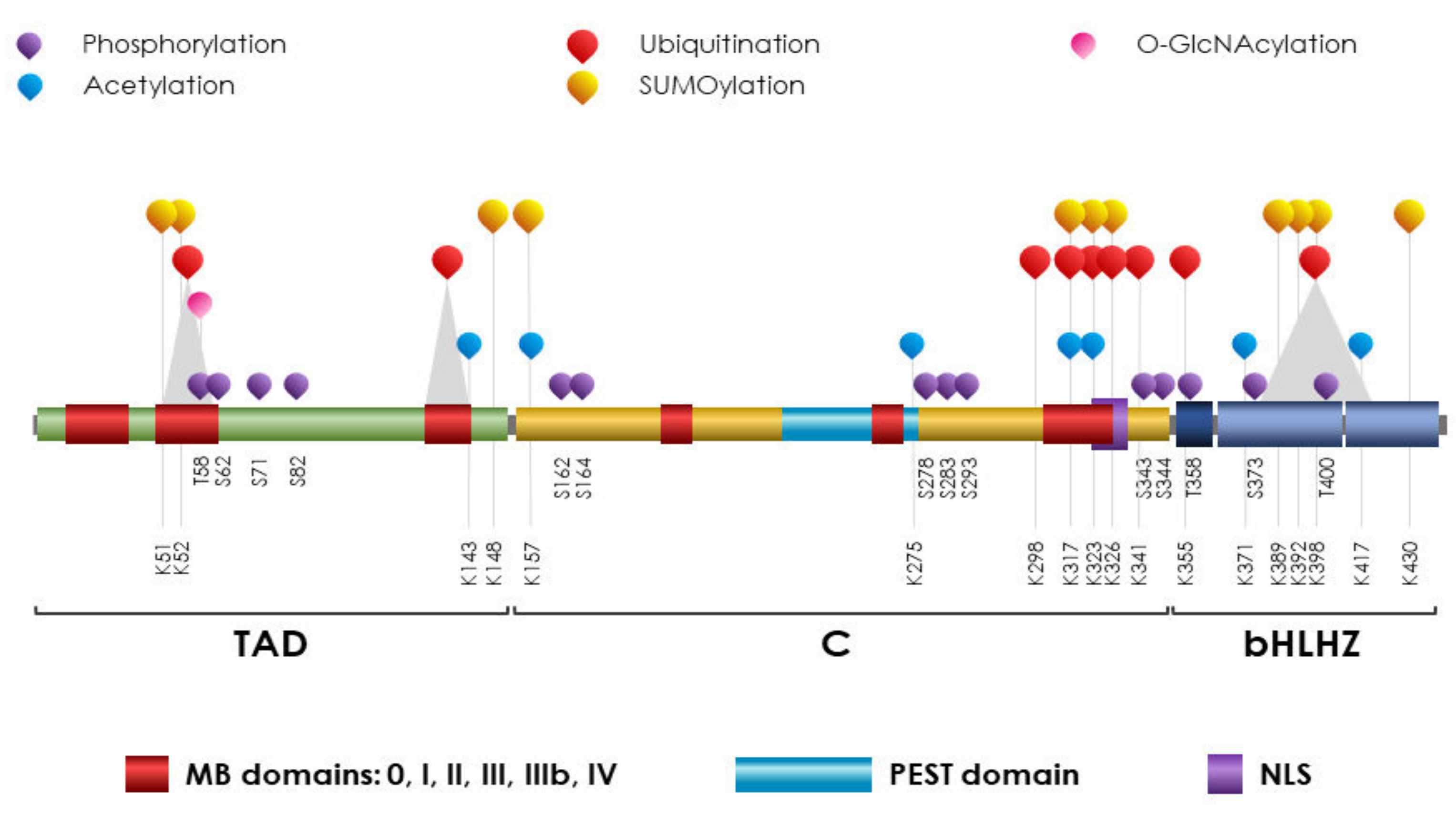
5. MYC
PTMs on MYC
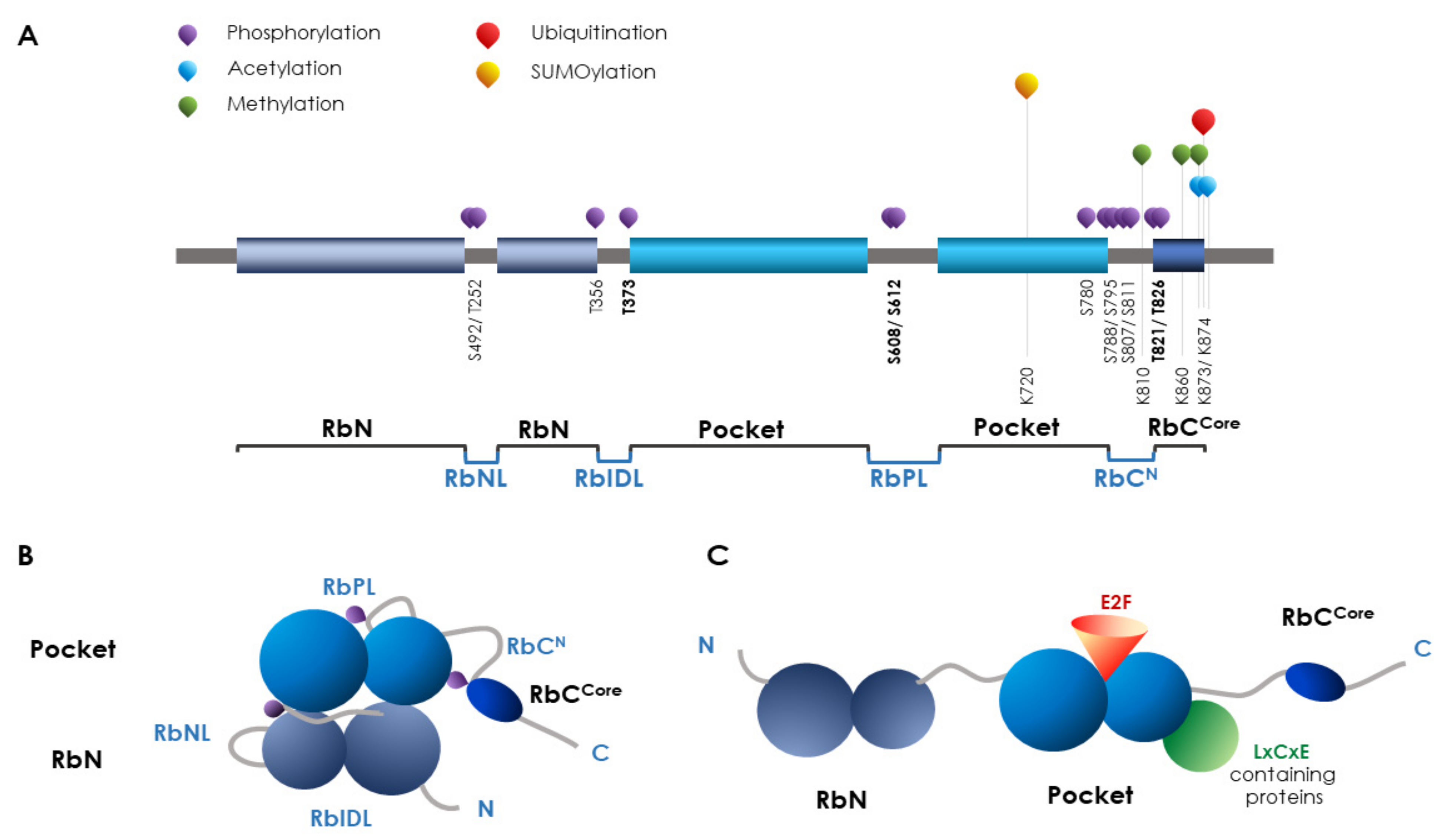
6. RB
PTMs on RB
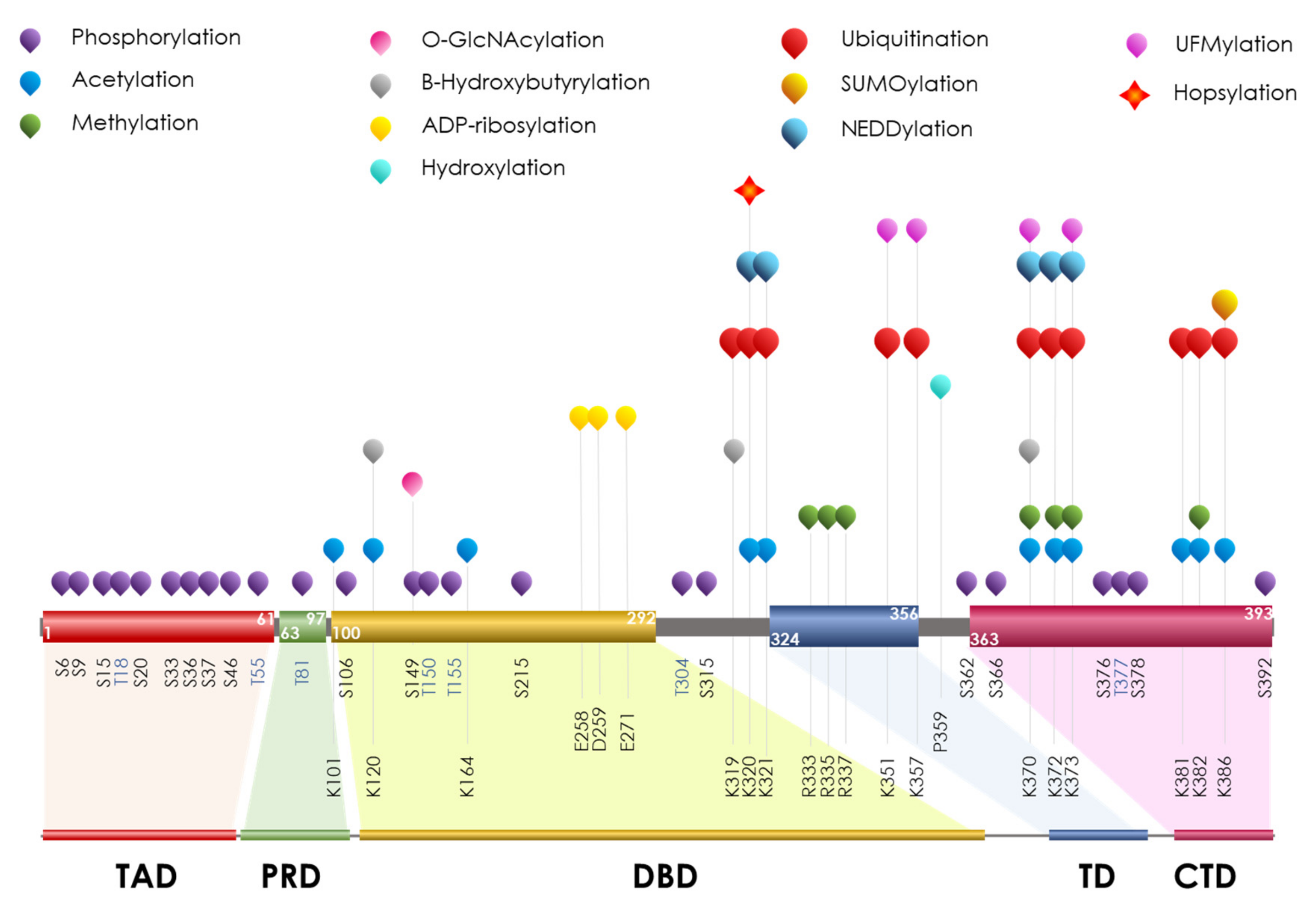
7. p53
PTMs on p53
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bishop, J.M.; Courtneidge, S.A.; Levinson, A.D.; Oppermann, H.; Quintrell, N.; Sheiness, D.K.; Weiss, S.R.; Varmus, H.E. Origin and Function of Avian Retrovirus Transforming Genes. Cold Spring Harb. Symp. Quant. Biol. 1980, 44, 919–930. [Google Scholar] [CrossRef]
- Spandidos, D.A.; Anderson, M.L.M. Oncogenes and onco-suppressor genes: Their involvement in cancer. J. Pathol. 1989, 157, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Liu, S.; Tao, Y. Regulating tumor suppressor genes: Post-translational modifications. Signal Transduct. Target. Ther. 2020, 5, 90. [Google Scholar] [CrossRef] [PubMed]
- Martincorena, I.; Campbell, P.J. Somatic mutation in cancer and normal cells. Science 2015, 349, 1483–1489. [Google Scholar] [CrossRef] [PubMed]
- Potapova, T.A.; Zhu, J.; Li, R. Aneuploidy and chromosomal instability: A vicious cycle driving cellular evolution and cancer genome chaos. Cancer Metastasis Rev. 2013, 32, 377–389. [Google Scholar] [CrossRef] [Green Version]
- Macaluso, M.; Paggi, M.G.; Giordano, A. Genetic and epigenetic alterations as hallmarks of the intricate road to cancer. Oncogene 2003, 22, 6472–6478. [Google Scholar] [CrossRef] [Green Version]
- Baxter, E.; Windloch, K.; Gannon, F.; Lee, J.S. Epigenetic regulation in cancer progression. Cell Biosci. 2014, 4, 45. [Google Scholar] [CrossRef] [Green Version]
- Laird, P.W.; Jaenisch, R. The Role of DNA Methylation in Cancer Genetics and Epigenetics. Annu. Rev. Genet. 1996, 30, 441–464. [Google Scholar] [CrossRef]
- Sutherland, J.E.; Costa, M. Epigenetics and the Environment. Ann. N. Y. Acad. Sci. 2003, 983, 151–160. [Google Scholar] [CrossRef]
- Muegge, K.; Young, H.; Ruscetti, F.; Mikovits, J. Epigenetic Control during Lymphoid Development and Immune Responses. Ann. N. Y. Acad. Sci. 2003, 983, 55–70. [Google Scholar] [CrossRef]
- Yasui, W.; Oue, N.; Ono, S.; Mitani, Y.; Ito, R.; Nakayama, H. Histone Acetylation and Gastrointestinal Carcinogenesis. Ann. N. Y. Acad. Sci. 2003, 983, 220–231. [Google Scholar] [CrossRef]
- Bird, A.P.; Wolffe, A.P. Methylation-Induced Repression— Belts, Braces, and Chromatin. Cell 1999, 99, 451–454. [Google Scholar] [CrossRef] [Green Version]
- Hayes, J.J.; Hansen, J.C. Nucleosomes and the chromatin fiber. Curr. Opin. Genet. Dev. 2001, 11, 124–129. [Google Scholar] [CrossRef]
- Jones, P.A.; Baylin, S.B. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 2002, 3, 415–428. [Google Scholar] [CrossRef]
- Nephew, K.P.; Huang, T.H.-M. Epigenetic gene silencing in cancer initiation and progression. Cancer Lett. 2002, 190, 125–133. [Google Scholar] [CrossRef]
- Brandman, O.; Hegde, R.S. Ribosome-associated protein quality control. Nat. Struct. Mol. Biol. 2016, 23, 7–15. [Google Scholar] [CrossRef] [Green Version]
- Ingolia, N.T.; Hussmann, J.A.; Weissman, J.S. Ribosome Profiling: Global Views of Translation. Cold Spring Harb. Perspect. Biol. 2018, 11, a032698. [Google Scholar] [CrossRef]
- Gloge, F.; Becker, A.H.; Kramer, G.; Bukau, B. Co-translational mechanisms of protein maturation. Curr. Opin. Struct. Biol. 2014, 24, 24–33. [Google Scholar] [CrossRef]
- Gudipaty, S.A.; Conner, C.M.; Rosenblatt, J.; Montell, D.J. Unconventional Ways to Live and Die: Cell Death and Survival in Development, Homeostasis, and Disease. Annu. Rev. Cell Dev. Biol. 2018, 34, 311–332. [Google Scholar] [CrossRef]
- Gong, Y.-N.; Crawford, J.C.; Heckmann, B.L.; Green, D.R. To the edge of cell death and back. FEBS J. 2019, 286, 430–440. [Google Scholar] [CrossRef]
- Han, Z.-J.; Feng, Y.-H.; Gu, B.-H.; Li, Y.-M.; Chen, H. The post-translational modification, SUMOylation, and cancer (Review). Int. J. Oncol. 2018, 52, 1081–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stram, A.R.; Payne, R.M. Post-translational modifications in mitochondria: Protein signaling in the powerhouse. Cell. Mol. Life Sci. 2016, 73, 4063–4073. [Google Scholar] [CrossRef] [Green Version]
- Lo, W.-S.; Trievel, R.C.; Rojas, J.R.; Duggan, L.; Hsu, J.-Y.; Allis, C.; Marmorstein, R.; Berger, S.L. Phosphorylation of Serine 10 in Histone H3 Is Functionally Linked In Vitro and In Vivo to Gcn5-Mediated Acetylation at Lysine 14. Mol. Cell 2000, 5, 917–926. [Google Scholar] [CrossRef]
- Fujiki, R.; Hashiba, W.; Sekine, H.; Yokoyama, A.; Chikanishi, T.; Ito, S.; Imai, Y.; Kim, J.; He, H.H.; Igarashi, K.; et al. GlcNAcylation of histone H2B facilitates its monoubiquitination. Nature 2011, 480, 557–560. [Google Scholar] [CrossRef] [PubMed]
- Minguez, P.; Parca, L.; Diella, F.; Mende, D.R.; Kumar, R.; Helmer-Citterich, M.; Gavin, A.-C.; van Noort, V.; Bork, P. Deciphering a global network of functionally associated post-translational modifications. Mol. Syst. Biol. 2012, 8, 599. [Google Scholar] [CrossRef]
- Huang, K.-Y.; Lee, T.-Y.; Kao, H.-J.; Ma, C.-T.; Lee, C.-C.; Lin, T.-H.; Chang, W.-C.; Huang, H.-D. dbPTM in 2019: Exploring disease association and cross-talk of post-translational modifications. Nucleic Acids Res. 2018, 47, D298–D308. [Google Scholar] [CrossRef] [Green Version]
- Vu, L.D.; Gevaert, K.; De Smet, I. Protein Language: Post-Translational Modifications Talking to Each Other. Trends Plant Sci. 2018, 23, 1068–1080. [Google Scholar] [CrossRef]
- Venne, A.S.; Kollipara, L.; Zahedi, R.P. The next level of complexity: Crosstalk of posttranslational modifications. Proteomics 2014, 14, 513–524. [Google Scholar] [CrossRef]
- Walsh, C.T.; Garneau-Tsodikova, S.; Gatto, G.J., Jr. Protein Posttranslational Modifications: The Chemistry of Proteome Diversifications. Angew. Chem. Int. Ed. 2005, 44, 7342–7372. [Google Scholar] [CrossRef]
- Horita, H.; Law, A.; Middleton, K. Utilizing Optimized Tools to Investigate PTM Crosstalk: Identifying Potential PTM Crosstalk of Acetylated Mitochondrial Proteins. Proteomes 2018, 6, 24. [Google Scholar] [CrossRef]
- Minguez, P.; Letunic, I.; Parca, L.; Garcia-Alonso, L.; Dopazo, J.; Huerta-Cepas, J.; Bork, P. PTMcode v2: A resource for functional associations of post-translational modifications within and between proteins. Nucleic Acids Res. 2014, 43, D494–D502. [Google Scholar] [CrossRef] [Green Version]
- Leutert, M.; Entwisle, S.W.; Villén, J. Decoding Post-Translational Modification Crosstalk with Proteomics. Mol. Cell. Proteom. 2021, 20, 100129. [Google Scholar] [CrossRef]
- Johnson, L.N. The regulation of protein phosphorylation. Biochem. Soc. Trans. 2009, 37, 627–641. [Google Scholar] [CrossRef]
- Boopathy, G.T.; Lynn, J.L.S.; Wee, S.; Gunaratne, J.; Hong, W. Phosphorylation of Mig6 negatively regulates the ubiquitination and degradation of EGFR mutants in lung adenocarcinoma cell lines. Cell. Signal. 2017, 43, 21–31. [Google Scholar] [CrossRef]
- Van der Laarse, S.A.; Leney, A.C.; Heck, A.J.R. Crosstalk between phosphorylation and O-GlcNAcylation: Friend or foe. FEBS J. 2018, 285, 3152–3167. [Google Scholar] [CrossRef] [Green Version]
- Leroy, C.; Shen, Q.; Strande, V.; Meyer, R.; E McLaughlin, M.; Lezan, E.; Bentires-Alj, M.; Voshol, H.; Bonenfant, D.; Gaither, L.A. CUB-domain-containing protein 1 overexpression in solid cancers promotes cancer cell growth by activating Src family kinases. Oncogene 2015, 34, 5593–5598. [Google Scholar] [CrossRef] [Green Version]
- Olivier-Van Stichelen, S.; Dehennaut, V.; Buzy, A.; Zachayus, J.-L.; Guinez, C.; Mir, A.-M.; El Yazidi-Belkoura, I.; Copin, M.-C.; Boureme, D.; Loyaux, D.; et al. O-GlcNAcylation stabilizes β-catenin through direct competition with phosphorylation at threonine 41. FASEB J. 2014, 28, 3325–3338. [Google Scholar] [CrossRef] [Green Version]
- Hunter, T. The Age of Crosstalk: Phosphorylation, Ubiquitination, and Beyond. Mol. Cell 2007, 28, 730–738. [Google Scholar] [CrossRef]
- Swaney, D.L.; Beltrao, P.; Starita, L.; Guo, A.; Rush, J.; Fields, S.; Krogan, N.J.; Villen, J. Global analysis of phosphorylation and ubiquitylation cross-talk in protein degradation. Nat. Methods 2013, 10, 676–682. [Google Scholar] [CrossRef]
- Wang, S.; Huang, X.; Sun, D.; Xin, X.; Pan, Q.; Peng, S.; Liang, Z.; Luo, C.; Yang, Y.; Jiang, H.; et al. Extensive Crosstalk between O-GlcNAcylation and Phosphorylation Regulates Akt Signaling. PLoS ONE 2012, 7, e37427. [Google Scholar] [CrossRef]
- Morrow, J.K.; Lin, H.-K.; Sun, S.-C.; Zhang, S. Targeting ubiquitination for cancer therapies. Futur. Med. Chem. 2015, 7, 2333–2350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bos, J.L. Ras Oncogenes in Human Cancer: A Review. Cancer Res. 1989, 49, 4682–4689. [Google Scholar] [PubMed]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A Comprehensive Survey of Ras Mutations in Cancer. Cancer Res 2012, 72, 2457–2467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, F.D.; Lopes, M.S.; Zhou, M.; Court, H.; Ponce, O.; Fiordalisi, J.J.; Gierut, J.J.; Cox, A.D.; Haigis, K.M.; Philips, M.R. K-Ras4A splice variant is widely expressed in cancer and uses a hybrid membrane-targeting motif. Proc. Natl. Acad. Sci. USA 2015, 112, 779–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlesinger, D.H.; Goldstein, G.; Niall, H.D. Complete amino acid sequence of ubiquitin, an adenylate cyclase stimulating polypeptide probably universal in living cells. Biochemistry 1975, 14, 2214–2218. [Google Scholar] [CrossRef]
- Goldstein, G.; Scheid, M.; Hammerling, U.; Schlesinger, D.H.; Niall, H.D.; A Boyse, E. Isolation of a polypeptide that has lymphocyte-differentiating properties and is probably represented universally in living cells. Proc. Natl. Acad. Sci. USA 1975, 72, 11–15. [Google Scholar] [CrossRef] [Green Version]
- Akutsu, M.; Dikic, I.; Bremm, A. Ubiquitin chain diversity at a glance. J. Cell Sci. 2016, 129, 875–880. [Google Scholar] [CrossRef] [Green Version]
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef]
- Scheffner, M.; Nuber, U.; Huibregtse, J.M. Protein ubiquitination involving an E1–E2–E3 enzyme ubiquitin thioester cascade. Nature 1995, 373, 81–83. [Google Scholar] [CrossRef] [Green Version]
- Schulman, B.A.; Harper, J.W. Ubiquitin-like protein activation by E1 enzymes: The apex for downstream signalling pathways. Nat. Rev. Mol. Cell Biol. 2009, 10, 319–331. [Google Scholar] [CrossRef]
- Van Wijk, S.J.L.; Timmers, H.T.M. The family of ubiquitin-conjugating enzymes (E2s): Deciding between life and death of proteins. FASEB J. 2009, 24, 981–993. [Google Scholar] [CrossRef]
- Ye, Y.; Rape, M. Building ubiquitin chains: E2 enzymes at work. Nat. Rev. Mol. Cell Biol. 2009, 10, 755–764. [Google Scholar] [CrossRef] [Green Version]
- David, Y.; Ziv, T.; Admon, A.; Navon, A. The E2 Ubiquitin-conjugating Enzymes Direct Polyubiquitination to Preferred Lysines. J. Biol. Chem. 2010, 285, 8595–8604. [Google Scholar] [CrossRef] [Green Version]
- Pao, K.-C.; Wood, N.T.; Knebel, A.; Rafie, K.; Stanley, M.; Mabbitt, P.D.; Sundaramoorthy, R.; Hofmann, K.; Van Aalten, D.M.F.; Virdee, S. Activity-based E3 ligase profiling uncovers an E3 ligase with esterification activity. Nat. Cell Biol. 2018, 556, 381–385. [Google Scholar] [CrossRef] [Green Version]
- Walden, H.; Rittinger, K. RBR ligase–mediated ubiquitin transfer: A tale with many twists and turns. Nat. Struct. Mol. Biol. 2018, 25, 440–445. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Argiles-Castillo, D.; Kane, E.I.; Zhou, A.; Spratt, D.E. HECT E3 ubiquitin ligases–Emerging insights into their biological roles and disease relevance. J. Cell Sci. 2020, 133, jcs228072. [Google Scholar] [CrossRef]
- Baek, K.; Scott, D.C.; A Schulman, B. NEDD8 and ubiquitin ligation by cullin-RING E3 ligases. Curr. Opin. Struct. Biol. 2020, 67, 101–109. [Google Scholar] [CrossRef]
- Deshaies, R.J.; Joazeiro, C.A. Ring Domain E3 Ubiquitin Ligases. Annu. Rev. Biochem. 2009, 78, 399–434. [Google Scholar] [CrossRef]
- Jevtić, P.; Haakonsen, D.L.; Rapé, M. An E3 ligase guide to the galaxy of small-molecule-induced protein degradation. Cell Chem. Biol. 2021, 28, 1000–1013. [Google Scholar] [CrossRef]
- Ohtake, F.; Saeki, Y.; Sakamoto, K.; Ohtake, K.; Nishikawa, H.; Tsuchiya, H.; Ohta, T.; Tanaka, K.; Kanno, J. Ubiquitin acetylation inhibits polyubiquitin chain elongation. EMBO Rep. 2014, 16, 192–201. [Google Scholar] [CrossRef]
- Ordureau, A.; Sarraf, S.A.; Duda, D.M.; Heo, J.-M.; Jedrychowski, M.P.; Sviderskiy, V.O.; Olszewski, J.L.; Koerber, J.T.; Xie, T.; Beausoleil, S.A.; et al. Quantitative Proteomics Reveal a Feedforward Mechanism for Mitochondrial PARKIN Translocation and Ubiquitin Chain Synthesis. Mol. Cell 2014, 56, 360–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wauer, T.; Swatek, K.N.; Wagstaff, J.L.; Gladkova, C.; Pruneda, J.N.; A Michel, M.; Gersch, M.; Johnson, C.M.; Freund, S.M.; Komander, D. Ubiquitin Ser65 phosphorylation affects ubiquitin structure, chain assembly and hydrolysis. EMBO J. 2014, 34, 307–325. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Hunter, T. Pink1, the first ubiquitin kinase. EMBO J. 2014, 33, 1621–1623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, L.P.; Philips, M.R. Thematic review series: Lipid Posttranslational Modifications CAAX modification and membrane targeting of Ras. J. Lipid Res. 2006, 47, 883–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arimura, S.; Nakata, H.; Tomiyama, K.; Watanabe, Y. Phosphorylation of H-ras Proteins by Protein Kinase A. Cell. Signal. 1997, 9, 37–40. [Google Scholar] [CrossRef]
- Ballester, R.; E Furth, M.; Rosen, O.M. Phorbol ester- and protein kinase C-mediated phosphorylation of the cellular Kirsten ras gene product. J. Biol. Chem. 1987, 262, 2688–2695. [Google Scholar] [CrossRef]
- Bivona, T.G.; Quatela, S.E.; Bodemann, B.O.; Ahearn, I.M.; Soskis, M.J.; Mor, A.; Miura, J.; Wiener, H.H.; Wright, L.; Saba, S.G.; et al. PKC Regulates a Farnesyl-Electrostatic Switch on K-Ras that Promotes its Association with Bcl-Xl on Mitochondria and Induces Apoptosis. Mol. Cell 2006, 21, 481–493. [Google Scholar] [CrossRef]
- Alvarez-Moya, B.; López-Alcalá, C.; Drosten, M.; Bachs, O.; Agell, N. K-Ras4B phosphorylation at Ser181 is inhibited by calmodulin and modulates K-Ras activity and function. Oncogene 2010, 29, 5911–5922. [Google Scholar] [CrossRef] [Green Version]
- Cho, K.-J.; Casteel, D.E.; Prakash, P.; Tan, L.; van der Hoeven, D.; Salim, A.A.; Kim, C.; Capon, R.J.; Lacey, E.; Cunha, S.R.; et al. AMPK and Endothelial Nitric Oxide Synthase Signaling Regulates K-Ras Plasma Membrane Interactions via Cyclic GMP-Dependent Protein Kinase 2. Mol. Cell. Biol. 2016, 36, 3086–3099. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.-T.; Holderfield, M.; Galeas, J.; Delrosario, R.; To, M.D.; Balmain, A.; McCormick, F. K-Ras Promotes Tumorigenicity through Suppression of Non-canonical Wnt Signaling. Cell 2015, 163, 1237–1251. [Google Scholar] [CrossRef]
- Bunda, S.; Heir, P.; Srikumar, T.; Cook, J.D.; Burrell, K.; Kano, Y.; Lee, J.E.; Zadeh, G.; Raught, B.; Ohh, M. Src promotes GTPase activity of Ras via tyrosine 32 phosphorylation. Proc. Natl. Acad. Sci. USA 2014, 111, E3785–E3794. [Google Scholar] [CrossRef] [Green Version]
- Buday, L.; Vas, V. Novel regulation of Ras proteins by direct tyrosine phosphorylation and dephosphorylation. Cancer Metastasis Rev. 2020, 39, 1067–1073. [Google Scholar] [CrossRef]
- Allfrey, V.G.; Faulkner, R.; Mirsky, A.E. Acetylation and methylation of histones and their possible role in the regulation of rna synthesis. Proc. Natl. Acad. Sci. USA 1964, 51, 786–794. [Google Scholar] [CrossRef] [Green Version]
- Gu, W.; Roeder, R.G. Activation of p53 Sequence-Specific DNA Binding by Acetylation of the p53 C-Terminal Domain. Cell 1997, 90, 595–606. [Google Scholar] [CrossRef] [Green Version]
- Lill, N.L.; Grossman, S.R.; Ginsberg, D.; DeCaprio, J.; Livingston, D.M. Binding and modulation of p53 by p300/CBP coactivators. Nature 1997, 387, 823–827. [Google Scholar] [CrossRef]
- Verdin, E.; Ott, M. 50 years of protein acetylation: From gene regulation to epigenetics, metabolism and beyond. Nat. Rev. Mol. Cell Biol. 2014, 16, 258–264. [Google Scholar] [CrossRef]
- Narita, T.; Weinert, B.T.; Choudhary, C. Functions and mechanisms of non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 2018, 20, 156–174. [Google Scholar] [CrossRef]
- Knyphausen, P.; Lang, F.; Baldus, L.; Extra, A.; Lammers, M. Insights into K-Ras 4B regulation by post-translational lysine acetylation. Biol. Chem. 2016, 397, 1071–1085. [Google Scholar] [CrossRef]
- Yang, M.H.; Nickerson, S.; Kim, E.T.; Liot, C.; Laurent, G.; Spang, R.; Philips, M.R.; Shan, Y.; Shaw, D.E.; Bar-Sagi, D.; et al. Regulation of RAS oncogenicity by acetylation. Proc. Natl. Acad. Sci. USA 2012, 109, 10843–10848. [Google Scholar] [CrossRef] [Green Version]
- Yin, G.; Kistler, S.; George, S.D.; Kuhlmann, N.; Garvey, L.; Huynh, M.; Bagni, R.K.; Lammers, M.; Der, C.J.; Campbell, S.L. A KRAS GTPase K104Q Mutant Retains Downstream Signaling by Offsetting Defects in Regulation. J. Biol. Chem. 2017, 292, 4446–4456. [Google Scholar] [CrossRef]
- Yoshino, H.; Yin, G.; Kawaguchi, R.; Popov, K.I.; Temple, B.; Sasaki, M.; Kofuji, S.; Wolfe, K.; Kofuji, K.; Okumura, K.; et al. Identification of lysine methylation in the core GTPase domain by GoMADScan. PLoS ONE 2019, 14, e0219436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, R.; Wilkerson, E.M.; Sumita, K.; Isom, D.G.; Sasaki, A.T.; Dohlman, H.G.; Campbell, S.L. Differences in the Regulation of K-Ras and H-Ras Isoforms by Monoubiquitination. J. Biol. Chem. 2013, 288, 36856–36862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, R.; Lewis, S.M.; Sasaki, A.T.; Wilkerson, E.M.; Locasale, J.W.; Cantley, L.C.; Kuhlman, B.; Dohlman, H.G.; Campbell, S. Site-specific monoubiquitination activates Ras by impeding GTPase-activating protein function. Nat. Struct. Mol. Biol. 2012, 20, 46–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, T.; Wang, Q.; Fu, J.; Lin, Q.; Bi, J.; Ding, W.; Qiao, Y.; Zhang, S.; Zhao, W.; Lin, H.; et al. Impeded Nedd4-1-Mediated Ras Degradation Underlies Ras-Driven Tumorigenesis. Cell Rep. 2014, 7, 871–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meluh, P.B.; Koshland, D. Evidence that the MIF2 gene of Saccharomyces cerevisiae encodes a centromere protein with homology to the mammalian centromere protein CENP-C. Mol. Biol. Cell 1995, 6, 793–807. [Google Scholar] [CrossRef]
- Matunis, M.; Coutavas, E.; Blobel, G. A novel ubiquitin-like modification modulates the partitioning of the Ran-GTPase-activating protein RanGAP1 between the cytosol and the nuclear pore complex. J. Cell Biol. 1996, 135, 1457–1470. [Google Scholar] [CrossRef]
- Bayer, P.; Arndt, A.; Metzger, S.; Mahajan, R.; Melchior, F.; Jaenicke, R.; Becker, J. Structure determination of the small ubiquitin-related modifier SUMO-1. J. Mol. Biol. 1998, 280, 275–286. [Google Scholar] [CrossRef]
- Owerbach, D.; McKay, E.M.; Yeh, E.T.; Gabbay, K.H.; Bohren, K.M. A proline-90 residue unique to SUMO-4 prevents maturation and sumoylation. Biochem. Biophys. Res. Commun. 2005, 337, 517–520. [Google Scholar] [CrossRef]
- Liang, Y.-C.; Lee, C.-C.; Yao, Y.-L.; Lai, C.-C.; Schmitz, L.; Yang, W.-M. SUMO5, a Novel Poly-SUMO Isoform, Regulates PML Nuclear Bodies. Sci. Rep. 2016, 6, 26509. [Google Scholar] [CrossRef] [Green Version]
- Hay, R.T. Protein modification by SUMO. Trends Biochem. Sci. 2001, 26, 332–333. [Google Scholar] [CrossRef]
- Rodriguez, M.S.; Dargemont, C.; Hay, R.T. SUMO-1 Conjugation in Vivo Requires Both a Consensus Modification Motif and Nuclear Targeting. J. Biol. Chem. 2001, 276, 12654–12659. [Google Scholar] [CrossRef] [Green Version]
- Sampson, D.A.; Wang, M.; Matunis, M.J. The Small Ubiquitin-like Modifier-1 (SUMO-1) Consensus Sequence Mediates Ubc9 Binding and Is Essential for SUMO-1 Modification. J. Biol. Chem. 2001, 276, 21664–21669. [Google Scholar] [CrossRef] [Green Version]
- Lascorz, J.; Codina-Fabra, J.; Reverter, D.; Torres-Rosell, J. SUMO-SIM interactions: From structure to biological functions. Semin. Cell Dev. Biol. 2022, 132, 193–202. [Google Scholar] [CrossRef]
- Celen, A.B.; Sahin, U. Sumoylation on its 25th anniversary: Mechanisms, pathology, and emerging concepts. FEBS J. 2020, 287, 3110–3140. [Google Scholar] [CrossRef]
- Tatham, M.; Jaffray, E.; Vaughan, O.A.; Desterro, J.; Botting, C.H.; Naismith, J.; Hay, R. Polymeric Chains of SUMO-2 and SUMO-3 Are Conjugated to Protein Substrates by SAE1/SAE2 and Ubc9. J. Biol. Chem. 2001, 276, 35368–35374. [Google Scholar] [CrossRef] [Green Version]
- Hendriks, I.A.; Vertegaal, A.C.O. A comprehensive compilation of SUMO proteomics. Nat. Rev. Mol. Cell Biol. 2016, 17, 581–595. [Google Scholar] [CrossRef]
- Matic, I.; Schimmel, J.; Hendriks, I.A.; van Santen, M.A.; van de Rijke, F.; van Dam, H.; Gnad, F.; Mann, M.; Vertegaal, A.C. Site-Specific Identification of SUMO-2 Targets in Cells Reveals an Inverted SUMOylation Motif and a Hydrophobic Cluster SUMOylation Motif. Mol. Cell 2010, 39, 641–652. [Google Scholar] [CrossRef]
- Hendriks, I.A.; Lyon, D.; Su, D.; Skotte, N.H.; Daniel, J.A.; Jensen, L.J.; Nielsen, M.L. Site-specific characterization of endogenous SUMOylation across species and organs. Nat. Commun. 2018, 9, 2456. [Google Scholar] [CrossRef] [Green Version]
- Yeh, E.T. SUMOylation and De-SUMOylation: Wrestling with Life’s Processes. J. Biol. Chem. 2009, 284, 8223–8227. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.-J.; Grégoire, S. A Recurrent Phospho-Sumoyl Switch in Transcriptional Repression and Beyond. Mol. Cell 2006, 23, 779–786. [Google Scholar] [CrossRef]
- Ullmann, R.; Chien, C.D.; Avantaggiati, M.L.; Muller, S. An Acetylation Switch Regulates SUMO-Dependent Protein Interaction Networks. Mol. Cell 2012, 46, 759–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gärtner, A.; Wagner, K.; Hölper, S.; Kunz, K.; Rodriguez, M.S.; Müller, S. Acetylation of SUMO 2 at lysine 11 favors the formation of non-canonical SUMO chains. EMBO Rep. 2018, 19, e46117. [Google Scholar] [CrossRef] [PubMed]
- Pichler, A.; Fatouros, C.; Lee, H.; Eisenhardt, N. SUMO conjugation–A mechanistic view. Biomol. Concepts 2017, 8, 13–36. [Google Scholar] [CrossRef] [PubMed]
- Eifler, K.; Vertegaal, A.C. Mapping the SUMO ylated landscape. FEBS J. 2015, 282, 3669–3680. [Google Scholar] [CrossRef] [PubMed]
- Dohlman, H.G.; Campbell, S.L. Regulation of large and small G proteins by ubiquitination. J. Biol. Chem. 2019, 294, 18613–18623. [Google Scholar] [CrossRef] [Green Version]
- Jura, N.; Scotto-Lavino, E.; Sobczyk, A.; Bar-Sagi, D. Differential Modification of Ras Proteins by Ubiquitination. Mol. Cell 2006, 21, 679–687. [Google Scholar] [CrossRef]
- Sasaki, A.T.; Carracedo, A.; Locasale, J.W.; Anastasiou, D.; Takeuchi, K.; Kahoud, E.R.; Haviv, S.; Asara, J.M.; Pandolfi, P.P.; Cantley, L.C. Ubiquitination of K-Ras Enhances Activation and Facilitates Binding to Select Downstream Effectors. Sci. Signal. 2011, 4, ra13. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.E.; Yoon, J.-Y.; Jeong, W.-J.; Jeon, S.-H.; Park, Y.N.; Yoon, J.-B.; Kim, H.; Choi, K.-Y. H-Ras is degraded by Wnt/β-catenin signaling via β-TrCP-mediated polyubiquitylation. J. Cell Sci. 2009, 122, 842–848. [Google Scholar] [CrossRef] [Green Version]
- Yan, H.; Jahanshahi, M.; Horvath, E.A.; Liu, H.-Y.; Pfleger, C.M. Rabex-5 Ubiquitin Ligase Activity Restricts Ras Signaling to Establish Pathway Homeostasis in Drosophila. Curr. Biol. 2010, 20, 1378–1382. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Lubkov, V.; Taylor, L.J.; Bar-Sagi, D. Feedback Regulation of Ras Signaling by Rabex-5-Mediated Ubiquitination. Curr. Biol. 2010, 20, 1372–1377. [Google Scholar] [CrossRef]
- Abe, T.; Umeki, I.; Kanno, S.-I.; Inoue, S.-I.; Niihori, T.; Aoki, Y. LZTR1 facilitates polyubiquitination and degradation of RAS-GTPases. Cell Death Differ. 2019, 27, 1023–1035. [Google Scholar] [CrossRef] [Green Version]
- Steklov, M.; Pandolfi, S.; Baietti, M.F.; Batiuk, A.; Carai, P.; Najm, P.; Zhang, M.; Jang, H.; Renzi, F.; Cai, Y.; et al. Mutations in LZTR1 drive human disease by dysregulating RAS ubiquitination. Science 2018, 362, 1177–1182. [Google Scholar] [CrossRef]
- Baietti, M.F.; Simicek, M.; Asbagh, L.A.; Radaelli, E.; Lievens, S.; Crowther, J.; Steklov, M.; Aushev, V.N.; García, D.M.; Tavernier, J.; et al. OTUB 1 triggers lung cancer development by inhibiting RAS monoubiquitination. EMBO Mol. Med. 2016, 8, 288–303. [Google Scholar] [CrossRef]
- Choi, B.H.; Chen, C.; Philips, M.; Dai, W. RAS GTPases are modified by SUMOylation. Oncotarget 2017, 9, 4440–4450. [Google Scholar] [CrossRef] [Green Version]
- Choi, B.H.; Philips, M.R.; Chen, Y.; Lu, L.; Dai, W. K-Ras Lys-42 is crucial for its signaling, cell migration, and invasion. J. Biol. Chem. 2018, 293, 17574–17581. [Google Scholar] [CrossRef] [Green Version]
- Xie, M.; Yu, J.; Ge, S.; Huang, J.; Fan, X. SUMOylation homeostasis in tumorigenesis. Cancer Lett. 2019, 469, 301–309. [Google Scholar] [CrossRef]
- Ahearn, I.; Zhou, M.; Philips, M.R. Posttranslational Modifications of RAS Proteins. Cold Spring Harb. Perspect. Med. 2018, 8, a031484. [Google Scholar] [CrossRef]
- Campbell, S.L.; Philips, M.R. Post-translational modification of RAS proteins. Curr. Opin. Struct. Biol. 2021, 71, 180–192. [Google Scholar] [CrossRef]
- Duesberg, P.H.; Vogt, P.K. Avian acute leukemia viruses MC29 and MH2 share specific RNA sequences: Evidence for a second class of transforming genes. Proc. Natl. Acad. Sci. USA 1979, 76, 1633–1637. [Google Scholar] [CrossRef] [Green Version]
- Sheiness, D.; Bishop, J.M. DNA and RNA from Uninfected Vertebrate Cells Contain Nucleotide Sequences Related to the Putative Transforming Gene of Avian Myelocytomatosis Virus. J. Virol. 1979, 31, 514–521. [Google Scholar] [CrossRef]
- Hu, S.S.; Lai, M.M.; Vogt, P.K. Genome of avian myelocytomatosis virus MC29: Analysis by heteroduplex mapping. Proc. Natl. Acad. Sci. USA 1979, 76, 1265–1268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vennström, B.; Bishop, J.M. Isolation and characterization of chicken DNA homologous to the two putative oncogenes of avian erythroblastosis virus. Cell 1982, 28, 135–143. [Google Scholar] [CrossRef]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingvarsson, S. The myc gene family proteins and their role in transformation and differentiation. Semin. Cancer Biol. 1990, 1, 359–369. [Google Scholar] [PubMed]
- Lee, W.-H.; Murphree, A.L.; Benedict, W.F. Expression and amplification of the N-myc gene in primary retinoblastoma. Nature 1984, 309, 458–460. [Google Scholar] [CrossRef]
- Nau, M.M.; Brooks, B.J.; Battey, J.F.; Sausville, E.; Gazdar, A.F.; Kirsch, I.R.; McBride, O.W.; Bertness, V.L.; Hollis, G.F.; Minna, J.D. L-myc, a new myc-related gene amplified and expressed in human small cell lung cancer. Nature 1985, 318, 69–73. [Google Scholar] [CrossRef]
- Grandori, C.; Cowley, S.M.; James, L.P.; Eisenman, R.N. The Myc/Max/Mad Network and the Transcriptional Control of Cell Behavior. Annu. Rev. Cell Dev. Biol. 2000, 16, 653–699. [Google Scholar] [CrossRef]
- Hatton, K.S.; Mahon, K.; Chin, L.; Chiu, F.C.; Lee, H.W.; Peng, D.; Morgenbesser, S.D.; Horner, J.; A DePinho, R. Expression and activity of L-Myc in normal mouse development. Mol. Cell. Biol. 1996, 16, 1794–1804. [Google Scholar] [CrossRef] [Green Version]
- Brodeur, G.M.; Seeger, R.C.; Schwab, M.; Varmus, H.E.; Bishop, J.M. Amplification of N- myc in Untreated Human Neuroblastomas Correlates with Advanced Disease Stage. Science 1984, 224, 1121–1124. [Google Scholar] [CrossRef]
- Liu, W.; Le, A.; Hancock, C.; Lane, A.N.; Dang, C.V.; Fan, T.W.-M.; Phang, J.M. Reprogramming of proline and glutamine metabolism contributes to the proliferative and metabolic responses regulated by oncogenic transcription factor c-MYC. Proc. Natl. Acad. Sci. USA 2012, 109, 8983–8988. [Google Scholar] [CrossRef]
- Hsieh, A.L.; Walton, Z.E.; Altman, B.; Stine, Z.E.; Dang, C.V. MYC and metabolism on the path to cancer. Semin. Cell Dev. Biol. 2015, 43, 11–21. [Google Scholar] [CrossRef] [Green Version]
- Tsai, W.-B.; Aiba, I.; Long, Y.; Lin, H.-K.; Feun, L.; Savaraj, N.; Kuo, M.T. Activation of Ras/PI3K/ERK Pathway Induces c-Myc Stabilization to Upregulate Argininosuccinate Synthetase, Leading to Arginine Deiminase Resistance in Melanoma Cells. Cancer Res. 2012, 72, 2622–2633. [Google Scholar] [CrossRef] [Green Version]
- Cole, M.D. The myc oncogene: Its role in transformation and differentiation. Annu. Rev. Genet. 1986, 20, 361–384. [Google Scholar] [CrossRef]
- Prendergast, G.C. Mechanisms of apoptosis by c-Myc. Oncogene 1999, 18, 2967–2987. [Google Scholar] [CrossRef] [Green Version]
- Dang, C.V. MYC on the Path to Cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef] [Green Version]
- Dang, C.V. c-Myc Target Genes Involved in Cell Growth, Apoptosis, and Metabolism. Mol. Cell. Biol. 1999, 19, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Meyer, N.; Penn, L.Z. Reflecting on 25 years with MYC. Nat. Rev. Cancer 2008, 8, 976–990. [Google Scholar] [CrossRef]
- Nesbit, C.E.; Tersak, J.M.; Prochownik, E.V. MYC oncogenes and human neoplastic disease. Oncogene 1999, 18, 3004–3016. [Google Scholar] [CrossRef] [Green Version]
- Yada, M.; Hatakeyama, S.; Kamura, T.; Nishiyama, M.; Tsunematsu, R.; Imaki, H.; Ishida, N.; Okumura, F.; Nakayama, K.; I Nakayama, K. Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. EMBO J. 2004, 23, 2116–2125. [Google Scholar] [CrossRef] [Green Version]
- Sears, R.; Leone, G.; DeGregori, J.; Nevins, J.R. Ras Enhances Myc Protein Stability. Mol. Cell 1999, 3, 169–179. [Google Scholar] [CrossRef]
- Sears, R.; Nuckolls, F.; Haura, E.; Taya, Y.; Tamai, K.; Nevins, J.R. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 2000, 14, 2501–2514. [Google Scholar] [CrossRef] [Green Version]
- Arnold, H.K.; Sears, R.C. Protein Phosphatase 2A Regulatory Subunit B56α Associates with c-Myc and Negatively Regulates c-Myc Accumulation. Mol. Cell. Biol. 2006, 26, 2832–2844. [Google Scholar] [CrossRef] [Green Version]
- Kamemura, K.; Hayes, B.K.; Comer, F.I.; Hart, G.W. Dynamic Interplay between O-Glycosylation and O-Phosphorylation of Nucleocytoplasmic Proteins. J. Biol. Chem. 2002, 277, 19229–19235. [Google Scholar] [CrossRef] [Green Version]
- Bahram, F.; Von Der Lehr, N.; Cetinkaya, C.; Larsson, L.-G. c-Myc hot spot mutations in lymphomas result in inefficient ubiquitination and decreased proteasome-mediated turnover. Blood 2000, 95, 2104–2110. [Google Scholar] [CrossRef]
- Uribesalgo, I.; Buschbeck, M.; Gutiérrez, A.; Teichmann, S.; Demajo, S.; Kuebler, B.; Nomdedeu, J.; Martín-Caballero, J.; Roma, G.; Benitah, S.A.; et al. E-box-independent regulation of transcription and differentiation by MYC. Nat. Cell Biol. 2011, 13, 1443–1449. [Google Scholar] [CrossRef]
- Lutterbach, B.; Hann, S.R. Overexpression of c-Myc and cell immortalization alters c-Myc phosphorylation. Oncogene 1997, 14, 967–975. [Google Scholar] [CrossRef] [Green Version]
- Adhikary, S.; Eilers, M. Transcriptional regulation and transformation by Myc proteins. Nat. Rev. Mol. Cell Biol. 2005, 6, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Vervoorts, J.; Lüscher-Firzlaff, J.M.; Rottmann, S.; Lilischkis, R.; Walsemann, G.; Dohmann, K.; Austen, M.; Lüscher, B. Stimulation of c-MYC transcriptional activity and acetylation by recruitment of the cofactor CBP. EMBO Rep. 2003, 4, 484–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, J.H.; Du, Y.; Ard, P.G.; Phillips, C.; Carella, B.; Chen, C.-J.; Rakowski, C.; Chatterjee, C.; Lieberman, P.M.; Lane, W.S.; et al. The c-MYC Oncoprotein Is a Substrate of the Acetyltransferases hGCN5/PCAF and TIP60. Mol. Cell. Biol. 2004, 24, 10826–10834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faiola, F.; Liu, X.; Lo, S.; Pan, S.; Zhang, K.; Lymar, E.; Farina, A.; Martinez, E. Dual Regulation of c-Myc by p300 via Acetylation-Dependent Control of Myc Protein Turnover and Coactivation of Myc-Induced Transcription. Mol. Cell. Biol. 2005, 25, 10220–10234. [Google Scholar] [CrossRef]
- Zhang, K.; Faiola, F.; Martinez, E. Six lysine residues on c-Myc are direct substrates for acetylation by p300. Biochem. Biophys. Res. Commun. 2005, 336, 274–280. [Google Scholar] [CrossRef]
- Zeng, L.; Zhou, M.-M. Bromodomain: An acetyl-lysine binding domain. FEBS Lett. 2002, 513, 124–128. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.; Minter-Dykhouse, K.; Lou, Z. A c-Myc–SIRT1 feedback loop regulates cell growth and transformation. J. Cell Biol. 2009, 185, 203–211. [Google Scholar] [CrossRef] [Green Version]
- Hann, S.R. Role of post-translational modifications in regulating c-Myc proteolysis, transcriptional activity and biological function. Semin. Cancer Biol. 2006, 16, 288–302. [Google Scholar] [CrossRef]
- Deshaies, R.J. SCF and Cullin/RING H2-Based Ubiquitin Ligases. Annu. Rev. Cell Dev. Biol. 1999, 15, 435–467. [Google Scholar] [CrossRef] [Green Version]
- Welcker, M.; Clurman, B.E. FBW7 ubiquitin ligase: A tumour suppressor at the crossroads of cell division, growth and differentiation. Nat. Rev. Cancer 2008, 8, 83–93. [Google Scholar] [CrossRef]
- Huber, A.-L.; Papp, S.J.; Chan, A.B.; Henriksson, E.; Jordan, S.D.; Kriebs, A.; Nguyen, M.; Wallace, M.; Li, Z.; Metallo, C.M.; et al. CRY2 and FBXL3 Cooperatively Degrade c-MYC. Mol. Cell 2016, 64, 774–789. [Google Scholar] [CrossRef] [Green Version]
- Popov, N.; Schülein, C.; Jaenicke, L.A.; Eilers, M. Ubiquitylation of the amino terminus of Myc by SCFβ-TrCP antagonizes SCFFbw7-mediated turnover. Nat. Cell Biol. 2010, 12, 973–981. [Google Scholar] [CrossRef]
- Frescas, D.; Pagano, M. Deregulated proteolysis by the F-box proteins SKP2 and β-TrCP: Tipping the scales of cancer. Nat. Rev. Cancer 2008, 8, 438–449. [Google Scholar] [CrossRef] [Green Version]
- Von Der Lehr, N.; Johansson, S.; Larsson, L.-G. Implication of the ubiquitin/proteasome system in Myc-regulated transcription. Cell Cycle 2003, 2, 402–406. [Google Scholar] [CrossRef]
- Von der Lehr, N.; Johansson, S.; Wu, S.; Bahram, F.; Castell, A.; Cetinkaya, C.; Hydbring, P.; Weidung, I.; Nakayama, K.; I Nakayama, K.; et al. The F-Box Protein Skp2 Participates in c-Myc Proteosomal Degradation and Acts as a Cofactor for c-Myc-Regulated Transcription. Mol. Cell 2003, 11, 1189–1200. [Google Scholar] [CrossRef]
- Fang, X.; Zhou, W.; Wu, Q.; Huang, Z.; Shi, Y.; Yang, K.; Chen, C.; Xie, Q.; Mack, S.C.; Wang, X.; et al. Deubiquitinase USP13 maintains glioblastoma stem cells by antagonizing FBXL14-mediated Myc ubiquitination. J. Exp. Med. 2016, 214, 245–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cepeda, D.; Ng, H.; Sharifi, H.R.; Mahmoudi, S.; Cerrato, V.S.; Fredlund, E.; Magnusson, K.; Nilsson, H.; Malyukova, A.; Rantala, J.; et al. CDK-mediated activation of the SCF FBXO 28 ubiquitin ligase promotes MYC-driven transcription and tumourigenesis and predicts poor survival in breast cancer. EMBO Mol. Med. 2013, 5, 1067–1086. [Google Scholar] [CrossRef] [PubMed]
- Geng, C.; Kaochar, S.; Li, M.; Rajapakshe, K.; Fiskus, W.; Dong, J.; Foley, C.; Dong, B.; Zhang, L.; Kwon, O.-J.; et al. SPOP regulates prostate epithelial cell proliferation and promotes ubiquitination and turnover of c-MYC oncoprotein. Oncogene 2017, 36, 4767–4777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hakem, A.; Bohgaki, M.; Lemmers, B.; Tai, E.; Salmena, L.; Matysiak-Zablocki, E.; Jung, Y.-S.; Karaskova, J.; Kaustov, L.; Duan, S.; et al. Role of Pirh2 in Mediating the Regulation of p53 and c-Myc. PLoS Genet. 2011, 7, e1002360. [Google Scholar] [CrossRef]
- Adhikary, S.; Marinoni, F.; Hock, A.; Hulleman, E.; Popov, N.; Beier, R.; Bernard, S.; Quarto, M.; Capra, M.; Goettig, S.; et al. The Ubiquitin Ligase HectH9 Regulates Transcriptional Activation by Myc and Is Essential for Tumor Cell Proliferation. Cell 2005, 123, 409–421. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Sun, X.-X.; Sears, R.C.; Dai, M.-S. Writing and erasing MYC ubiquitination and SUMOylation. Genes Dis. 2019, 6, 359–371. [Google Scholar] [CrossRef]
- Sabò, A.; Doni, M.; Amati, B. SUMOylation of Myc-Family Proteins. PLoS ONE 2014, 9, e91072. [Google Scholar] [CrossRef]
- Kalkat, M.; Chan, P.-K.; Wasylishen, A.R.; Srikumar, T.; Kim, S.S.; Ponzielli, R.; Bazett-Jones, D.P.; Raught, B.; Penn, L.Z. Identification of c-MYC SUMOylation by Mass Spectrometry. PLoS ONE 2014, 9, e115337. [Google Scholar] [CrossRef] [Green Version]
- González-Prieto, R.; Cuijpers, S.A.; Kumar, R.; Hendriks, I.A.; Vertegaal, A.C. c-Myc is targeted to the proteasome for degradation in a SUMOylation-dependent manner, regulated by PIAS1, SENP7 and RNF4. Cell Cycle 2015, 14, 1859–1872. [Google Scholar] [CrossRef]
- Rabellino, A.; Melegari, M.; Tompkins, V.S.; Chen, W.; Van Ness, B.G.; Teruya-Feldstein, J.; Conacci-Sorrell, M.; Janz, S.; Scaglioni, P.P. PIAS1 Promotes Lymphomagenesis through MYC Upregulation. Cell Rep. 2016, 15, 2266–2278. [Google Scholar] [CrossRef] [Green Version]
- Knudson, A.G., Jr. Mutation and Cancer: Statistical Study of Retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef] [Green Version]
- Thomas, D.M.; A Carty, S.; Piscopo, D.M.; Lee, J.-S.; Wang, W.-F.; Forrester, W.C.; Hinds, P.W. The Retinoblastoma Protein Acts as a Transcriptional Coactivator Required for Osteogenic Differentiation. Mol. Cell 2001, 8, 303–316. [Google Scholar] [CrossRef]
- Ianari, A.; Natale, T.; Calo, E.; Ferretti, E.; Alesse, E.; Screpanti, I.; Haigis, K.; Gulino, A.; Lees, J.A. Proapoptotic Function of the Retinoblastoma Tumor Suppressor Protein. Cancer Cell 2009, 15, 184–194. [Google Scholar] [CrossRef] [Green Version]
- Calo, E.; Quintero-Estades, J.A.; Danielian, P.S.; Nedelcu, S.; Berman, S.D.; Lees, J.A. Rb regulates fate choice and lineage commitment in vivo. Nature 2010, 466, 1110–1114. [Google Scholar] [CrossRef] [Green Version]
- Wells, J.; Yan, P.S.; Cechvala, M.; Huang, T.; Farnham, P.J. Identification of novel pRb binding sites using CpG microarrays suggests that E2F recruits pRb to specific genomic sites during S phase. Oncogene 2003, 22, 1445–1460. [Google Scholar] [CrossRef] [Green Version]
- Chicas, A.; Wang, X.; Zhang, C.; McCurrach, M.; Zhao, Z.; Mert, O.; Dickins, R.A.; Narita, M.; Zhang, M.; Lowe, S.W. Dissecting the Unique Role of the Retinoblastoma Tumor Suppressor during Cellular Senescence. Cancer Cell 2010, 17, 376–387. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, R.; Gou, D.; Jawdekar, G.; Johnson, S.A.; Nava, M.; Su, T.; Yousef, A.F.; Zemke, N.R.; Pellegrini, M.; Kurdistani, S.K.; et al. Adenovirus Small E1A Employs the Lysine Acetylases p300/CBP and Tumor Suppressor Rb to Repress Select Host Genes and Promote Productive Virus Infection. Cell Host Microbe 2014, 16, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Kareta, M.S.; Gorges, L.L.; Hafeez, S.; Benayoun, B.A.; Marro, S.; Zmoos, A.-F.; Cecchini, M.J.; Spacek, D.; Batista, L.F.; O’Brien, M.; et al. Inhibition of Pluripotency Networks by the Rb Tumor Suppressor Restricts Reprogramming and Tumorigenesis. Cell Stem Cell 2014, 16, 39–50. [Google Scholar] [CrossRef] [Green Version]
- Longworth, M.S.; Herr, A.; Ji, J.-Y.; Dyson, N.J. RBF1 promotes chromatin condensation through a conserved interaction with the Condensin II protein dCAP-D3. Genes Dev. 2008, 22, 1011–1024. [Google Scholar] [CrossRef]
- Manning, A.L.; Longworth, M.S.; Dyson, N.J. Loss of pRB causes centromere dysfunction and chromosomal instability. Genes Dev. 2010, 24, 1364–1376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coschi, C.H.; Martens, A.L.; Ritchie, K.; Francis, S.M.; Chakrabarti, S.; Berube, N.G.; Dick, F.A. Mitotic chromosome condensation mediated by the retinoblastoma protein is tumor-suppressive. Genes Dev. 2010, 24, 1351–1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coschi, C.H.; Ishak, C.A.; Gallo, D.; Marshall, A.; Talluri, S.; Wang, J.; Cecchini, M.J.; Martens, A.L.; Percy, V.; Welch, I.; et al. Haploinsufficiency of an RB–E2F1–Condensin II Complex Leads to Aberrant Replication and Aneuploidy. Cancer Discov. 2014, 4, 840–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchida, C.; Hattori, T.; Takahashi, H.; Yamamoto, N.; Kitagawa, M.; Taya, Y. Interaction between RB protein and NuMA is required for proper alignment of spindle microtubules. Genes Cells 2013, 19, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Dick, F.A.; Rubin, S.M. Molecular mechanisms underlying RB protein function. Nat. Rev. Mol. Cell Biol. 2013, 14, 297–306. [Google Scholar] [CrossRef] [Green Version]
- Knudsen, E.S.; Wang, J.Y.J. Differential Regulation of Retinoblastoma Protein Function by Specific Cdk Phosphorylation Sites. J. Biol. Chem. 1996, 271, 8313–8320. [Google Scholar] [CrossRef] [Green Version]
- Zarkowska, T.; Mittnacht, S. Differential Phosphorylation of the Retinoblastoma Protein by G1/S Cyclin-dependent Kinases. J. Biol. Chem. 1997, 272, 12738–12746. [Google Scholar] [CrossRef] [Green Version]
- Chan, H.M.; Krstic-Demonacos, M.; Smith, L.; Demonacos, C.; La Thangue, N.B. Acetylation control of the retinoblastoma tumour-suppressor protein. Nat. Cell Biol. 2001, 3, 667–674. [Google Scholar] [CrossRef]
- Nguyen, D.X.; A Baglia, L.; Huang, S.-M.; Baker, C.M.; McCance, D.J. Acetylation regulates the differentiation-specific functions of the retinoblastoma protein. EMBO J. 2004, 23, 1609–1618. [Google Scholar] [CrossRef] [Green Version]
- Takaki, T.; Fukasawa, K.; Suzuki-Takahashi, I.; Hirai, H. Cdk-mediated phosphorylation of pRB regulates HDAC binding in vitro. Biochem. Biophys. Res. Commun. 2004, 316, 252–255. [Google Scholar] [CrossRef]
- Rubin, S.M. Deciphering the retinoblastoma protein phosphorylation code. Trends Biochem. Sci. 2013, 38, 12–19. [Google Scholar] [CrossRef] [Green Version]
- Morris, E.J.; Dyson, N.J. Retinoblastoma protein partners. Adv. Cancer Res. 2001, 82, 1–54. [Google Scholar] [CrossRef]
- Weinberg, R.A. The retinoblastoma protein and cell cycle control. Cell 1995, 81, 323–330. [Google Scholar] [CrossRef] [Green Version]
- Munro, S.; Carr, S.M.; La Thangue, N.B. Diversity within the pRb pathway: Is there a code of conduct? Oncogene 2012, 31, 4343–4352. [Google Scholar] [CrossRef] [Green Version]
- Hassler, M.; Singh, S.; Yue, W.W.; Luczynski, M.; Lakbir, R.; Sanchez-Sanchez, F.; Bader, T.; Pearl, L.H.; Mittnacht, S. Crystal Structure of the Retinoblastoma Protein N Domain Provides Insight into Tumor Suppression, Ligand Interaction, and Holoprotein Architecture. Mol. Cell 2007, 28, 371–385. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-O.; Russo, A.A.; Pavletich, N.P. Structure of the retinoblastoma tumour-suppressor pocket domain bound to a peptide from HPV E7. Nature 1998, 391, 859–865. [Google Scholar] [CrossRef]
- Lee, C.; Chang, J.H.; Lee, H.S.; Cho, Y. Structural basis for the recognition of the E2F transactivation domain by the retinoblastoma tumor suppressor. Genes Dev. 2002, 16, 3199–3212. [Google Scholar] [CrossRef] [Green Version]
- Xiao, B.; Spencer, J.; Clements, A.; Ali-Khan, N.; Mittnacht, S.; Broceño, C.; Burghammer, M.; Perrakis, A.; Marmorstein, R.; Gamblin, S.J. Crystal structure of the retinoblastoma tumor suppressor protein bound to E2F and the molecular basis of its regulation. Proc. Natl. Acad. Sci. USA 2003, 100, 2363–2368. [Google Scholar] [CrossRef] [Green Version]
- Rubin, S.M.; Gall, A.-L.; Zheng, N.; Pavletich, N.P. Structure of the Rb C-Terminal Domain Bound to E2F1-DP1: A Mechanism for Phosphorylation-Induced E2F Release. Cell 2005, 123, 1093–1106. [Google Scholar] [CrossRef] [Green Version]
- Burke, J.R.; Deshong, A.J.; Pelton, J.G.; Rubin, S.M. Phosphorylation-induced Conformational Changes in the Retinoblastoma Protein Inhibit E2F Transactivation Domain Binding. J. Biol. Chem. 2010, 285, 16286–16293. [Google Scholar] [CrossRef]
- Burke, J.R.; Hura, G.L.; Rubin, S.M. Structures of inactive retinoblastoma protein reveal multiple mechanisms for cell cycle control. Genes Dev. 2012, 26, 1156–1166. [Google Scholar] [CrossRef] [Green Version]
- Ezhevsky, S.A.; Ho, A.; Becker-Hapak, M.; Davis, P.K.; Dowdy, S.F. Differential Regulation of Retinoblastoma Tumor Suppressor Protein by G(1) Cyclin-Dependent Kinase Complexes In Vivo. Mol. Cell. Biol. 2001, 21, 4773–4784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giacinti, C.; Giordano, A. RB and cell cycle progression. Oncogene 2006, 25, 5220–5227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ezhevsky, S.A.; Nagahara, H.; Vocero-Akbani, A.M.; Gius, D.R.; Wei, M.C.; Dowdy, S.F. Hypo-phosphorylation of the retinoblastoma protein (pRb) by cyclin D: Cdk4/6 complexes results in active pRb. Proc. Natl. Acad. Sci. USA 1997, 94, 10699–10704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzolio, F.; Lucchetti, C.; Caligiuri, I.; Marchesi, I.; Caputo, M.; Klein-Szanto, A.J.; Bagella, L.; Castronovo, M.; Giordano, A. Retinoblastoma tumor-suppressor protein phosphorylation and inactivation depend on direct interaction with Pin1. Cell Death Differ. 2012, 19, 1152–1161. [Google Scholar] [CrossRef] [Green Version]
- Harbour, J.W.; Luo, R.X.; Santi, A.D.; Postigo, A.A.; Dean, D.C. Cdk Phosphorylation Triggers Sequential Intramolecular Interactions that Progressively Block Rb Functions as Cells Move through G1. Cell 1999, 98, 859–869. [Google Scholar] [CrossRef] [Green Version]
- Mittnacht, S. Control of pRB phosphorylation. Curr. Opin. Genet. Dev. 1998, 8, 21–27. [Google Scholar] [CrossRef]
- Kolupaeva, V.; Janssens, V. PP1 and PP2A phosphatases-cooperating partners in modulating retinoblastoma protein activation. FEBS J. 2012, 280, 627–643. [Google Scholar] [CrossRef]
- Rubin, E.; Mittnacht, S.; Villa-Moruzzi, E.; Ludlow, J.W. Site-specific and temporally-regulated retinoblastoma protein dephosphorylation by protein phosphatase type 1. Oncogene 2001, 20, 3776–3785. [Google Scholar] [CrossRef] [Green Version]
- Lentine, B.; Antonucci, L.; Hunce, R.; Edwards, J.; Marallano, V.; Krucher, N.A. Dephosphorylation of threonine-821 of the retinoblastoma tumor suppressor protein (Rb) is required for apoptosis induced by UV and Cdk inhibition. Cell Cycle 2012, 11, 3324–3330. [Google Scholar] [CrossRef]
- Zhang, H.; A Postigo, A.; Dean, D.C. Active Transcriptional Repression by the Rb–E2F Complex Mediates G1 Arrest Triggered by p16INK4a, TGFβ, and Contact Inhibition. Cell 1999, 97, 53–61. [Google Scholar] [CrossRef] [Green Version]
- Beasley, M.B.; Lantuejoul, S.; Abbondanzo, S.; Chu, W.-S.; Hasleton, P.S.; Travis, W.D.; Brambilla, E. The P16/cyclin D1/Rb pathway in neuroendocrine tumors of the lung. Hum. Pathol. 2003, 34, 136–142. [Google Scholar] [CrossRef]
- Carr, S.M.; Munro, S.; Kessler, B.; Oppermann, U.; La Thangue, N.B. Interplay between lysine methylation and Cdk phosphorylation in growth control by the retinoblastoma protein. EMBO J. 2010, 30, 317–327. [Google Scholar] [CrossRef] [Green Version]
- Markham, D.; Munro, S.; Soloway, J.; O’Connor, D.; La Thangue, N.B. DNA-damage-responsive acetylation of pRb regulates binding to E2F-1. EMBO Rep. 2006, 7, 192–198. [Google Scholar] [CrossRef]
- Munro, S.; Khaire, N.; Inche, A.; Carr, S.; La Thangue, N.B. Lysine methylation regulates the pRb tumour suppressor protein. Oncogene 2010, 29, 2357–2367. [Google Scholar] [CrossRef] [Green Version]
- Adams, P.D.; Li, X.; Sellers, W.R.; Baker, K.B.; Leng, X.; Harper, J.W.; Taya, Y.; Kaelin, W.G. Retinoblastoma Protein Contains a C-terminal Motif That Targets It for Phosphorylation by Cyclin-cdk Complexes. Mol. Cell. Biol. 1999, 19, 1068–1080. [Google Scholar] [CrossRef] [Green Version]
- Dick, F.A.; Goodrich, D.W.; Sage, J.; Dyson, N.J. Non-canonical functions of the RB protein in cancer. Nat. Cancer 2018, 18, 442–451. [Google Scholar] [CrossRef]
- Jiang, Z.; Zacksenhaus, E.; Gallie, B.L.; A Phillips, R. The retinoblastoma gene family is differentially expressed during embryogenesis. Oncogene 1997, 14, 1789–1797. [Google Scholar] [CrossRef] [Green Version]
- Boyer, S.N.; Wazer, D.E.; Band, V. E7 protein of human papilloma virus-16 induces degradation of reti-noblastoma protein through the ubiquitin-proteasome pathway. Cancer Res. 1996, 56, 4620–4624. Available online: http://www.ncbi.nlm.nih.gov/pubmed/8840974 (accessed on 3 February 2022).
- Wang, J.; Sampath, A.; Raychaudhuri, P.; Bagchi, S. Both Rb and E7 are regulated by the ubiquitin proteasome pathway in HPV-containing cervical tumor cells. Oncogene 2001, 20, 4740–4749. [Google Scholar] [CrossRef] [Green Version]
- Kalejta, R.F.; Shenk, T. Proteasome-dependent, ubiquitin-independent degradation of the Rb family of tumor suppressors by the human cytomegalovirus pp71 protein. Proc. Natl. Acad. Sci. USA 2003, 100, 3263–3268. [Google Scholar] [CrossRef] [PubMed]
- Knight, J.S.; Sharma, N.; Robertson, E.S. Epstein–Barr virus latent antigen 3C can mediate the degradation of the retinoblastoma protein through an SCF cellular ubiquitin ligase. Proc. Natl. Acad. Sci. USA 2005, 102, 18562–18566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munakata, T.; Nakamura, M.; Liang, Y.; Li, K.; Lemon, S.M. Down-regulation of the retinoblastoma tumor suppressor by the hepatitis C virus NS5B RNA-dependent RNA polymerase. Proc. Natl. Acad. Sci. USA 2005, 102, 18159–18164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, S.L.; Stremlau, M.; He, X.; Basile, J.R.; Münger, K. Degradation of the Retinoblastoma Tumor Suppressor by the Human Papillomavirus Type 16 E7 Oncoprotein Is Important for Functional Inactivation and Is Separable from Proteasomal Degradation of E7. J. Virol. 2001, 75, 7583–7591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchida, C.; Miwa, S.; Kitagawa, K.; Hattori, T.; Isobe, T.; Otani, S.; Oda, T.; Sugimura, H.; Kamijo, T.; Ookawa, K.; et al. Enhanced Mdm2 activity inhibits pRB function via ubiquitin-dependent degradation. EMBO J. 2004, 24, 160–169. [Google Scholar] [CrossRef] [Green Version]
- Gareau, J.R.; Lima, C.D. The SUMO pathway: Emerging mechanisms that shape specificity, conjugation and recognition. Nat. Rev. Mol. Cell Biol. 2010, 11, 861–871. [Google Scholar] [CrossRef] [Green Version]
- Yao, Q.; Li, H.; Liu, B.-Q.; Huang, X.-Y.; Guo, L. SUMOylation-regulated Protein Phosphorylation, Evidence from Quantitative Phosphoproteomics Analyses. J. Biol. Chem. 2011, 286, 27342–27349. [Google Scholar] [CrossRef] [Green Version]
- Geiss-Friedlander, R.; Melchior, F. Concepts in sumoylation: A decade on. Nat. Rev. Mol. Cell Biol. 2007, 8, 947–956. [Google Scholar] [CrossRef]
- Meng, F.; Qian, J.; Yue, H.; Li, X.; Xue, K. SUMOylation of Rb enhances its binding with CDK2 and phosphorylation at early G1 phase. Cell Cycle 2016, 15, 1724–1732. [Google Scholar] [CrossRef]
- Girdwood, D.; Bumpass, D.; Vaughan, O.A.; Thain, A.; Anderson, L.A.; Snowden, A.W.; Garcia-Wilson, E.; Perkins, N.D.; Hay, R.T. p300 Transcriptional Repression Is Mediated by SUMO Modification. Mol. Cell 2003, 11, 1043–1054. [Google Scholar] [CrossRef]
- Yang, S.-H.; Sharrocks, A.D. SUMO Promotes HDAC-Mediated Transcriptional Repression. Mol. Cell 2004, 13, 611–617. [Google Scholar] [CrossRef]
- De Luca, A.; MacLachlan, T.K.; Bagella, L.; Dean, C.; Howard, C.; Claudio, P.P.; Baldi, A.; Khalili, K.; Giordano, A. A Unique Domain of pRb2/p130 Acts as an Inhibitor of Cdk2 Kinase Activity. J. Biol. Chem. 1997, 272, 20971–20974. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Gavin, M.; Dahiya, A.; Postigo, A.; Ma, D.; Luo, R.X.; Harbour, J.; Dean, D.C. Exit from G1 and S Phase of the Cell Cycle Is Regulated by Repressor Complexes Containing HDAC-Rb-hSWI/SNF and Rb-hSWI/SNF. Cell 2000, 101, 79–89. [Google Scholar] [CrossRef] [Green Version]
- Hickey, C.M.; Wilson, N.R.; Hochstrasser, M. Function and regulation of SUMO proteases. Nat. Rev. Mol. Cell Biol. 2012, 13, 755–766. [Google Scholar] [CrossRef] [Green Version]
- Ledl, A.; Schmidt, D.; Müller, S. Viral oncoproteins E1A and E7 and cellular LxCxE proteins repress SUMO modification of the retinoblastoma tumor suppressor. Oncogene 2005, 24, 3810–3818. [Google Scholar] [CrossRef] [Green Version]
- Kruiswijk, F.; Labuschagne, C.F.; Vousden, K.H. p53 in survival, death and metabolic health: A lifeguard with a licence to kill. Nat. Rev. Mol. Cell Biol. 2015, 16, 393–405. [Google Scholar] [CrossRef]
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Mao, Y.; Zhao, L.; Li, L.; Wu, J.; Zhao, M.; Du, W.; Yu, L.; Jiang, P. p53 regulation of ammonia metabolism through urea cycle controls polyamine biosynthesis. Nature 2019, 567, 253–256, Publisher Correction in Nature 2019, 569, E10. [Google Scholar] [CrossRef]
- Liu, Y.; Gu, W. The complexity of p53-mediated metabolic regulation in tumor suppression. Semin. Cancer Biol. 2021, 85, 4–32. [Google Scholar] [CrossRef]
- Liu, Y.; Tavana, O.; Gu, W. p53 modifications: Exquisite decorations of the powerful guardian. J. Mol. Cell Biol. 2019, 11, 564–577. [Google Scholar] [CrossRef] [Green Version]
- Lane, D.P. p53, guardian of the genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef] [PubMed]
- Hafner, A.; Bulyk, M.L.; Jambhekar, A.; Lahav, G. The multiple mechanisms that regulate p53 activity and cell fate. Nat. Rev. Mol. Cell Biol. 2019, 20, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Joerger, A.C.; Fersht, A.R. Structural Biology of the Tumor Suppressor p53. Annu. Rev. Biochem. 2008, 77, 557–582. [Google Scholar] [CrossRef] [PubMed]
- Katayama, H.; Sasai, K.; Kawai, H.; Yuan, Z.-M.; Bondaruk, J.; Suzuki, F.; Fujii, S.; Arlinghaus, R.B.; A Czerniak, B.; Sen, S. Phosphorylation by aurora kinase A induces Mdm2-mediated destabilization and inhibition of p53. Nat. Genet. 2003, 36, 55–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, Y.; Padre, R.C.; De Mendoza, T.H.; Bottero, V.; Tergaonkar, V.B.; Verma, I.M. Phosphorylation of p53 by IκB kinase 2 promotes its degradation by β-TrCP. Proc. Natl. Acad. Sci. USA 2009, 106, 2629–2634. [Google Scholar] [CrossRef] [Green Version]
- Li, H.-H.; Cai, X.; Shouse, G.; Piluso, L.G.; Liu, X. A specific PP2A regulatory subunit, B56γ, mediates DNA damage-induced dephosphorylation of p53 at Thr55. EMBO J. 2007, 26, 402–411. [Google Scholar] [CrossRef]
- Wu, Y.; Lin, J.C.; Piluso, L.G.; Dhahbi, J.M.; Bobadilla, S.; Spindler, S.R.; Liu, X. Phosphorylation of p53 by TAF1 Inactivates p53-Dependent Transcription in the DNA Damage Response. Mol. Cell 2013, 53, 63–74. [Google Scholar] [CrossRef] [Green Version]
- Waterman, M.J.F.; Stavridi, E.S.; Waterman, J.L.F.; Halazonetis, T.D. ATM-dependent activation of p53 involves dephosphorylation and association with 14-3-3 proteins. Nat. Genet. 1998, 19, 175–178. [Google Scholar] [CrossRef]
- Rajagopalan, S.; Jaulent, A.M.; Wells, M.; Veprintsev, D.; Fersht, A.R. 14-3-3 activation of DNA binding of p53 by enhancing its association into tetramers. Nucleic Acids Res. 2008, 36, 5983–5991. [Google Scholar] [CrossRef] [Green Version]
- Shieh, S.-Y.; Ikeda, M.; Taya, Y.; Prives, C. DNA Damage-Induced Phosphorylation of p53 Alleviates Inhibition by MDM2. Cell 1997, 91, 325–334. [Google Scholar] [CrossRef] [Green Version]
- Shieh, S.Y.; Ahn, J.; Tamai, K.; Taya, Y.; Prives, C. The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes Dev. 2000, 14, 289–300. Available online: http://www.ncbi.nlm.nih.gov/pubmed/10673501 (accessed on 1 February 2022). [CrossRef]
- Takekawa, M.; Adachi, M.; Nakahata, A.; Nakayama, I.; Itoh, F.; Tsukuda, H.; Taya, Y.; Imai, K. p53-inducible Wip1 phosphatase mediates a negative feedback regulation of p38 MAPK-p53 signaling in response to UV radiation. EMBO J. 2000, 19, 6517–6526. [Google Scholar] [CrossRef]
- Buschmann, T.; Potapova, O.; Bar-Shira, A.; Ivanov, V.N.; Fuchs, S.Y.; Henderson, S.; Fried, V.A.; Minamoto, T.; Alarcon-Vargas, D.; Pincus, M.R.; et al. Jun NH2-Terminal Kinase Phosphorylation of p53 on Thr-81 Is Important for p53 Stabilization and Transcriptional Activities in Response to Stress. Mol. Cell. Biol. 2001, 21, 2743–2754. [Google Scholar] [CrossRef] [Green Version]
- Keller, D.M.; Zeng, X.; Wang, Y.; Zhang, Q.H.; Kapoor, M.; Shu, H.; Goodman, R.; Lozano, G.; Zhao, Y.; Lu, H. A DNA Damage–Induced p53 Serine 392 Kinase Complex Contains CK2, hSpt16, and SSRP1. Mol. Cell 2001, 7, 283–292. [Google Scholar] [CrossRef]
- D’Orazi, G.; Cecchinelli, B.; Bruno, T.; Manni, I.; Higashimoto, Y.; Saito, S.; Gostissa, M.; Coen, S.; Marchetti, A.; Del Sal, G.; et al. Homeodomain-interacting protein kinase-2 phosphorylates p53 at Ser 46 and mediates apoptosis. Nat. Cell Biol. 2001, 4, 11–19. [Google Scholar] [CrossRef]
- Saito, S.; Yamaguchi, H.; Higashimoto, Y.; Chao, C.; Xu, Y.; Fornace, A.J., Jr.; Appella, E.; Anderson, C.W. Phosphorylation Site Interdependence of Human p53 Post-translational Modifications in Response to Stress. J. Biol. Chem. 2003, 278, 37536–37544. [Google Scholar] [CrossRef] [Green Version]
- Li, D.W.-C.; Liu, J.-P.; Schmid, P.C.; Schlosser, R.; Feng, H.; Liu, W.-B.; Yan, Q.; Gong, L.; Sun, S.-M.; Deng, M.; et al. Protein serine/threonine phosphatase-1 dephosphorylates p53 at Ser-15 and Ser-37 to modulate its transcriptional and apoptotic activities. Oncogene 2006, 25, 3006–3022. [Google Scholar] [CrossRef] [Green Version]
- Teufel, D.P.; Bycroft, M.; Fersht, A.R. Regulation by phosphorylation of the relative affinities of the N-terminal transactivation domains of p53 for p300 domains and Mdm2. Oncogene 2009, 28, 2112–2118. [Google Scholar] [CrossRef] [Green Version]
- Hsueh, K.-W.; Fu, S.-L.; Chang, C.-B.; Chang, Y.-L.; Lin, C.-H. A novel Aurora-A-mediated phosphorylation of p53 inhibits its interaction with MDM2. Biochim. Biophys. Acta 2013, 1834, 508–515. [Google Scholar] [CrossRef]
- Xu, H.-T.; Lai, W.-L.; Liu, H.-F.; Wong, L.L.-Y.; Ng, I.O.-L.; Ching, Y.P. PAK4 Phosphorylates p53 at Serine 215 to Promote Liver Cancer Metastasis. Cancer Res. 2016, 76, 5732–5742. [Google Scholar] [CrossRef] [Green Version]
- Gabra, M.B.I.; Yang, Y.; Lowman, X.H.; Reid, M.; Tran, T.Q.; Kong, M. IKKβ activates p53 to promote cancer cell adaptation to glutamine deprivation. Oncogenesis 2018, 7, 93. [Google Scholar] [CrossRef] [PubMed]
- Zacchi, P.; Gostissa, M.; Uchida, T.; Salvagno, C.; Avolio, F.; Volinia, S.; Ronai, Z.; Blandino, G.; Schneider, C.; Del Sal, G. The prolyl isomerase Pin1 reveals a mechanism to control p53 functions after genotoxic insults. Nature 2002, 419, 853–857. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; You, H.; Zhou, X.Z.; Murray, S.A.; Uchida, T.; Wulf, G.; Gu, L.; Tang, X.; Lu, K.P.; Xiao, Z.-X.J. The prolyl isomerase Pin1 is a regulator of p53 in genotoxic response. Nature 2002, 419, 849–853. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Lin, J.; Said, A.B.; Yau, Y.H.; Shochat, S.G.; Ruiz-Carrillo, D.; Sun, K.; Chandrasekaran, R.; Sze, S.K.; Lescar, J.; et al. Pellino1 specifically binds to phospho-Thr18 of p53 and is recruited to sites of DNA damage. Biochem. Biophys. Res. Commun. 2019, 513, 714–720. [Google Scholar] [CrossRef] [PubMed]
- Castrogiovanni, C.; Waterschoot, B.; De Backer, O.; Dumont, P. Serine 392 phosphorylation modulates p53 mitochondrial translocation and transcription-independent apoptosis. Cell Death Differ. 2017, 25, 190–203. [Google Scholar] [CrossRef]
- Hammond, E.M.; Denko, N.C.; Dorie, M.J.; Abraham, R.T.; Giaccia, A.J. Hypoxia Links ATR and p53 through Replication Arrest. Mol. Cell. Biol. 2002, 22, 1834–1843. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.-J.; Lim, C.-J.; Min, J.-K.; Lee, J.-K.; Kim, Y.-M.; Lee, J.-Y.; Won, M.-H.; Kwon, Y.-G. Protein phosphatase 1 nuclear targeting subunit is a hypoxia inducible gene: Its role in post-translational modification of p53 and MDM2. Cell Death Differ. 2007, 14, 1106–1116. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Su, F.; Chen, D.; Shiloh, A.; Gu, W. Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature 2000, 408, 377–381. [Google Scholar] [CrossRef]
- Vaziri, H.; Dessain, S.K.; Eaton, E.N.; Imai, S.-I.; Frye, R.A.; Pandita, T.K.; Guarente, L.; Weinberg, R.A. hSIR2SIRT1 Functions as an NAD-Dependent p53 Deacetylase. Cell 2001, 107, 149–159. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Li, M.; Tang, Y.; Laszkowska, M.; Roeder, R.G.; Gu, W. Acetylation of p53 augments its site-specific DNA binding both in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2004, 101, 2259–2264. [Google Scholar] [CrossRef] [Green Version]
- Reed, S.M.; Quelle, D.E. p53 Acetylation: Regulation and Consequences. Cancers 2014, 7, 30–69. [Google Scholar] [CrossRef]
- Tang, Y.; Zhao, W.; Chen, Y.; Zhao, Y.; Gu, W. Acetylation Is Indispensable for p53 Activation. Cell 2008, 133, 612–626. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.-J.; Li, D.; Ou, Y.; Jiang, L.; Chen, Y.; Zhao, Y.; Gu, W. Acetylation Is Crucial for p53-Mediated Ferroptosis and Tumor Suppression. Cell Rep. 2016, 17, 366–373. [Google Scholar] [CrossRef] [Green Version]
- Sykes, S.M.; Mellert, H.S.; Holbert, M.A.; Li, K.; Marmorstein, R.; Lane, W.S.; McMahon, S.B. Acetylation of the p53 DNA-Binding Domain Regulates Apoptosis Induction. Mol. Cell 2006, 24, 841–851. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Luo, J.; Zhang, W.; Gu, W. Tip60-Dependent Acetylation of p53 Modulates the Decision between Cell-Cycle Arrest and Apoptosis. Mol. Cell 2006, 24, 827–839. [Google Scholar] [CrossRef]
- Li, X.; Wu, L.; Corsa, C.A.S.; Kunkel, S.; Dou, Y. Two Mammalian MOF Complexes Regulate Transcription Activation by Distinct Mechanisms. Mol. Cell 2009, 36, 290–301. [Google Scholar] [CrossRef] [Green Version]
- Rokudai, S.; Aikawa, Y.; Tagata, Y.; Tsuchida, N.; Taya, Y.; Kitabayashi, I. Monocytic Leukemia Zinc Finger (MOZ) Interacts with p53 to Induce p21 Expression and Cell-cycle Arrest. J. Biol. Chem. 2009, 284, 237–244. [Google Scholar] [CrossRef] [Green Version]
- Rokudai, S.; Laptenko, O.; Arnal, S.M.; Taya, Y.; Kitabayashi, I.; Prives, C. MOZ increases p53 acetylation and premature senescence through its complex formation with PML. Proc. Natl. Acad. Sci. USA 2013, 110, 3895–3900. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Kon, N.; Jiang, L.; Tan, M.; Ludwig, T.; Zhao, Y.; Baer, R.; Gu, W. Tumor Suppression in the Absence of p53-Mediated Cell-Cycle Arrest, Apoptosis, and Senescence. Cell 2012, 149, 1269–1283. [Google Scholar] [CrossRef] [Green Version]
- Knights, C.D.; Catania, J.; Di Giovanni, S.; Muratoglu, S.; Perez, R.; Swartzbeck, A.; Quong, A.A.; Zhang, X.; Beerman, T.; Pestell, R.G.; et al. Distinct p53 acetylation cassettes differentially influence gene-expression patterns and cell fate. J. Cell Biol. 2006, 173, 533–544. [Google Scholar] [CrossRef]
- Marmorstein, R.; Zhou, M.-M. Writers and Readers of Histone Acetylation: Structure, Mechanism, and Inhibition. Cold Spring Harb. Perspect. Biol. 2014, 6, a018762. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, T.; Filippakopoulos, T.F.P. Functions of bromodomain-containing proteins and their roles in homeostasis and cancer. Nat. Rev. Mol. Cell Biol. 2017, 18, 246–262. [Google Scholar] [CrossRef] [PubMed]
- Mujtaba, S.; Zhou, M.-M. Use of Nuclear Magnetic Resonance Spectroscopy to Study Structure-Function of Bromodomains. Methods Enzymol. 2003, 376, 119–130. [Google Scholar] [CrossRef]
- Chuikov, S.; Kurash, J.K.; Wilson, J.R.; Xiao, B.; Justin, N.; Ivanov, G.S.; McKinney, K.; Tempst, P.; Prives, C.; Gamblin, S.J.; et al. Regulation of p53 activity through lysine methylation. Nature 2004, 432, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Kachirskaia, I.; Yamaguchi, H.; West, L.E.; Wen, H.; Wang, E.W.; Dutta, S.; Appella, E.; Gozani, O. Modulation of p53 Function by SET8-Mediated Methylation at Lysine 382. Mol. Cell 2007, 27, 636–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Perez-Burgos, L.; Placek, B.J.; Sengupta, R.; Richter, M.; Dorsey, J.A.; Kubicek, S.; Opravil, S.; Jenuwein, T.; Berger, S.L. Repression of p53 activity by Smyd2-mediated methylation. Nature 2006, 444, 629–632. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Dorsey, J.; Chuikov, S.; Zhang, X.; Jenuwein, T.; Reinberg, D.; Berger, S. G9a and Glp Methylate Lysine 373 in the Tumor Suppressor p53. J. Biol. Chem. 2010, 285, 9636–9641. [Google Scholar] [CrossRef] [Green Version]
- Jansson, M.; Durant, S.T.; Cho, E.-C.; Sheahan, S.; Edelmann, M.; Kessler, B.; La Thangue, N.B. Arginine methylation regulates the p53 response. Nat. Cell Biol. 2008, 10, 1431–1439. [Google Scholar] [CrossRef]
- Maurer-Stroh, S.; Dickens, N.J.; Hughes-Davies, L.; Kouzarides, T.; Eisenhaber, F.; Ponting, C.P. The Tudor domain ‘Royal Family’: Tudor, plant Agenet, Chromo, PWWP and MBT domains. Trends Biochem. Sci. 2003, 28, 69–74. [Google Scholar] [CrossRef]
- Patel, D.J. A Structural Perspective on Readout of Epigenetic Histone and DNA Methylation Marks. Cold Spring Harb. Perspect. Biol. 2016, 8, a018754. [Google Scholar] [CrossRef] [Green Version]
- Scheffner, M.; Huibregtse, J.M.; Vierstra, R.D.; Howley, P.M. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 1993, 75, 495–505. [Google Scholar] [CrossRef]
- Kwon, S.-K.; Saindane, M.; Baek, K.-H. p53 stability is regulated by diverse deubiquitinating enzymes. Biochim. Biophys. Acta 2017, 1868, 404–411. [Google Scholar] [CrossRef]
- Bang, S.; Kaur, S.; Kurokawa, M. Regulation of the p53 Family Proteins by the Ubiquitin Proteasomal Pathway. Int. J. Mol. Sci. 2019, 21, 261. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Brooks, C.L.; Wu-Baer, F.; Chen, D.; Baer, R.; Gu, W. Mono- Versus Polyubiquitination: Differential Control of p53 Fate by Mdm2. Science 2003, 302, 1972–1975. [Google Scholar] [CrossRef] [Green Version]
- Green, D.R.; Kroemer, G. Cytoplasmic functions of the tumour suppressor p53. Nature 2009, 458, 1127–1130. [Google Scholar] [CrossRef] [Green Version]
- Comel, A.; Sorrentino, G.; Capaci, V.; Del Sal, G. The cytoplasmic side of p53’s oncosuppressive activities. FEBS Lett. 2014, 588, 2600–2609. [Google Scholar] [CrossRef] [Green Version]
- Marchenko, N.D.; Wolff, S.; Erster, S.; Becker, K.; Moll, U.M. Monoubiquitylation promotes mitochondrial p53 translocation. EMBO J. 2007, 26, 923–934. [Google Scholar] [CrossRef]
- Rodriguez, M.S.; Desterro, J.M.P.; Lain, S.; Lane, D.; Hay, R.T. Multiple C-Terminal Lysine Residues Target p53 for Ubiquitin-Proteasome-Mediated Degradation. Mol. Cell. Biol. 2000, 20, 8458–8467. [Google Scholar] [CrossRef] [Green Version]
- Brooks, C.L.; Gu, W. p53 Ubiquitination: Mdm2 and Beyond. Mol. Cell 2006, 21, 307–315. [Google Scholar] [CrossRef] [Green Version]
- Lu, H. p53 and MDM2: Their Yin-Yang intimacy. J. Mol. Cell Biol. 2017, 9, 1–2. [Google Scholar] [CrossRef]
- Zhou, X.; Cao, B.; Lu, H. Negative auto-regulators trap p53 in their web. J. Mol. Cell Biol. 2017, 9, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Laine, A.; Ronai, Z. Regulation of p53 localization and transcription by the HECT domain E3 ligase WWP1. Oncogene 2006, 26, 1477–1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruse, J.-P.; Gu, W. MSL2 Promotes Mdm2-independent Cytoplasmic Localization of p53. J. Biol. Chem. 2009, 284, 3250–3263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Cam, L.; Linares, L.K.; Paul, C.; Julien, E.; Lacroix, M.; Hatchi, E.; Triboulet, R.; Bossis, G.; Shmueli, A.; Rodriguez, M.S.; et al. E4F1 Is an Atypical Ubiquitin Ligase that Modulates p53 Effector Functions Independently of Degradation. Cell 2006, 127, 775–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.; Ji, Z.; Wang, Y.; Li, J.; Cao, H.; Zhu, H.H.; Gao, W.-Q. TRIM59 Is Up-regulated in Gastric Tumors, Promoting Ubiquitination and Degradation of p53. Gastroenterology 2014, 147, 1043–1054. [Google Scholar] [CrossRef]
- Shen, J.; Li, P.; Shao, X.; Yang, Y.; Liu, X.; Feng, M.; Yu, Q.; Hu, R.; Wang, Z. The E3 Ligase RING1 Targets p53 for Degradation and Promotes Cancer Cell Proliferation and Survival. Cancer Res. 2018, 78, 359–371. [Google Scholar] [CrossRef] [Green Version]
- Kwek, S.S.; Derry, J.; Tyner, A.L.; Shen, Z.; Gudkov, A.V. Functional analysis and intracellular localization of p53 modified by SUMO-1. Oncogene 2001, 20, 2587–2599. [Google Scholar] [CrossRef] [Green Version]
- Stehmeier, P.; Müller, S. Regulation of p53 family members by the ubiquitin-like SUMO system. DNA Repair 2009, 8, 491–498. [Google Scholar] [CrossRef]
- Carter, S.; Bischof, O.; Dejean, A.; Vousden, K.H. C-terminal modifications regulate MDM2 dissociation and nuclear export of p53. Nature 2007, 9, 428–435. [Google Scholar] [CrossRef]
- Batuello, C.N.; Hauck, P.M.; Gendron, J.M.; Lehman, J.A.; Mayo, L.D. Src phosphorylation converts Mdm2 from a ubiquitinating to a neddylating E3 ligase. Proc. Natl. Acad. Sci. USA 2015, 112, 1749–1754. [Google Scholar] [CrossRef] [Green Version]
- Abida, W.M.; Nikolaev, A.; Zhao, W.; Zhang, W.; Gu, W.; Abida, W.M.; Nikolaev, A.; Zhao, W.; Zhang, W.; Gu, W. FBXO11 Promotes the Neddylation of p53 and Inhibits Its Transcriptional Activity. J. Biol. Chem. 2007, 282, 1797–1804. [Google Scholar] [CrossRef]
- Liu, G.; Xirodimas, D.P. NUB1 promotes cytoplasmic localization of p53 through cooperation of the NEDD8 and ubiquitin pathways. Oncogene 2010, 29, 2252–2261. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Santockyte, R.; Shen, R.-F.; Tekle, E.; Wang, G.; Yang, D.C.H.; Chock, P.B. Expression of SUMO-2/3 Induced Senescence through p53- and pRB-mediated Pathways. J. Biol. Chem. 2006, 281, 36221–36227. [Google Scholar] [CrossRef] [Green Version]
- Bischof, O.; Schwamborn, K.; Martin, N.; Werner, A.; Sustmann, C.; Grosschedl, R.; Dejean, A. RETRACTED: The E3 SUMO Ligase PIASy Is a Regulator of Cellular Senescence and Apoptosis. Mol. Cell 2006, 22, 783–794. [Google Scholar] [CrossRef]
- Wu, S.-Y.; Chiang, C.-M. Crosstalk between sumoylation and acetylation regulates p53-dependent chromatin transcription and DNA binding. EMBO J. 2009, 28, 1246–1259. [Google Scholar] [CrossRef] [Green Version]
- Santiago, A.; Li, D.; Zhao, L.Y.; Godsey, A.; Liao, D. p53 SUMOylation promotes its nuclear export by facilitating its release from the nuclear export receptor CRM1. Mol. Biol. Cell 2013, 24, 2739–2752. [Google Scholar] [CrossRef]
- Chauhan, K.M.; Chen, Y.; Chen, Y.; Liu, A.T.; Sun, X.; Dai, M. The SUMO-specific protease SENP1 deSUMOylates p53 and regulates its activity. J. Cell. Biochem. 2020, 122, 189–197. [Google Scholar] [CrossRef]
- Della-Fazia, M.A.; Castelli, M.; Piobbico, D.; Pieroni, S.; Servillo, G. The Ins and Outs of HOPS/TMUB1 in biology and pathology. FEBS J. 2020, 288, 2773–2783. [Google Scholar] [CrossRef]
- Castelli, M.; Piobbico, D.; Chiacchiaretta, M.; Brunacci, C.; Pieroni, S.; Bartoli, D.; Gargaro, M.; Fallarino, F.; Puccetti, P.; Soddu, S.; et al. HOPS/TMUB1 retains p53 in the cytoplasm and sustains p53-dependent mitochondrial apoptosis. EMBO Rep. 2019, 21, e48073. [Google Scholar] [CrossRef]
- Castelli, M.; Pieroni, S.; Brunacci, C.; Piobbico, D.; Bartoli, D.; Bellet, M.M.; Colombo, E.; Pelicci, P.G.; A Della Fazia, M.; Servillo, G. Hepatocyte odd protein shuttling (HOPS) is a bridging protein in the nucleophosmin-p19Arf network. Oncogene 2012, 32, 3350–3358. [Google Scholar] [CrossRef] [Green Version]
- Della-Fazia, M.A.; Castelli, M.; Piobbico, D.; Pieroni, S.; Servillo, G. HOPS and p53: Thick as thieves in life and death. Cell Cycle 2020, 19, 2996–3003. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Chiba, T.; Tatsumi, K.; Iemura, S.-I.; Tanida, I.; Okazaki, N.; Ueno, T.; Kominami, E.; Natsume, T.; Tanaka, K. A novel protein-conjugating system for Ufm1, a ubiquitin-fold modifier. EMBO J. 2004, 23, 1977–1986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, S.; Kumar, M.; Wiener, R. Decrypting UFMylation: How Proteins Are Modified with UFM1. Biomolecules 2020, 10, 1442. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Guan, D.; Dong, M.; Yang, J.; Wei, H.; Liang, Q.; Song, L.; Xu, L.; Bai, J.; Liu, C.; et al. UFMylation maintains tumour suppressor p53 stability by antagonizing its ubiquitination. Nat. Cell Biol. 2020, 22, 1056–1063. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Description | Oncogenic Alteration | Related Cancers | |
|---|---|---|---|---|
| RAS | -KRAS | RAS Proto-Oncogenes GTPase activity Signal transduction | Mutation Amplification Deep deletion | Pancreatic Colorectal Lung NSCLC |
| -NRAS | Mutation Amplification Deep deletion | Melanoma AML Endometrial NSCLC | ||
| -HRAS | Amplification Deep deletion Mutation | Lung Head and Neck Soft tissue Esophagogastric | ||
| MYC | MYC Proto-Oncogene BHLH Transcription Factor | Amplification | Endometrial Ovarian Brest Head and Neck | |
| RB | Transcriptional Corepressor 1 Tumor suppressor Cell proliferation | Amplification Deep deletion Mutation | Bladder Colorectal Esophagogastric Soft tissue | |
| TP53 | Tumor suppressor growth arrest apoptosis induction cell cycle regulation | Mutation | Ovarian Endometrial NSCLC Esophagogastric | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pieroni, S.; Castelli, M.; Piobbico, D.; Ferracchiato, S.; Scopetti, D.; Di-Iacovo, N.; Della-Fazia, M.A.; Servillo, G. The Four Homeostasis Knights: In Balance upon Post-Translational Modifications. Int. J. Mol. Sci. 2022, 23, 14480. https://doi.org/10.3390/ijms232214480
Pieroni S, Castelli M, Piobbico D, Ferracchiato S, Scopetti D, Di-Iacovo N, Della-Fazia MA, Servillo G. The Four Homeostasis Knights: In Balance upon Post-Translational Modifications. International Journal of Molecular Sciences. 2022; 23(22):14480. https://doi.org/10.3390/ijms232214480
Chicago/Turabian StylePieroni, Stefania, Marilena Castelli, Danilo Piobbico, Simona Ferracchiato, Damiano Scopetti, Nicola Di-Iacovo, Maria Agnese Della-Fazia, and Giuseppe Servillo. 2022. "The Four Homeostasis Knights: In Balance upon Post-Translational Modifications" International Journal of Molecular Sciences 23, no. 22: 14480. https://doi.org/10.3390/ijms232214480
APA StylePieroni, S., Castelli, M., Piobbico, D., Ferracchiato, S., Scopetti, D., Di-Iacovo, N., Della-Fazia, M. A., & Servillo, G. (2022). The Four Homeostasis Knights: In Balance upon Post-Translational Modifications. International Journal of Molecular Sciences, 23(22), 14480. https://doi.org/10.3390/ijms232214480