

S-Glutathionylation and S-Nitrosylation in Mitochondria: Focus on Homeostasis and Neurodegenerative Diseases


Abstract
:1. Introduction
2. Protein S-Glutathionylation
2.1. Regulation of OXPHOS System by S-Glutathionylation
2.2. Regulation of Nutrient Metabolism by S-Glutathionylation
2.3. Regulation of Mitochondrial Permeability Transition Pore and Apoptosis by S-Glutathionylation
2.4. Regulation of Mitochondria Morphology by S-Glutathionylation
3. Protein S-Nitrosylation
3.1. Regulation of the OXPHOS System by S-Nitrosylation
3.2. Regulation of Metabolic Enzymes by S-Nitrosylation
3.3. Regulation of Apoptosis by S-Nitrosylation
- Mitochondrial permeability transition pores (MPTPs) are formed under oxidative stress and modulate the redox potential. Under oxidative stress, MPTP increases the permeability relative to high molecular weight macromolecules, leading to mitochondria swelling and necroptosis [100]. Under the basal levels of NO, cyclophilin D (CYPD), critical for an MPTP opening, can be S-nitrosylated [101]. This inhibits its interaction with MPTPs, preventing pore opening and protecting cells during stress. Conversely, under the increased production of NO, peroxynitrite is being produced by excessive ROS, which leads to the opening of MPTPs, the ablation of ATP production and necrosis [102].
- VDAC has multiple roles. Its localization at the mitochondrial outer membrane allows VDAC to regulate the fluxes of metabolites. At the same time, VDAC modulates cytochrome c, which activates caspase-dependent apoptotic cell death [103]. Similarly to MPTP regulation by NO, VDAC S-nitrosylation at basal levels of NO inhibits its function, protecting cells from apoptosis, whereas elevated levels of NO upregulate VDAC functions [104].
- Crucial to their role in apoptosis, caspase-3 and caspase-9 are S-nitrosylated in the absence of apoptotic triggers, while their denitrosylation leads to their activation upon the activation of the Fas receptor [105].
- Another component of the outer mitochondrial membrane with an anti-apoptotic role involves BCL2 family proteins, which interfere with apoptosis by regulating the release of cytochrome c [106]. Upon apoptotic signaling, the S-nitrosylation of BCL-2 inhibits its ubiquitination and degradation, allowing it to exert anti-apoptotic protection [107].
3.4. Regulation of Fission by S-Nitrosylation
4. Redox PTMs in Alzheimer’s Disease
4.1. S-Glutathionylation in Alzheimer’s Disease
4.1.1. S-Glutathionylation/Glutathione as Potential Biomarkers for Alzheimer’s Disease
4.1.2. Redox and Metabolic Enzymes in Alzheimer’s Disease
4.1.3. Role of Glutathione in Aβ and Tau Accumulation in Alzheimer’s Disease
4.1.4. GSTs Diverse Roles in Alzheimer’s Disease
4.2. S-Nitrosylation in Alzheimer’s Disease
5. Redox PTMs in Parkinson’s Disease
5.1. S-Glutathionylation in Parkinson’s Disease
5.1.1. S-Glutathionylation/Glutathione in Parkinson’s Disease
5.1.2. The Multifaceted Roles of GRX1 in Parkinson’s Disease
5.2. S-Nitrosylation in Parkinson’s Disease
Mitochondria Biogenesis and Bioenergetics
6. Redox PTMs in Amyotrophic Lateral Sclerosis
6.1. S-Glutathionylation in Amyotrophic Lateral Sclerosis
6.1.1. SOD1 S-Glutathionylation
6.1.2. SOD1 Role in Mitochondria
- Misfolded SOD1 events in the outer mitochondrial membrane: One hypothesis is that SOD1 small misfolded species trigger mitochondrial cytochrome c release and caspase-dependent-programmed cell death. In both in vitro (G93A, G85R) and mutant SOD1 murine models (G93A, G37R), the accumulation of misfolded mutant SOD1 oligomers on the outer mitochondrial membrane has been proposed to trigger apoptosis [238]. This misfolded SOD1 localization has been shown to be highly BCL2-dependent in cell cultures, mutant SOD1 murine models and SOD1-linked familial ALS patients [239,240]. BCL2 halts the release of pro-apoptotic factors from mitochondria, such as cytochrome c, preventing caspase activation and apoptosis under physiological conditions. Pedrini et al. showed that misfolded SOD1 binding triggers conformational changes in BCL2, resulting in the exposure of its toxic BH3 domain and, thus, triggering cytochrome c release and eventually apoptosis [239]. Importantly, GRX1 and GRX2 can reduce disulfides to protein thiols that prevent mutated SOD1 aggregation and rescue mitochondria function while preventing neuronal cell apoptosis [241]. In addition, it has been shown that misfolded SOD1/BCL2 interactions decrease the mitochondrial membrane’s permeability relative to ADP by direct inhibition of the outer mitochondrial porin voltage-dependent anion channel 1 (VDAC1), which regulates mitochondrial ATP production and export [242]. This toxic conformation of BCL2 triggered by misfolded SOD1 can lead to bioenergetic defects, increased levels of ROS and calcium homeostasis deregulation, as has been shown in the motor neurons of pre-symptomatic G93A mice [239,240]. Interestingly, oxidized wild-type SOD1 recapitulates the same toxic behavior, pinpointing the possible common pathogenic mechanism and, thus, potential therapeutic targets between mutated SOD1-related fALS and sporadic disorders exhibiting oxidized wild-type SOD1, including sporadic ALS and Alzheimer’s and Parkinson’s diseases [163].
- Misfolded SOD1 events inside mitochondria: As mentioned above, apoSOD1 and CCS can translocate within the IMS, mitigating superoxide emission and triggering SOD2-mediated superoxide detoxification within mitochondria. However, if apoSOD1 becomes misfolded inside the mitochondria, a substantial accumulation of SOD1 aggregates can happen within this sub-compartment, which is associated with electron transport chain defects and increased ROS production. In this case, it has been suggested that the reduced electron transport chain function is attributed to the preference for the delivery of Cu to SOD1 at the expanse of mitochondrial cytochrome c oxidase [227]. Banks et al. were the first to identify that acylation on SOD1 modifies its capability to suppress mitochondrial respiration [243]. The authors showed that increased levels of sirtuin activity deacylated SOD1, which in turn, activates SOD1 respiration-suppression activity at the CI of ETC. Sirtuin activities are linked to NAD+ levels, which are linked to the overall metabolic cellular status, suggesting that the acylation of SOD1 might act as a sensor link between nutrient metabolism and the SOD1-mediated suppression of respiration [243]. While this identification may add an additional role to SOD1, the mechanistic pathway with which this SOD1-mediated suppression of respiration correlates to SOD1-mediated cell survival is still uncertain.
- Misfolded SOD1 and mitochondrial integrity: The impact of SOD1 gene mutations on mitochondrial integrity has been uncovered by utilizing human iPSC-derived spinal cord motor neurons (MNs) of three quintessential SOD1 gene mutations [244]. It has been shown that mitochondria integrity defects precede DNA damage in neurons from patients with SOD1 mutations, suggesting that a mitochondrial homeostasis deregulation is an upstream event in SOD1-related ALS. In particular, MNs expressing SOD1 R115G and D90A showed elongated mitochondria, highly reduced membrane potential and intracellular ATP levels and a higher fraction of moving mitochondria. In contrast, MNs expressing SOD1 A4V, while they did not show those morphological/motility alterations, had mitochondrial membrane potentials that were highly reduced [244]. The observation of mitochondria elongation in SOD1 D90A and R115G could be explained by protective mitofusion or hyperfusion that can act as a protective mechanism for mitochondria dysfunction against mitochondrial fission and macro-autophagy [244]. The misexpression of mitochondrial dynamics genes has been associated with mutant SOD1 in ALS [245]. Excessive mitochondrial fission and increased mitochondrial fragmentation have been reported in both ALS-patient-derived fibroblasts and the motor neuron cultures of multiple familial forms of mutated SOD1 ALS expression [246]. Interestingly, the inhibition of DRP1/FIS1 by a selective peptide inhibitor, P110, led to a significant improvement of mitochondria structure and function in mice expressing G93A SOD1 mutations [246].
6.2. S-Nitrosylation in Amyotrophic Lateral Sclerosis
7. Redox PTMs in Friedreich’s Ataxia
7.1. S-Glutathionylation in Friedreich’s Ataxia
7.2. S-Nitrosylation in Friedrich’s Ataxia
8. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Weidinger, A.; Kozlov, A.V. Biological Activities of Reactive Oxygen and Nitrogen Species: Oxidative Stress versus Signal Transduction. Biomolecules 2015, 5, 472–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, A.L.; Lindner, A.B. Protein Posttranslational Modifications: Roles in Aging and Age-Related Disease. Oxidative Med. Cell Longev. 2017, 2017, 5716409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beckhauser, T.F.; Francis-Oliveira, J.; De Pasquale, R. Reactive Oxygen Species: Physiological and Physiopathological Effects on Synaptic Plasticity. J. Exp. Neurosci. 2016, 10, 23–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, D.P. Redefining oxidative stress. Antioxid. Redox. Signal. 2006, 8, 1865–1879. [Google Scholar] [CrossRef]
- Ray, P.D.; Huang, B.W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Lambeth, J.D.; Neish, A.S. Nox enzymes and new thinking on reactive oxygen: A double-edged sword revisited. Annu. Rev. Pathol. 2014, 9, 119–145. [Google Scholar] [CrossRef]
- Ma, M.W.; Wang, J.; Zhang, Q.; Wang, R.; Dhandapani, K.M.; Vadlamudi, R.K.; Brann, D.W. NADPH oxidase in brain injury and neurodegenerative disorders. Mol. Neurodegener. 2017, 12, 7. [Google Scholar] [CrossRef] [Green Version]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxid. Med. Cell Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef]
- Moldogazieva, N.T.; Mokhosoev, I.M.; Feldman, N.B.; Lutsenko, S.V. ROS and RNS signalling: Adaptive redox switches through oxidative/nitrosative protein modifications. Free Radic. Res. 2018, 52, 507–543. [Google Scholar] [CrossRef] [PubMed]
- Paul, B.D.; Sbodio, J.I.; Snyder, S.H. Cysteine Metabolism in Neuronal Redox Homeostasis. Trends. Pharm. Sci. 2018, 39, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Sbodio, J.I.; Snyder, S.H.; Paul, B.D. Redox Mechanisms in Neurodegeneration: From Disease Outcomes to Therapeutic Opportunities. Antioxid. Redox. Signal. 2019, 30, 1450–1499. [Google Scholar] [CrossRef] [PubMed]
- Stadtman, E.R.; Levine, R.L. Free radical-mediated oxidation of free amino acids and amino acid residues in proteins. Amino. Acids. 2003, 25, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.J. Protein oxidation and peroxidation. Biochem. J. 2016, 473, 805–825. [Google Scholar] [CrossRef] [Green Version]
- Wall, S.B.; Oh, J.Y.; Diers, A.R.; Landar, A. Oxidative modification of proteins: An emerging mechanism of cell signaling. Front. Physiol. 2012, 3, 369. [Google Scholar] [CrossRef] [Green Version]
- Wani, R.; Nagata, A.; Murray, B.W. Protein redox chemistry: Post-translational cysteine modifications that regulate signal transduction and drug pharmacology. Front. Pharmacol. 2014, 5, 224. [Google Scholar] [CrossRef] [Green Version]
- Halloran, M.; Parakh, S.; Atkin, J.D. The role of s-nitrosylation and s-glutathionylation of protein disulphide isomerase in protein misfolding and neurodegeneration. Int. J. Cell Biol. 2013, 2013, 797914. [Google Scholar] [CrossRef] [Green Version]
- Chung, H.S.; Wang, S.B.; Venkatraman, V.; Murray, C.I.; Van Eyk, J.E. Cysteine oxidative posttranslational modifications: Emerging regulation in the cardiovascular system. Circ. Res. 2013, 112, 382–392. [Google Scholar] [CrossRef] [Green Version]
- Fra, A.; Yoboue, E.D.; Sitia, R. Cysteines as Redox Molecular Switches and Targets of Disease. Front. Mol. Neurosci. 2017, 10, 167. [Google Scholar] [CrossRef]
- Davies, M.J. The oxidative environment and protein damage. Biochim. Biophys. Acta 2005, 1703, 93–109. [Google Scholar] [CrossRef] [PubMed]
- Pajares, M.; Jimenez-Moreno, N.; Dias, I.H.K.; Debelec, B.; Vucetic, M.; Fladmark, K.E.; Basaga, H.; Ribaric, S.; Milisav, I.; Cuadrado, A. Redox control of protein degradation. Redox. Biol. 2015, 6, 409–420. [Google Scholar] [CrossRef] [Green Version]
- Song, H.; Liu, B.; Huai, W.; Yu, Z.; Wang, W.; Zhao, J.; Han, L.; Jiang, G.; Zhang, L.; Gao, C.; et al. The E3 ubiquitin ligase TRIM31 attenuates NLRP3 inflammasome activation by promoting proteasomal degradation of NLRP3. Nat. Commun. 2016, 7, 13727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muronetz, V.I.; Medvedeva, M.V.; Sevostyanova, I.A.; Schmalhausen, E.V. Modification of Glyceraldehyde-3-Phosphate Dehydrogenase with Nitric Oxide: Role in Signal Transduction and Development of Apoptosis. Biomolecules 2021, 11, 1656. [Google Scholar] [CrossRef]
- Cha, S.J.; Kim, H.; Choi, H.J.; Lee, S.; Kim, K. Protein Glutathionylation in the Pathogenesis of Neurodegenerative Diseases. Oxid. Med. Cell Longev. 2017, 2017, 2818565. [Google Scholar] [CrossRef] [Green Version]
- Montagna, C.; Cirotti, C.; Rizza, S.; Filomeni, G. When S-Nitrosylation Gets to Mitochondria: From Signaling to Age-Related Diseases. Antioxid. Redox. Signal. 2020, 32, 884–905. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Tu, S.; Akhtar, M.W.; Sunico, C.R.; Okamoto, S.; Lipton, S.A. Aberrant protein s-nitrosylation in neurodegenerative diseases. Neuron 2013, 78, 596–614. [Google Scholar] [CrossRef] [Green Version]
- Mailloux, R.J.; Treberg, J.R. Protein S-glutathionlyation links energy metabolism to redox signaling in mitochondria. Redox. Biol. 2016, 8, 110–118. [Google Scholar] [CrossRef] [Green Version]
- Rizza, S.; Cardaci, S.; Montagna, C.; Di Giacomo, G.; De Zio, D.; Bordi, M.; Maiani, E.; Campello, S.; Borreca, A.; Puca, A.A.; et al. S-nitrosylation drives cell senescence and aging in mammals by controlling mitochondrial dynamics and mitophagy. Proc. Natl. Acad. Sci. USA 2018, 115, E3388–E3397. [Google Scholar] [CrossRef] [Green Version]
- Zahid, S.; Khan, R.; Oellerich, M.; Ahmed, N.; Asif, A.R. Differential S-nitrosylation of proteins in Alzheimer’s disease. Neuroscience 2014, 256, 126–136. [Google Scholar] [CrossRef]
- Sultana, R.; Butterfield, D.A. Oxidative modification of brain proteins in Alzheimer’s disease: Perspective on future studies based on results of redox proteomics studies. J. Alzheimers Dis. 2013, 33 (Suppl. S1), S243–S251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabens Liedhegner, E.A.; Gao, X.H.; Mieyal, J.J. Mechanisms of altered redox regulation in neurodegenerative diseases--focus on S--glutathionylation. Antioxid. Redox. Signal. 2012, 16, 543–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, T.; Lipton, S.A. Emerging role of protein-protein transnitrosylation in cell signaling pathways. Antioxid. Redox. Signal. 2013, 18, 239–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moosmann, B.; Behl, C. Antioxidants as treatment for neurodegenerative disorders. Expert Opin. Investig. Drugs 2002, 11, 1407–1435. [Google Scholar] [CrossRef]
- Mailloux, R.J.; Jin, X.; Willmore, W.G. Redox regulation of mitochondrial function with emphasis on cysteine oxidation reactions. Redox. Biol. 2014, 2, 123–139. [Google Scholar] [CrossRef] [Green Version]
- Dalle-Donne, I.; Rossi, R.; Giustarini, D.; Colombo, R.; Milzani, A. S-glutathionylation in protein redox regulation. Free Radic. Biol. Med. 2007, 43, 883–898. [Google Scholar] [CrossRef]
- Kang, P.T.; Zhang, L.; Chen, C.L.; Chen, J.; Green, K.B.; Chen, Y.R. Protein thiyl radical mediates S-glutathionylation of complex I. Free Radic. Biol. Med. 2012, 53, 962–973. [Google Scholar] [CrossRef] [Green Version]
- Ziegler, D.M. Role of reversible oxidation-reduction of enzyme thiols-disulfides in metabolic regulation. Annu. Rev. Biochem. 1985, 54, 305–329. [Google Scholar] [CrossRef]
- Gallogly, M.M.; Starke, D.W.; Mieyal, J.J. Mechanistic and kinetic details of catalysis of thiol-disulfide exchange by glutaredoxins and potential mechanisms of regulation. Antioxid. Redox. Signal. 2009, 11, 1059–1081. [Google Scholar] [CrossRef] [Green Version]
- Stroher, E.; Millar, A.H. The biological roles of glutaredoxins. Biochem. J. 2012, 446, 333–348. [Google Scholar] [CrossRef]
- Mitra, S.; Elliott, S.J. Oxidative disassembly of the [2Fe-2S] cluster of human Grx2 and redox regulation in the mitochondria. Biochemistry 2009, 48, 3813–3815. [Google Scholar] [CrossRef] [PubMed]
- Beer, S.M.; Taylor, E.R.; Brown, S.E.; Dahm, C.C.; Costa, N.J.; Runswick, M.J.; Murphy, M.P. Glutaredoxin 2 catalyzes the reversible oxidation and glutathionylation of mitochondrial membrane thiol proteins: Implications for mitochondrial redox regulation and antioxidant DEFENSE. J. Biol. Chem. 2004, 279, 47939–47951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mailloux, R.J.; Xuan, J.Y.; Beauchamp, B.; Jui, L.; Lou, M.; Harper, M.E. Glutaredoxin-2 is required to control proton leak through uncoupling protein-3. J. Biol. Chem. 2013, 288, 8365–8379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klaus, A.; Zorman, S.; Berthier, A.; Polge, C.; Ramirez, S.; Michelland, S.; Seve, M.; Vertommen, D.; Rider, M.; Lentze, N.; et al. Glutathione S-transferases interact with AMP-activated protein kinase: Evidence for S-glutathionylation and activation in vitro. PLoS ONE 2013, 8, e62497. [Google Scholar] [CrossRef]
- Raza, H. Dual localization of glutathione S-transferase in the cytosol and mitochondria: Implications in oxidative stress, toxicity and disease. FEBS J. 2011, 278, 4243–4251. [Google Scholar] [CrossRef] [Green Version]
- Park, J.W.; Mieyal, J.J.; Rhee, S.G.; Chock, P.B. Deglutathionylation of 2-Cys peroxiredoxin is specifically catalyzed by sulfiredoxin. J. Biol. Chem. 2009, 284, 23364–23374. [Google Scholar] [CrossRef] [Green Version]
- Aon, M.A.; Cortassa, S.; Maack, C.; O’Rourke, B. Sequential opening of mitochondrial ion channels as a function of glutathione redox thiol status. J. Biol. Chem. 2007, 282, 21889–21900. [Google Scholar] [CrossRef] [Green Version]
- Hurd, T.R.; Requejo, R.; Filipovska, A.; Brown, S.; Prime, T.A.; Robinson, A.J.; Fearnley, I.M.; Murphy, M.P. Complex I within oxidatively stressed bovine heart mitochondria is glutathionylated on Cys-531 and Cys-704 of the 75-kDa subunit: Potential role of CYS residues in decreasing oxidative damage. J. Biol. Chem. 2008, 283, 24801–24815. [Google Scholar]
- Chen, C.L.; Zhang, L.; Yeh, A.; Chen, C.A.; Green-Church, K.B.; Zweier, J.L.; Chen, Y.R. Site-specific S-glutathiolation of mitochondrial NADH ubiquinone reductase. Biochemistry 2007, 46, 5754–5765. [Google Scholar] [CrossRef] [Green Version]
- Diotte, N.M.; Xiong, Y.; Gao, J.; Chua, B.H.; Ho, Y.S. Attenuation of doxorubicin-induced cardiac injury by mitochondrial glutaredoxin 2. Biochim. Biophys. Acta 2009, 1793, 427–438. [Google Scholar] [CrossRef] [Green Version]
- Taylor, E.R.; Hurrell, F.; Shannon, R.J.; Lin, T.K.; Hirst, J.; Murphy, M.P. Reversible glutathionylation of complex I increases mitochondrial superoxide formation. J. Biol. Chem. 2003, 278, 19603–19610. [Google Scholar] [CrossRef] [PubMed]
- Tretter, L.; Adam-Vizi, V. Inhibition of Krebs cycle enzymes by hydrogen peroxide: A key role of [alpha]-ketoglutarate dehydrogenase in limiting NADH production under oxidative stress. J. Neurosci. 2000, 20, 8972–8979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, J.; Han, D.; Sancheti, H.; Yap, L.P.; Kaplowitz, N.; Cadenas, E. Regulation of mitochondrial glutathione redox status and protein glutathionylation by respiratory substrates. J. Biol. Chem. 2010, 285, 39646–39654. [Google Scholar] [CrossRef] [Green Version]
- Korshunov, S.S.; Skulachev, V.P.; Starkov, A.A. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997, 416, 15–18. [Google Scholar] [CrossRef] [Green Version]
- Bulteau, A.L.; Lundberg, K.C.; Ikeda-Saito, M.; Isaya, G.; Szweda, L.I. Reversible redox-dependent modulation of mitochondrial aconitase and proteolytic activity during in vivo cardiac ischemia/reperfusion. Proc. Natl. Acad. Sci. USA 2005, 102, 5987–5991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kil, I.S.; Park, J.W. Regulation of mitochondrial NADP+-dependent isocitrate dehydrogenase activity by glutathionylation. J. Biol. Chem. 2005, 280, 10846–10854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLain, A.L.; Szweda, P.A.; Szweda, L.I. alpha-Ketoglutarate dehydrogenase: A mitochondrial redox sensor. Free Radic. Res. 2011, 45, 29–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, M.; Chalker, J.; Slade, L.; Gardiner, D.; Mailloux, R.J. Protein S-glutathionylation alters superoxide/hydrogen peroxide emission from pyruvate dehydrogenase complex. Free Radic. Biol. Med. 2017, 106, 302–314. [Google Scholar] [CrossRef]
- Mailloux, R.J.; Xuan, J.Y.; McBride, S.; Maharsy, W.; Thorn, S.; Holterman, C.E.; Kennedy, C.R.; Rippstein, P.; de Kemp, R.; da Silva, J.; et al. Glutaredoxin-2 is required to control oxidative phosphorylation in cardiac muscle by mediating deglutathionylation reactions. J. Biol. Chem. 2014, 289, 14812–14828. [Google Scholar] [CrossRef] [Green Version]
- Muoio, D.M. Metabolic inflexibility: When mitochondrial indecision leads to metabolic gridlock. Cell 2014, 159, 1253–1262. [Google Scholar] [CrossRef] [Green Version]
- Giangregorio, N.; Palmieri, F.; Indiveri, C. Glutathione controls the redox state of the mitochondrial carnitine/acylcarnitine carrier Cys residues by glutathionylation. Biochim. Biophys. Acta 2013, 1830, 5299–5304. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ye, Z.W.; Singh, S.; Townsend, D.M.; Tew, K.D. An evolving understanding of the S-glutathionylation cycle in pathways of redox regulation. Free Radic. Biol. Med. 2018, 120, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Cadenas, S. Mitochondrial uncoupling, ROS generation and cardioprotection. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 940–950. [Google Scholar] [CrossRef] [PubMed]
- Bouillaud, F.; Alves-Guerra, M.C.; Ricquier, D. UCPs, at the interface between bioenergetics and metabolism. Biochim. Biophys. Acta 2016, 1863, 2443–2456. [Google Scholar] [CrossRef] [PubMed]
- Mailloux, R.J.; Seifert, E.L.; Bouillaud, F.; Aguer, C.; Collins, S.; Harper, M.E. Glutathionylation acts as a control switch for uncoupling proteins UCP2 and UCP3. J. Biol. Chem. 2011, 286, 21865–21875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [Green Version]
- Kowaltowski, A.J.; Netto, L.E.; Vercesi, A.E. The thiol-specific antioxidant enzyme prevents mitochondrial permeability transition. Evidence for the participation of reactive oxygen species in this mechanism. J. Biol. Chem. 1998, 273, 12766–12769. [Google Scholar] [CrossRef] [Green Version]
- Queiroga, C.S.; Almeida, A.S.; Martel, C.; Brenner, C.; Alves, P.M.; Vieira, H.L. Glutathionylation of adenine nucleotide translocase induced by carbon monoxide prevents mitochondrial membrane permeabilization and apoptosis. J. Biol. Chem. 2010, 285, 17077–17088. [Google Scholar] [CrossRef] [Green Version]
- Skulachev, V.P. Bioenergetic aspects of apoptosis, necrosis and mitoptosis. Apoptosis 2006, 11, 473–485. [Google Scholar] [CrossRef]
- Itani, H.A.; Dikalova, A.E.; McMaster, W.G.; Nazarewicz, R.R.; Bikineyeva, A.T.; Harrison, D.G.; Dikalov, S.I. Mitochondrial Cyclophilin D in Vascular Oxidative Stress and Hypertension. Hypertension 2016, 67, 1218–1227. [Google Scholar] [CrossRef]
- Thiriveedi, V.R.; Mattam, U.; Pattabhi, P.; Bisoyi, V.; Talari, N.K.; Krishnamoorthy, T.; Sepuri, N.B.V. Glutathionylated and Fe-S cluster containing hMIA40 (CHCHD4) regulates ROS and mitochondrial complex III and IV activities of the electron transport chain. Redox. Biol. 2020, 37, 101725. [Google Scholar] [CrossRef] [PubMed]
- Liesa, M.; Shirihai, O.S. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013, 17, 491–506. [Google Scholar] [CrossRef] [Green Version]
- Willems, P.H.; Rossignol, R.; Dieteren, C.E.; Murphy, M.P.; Koopman, W.J. Redox Homeostasis and Mitochondrial Dynamics. Cell Metab. 2015, 22, 207–218. [Google Scholar] [CrossRef] [Green Version]
- Shutt, T.; Geoffrion, M.; Milne, R.; McBride, H.M. The intracellular redox state is a core determinant of mitochondrial fusion. EMBO Rep. 2012, 13, 909–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Che, L.; Yang, C.L.; Chen, Y.; Wu, Z.L.; Du, Z.B.; Wu, J.S.; Gan, C.L.; Yan, S.P.; Huang, J.; Guo, N.J.; et al. Mitochondrial redox-driven mitofusin 2 S-glutathionylation promotes neuronal necroptosis via disrupting ER-mitochondria crosstalk in cadmium-induced neurotoxicity. Chemosphere 2021, 262, 127878. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Ruiz, A.; Araujo, I.M.; Izquierdo-Alvarez, A.; Hernansanz-Agustin, P.; Lamas, S.; Serrador, J.M. Specificity in S-nitrosylation: A short-range mechanism for NO signaling? Antioxid. Redox. Signal. 2013, 19, 1220–1235. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Ruiz, A.; Lamas, S. Signalling by NO-induced protein S-nitrosylation and S-glutathionylation: Convergences and divergences. Cardiovasc. Res. 2007, 75, 220–228. [Google Scholar] [CrossRef] [Green Version]
- Fernando, V.; Zheng, X.; Walia, Y.; Sharma, V.; Letson, J.; Furuta, S. S-Nitrosylation: An Emerging Paradigm of Redox Signaling. Antioxidants 2019, 8, 404. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Steenbergen, C.; Murphy, E. S-nitrosylation: NO-related redox signaling to protect against oxidative stress. Antioxid. Redox. Signal. 2006, 8, 1693–1705. [Google Scholar] [CrossRef] [Green Version]
- Lancaster, J.R., Jr. Nitric oxide: A brief overview of chemical and physical properties relevant to therapeutic applications. Future Sci. OA 2015, 1, FSO59. [Google Scholar] [CrossRef] [Green Version]
- Beltran, B.; Orsi, A.; Clementi, E.; Moncada, S. Oxidative stress and S-nitrosylation of proteins in cells. Br. J. Pharm. 2000, 129, 953–960. [Google Scholar] [CrossRef] [PubMed]
- Whiteman, M.; Chua, Y.L.; Zhang, D.; Duan, W.; Liou, Y.C.; Armstrong, J.S. Nitric oxide protects against mitochondrial permeabilization induced by glutathione depletion: Role of S-nitrosylation? Biochem. Biophys. Res. Commun. 2006, 339, 255–262. [Google Scholar] [CrossRef]
- Stomberski, C.T.; Hess, D.T.; Stamler, J.S. Protein S-Nitrosylation: Determinants of Specificity and Enzymatic Regulation of S-Nitrosothiol-Based Signaling. Antioxid. Redox. Signal. 2019, 30, 1331–1351. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wu, K.; Su, W.; Zhang, D.F.; Wang, P.; Qiao, X.; Yao, Q.; Yuan, Z.; Yao, Y.G.; Liu, G.; et al. Increased GSNOR Expression during Aging Impairs Cognitive Function and Decreases S-Nitrosation of CaMKIIalpha. J. Neurosci. 2017, 37, 9741–9758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, T.; Lipton, S.A. Cell death: Protein misfolding and neurodegenerative diseases. Apoptosis 2009, 14, 455–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozawa, K.; Komatsubara, A.T.; Nishimura, Y.; Sawada, T.; Kawafune, H.; Tsumoto, H.; Tsuji, Y.; Zhao, J.; Kyotani, Y.; Tanaka, T.; et al. S-nitrosylation regulates mitochondrial quality control via activation of parkin. Sci. Rep. 2013, 3, 2202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piantadosi, C.A. Regulation of mitochondrial processes by protein S-nitrosylation. Biochim. Biophys. Acta 2012, 1820, 712–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghafourifar, P.; Cadenas, E. Mitochondrial nitric oxide synthase. Trends. Pharm. Sci. 2005, 26, 190–195. [Google Scholar] [CrossRef]
- Zhang, J.; Jin, B.; Li, L.; Block, E.R.; Patel, J.M. Nitric oxide-induced persistent inhibition and nitrosylation of active site cysteine residues of mitochondrial cytochrome-c oxidase in lung endothelial cells. Am. J. Physiol. Cell Physiol. 2005, 288, C840–C849. [Google Scholar] [CrossRef] [Green Version]
- Brown, G.C.; Borutaite, V. Inhibition of mitochondrial respiratory complex I by nitric oxide, peroxynitrite and S-nitrosothiols. Biochim. Biophys. Acta 2004, 1658, 44–49. [Google Scholar] [CrossRef] [Green Version]
- Hagen, T.; Taylor, C.T.; Lam, F.; Moncada, S. Redistribution of intracellular oxygen in hypoxia by nitric oxide: Effect on HIF1alpha. Science 2003, 302, 1975–1978. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.H.; Sancheti, H.; Garcia, J.; Kaplowitz, N.; Cadenas, E.; Han, D. Respiratory substrates regulate S-nitrosylation of mitochondrial proteins through a thiol-dependent pathway. Chem. Res. Toxicol. 2014, 27, 794–804. [Google Scholar] [CrossRef] [PubMed]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial membrane potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Keszler, A.; Azarova, N.A.; Nwanze, N.; Perlegas, A.; Shiva, S.; Broniowska, K.A.; Hogg, N.; Kim-Shapiro, D.B. A novel role for cytochrome c: Efficient catalysis of S-nitrosothiol formation. Free Radic. Biol. Med. 2010, 48, 255–263. [Google Scholar] [CrossRef] [Green Version]
- Schonhoff, C.M.; Gaston, B.; Mannick, J.B. Nitrosylation of cytochrome c during apoptosis. J. Biol. Chem. 2003, 278, 18265–18270. [Google Scholar] [CrossRef] [Green Version]
- Speijer, D.; Manjeri, G.R.; Szklarczyk, R. How to deal with oxygen radicals stemming from mitochondrial fatty acid oxidation. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130446. [Google Scholar] [CrossRef] [Green Version]
- Quijano, C.; Trujillo, M.; Castro, L.; Trostchansky, A. Interplay between oxidant species and energy metabolism. Redox. Biol. 2016, 8, 28–42. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, C.L.; Perevoshchikova, I.V.; Hey-Mogensen, M.; Orr, A.L.; Brand, M.D. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox. Biol. 2013, 1, 304–312. [Google Scholar] [CrossRef] [Green Version]
- Seim, G.L.; John, S.V.; Arp, N.L.; Fang, Z.; Pagliarini, D.J.; Fan, J. Nitric oxide-driven modifications of lipoic arm inhibit alpha-ketoacid dehydrogenases. Nat. Chem. Biol. 2022. Online ahead of print. [Google Scholar] [CrossRef]
- Lemasters, J.J.; Theruvath, T.P.; Zhong, Z.; Nieminen, A.L. Mitochondrial calcium and the permeability transition in cell death. Biochim. Biophys. Acta 2009, 1787, 1395–1401. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.T.; Stevens, M.V.; Kohr, M.; Steenbergen, C.; Sack, M.N.; Murphy, E. Cysteine 203 of cyclophilin D is critical for cyclophilin D activation of the mitochondrial permeability transition pore. J. Biol. Chem. 2011, 286, 40184–40192. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, H.; Katoh, H.; Tanaka, T.; Saotome, M.; Urushida, T.; Satoh, H.; Hayashi, H. Effects of nitric oxide on mitochondrial permeability transition pore and thiol-mediated responses in cardiac myocytes. Nitric. Oxide. 2012, 26, 95–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoshan-Barmatz, V.; De Pinto, V.; Zweckstetter, M.; Raviv, Z.; Keinan, N.; Arbel, N. VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol. Asp. Med. 2010, 31, 227–285. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Sedlic, F.; Pravdic, D.; Bosnjak, Z.J.; Kwok, W.M. Biphasic effect of nitric oxide on the cardiac voltage-dependent anion channel. FEBS Lett. 2011, 585, 328–334. [Google Scholar] [CrossRef] [Green Version]
- Mannick, J.B.; Schonhoff, C.; Papeta, N.; Ghafourifar, P.; Szibor, M.; Fang, K.; Gaston, B. S-Nitrosylation of mitochondrial caspases. J. Cell Biol. 2001, 154, 1111–1116. [Google Scholar] [CrossRef] [Green Version]
- Crow, M.T.; Mani, K.; Nam, Y.J.; Kitsis, R.N. The mitochondrial death pathway and cardiac myocyte apoptosis. Circ. Res. 2004, 95, 957–970. [Google Scholar] [CrossRef] [Green Version]
- Azad, N.; Vallyathan, V.; Wang, L.; Tantishaiyakul, V.; Stehlik, C.; Leonard, S.S.; Rojanasakul, Y. S-nitrosylation of Bcl-2 inhibits its ubiquitin-proteasomal degradation. A novel antiapoptotic mechanism that suppresses apoptosis. J. Biol. Chem. 2006, 281, 34124–34134. [Google Scholar] [CrossRef] [Green Version]
- Gu, Z.; Nakamura, T.; Lipton, S.A. Redox reactions induced by nitrosative stress mediate protein misfolding and mitochondrial dysfunction in neurodegenerative diseases. Mol. Neurobiol. 2010, 41, 55–72. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, T.; Cieplak, P.; Cho, D.H.; Godzik, A.; Lipton, S.A. S-nitrosylation of Drp1 links excessive mitochondrial fission to neuronal injury in neurodegeneration. Mitochondrion 2010, 10, 573–578. [Google Scholar] [CrossRef] [Green Version]
- Bertram, L.; Lill, C.M.; Tanzi, R.E. The genetics of Alzheimer disease: Back to the future. Neuron 2010, 68, 270–281. [Google Scholar] [CrossRef] [Green Version]
- Matej, R.; Tesar, A.; Rusina, R. Alzheimer’s disease and other neurodegenerative dementias in comorbidity: A clinical and neuropathological overview. Clin. Biochem. 2019, 73, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, G.; Chakrabarti, S.; Chatterjee, U.; Saso, L. Proteinopathy, oxidative stress and mitochondrial dysfunction: Cross talk in Alzheimer’s disease and Parkinson’s disease. Drug Des. Devel. Ther. 2017, 11, 797–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Protter, D.; Lang, C.; Cooper, A.A. alphaSynuclein and Mitochondrial Dysfunction: A Pathogenic Partnership in Parkinson’s Disease? Park. Dis. 2012, 2012, 829207. [Google Scholar]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging. 2018, 13, 757–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenough, M.A.; Camakaris, J.; Bush, A.I. Metal dyshomeostasis and oxidative stress in Alzheimer’s disease. Neurochem. Int. 2013, 62, 540–555. [Google Scholar] [CrossRef] [PubMed]
- Bermejo, P.; Martin-Aragon, S.; Benedi, J.; Susin, C.; Felici, E.; Gil, P.; Ribera, J.M.; Villar, A.M. Peripheral levels of glutathione and protein oxidation as markers in the development of Alzheimer’s disease from Mild Cognitive Impairment. Free Radic. Res. 2008, 42, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Vina, J.; Lloret, A.; Orti, R.; Alonso, D. Molecular bases of the treatment of Alzheimer’s disease with antioxidants: Prevention of oxidative stress. Mol. Asp. Med. 2004, 25, 117–123. [Google Scholar] [CrossRef]
- Mandal, P.K.; Saharan, S.; Tripathi, M.; Murari, G. Brain glutathione levels--a novel biomarker for mild cognitive impairment and Alzheimer’s disease. Biol. Psychiatry 2015, 78, 702–710. [Google Scholar] [CrossRef]
- Torres, L.L.; Quaglio, N.B.; de Souza, G.T.; Garcia, R.T.; Dati, L.M.; Moreira, W.L.; Loureiro, A.P.; de Souza-Talarico, J.N.; Smid, J.; Porto, C.S.; et al. Peripheral oxidative stress biomarkers in mild cognitive impairment and Alzheimer’s disease. J. Alzheimer’s Dis. 2011, 26, 59–68. [Google Scholar] [CrossRef]
- Newman, S.F.; Sultana, R.; Perluigi, M.; Coccia, R.; Cai, J.; Pierce, W.M.; Klein, J.B.; Turner, D.M.; Butterfield, D.A. An increase in S-glutathionylated proteins in the Alzheimer’s disease inferior parietal lobule, a proteomics approach. J. Neurosci. Res. 2007, 85, 1506–1514. [Google Scholar] [CrossRef]
- Baldeiras, I.; Santana, I.; Proenca, M.T.; Garrucho, M.H.; Pascoal, R.; Rodrigues, A.; Duro, D.; Oliveira, C.R. Peripheral oxidative damage in mild cognitive impairment and mild Alzheimer’s disease. J. Alzheimer’s Dis. 2008, 15, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Rodriguez, C.; Circu, M.L.; Aw, T.Y.; Feng, J. S-Glutathionyl quantification in the attomole range using glutaredoxin-3-catalyzed cysteine derivatization and capillary gel electrophoresis with laser-induced fluorescence detection. Anal. Bioanal. Chem. 2011, 401, 2165–2175. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Kuo, C.C.; Chiu, A.W.; Feng, J. Prediction of S-glutathionylated proteins progression in Alzheimer’s transgenic mouse model using principle component analysis. J. Alzheimer’s Dis. 2012, 30, 919–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prendecki, M.; Florczak-Wyspianska, J.; Kowalska, M.; Ilkowski, J.; Grzelak, T.; Bialas, K.; Wiszniewska, M.; Kozubski, W.; Dorszewska, J. Biothiols and oxidative stress markers and polymorphisms of TOMM40 and APOC1 genes in Alzheimer’s disease patients. Oncotarget 2018, 9, 35207–35225. [Google Scholar] [CrossRef] [Green Version]
- Ribas, V.; Garcia-Ruiz, C.; Fernandez-Checa, J.C. Glutathione and mitochondria. Front. Pharmacol. 2014, 5, 151. [Google Scholar] [CrossRef] [Green Version]
- Ruszkiewicz, J.; Albrecht, J. Changes in the mitochondrial antioxidant systems in neurodegenerative diseases and acute brain disorders. Neurochem. Int. 2015, 88, 66–72. [Google Scholar] [CrossRef]
- Melin, J.; Schulz, C.; Wrobel, L.; Bernhard, O.; Chacinska, A.; Jahn, O.; Schmidt, B.; Rehling, P. Presequence recognition by the tom40 channel contributes to precursor translocation into the mitochondrial matrix. Mol. Cell Biol. 2014, 34, 3473–3485. [Google Scholar] [CrossRef] [Green Version]
- Dorszewska, J.; Kempisty, B.; Jaroszewska-Kolecka, J.; Rozycka, A.; Florczak, J.; Lianeri, M.; Jagodzinski, P.P.; Kozubski, W. Expression and polymorphisms of gene 8-oxoguanine glycosylase 1 and the level of oxidative DNA damage in peripheral blood lymphocytes of patients with Alzheimer’s disease. DNA Cell Biol. 2009, 28, 579–588. [Google Scholar] [CrossRef]
- Dalle-Donne, I.; Giustarini, D.; Rossi, R.; Colombo, R.; Milzani, A. Reversible S-glutathionylation of Cys 374 regulates actin filament formation by inducing structural changes in the actin molecule. Free Radic. Biol. Med. 2003, 34, 23–32. [Google Scholar]
- Gibson, G.E.; Zhang, H.; Sheu, K.F.; Bogdanovich, N.; Lindsay, J.G.; Lannfelt, L.; Vestling, M.; Cowburn, R.F. Alpha-ketoglutarate dehydrogenase in Alzheimer brains bearing the APP670/671 mutation. Ann. Neurol. 1998, 44, 676–681. [Google Scholar]
- Nulton-Persson, A.C.; Starke, D.W.; Mieyal, J.J.; Szweda, L.I. Reversible inactivation of alpha-ketoglutarate dehydrogenase in response to alterations in the mitochondrial glutathione status. Biochemistry 2003, 42, 4235–4242. [Google Scholar] [CrossRef] [PubMed]
- Arodin, L.; Lamparter, H.; Karlsson, H.; Nennesmo, I.; Bjornstedt, M.; Schroder, J.; Fernandes, A.P. Alteration of thioredoxin and glutaredoxin in the progression of Alzheimer’s disease. J. Alzheimer’s Dis. 2014, 39, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Akterin, S.; Cowburn, R.F.; Miranda-Vizuete, A.; Jimenez, A.; Bogdanovic, N.; Winblad, B.; Cedazo-Minguez, A. Involvement of glutaredoxin-1 and thioredoxin-1 in beta-amyloid toxicity and Alzheimer’s disease. Cell Death Differ. 2006, 13, 1454–1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kommaddi, R.P.; Tomar, D.S.; Karunakaran, S.; Bapat, D.; Nanguneri, S.; Ray, A.; Schneider, B.L.; Nair, D.; Ravindranath, V. Glutaredoxin1 Diminishes Amyloid Beta-Mediated Oxidation of F-Actin and Reverses Cognitive Deficits in an Alzheimer’s Disease Mouse Model. Antioxid. Redox. Signal. 2019, 31, 1321–1338. [Google Scholar] [CrossRef] [PubMed]
- Mandal, P.K.; Roy, R.G.; Samkaria, A. Oxidative Stress: Glutathione and Its Potential to Protect Methionine-35 of Abeta Peptide from Oxidation. ACS Omega 2022, 7, 27052–27061. [Google Scholar] [CrossRef]
- Chen, J.X.; Yan, S.D. Amyloid-beta-induced mitochondrial dysfunction. J. Alzheimer’s Dis. 2007, 12, 177–184. [Google Scholar] [CrossRef]
- El Hindy, M.; Hezwani, M.; Corry, D.; Hull, J.; El Amraoui, F.; Harris, M.; Lee, C.; Forshaw, T.; Wilson, A.; Mansbridge, A.; et al. The branched-chain aminotransferase proteins: Novel redox chaperones for protein disulfide isomerase--implications in Alzheimer’s disease. Antioxid. Redox. Signal. 2014, 20, 2497–2513. [Google Scholar] [CrossRef] [Green Version]
- Hutson, S.M.; Wallin, R.; Hall, T.R. Identification of mitochondrial branched chain aminotransferase and its isoforms in rat tissues. J. Biol. Chem. 1992, 267, 15681–15686. [Google Scholar] [CrossRef]
- Dinoto, L.; Deture, M.A.; Purich, D.L. Structural insights into Alzheimer filament assembly pathways based on site-directed mutagenesis and S-glutathionylation of three-repeat neuronal Tau protein. Microsc. Res. Tech. 2005, 67, 156–163. [Google Scholar] [CrossRef]
- Samluk, L.; Ostapczuk, P.; Dziembowska, M. Long-term mitochondrial stress induces early steps of Tau aggregation by increasing reactive oxygen species levels and affecting cellular proteostasis. Mol. Biol. Cell 2022, 33, ar67. [Google Scholar] [CrossRef]
- Strange, R.C.; Spiteri, M.A.; Ramachandran, S.; Fryer, A.A. Glutathione-S-transferase family of enzymes. Mutat. Res. 2001, 482, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Ryu, H.; Kowall, N.W. Differential regulation of neuronal and inducible nitric oxide synthase (NOS) in the spinal cord of mutant SOD1 (G93A) ALS mice. Biochem. Biophys. Res. Commun. 2009, 387, 202–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abrams, A.J.; Farooq, A.; Wang, G. S-nitrosylation of ApoE in Alzheimer’s disease. Biochemistry 2011, 50, 3405–3407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, G.E. Interactions of oxidative stress with cellular calcium dynamics and glucose metabolism in Alzheimer’s disease. Free Radic. Biol. Med. 2002, 32, 1061–1070. [Google Scholar] [CrossRef] [PubMed]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 2008, 104, 1433–1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barsoum, M.J.; Yuan, H.; Gerencser, A.A.; Liot, G.; Kushnareva, Y.; Graber, S.; Kovacs, I.; Lee, W.D.; Waggoner, J.; Cui, J.; et al. Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J. 2006, 25, 3900–3911. [Google Scholar] [CrossRef]
- Qu, J.; Nakamura, T.; Cao, G.; Holland, E.A.; McKercher, S.R.; Lipton, S.A. S-Nitrosylation activates Cdk5 and contributes to synaptic spine loss induced by beta-amyloid peptide. Proc. Natl. Acad. Sci. USA 2011, 108, 14330–14335. [Google Scholar] [CrossRef] [Green Version]
- Bloem, B.R.; Okun, M.S.; Klein, C. Parkinson’s disease. Lancet 2021, 397, 2284–2303. [Google Scholar] [CrossRef]
- Milanese, C.; Tapias, V.; Gabriels, S.; Cerri, S.; Levandis, G.; Blandini, F.; Tresini, M.; Shiva, S.; Greenamyre, J.T.; Gladwin, M.T.; et al. Mitochondrial Complex I Reversible S-Nitrosation Improves Bioenergetics and Is Protective in Parkinson’s Disease. Antioxid. Redox. Signal. 2018, 28, 44–61. [Google Scholar] [CrossRef]
- Yao, D.; Gu, Z.; Nakamura, T.; Shi, Z.Q.; Ma, Y.; Gaston, B.; Palmer, L.A.; Rockenstein, E.M.; Zhang, Z.; Masliah, E.; et al. Nitrosative stress linked to sporadic Parkinson’s disease: S-nitrosylation of parkin regulates its E3 ubiquitin ligase activity. Proc. Natl. Acad. Sci. USA 2004, 101, 10810–10814. [Google Scholar] [CrossRef] [Green Version]
- Chung, K.K.; Thomas, B.; Li, X.; Pletnikova, O.; Troncoso, J.C.; Marsh, L.; Dawson, V.L.; Dawson, T.M. S-nitrosylation of parkin regulates ubiquitination and compromises parkin’s protective function. Science 2004, 304, 1328–1331. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Kang, U.J. Characterization of PINK1 processing, stability, and subcellular localization. J. Neurochem. 2008, 106, 464–474. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Castelao, B.; Munoz, C.; Sanchez, I.; Goethals, M.; Vandekerckhove, J.; Castano, J.G. Reduced protein stability of human DJ-1/PARK7 L166P, linked to autosomal recessive Parkinson disease, is due to direct endoproteolytic cleavage by the proteasome. Biochim. Biophys. Acta 2012, 1823, 524–533. [Google Scholar] [CrossRef] [PubMed]
- Guzman, J.N.; Sanchez-Padilla, J.; Wokosin, D.; Kondapalli, J.; Ilijic, E.; Schumacker, P.T.; Surmeier, D.J. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature 2010, 468, 696–700. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W.M.; Golczak, M.; Choe, K.; Curran, P.L.; Miller, O.G.; Yao, C.; Wang, W.; Lin, J.; Milkovic, N.M.; Ray, A.; et al. Regulation of DJ-1 by Glutaredoxin 1 in Vivo: Implications for Parkinson’s Disease. Biochemistry 2016, 55, 4519–4532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, W.M.; Yao, C.; Siedlak, S.L.; Wang, W.; Zhu, X.; Caldwell, G.A.; Wilson-Delfosse, A.L.; Mieyal, J.J.; Chen, S.G. Glutaredoxin deficiency exacerbates neurodegeneration in C. elegans models of Parkinson’s disease. Hum. Mol. Genet. 2015, 24, 1322–1335. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Liu, N.; Lu, B. Mechanisms and roles of mitophagy in neurodegenerative diseases. CNS Neurosci. Ther. 2019, 25, 859–875. [Google Scholar] [CrossRef]
- Chinta, S.J.; Andersen, J.K. Nitrosylation and nitration of mitochondrial complex I in Parkinson’s disease. Free Radic. Res. 2011, 45, 53–58. [Google Scholar] [CrossRef]
- Danielson, S.R.; Held, J.M.; Schilling, B.; Oo, M.; Gibson, B.W.; Andersen, J.K. Preferentially increased nitration of alpha-synuclein at tyrosine-39 in a cellular oxidative model of Parkinson’s disease. Anal. Chem. 2009, 81, 7823–7828. [Google Scholar] [CrossRef] [Green Version]
- Fang, J.; Nakamura, T.; Cho, D.H.; Gu, Z.; Lipton, S.A. S-nitrosylation of peroxiredoxin 2 promotes oxidative stress-induced neuronal cell death in Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2007, 104, 18742–18747. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Y.; Li, X.; Du, X.; Bi, M.; Ma, F.; Xie, J.; Jiang, H. The S-nitrosylation of parkin attenuated the ubiquitination of divalent metal transporter 1 in MPP(+)-treated SH-SY5Y cells. Sci. Rep. 2020, 10, 15542. [Google Scholar] [CrossRef] [PubMed]
- Vicente Miranda, H.; Cassio, R.; Correia-Guedes, L.; Gomes, M.A.; Chegao, A.; Miranda, E.; Soares, T.; Coelho, M.; Rosa, M.M.; Ferreira, J.J.; et al. Posttranslational modifications of blood-derived alpha-synuclein as biochemical markers for Parkinson’s disease. Sci. Rep. 2017, 7, 13713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fay, J.M.; Zhu, C.; Proctor, E.A.; Tao, Y.; Cui, W.; Ke, H.; Dokholyan, N.V. A Phosphomimetic Mutation Stabilizes SOD1 and Rescues Cell Viability in the Context of an ALS-Associated Mutation. Structure 2016, 24, 1898–1906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Wing, S.S.; Ponka, P. S-nitrosylation of IRP2 regulates its stability via the ubiquitin-proteasome pathway. Mol. Cell Biol. 2004, 24, 330–337. [Google Scholar] [CrossRef] [PubMed]
- LaVaute, T.; Smith, S.; Cooperman, S.; Iwai, K.; Land, W.; Meyron-Holtz, E.; Drake, S.K.; Miller, G.; Abu-Asab, M.; Tsokos, M.; et al. Targeted deletion of the gene encoding iron regulatory protein-2 causes misregulation of iron metabolism and neurodegenerative disease in mice. Nat. Genet. 2001, 27, 209–214. [Google Scholar] [CrossRef]
- Garcia-Gimenez, J.L.; Gimeno, A.; Gonzalez-Cabo, P.; Dasi, F.; Bolinches-Amoros, A.; Molla, B.; Palau, F.; Pallardo, F.V. Differential expression of PGC-1alpha and metabolic sensors suggest age-dependent induction of mitochondrial biogenesis in Friedreich ataxia fibroblasts. PLoS ONE 2011, 6, e20666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, S.H.; Jung, I.S.; Han, G.Y.; Kim, N.H.; Kim, H.J.; Kim, C.W. Proteomic profiling of brain cortex tissues in a Tau transgenic mouse model of Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2013, 430, 670–675. [Google Scholar] [CrossRef]
- Kolsch, H.; Linnebank, M.; Lutjohann, D.; Jessen, F.; Wullner, U.; Harbrecht, U.; Thelen, K.M.; Kreis, M.; Hentschel, F.; Schulz, A.; et al. Polymorphisms in glutathione S-transferase omega-1 and AD, vascular dementia, and stroke. Neurology 2004, 63, 2255–2260. [Google Scholar] [CrossRef]
- Pinhel, M.A.; Nakazone, M.A.; Cacao, J.C.; Piteri, R.C.; Dantas, R.T.; Godoy, M.F.; Godoy, M.R.; Tognola, W.A.; Conforti-Froes, N.D.; Souza, D. Glutathione S-transferase variants increase susceptibility for late-onset Alzheimer’s disease: Association study and relationship with apolipoprotein E epsilon4 allele. Clin. Chem. Lab. Med. 2008, 46, 439–445. [Google Scholar] [CrossRef]
- Jafarian, Z.; Saliminejad, K.; Kamali, K.; Ohadi, M.; Kowsari, A.; Nasehi, L.; Khorram Khorshid, H.R. Association of glutathione S-transferases M1, P1 and T1 variations and risk of late-onset Alzheimer’s disease. Neurol. Res. 2018, 40, 41–44. [Google Scholar] [CrossRef]
- Bierman, E.J.; Comijs, H.C.; Jonker, C.; Beekman, A.T. Symptoms of anxiety and depression in the course of cognitive decline. Dement. Geriatr. Cogn. Disord. 2007, 24, 213–219. [Google Scholar] [CrossRef]
- Bouayed, J.; Rammal, H.; Soulimani, R. Oxidative stress and anxiety: Relationship and cellular pathways. Oxid. Med. Cell Longev. 2009, 2, 63–67. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.C.; Wang, Z.F.; Wang, Q.; Wang, Y.P.; Wang, J.Z. Melatonin attenuates beta-amyloid-induced inhibition of neurofilament expression. Acta. Pharm. Sin. 2004, 25, 447–451. [Google Scholar]
- Garcia-Mesa, Y.; Gimenez-Llort, L.; Lopez, L.C.; Venegas, C.; Cristofol, R.; Escames, G.; Acuna-Castroviejo, D.; Sanfeliu, C. Melatonin plus physical exercise are highly neuroprotective in the 3xTg-AD mouse. Neurobiol. Aging 2012, 33, 1124.e13-29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nie, L.; Wei, G.; Peng, S.; Qu, Z.; Yang, Y.; Yang, Q.; Huang, X.; Liu, J.; Zhuang, Z.; Yang, X. Melatonin ameliorates anxiety and depression-like behaviors and modulates proteomic changes in triple transgenic mice of Alzheimer’s disease. Biofactors 2017, 43, 593–611. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.A.; Lin, T.H.; Sheehan, A.E.; Van der Goes van Naters, W.; Neukomm, L.J.; Graves, H.K.; Bis-Brewer, D.M.; Zuchner, S.; Freeman, M.R. Glutathione S-Transferase Regulates Mitochondrial Populations in Axons through Increased Glutathione Oxidation. Neuron 2019, 103, 52–65 e56. [Google Scholar] [CrossRef] [Green Version]
- Dyer, R.R.; Ford, K.I.; Robinson, R.A.S. The roles of S-nitrosylation and S-glutathionylation in Alzheimer’s disease. Methods Enzym. 2019, 626, 499–538. [Google Scholar]
- Spiers, J.G.; Chen, H.C.; Bourgognon, J.M.; Steinert, J.R. Dysregulation of stress systems and nitric oxide signaling underlies neuronal dysfunction in Alzheimer’s disease. Free Radic. Biol. Med. 2019, 134, 468–483. [Google Scholar] [CrossRef]
- Manczak, M.; Reddy, P.H. Abnormal interaction of VDAC1 with amyloid beta and phosphorylated tau causes mitochondrial dysfunction in Alzheimer’s disease. Hum. Mol. Genet. 2012, 21, 5131–5146. [Google Scholar] [CrossRef]
- Colombini, M. VDAC structure, selectivity, and dynamics. Biochim. Biophys. Acta 2012, 1818, 1457–1465. [Google Scholar] [CrossRef] [Green Version]
- Fukai, T.; Ushio-Fukai, M. Superoxide dismutases: Role in redox signaling, vascular function, and diseases. Antioxid. Redox. Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef] [Green Version]
- Qu, J.; Nakamura, T.; Holland, E.A.; McKercher, S.R.; Lipton, S.A. S-nitrosylation of Cdk5: Potential implications in amyloid-beta-related neurotoxicity in Alzheimer disease. Prion 2012, 6, 364–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leroy, K.; Yilmaz, Z.; Brion, J.P. Increased level of active GSK-3beta in Alzheimer’s disease and accumulation in argyrophilic grains and in neurones at different stages of neurofibrillary degeneration. Neuropathol. Appl. Neurobiol. 2007, 33, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Seneviratne, U.; Nott, A.; Bhat, V.B.; Ravindra, K.C.; Wishnok, J.S.; Tsai, L.H.; Tannenbaum, S.R. S-nitrosation of proteins relevant to Alzheimer’s disease during early stages of neurodegeneration. Proc. Natl. Acad. Sci. USA 2016, 113, 4152–4157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, D.H.; Nakamura, T.; Fang, J.; Cieplak, P.; Godzik, A.; Gu, Z.; Lipton, S.A. S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science 2009, 324, 102–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, L.; Robinson, R.A. High-throughput endogenous measurement of S-nitrosylation in Alzheimer’s disease using oxidized cysteine-selective cPILOT. Analyst 2016, 141, 3904–3915. [Google Scholar] [CrossRef]
- Akhtar, M.W.; Sanz-Blasco, S.; Dolatabadi, N.; Parker, J.; Chon, K.; Lee, M.S.; Soussou, W.; McKercher, S.R.; Ambasudhan, R.; Nakamura, T.; et al. Elevated glucose and oligomeric beta-amyloid disrupt synapses via a common pathway of aberrant protein S-nitrosylation. Nat. Commun. 2016, 7, 10242. [Google Scholar] [CrossRef] [Green Version]
- Wijasa, T.S.; Sylvester, M.; Brocke-Ahmadinejad, N.; Schwartz, S.; Santarelli, F.; Gieselmann, V.; Klockgether, T.; Brosseron, F.; Heneka, M.T. Quantitative proteomics of synaptosome S-nitrosylation in Alzheimer’s disease. J. Neurochem. 2020, 152, 710–726. [Google Scholar] [CrossRef] [Green Version]
- Klingelhoefer, L.; Reichmann, H. Parkinson’s disease as a multisystem disorder. J. Neural. Transm. 2017, 124, 709–713. [Google Scholar] [CrossRef]
- Shadrina, M.I.; Slominsky, P.A. Molecular genetics of Parkinson’s disease. Russ. J. Genet. 2006, 42, 858–871. [Google Scholar] [CrossRef]
- Schiesling, C.; Kieper, N.; Seidel, K.; Kruger, R. Review: Familial Parkinson’s disease--genetics, clinical phenotype and neuropathology in relation to the common sporadic form of the disease. Neuropathol. Appl. Neurobiol. 2008, 34, 255–271. [Google Scholar] [CrossRef]
- Schapira, A.H.; Gegg, M. Mitochondrial contribution to Parkinson’s disease pathogenesis. Park. Dis. 2011, 2011, 159160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, T.N.; Greenamyre, J.T. Toxin models of mitochondrial dysfunction in Parkinson’s disease. Antioxid. Redox. Signal. 2012, 16, 920–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weng, M.; Xie, X.; Liu, C.; Lim, K.L.; Zhang, C.W.; Li, L. The Sources of Reactive Oxygen Species and Its Possible Role in the Pathogenesis of Parkinson’s Disease. Park. Dis. 2018, 2018, 9163040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vali, S.; Mythri, R.B.; Jagatha, B.; Padiadpu, J.; Ramanujan, K.S.; Andersen, J.K.; Gorin, F.; Bharath, M.M. Integrating glutathione metabolism and mitochondrial dysfunction with implications for Parkinson’s disease: A dynamic model. Neuroscience 2007, 149, 917–930. [Google Scholar] [CrossRef]
- Chinta, S.J.; Kumar, J.M.; Zhang, H.; Forman, H.J.; Andersen, J.K. Up-regulation of gamma-glutamyl transpeptidase activity following glutathione depletion has a compensatory rather than an inhibitory effect on mitochondrial complex I activity: Implications for Parkinson’s disease. Free Radic. Biol. Med. 2006, 40, 1557–1563. [Google Scholar] [CrossRef]
- Perry, T.L.; Yong, V.W. Idiopathic Parkinson’s disease, progressive supranuclear palsy and glutathione metabolism in the substantia nigra of patients. Neurosci. Lett. 1986, 67, 269–274. [Google Scholar] [CrossRef]
- Mischley, L.K.; Standish, L.J.; Weiss, N.S.; Padowski, J.M.; Kavanagh, T.J.; White, C.C.; Rosenfeld, M.E. Glutathione as a Biomarker in Parkinson’s Disease: Associations with Aging and Disease Severity. Oxid. Med. Cell Longev. 2016, 2016, 9409363. [Google Scholar] [CrossRef] [Green Version]
- Wei, H.J.; Du, M.; Bai, H.Y. Correlations of Melatonin and Glutathione Levels with Oxidative Stress Mechanism in Parkinson’s Disease. Zhongguo Yi Xue Ke Xue Yuan Xue Bao Acta Acad. Med. Sin. 2019, 41, 183–187. [Google Scholar]
- Tokarew, J.M.; El-Kodsi, D.N.; Lengacher, N.A.; Fehr, T.K.; Nguyen, A.P.; Shutinoski, B.; O’Nuallain, B.; Jin, M.; Khan, J.M.; Ng, A.C.H.; et al. Age-associated insolubility of parkin in human midbrain is linked to redox balance and sequestration of reactive dopamine metabolites. Acta Neuropathol. 2021, 141, 725–754. [Google Scholar] [CrossRef]
- Verma, A.; Ray, A.; Bapat, D.; Diwakar, L.; Kommaddi, R.P.; Schneider, B.L.; Hirsch, E.C.; Ravindranath, V. Glutaredoxin 1 Downregulation in the Substantia Nigra Leads to Dopaminergic Degeneration in Mice. Mov. Disord. 2020, 35, 1843–1853. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Rocha, H.; Garcia Garcia, A.; Zavala-Flores, L.; Li, S.; Madayiputhiya, N.; Franco, R. Glutaredoxin 1 protects dopaminergic cells by increased protein glutathionylation in experimental Parkinson’s disease. Antioxid. Redox. Signal. 2012, 17, 1676–1693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.; Kim, S.H.; Kim, J.; Kim, H.; Yim, J. Glutathione s-transferase omega 1 activity is sufficient to suppress neurodegeneration in a Drosophila model of Parkinson disease. J. Biol. Chem. 2012, 287, 6628–6641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taira, T.; Saito, Y.; Niki, T.; Iguchi-Ariga, S.M.; Takahashi, K.; Ariga, H. DJ-1 has a role in antioxidative stress to prevent cell death. EMBO Rep. 2004, 5, 213–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolgacheva, L.P.; Berezhnov, A.V.; Fedotova, E.I.; Zinchenko, V.P.; Abramov, A.Y. Role of DJ-1 in the mechanism of pathogenesis of Parkinson’s disease. J. Bioenerg. Biomembr. 2019, 51, 175–188. [Google Scholar] [CrossRef] [Green Version]
- Thomas, K.J.; McCoy, M.K.; Blackinton, J.; Beilina, A.; van der Brug, M.; Sandebring, A.; Miller, D.; Maric, D.; Cedazo-Minguez, A.; Cookson, M.R. DJ-1 acts in parallel to the PINK1/parkin pathway to control mitochondrial function and autophagy. Hum. Mol. Genet. 2011, 20, 40–50. [Google Scholar] [CrossRef] [Green Version]
- Junn, E.; Jang, W.H.; Zhao, X.; Jeong, B.S.; Mouradian, M.M. Mitochondrial localization of DJ-1 leads to enhanced neuroprotection. J. Neurosci. Res. 2009, 87, 123–129. [Google Scholar] [CrossRef]
- Hayashi, T.; Ishimori, C.; Takahashi-Niki, K.; Taira, T.; Kim, Y.C.; Maita, H.; Maita, C.; Ariga, H.; Iguchi-Ariga, S.M. DJ-1 binds to mitochondrial complex I and maintains its activity. Biochem. Biophys. Res. Commun. 2009, 390, 667–672. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.W.; Kaur, D.; Chinta, S.J.; Rajagopalan, S.; Andersen, J.K. A disruption in iron-sulfur center biogenesis via inhibition of mitochondrial dithiol glutaredoxin 2 may contribute to mitochondrial and cellular iron dysregulation in mammalian glutathione-depleted dopaminergic cells: Implications for Parkinson’s disease. Antioxid. Redox. Signal. 2009, 11, 2083–2094. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Liu, L.; Jiang, X.; Zhai, S.; Xing, D. The Essential Role of Drp1 and Its Regulation by S-Nitrosylation of Parkin in Dopaminergic Neurodegeneration: Implications for Parkinson’s Disease. Antioxid. Redox. Signal. 2016, 25, 609–622. [Google Scholar] [CrossRef] [Green Version]
- Meng, F.; Yao, D.; Shi, Y.; Kabakoff, J.; Wu, W.; Reicher, J.; Ma, Y.; Moosmann, B.; Masliah, E.; Lipton, S.A.; et al. Oxidation of the cysteine-rich regions of parkin perturbs its E3 ligase activity and contributes to protein aggregation. Mol. Neurodegener. 2011, 6, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipton, S.A.; Nakamura, T.; Yao, D.; Shi, Z.Q.; Uehara, T.; Gu, Z. Comment on “S-nitrosylation of parkin regulates ubiquitination and compromises parkin’s protective function”. Science 2005, 308, 1870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkaniec, A.; Lenkiewicz, A.M.; Czapski, G.A.; Jesko, H.M.; Hilgier, W.; Brodzik, R.; Gassowska-Dobrowolska, M.; Culmsee, C.; Adamczyk, A. Extracellular Alpha-Synuclein Oligomers Induce Parkin S-Nitrosylation: Relevance to Sporadic Parkinson’s Disease Etiopathology. Mol. Neurobiol. 2019, 56, 125–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kane, L.A.; Lazarou, M.; Fogel, A.I.; Li, Y.; Yamano, K.; Sarraf, S.A.; Banerjee, S.; Youle, R.J. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol. 2014, 205, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Pickrell, A.M.; Youle, R.J. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, C.K.; Sultan, A.; Platzer, J.; Dolatabadi, N.; Soldner, F.; McClatchy, D.B.; Diedrich, J.K.; Yates, J.R., 3rd; Ambasudhan, R.; Nakamura, T.; et al. S-Nitrosylation of PINK1 Attenuates PINK1/Parkin-Dependent Mitophagy in hiPSC-Based Parkinson’s Disease Models. Cell Rep. 2017, 21, 2171–2182. [Google Scholar] [CrossRef] [Green Version]
- Ozawa, K.; Tsumoto, H.; Miura, Y.; Yamaguchi, J.; Iguchi-Ariga, S.M.M.; Sakuma, T.; Yamamoto, T.; Uchiyama, Y. DJ-1 is indispensable for the S-nitrosylation of Parkin, which maintains function of mitochondria. Sci. Rep. 2020, 10, 4377. [Google Scholar] [CrossRef]
- Ryan, S.D.; Dolatabadi, N.; Chan, S.F.; Zhang, X.; Akhtar, M.W.; Parker, J.; Soldner, F.; Sunico, C.R.; Nagar, S.; Talantova, M.; et al. Isogenic human iPSC Parkinson’s model shows nitrosative stress-induced dysfunction in MEF2-PGC1alpha transcription. Cell 2013, 155, 1351–1364. [Google Scholar] [CrossRef] [Green Version]
- Lundberg, J.O.; Gladwin, M.T.; Weitzberg, E. Strategies to increase nitric oxide signalling in cardiovascular disease. Nat. Rev. Drug Discov. 2015, 14, 623–641. [Google Scholar] [CrossRef]
- Qu, Y.; Konrad, C.; Anderson, C.; Qian, L.; Yin, T.; Manfredi, G.; Iadecola, C.; Zhou, P. Prohibitin S-Nitrosylation Is Required for the Neuroprotective Effect of Nitric Oxide in Neuronal Cultures. J. Neurosci. 2020, 40, 3142–3151. [Google Scholar] [CrossRef]
- Dutta, D.; Ali, N.; Banerjee, E.; Singh, R.; Naskar, A.; Paidi, R.K.; Mohanakumar, K.P. Low Levels of Prohibitin in Substantia Nigra Makes Dopaminergic Neurons Vulnerable in Parkinson’s Disease. Mol. Neurobiol. 2018, 55, 804–821. [Google Scholar] [CrossRef] [PubMed]
- Burai, R.; Ait-Bouziad, N.; Chiki, A.; Lashuel, H.A. Elucidating the Role of Site-Specific Nitration of alpha-Synuclein in the Pathogenesis of Parkinson’s Disease via Protein Semisynthesis and Mutagenesis. J. Am. Chem. Soc. 2015, 137, 5041–5052. [Google Scholar] [CrossRef] [PubMed]
- Byrne, S.; Walsh, C.; Lynch, C.; Bede, P.; Elamin, M.; Kenna, K.; McLaughlin, R.; Hardiman, O. Rate of familial amyotrophic lateral sclerosis: A systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry 2011, 82, 623–627. [Google Scholar] [CrossRef] [Green Version]
- Andersen, P.M. Extensive heterogeneity in patients with ALS with mutations in SOD1 in France. J. Neurol. Neurosurg. Psychiatry 2021, 92, 914. [Google Scholar] [CrossRef] [PubMed]
- Andersen, P.M.; Al-Chalabi, A. Clinical genetics of amyotrophic lateral sclerosis: What do we really know? Nat. Rev. Neurol. 2011, 7, 603–615. [Google Scholar] [CrossRef]
- Trist, B.G.; Hilton, J.B.; Hare, D.J.; Crouch, P.J.; Double, K.L. Superoxide Dismutase 1 in Health and Disease: How a Frontline Antioxidant Becomes Neurotoxic. Angew. Chem. Int. Ed. Engl. 2021, 60, 9215–9246. [Google Scholar] [CrossRef] [PubMed]
- Proctor, E.A.; Fee, L.; Tao, Y.; Redler, R.L.; Fay, J.M.; Zhang, Y.; Lv, Z.; Mercer, I.P.; Deshmukh, M.; Lyubchenko, Y.L.; et al. Nonnative SOD1 trimer is toxic to motor neurons in a model of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2016, 113, 614–619. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Beck, M.V.; Griffith, J.D.; Deshmukh, M.; Dokholyan, N.V. Large SOD1 aggregates, unlike trimeric SOD1, do not impact cell viability in a model of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2018, 115, 4661–4665. [Google Scholar] [CrossRef] [Green Version]
- Coelho, F.R.; Iqbal, A.; Linares, E.; Silva, D.F.; Lima, F.S.; Cuccovia, I.M.; Augusto, O. Oxidation of the tryptophan 32 residue of human superoxide dismutase 1 caused by its bicarbonate-dependent peroxidase activity triggers the non-amyloid aggregation of the enzyme. J. Biol. Chem. 2014, 289, 30690–30701. [Google Scholar] [CrossRef] [Green Version]
- Redler, R.L.; Wilcox, K.C.; Proctor, E.A.; Fee, L.; Caplow, M.; Dokholyan, N.V. Glutathionylation at Cys-111 induces dissociation of wild type and FALS mutant SOD1 dimers. Biochemistry 2011, 50, 7057–7066. [Google Scholar] [CrossRef] [Green Version]
- Auclair, J.R.; Johnson, J.L.; Liu, Q.; Salisbury, J.P.; Rotunno, M.S.; Petsko, G.A.; Ringe, D.; Brown, R.H., Jr.; Bosco, D.A.; Agar, J.N. Post-translational modification by cysteine protects Cu/Zn-superoxide dismutase from oxidative damage. Biochemistry 2013, 52, 6137–6144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karnati, S.; Luers, G.; Pfreimer, S.; Baumgart-Vogt, E. Mammalian SOD2 is exclusively located in mitochondria and not present in peroxisomes. Histochem. Cell Biol. 2013, 140, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Oury, T.D.; Day, B.J.; Crapo, J.D. Extracellular superoxide dismutase in vessels and airways of humans and baboons. Free Radic. Biol. Med. 1996, 20, 957–965. [Google Scholar] [CrossRef] [PubMed]
- Kawamata, H.; Manfredi, G. Import, maturation, and function of SOD1 and its copper chaperone CCS in the mitochondrial intermembrane space. Antioxid. Redox. Signal. 2010, 13, 1375–1384. [Google Scholar] [CrossRef] [Green Version]
- Mesecke, N.; Terziyska, N.; Kozany, C.; Baumann, F.; Neupert, W.; Hell, K.; Herrmann, J.M. A disulfide relay system in the intermembrane space of mitochondria that mediates protein import. Cell 2005, 121, 1059–1069. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Wang, X.; Huo, Z.; Chen, Y.; Liu, J.; Zhao, Z.; Meng, F.; Su, Q.; Bao, W.; Zhang, L.; et al. The Impact of Mitochondrial Dysfunction in Amyotrophic Lateral Sclerosis. Cells 2022, 11, 2049. [Google Scholar] [CrossRef]
- Cozzolino, M.; Pesaresi, M.G.; Amori, I.; Crosio, C.; Ferri, A.; Nencini, M.; Carri, M.T. Oligomerization of mutant SOD1 in mitochondria of motoneuronal cells drives mitochondrial damage and cell toxicity. Antioxid. Redox. Signal. 2009, 11, 1547–1558. [Google Scholar] [CrossRef]
- Pedrini, S.; Sau, D.; Guareschi, S.; Bogush, M.; Brown, R.H., Jr.; Naniche, N.; Kia, A.; Trotti, D.; Pasinelli, P. ALS-linked mutant SOD1 damages mitochondria by promoting conformational changes in Bcl-2. Hum. Mol. Genet. 2010, 19, 2974–2986. [Google Scholar] [CrossRef] [Green Version]
- Tan, W.; Naniche, N.; Bogush, A.; Pedrini, S.; Trotti, D.; Pasinelli, P. Small peptides against the mutant SOD1/Bcl-2 toxic mitochondrial complex restore mitochondrial function and cell viability in mutant SOD1-mediated ALS. J. Neurosci. 2013, 33, 11588–11598. [Google Scholar] [CrossRef] [Green Version]
- Ferri, A.; Fiorenzo, P.; Nencini, M.; Cozzolino, M.; Pesaresi, M.G.; Valle, C.; Sepe, S.; Moreno, S.; Carri, M.T. Glutaredoxin 2 prevents aggregation of mutant SOD1 in mitochondria and abolishes its toxicity. Hum. Mol. Genet. 2010, 19, 4529–4542. [Google Scholar] [CrossRef] [Green Version]
- Israelson, A.; Arbel, N.; Da Cruz, S.; Ilieva, H.; Yamanaka, K.; Shoshan-Barmatz, V.; Cleveland, D.W. Misfolded mutant SOD1 directly inhibits VDAC1 conductance in a mouse model of inherited ALS. Neuron 2010, 67, 575–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banks, C.J.; Rodriguez, N.W.; Gashler, K.R.; Pandya, R.R.; Mortenson, J.B.; Whited, M.D.; Soderblom, E.J.; Thompson, J.W.; Moseley, M.A.; Reddi, A.R.; et al. Acylation of Superoxide Dismutase 1 (SOD1) at K122 Governs SOD1-Mediated Inhibition of Mitochondrial Respiration. Mol. Cell Biol. 2017, 37, e00354-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Günther, R.; Pal, A.; Williams, C.; Zimyanin, V.L.; Liehr, M.; von Neubeck, C.; Krause, M.; Parab, M.G.; Petri, S.; Kalmbach, N.; et al. Alteration of Mitochondrial Integrity as Upstream Event in the Pathophysiology of SOD1-ALS. Cells 2022, 11, 1246. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, S.; Iwata, M. Mitochondrial alterations in the spinal cord of patients with sporadic amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2007, 66, 10–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, A.U.; Saw, N.L.; Vogel, H.; Cunnigham, A.D.; Shamloo, M.; Mochly-Rosen, D. Inhibition of Drp1/Fis1 interaction slows progression of amyotrophic lateral sclerosis. EMBO Mol. Med. 2018, 10, e8166. [Google Scholar] [CrossRef] [PubMed]
- Schonhoff, C.M.; Matsuoka, M.; Tummala, H.; Johnson, M.A.; Estevez, A.G.; Wu, R.; Kamaid, A.; Ricart, K.C.; Hashimoto, Y.; Gaston, B.; et al. S-nitrosothiol depletion in amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2006, 103, 2404–2409. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Hausladen, A.; Zeng, M.; Que, L.; Heitman, J.; Stamler, J.S. A metabolic enzyme for S-nitrosothiol conserved from bacteria to humans. Nature 2001, 410, 490–494. [Google Scholar] [CrossRef]
- Takeuchi, H.; Kobayashi, Y.; Ishigaki, S.; Doyu, M.; Sobue, G. Mitochondrial localization of mutant superoxide dismutase 1 triggers caspase-dependent cell death in a cellular model of familial amyotrophic lateral sclerosis. J. Biol. Chem. 2002, 277, 50966–50972. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Lillo, C.; Jonsson, P.A.; Vande Velde, C.; Ward, C.M.; Miller, T.M.; Subramaniam, J.R.; Rothstein, J.D.; Marklund, S.; Andersen, P.M.; et al. Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron 2004, 43, 5–17. [Google Scholar] [CrossRef] [Green Version]
- Pasinelli, P.; Belford, M.E.; Lennon, N.; Bacskai, B.J.; Hyman, B.T.; Trotti, D.; Brown, R.H., Jr. Amyotrophic lateral sclerosis-associated SOD1 mutant proteins bind and aggregate with Bcl-2 in spinal cord mitochondria. Neuron 2004, 43, 19–30. [Google Scholar] [CrossRef] [Green Version]
- Kong, J.; Xu, Z. Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. J. Neurosci. 1998, 18, 3241–3250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furukawa, Y.; O’Halloran, T.V. Amyotrophic lateral sclerosis mutations have the greatest destabilizing effect on the apo- and reduced form of SOD1, leading to unfolding and oxidative aggregation. J. Biol. Chem. 2005, 280, 17266–17274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeon, G.S.; Nakamura, T.; Lee, J.-S.; Choi, W.-J.; Ahn, S.-W.; Lee, K.-W.; Sung, J.-J.; Lipton, S.A. Potential Effect of S-Nitrosylated Protein Disulfide Isomerase on Mutant SOD1 Aggregation and Neuronal Cell Death in Amyotrophic Lateral Sclerosis. Mol. Neurobiol. 2014, 49, 796–807. [Google Scholar] [CrossRef] [PubMed]
- Lacza, Z.; Snipes, J.A.; Zhang, J.; Horvath, E.M.; Figueroa, J.P.; Szabo, C.; Busija, D.W. Mitochondrial nitric oxide synthase is not eNOS, nNOS or iNOS. Free Radic. Biol. Med. 2003, 35, 1217–1228. [Google Scholar] [CrossRef]
- Giulivi, C. Characterization and function of mitochondrial nitric-oxide synthase. Free Radic. Biol. Med. 2003, 34, 397–408. [Google Scholar] [CrossRef]
- Chen, K.; Northington, F.J.; Martin, L.J. Inducible nitric oxide synthase is present in motor neuron mitochondria and Schwann cells and contributes to disease mechanisms in ALS mice. Brain. Struct. Funct. 2010, 214, 219–234. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, T.; Oh, C.K.; Zhang, X.; Lipton, S.A. Protein S-nitrosylation and oxidation contribute to protein misfolding in neurodegeneration. Free Radic. Biol. Med. 2021, 172, 562–577. [Google Scholar] [CrossRef]
- Delatycki, M.B.; Williamson, R.; Forrest, S.M. Friedreich ataxia: An overview. J. Med. Genet. 2000, 37, 1–8. [Google Scholar] [CrossRef]
- Campuzano, V.; Montermini, L.; Molto, M.D.; Pianese, L.; Cossee, M.; Cavalcanti, F.; Monros, E.; Rodius, F.; Duclos, F.; Monticelli, A.; et al. Friedreich’s ataxia: Autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 1996, 271, 1423–1427. [Google Scholar] [CrossRef]
- Petrillo, S.; Santoro, M.; La Rosa, P.; Perna, A.; Gallo, M.G.; Bertini, E.S.; Silvestri, G.; Piemonte, F. Nuclear Factor Erythroid 2-Related Factor 2 Activation Might Mitigate Clinical Symptoms in Friedreich’s Ataxia: Clues of an “Out-Brain Origin” of the Disease From a Family Study. Front. Neurosci. 2021, 15, 638810. [Google Scholar] [CrossRef]
- Piemonte, F.; Pastore, A.; Tozzi, G.; Tagliacozzi, D.; Santorelli, F.M.; Carrozzo, R.; Casali, C.; Damiano, M.; Federici, G.; Bertini, E. Glutathione in blood of patients with Friedreich’s ataxia. Eur. J. Clin. Invest. 2001, 31, 1007–1011. [Google Scholar] [CrossRef] [PubMed]
- Durr, A.; Cossee, M.; Agid, Y.; Campuzano, V.; Mignard, C.; Penet, C.; Mandel, J.L.; Brice, A.; Koenig, M. Clinical and genetic abnormalities in patients with Friedreich’s ataxia. N. Engl. J. Med. 1996, 335, 1169–1175. [Google Scholar] [CrossRef] [PubMed]
- Cavadini, P.; O’Neill, H.A.; Benada, O.; Isaya, G. Assembly and iron-binding properties of human frataxin, the protein deficient in Friedreich ataxia. Hum. Mol. Genet. 2002, 11, 217–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastore, C.; Franzese, M.; Sica, F.; Temussi, P.; Pastore, A. Understanding the binding properties of an unusual metal-binding protein--a study of bacterial frataxin. FEBS J. 2007, 274, 4199–4210. [Google Scholar] [CrossRef] [Green Version]
- Prischi, F.; Pastore, A. Hybrid Methods in Iron-Sulfur Cluster Biogenesis. Front. Mol. Biosci. 2017, 4, 12. [Google Scholar] [CrossRef] [Green Version]
- Adinolfi, S.; Iannuzzi, C.; Prischi, F.; Pastore, C.; Iametti, S.; Martin, S.R.; Bonomi, F.; Pastore, A. Bacterial frataxin CyaY is the gatekeeper of iron-sulfur cluster formation catalyzed by IscS. Nat. Struct. Mol. Biol. 2009, 16, 390–396. [Google Scholar] [CrossRef]
- Gerber, J.; Muhlenhoff, U.; Lill, R. An interaction between frataxin and Isu1/Nfs1 that is crucial for Fe/S cluster synthesis on Isu1. EMBO Rep. 2003, 4, 906–911. [Google Scholar] [CrossRef]
- Bradley, J.L.; Blake, J.C.; Chamberlain, S.; Thomas, P.K.; Cooper, J.M.; Schapira, A.H. Clinical, biochemical and molecular genetic correlations in Friedreich’s ataxia. Hum. Mol. Genet. 2000, 9, 275–282. [Google Scholar] [CrossRef] [Green Version]
- Calabrese, V.; Lodi, R.; Tonon, C.; D’Agata, V.; Sapienza, M.; Scapagnini, G.; Mangiameli, A.; Pennisi, G.; Stella, A.M.; Butterfield, D.A. Oxidative stress, mitochondrial dysfunction and cellular stress response in Friedreich’s ataxia. J. Neurol. Sci. 2005, 233, 145–162. [Google Scholar] [CrossRef]
- Puccio, H.; Simon, D.; Cossee, M.; Criqui-Filipe, P.; Tiziano, F.; Melki, J.; Hindelang, C.; Matyas, R.; Rustin, P.; Koenig, M. Mouse models for Friedreich ataxia exhibit cardiomyopathy, sensory nerve defect and Fe-S enzyme deficiency followed by intramitochondrial iron deposits. Nat. Genet. 2001, 27, 181–186. [Google Scholar] [CrossRef]
- Pastore, A.; Tozzi, G.; Gaeta, L.M.; Bertini, E.; Serafini, V.; Di Cesare, S.; Bonetto, V.; Casoni, F.; Carrozzo, R.; Federici, G.; et al. Actin glutathionylation increases in fibroblasts of patients with Friedreich’s ataxia: A potential role in the pathogenesis of the disease. J. Biol. Chem. 2003, 278, 42588–42595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Rosa, P.; Bertini, E.S.; Piemonte, F. The NRF2 Signaling Network Defines Clinical Biomarkers and Therapeutic Opportunity in Friedreich’s Ataxia. Int. J. Mol. Sci. 2020, 21, 916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Rosa, P.; Petrillo, S.; Turchi, R.; Berardinelli, F.; Schirinzi, T.; Vasco, G.; Lettieri-Barbato, D.; Fiorenza, M.T.; Bertini, E.S.; Aquilano, K.; et al. The Nrf2 induction prevents ferroptosis in Friedreich’s Ataxia. Redox. Biol. 2021, 38, 101791. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.; Colman, M.J.; Gomez-Casati, D.F.; Lamattina, L.; Zabaleta, E.J. Nitric oxide accumulation is required to protect against iron-mediated oxidative stress in frataxin-deficient Arabidopsis plants. FEBS Lett. 2009, 583, 542–548. [Google Scholar] [CrossRef] [Green Version]
- Alsina, D.; Ros, J.; Tamarit, J. Nitric oxide prevents Aft1 activation and metabolic remodeling in frataxin-deficient yeast. Redox. Biol. 2018, 14, 131–141. [Google Scholar] [CrossRef]
- Tortora, V.; Quijano, C.; Freeman, B.; Radi, R.; Castro, L. Mitochondrial aconitase reaction with nitric oxide, S-nitrosoglutathione, and peroxynitrite: Mechanisms and relative contributions to aconitase inactivation. Free Radic. Biol. Med. 2007, 42, 1075–1088. [Google Scholar] [CrossRef]
- Beckman, J.S.; Beckman, T.W.; Chen, J.; Marshall, P.A.; Freeman, B.A. Apparent hydroxyl radical production by peroxynitrite: Implications for endothelial injury from nitric oxide and superoxide. Proc. Natl. Acad. Sci. USA 1990, 87, 1620–1624. [Google Scholar] [CrossRef]
- Dey, R.; Kemp, K.; Gray, E.; Rice, C.; Scolding, N.; Wilkins, A. Human mesenchymal stem cells increase anti-oxidant defences in cells derived from patients with Friedreich’s ataxia. Cerebellum 2012, 11, 861–871. [Google Scholar] [CrossRef]
- Lipton, S.A.; Choi, Y.B.; Takahashi, H.; Zhang, D.; Li, W.; Godzik, A.; Bankston, L.A. Cysteine regulation of protein function--as exemplified by NMDA-receptor modulation. Trends. Neurosci. 2002, 25, 474–480. [Google Scholar] [CrossRef]
- Nakamura, T.; Lipton, S.A. Protein S-Nitrosylation as a Therapeutic Target for Neurodegenerative Diseases. Trends. Pharm. Sci. 2016, 37, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Waldmeier, P.C.; Boulton, A.A.; Cools, A.R.; Kato, A.C.; Tatton, W.G. Neurorescuing effects of the GAPDH ligand CGP 3466B. J. Neural. Transm. Suppl. 2000, 60, 197–214. [Google Scholar]
- Sen, T.; Saha, P.; Sen, N. Nitrosylation of GAPDH augments pathological tau acetylation upon exposure to amyloid-beta. Sci. Signal. 2018, 11, eaao6765. [Google Scholar] [CrossRef] [Green Version]
- Erb, M.; Meinen, S.; Barzaghi, P.; Sumanovski, L.T.; Courdier-Fruh, I.; Ruegg, M.A.; Meier, T. Omigapil ameliorates the pathology of muscle dystrophy caused by laminin-alpha2 deficiency. J. Pharm. Exp. Ther. 2009, 331, 787–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harraz, M.M.; Snyder, S.H. Nitric Oxide-GAPDH Transcriptional Signaling Mediates Behavioral Actions of Cocaine. CNS Neurol. Disord. Drug Targets 2015, 14, 757–763. [Google Scholar] [CrossRef] [PubMed]
- Liang, F.; Shi, L.; Zheng, J.; Chen, S.; Wang, Y.; Zhang, J. Neuroprotective Effects of CGP3466B on Apoptosis Are Modulated by Protein-L-isoaspartate (D-aspartate) O-methyltransferase/Mst1 Pathways after Traumatic Brain Injury in Rats. Sci. Rep. 2017, 7, 9201. [Google Scholar] [CrossRef] [Green Version]
- Ahmari, M.; Sharafi, A.; Mahmoudi, J.; Jafari-Anarkoli, I.; Gharbavi, M.; Hosseini, M.J. Selegiline (L-Deprenyl) Mitigated Oxidative Stress, Cognitive Abnormalities, and Histopathological Change in Rats: Alternative Therapy in Transient Global Ischemia. J. Mol. Neurosci. 2020, 70, 1639–1648. [Google Scholar] [CrossRef]
- Hara, M.R.; Thomas, B.; Cascio, M.B.; Bae, B.I.; Hester, L.D.; Dawson, V.L.; Dawson, T.M.; Sawa, A.; Snyder, S.H. Neuroprotection by pharmacologic blockade of the GAPDH death cascade. Proc. Natl. Acad. Sci. USA 2006, 103, 3887–3889. [Google Scholar] [CrossRef]









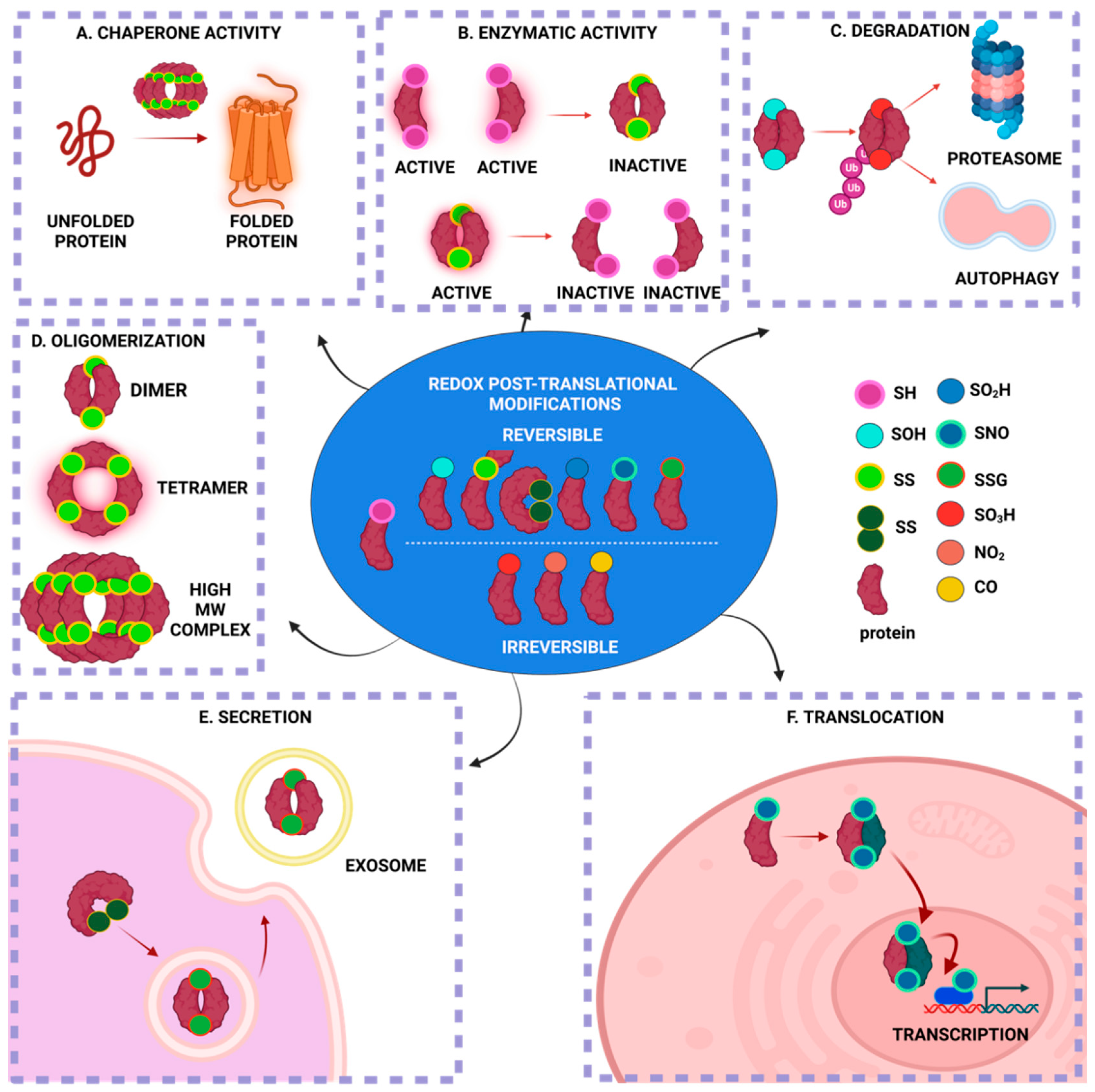{kind=link}
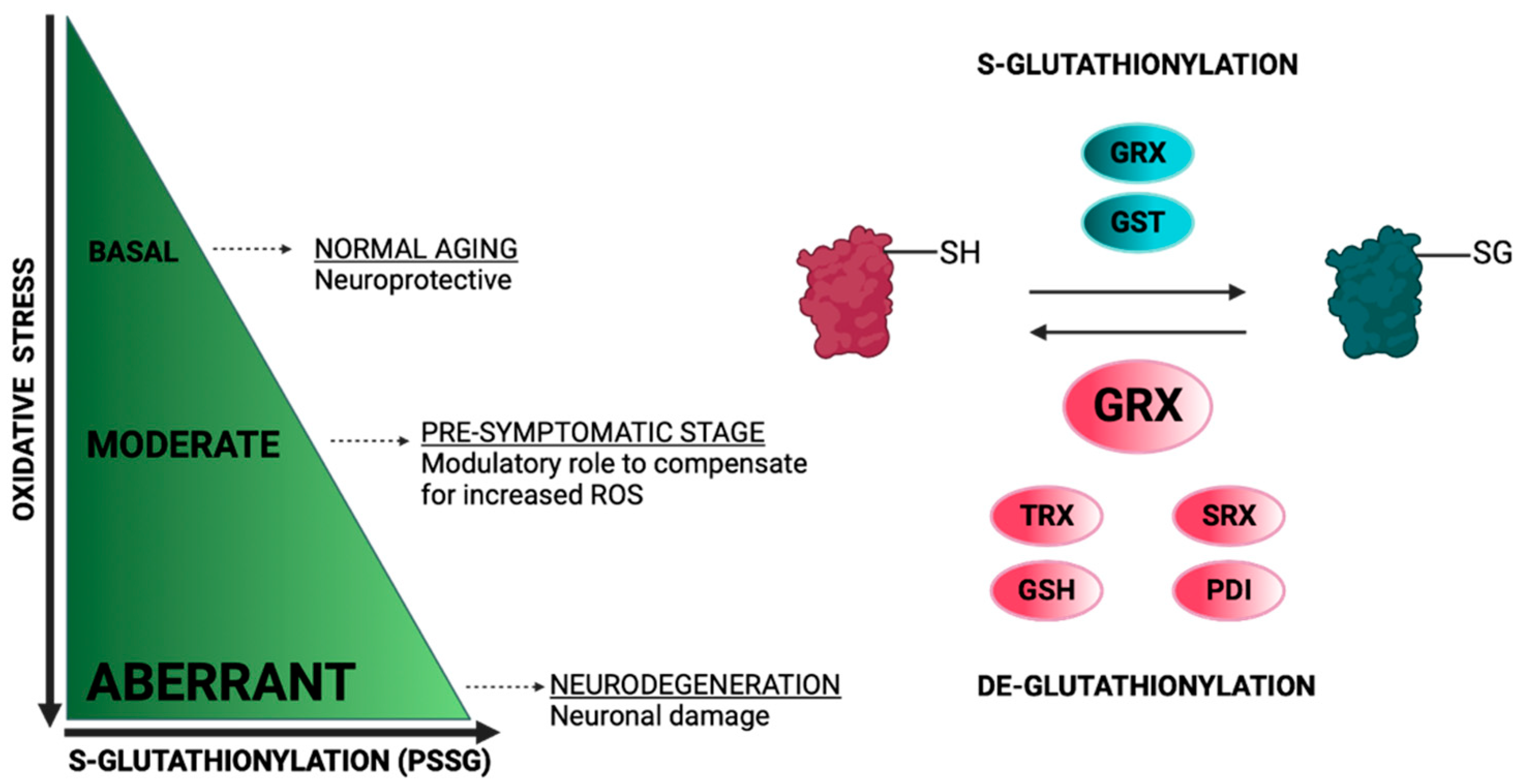{kind=link}
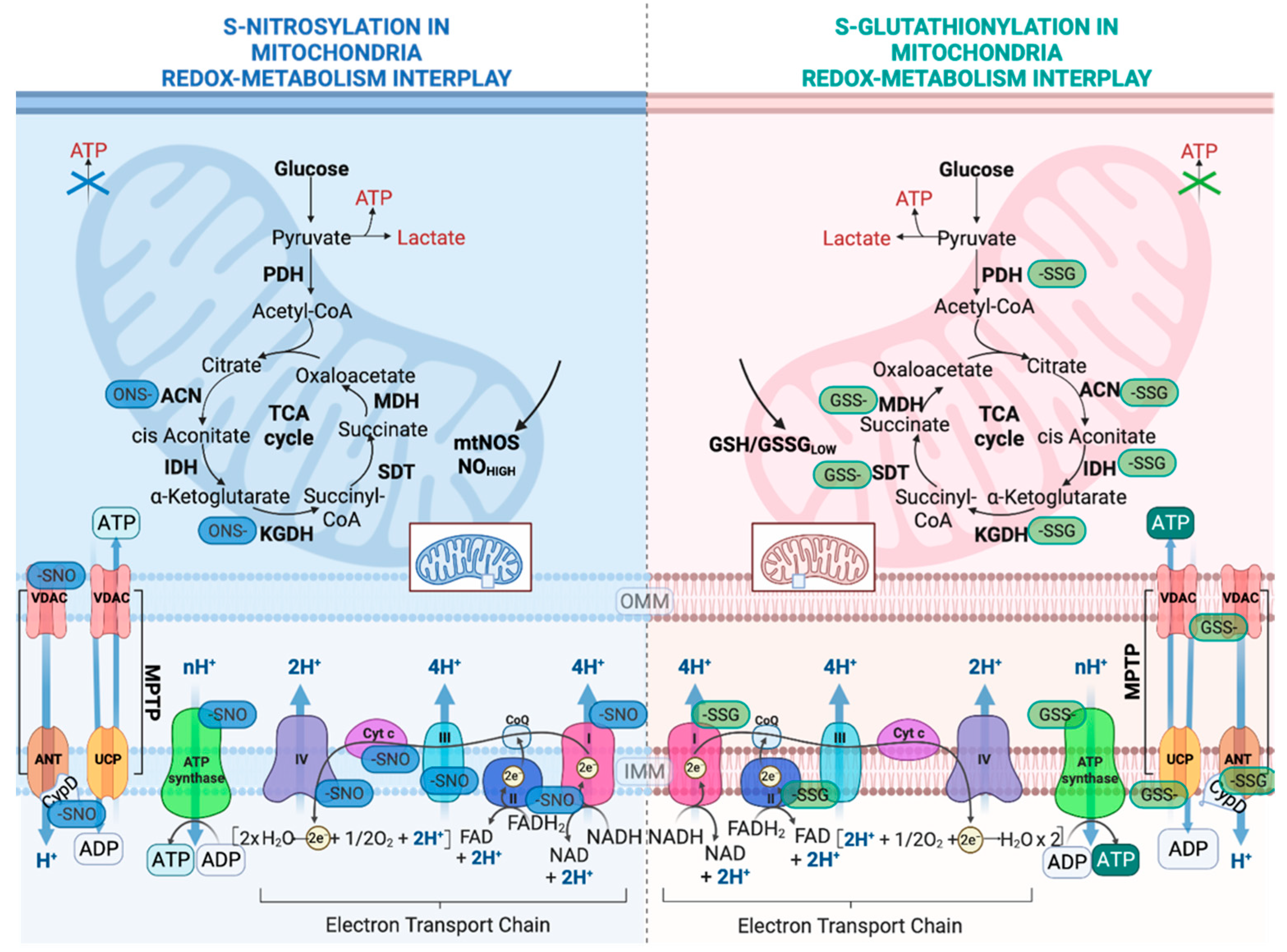{kind=link}
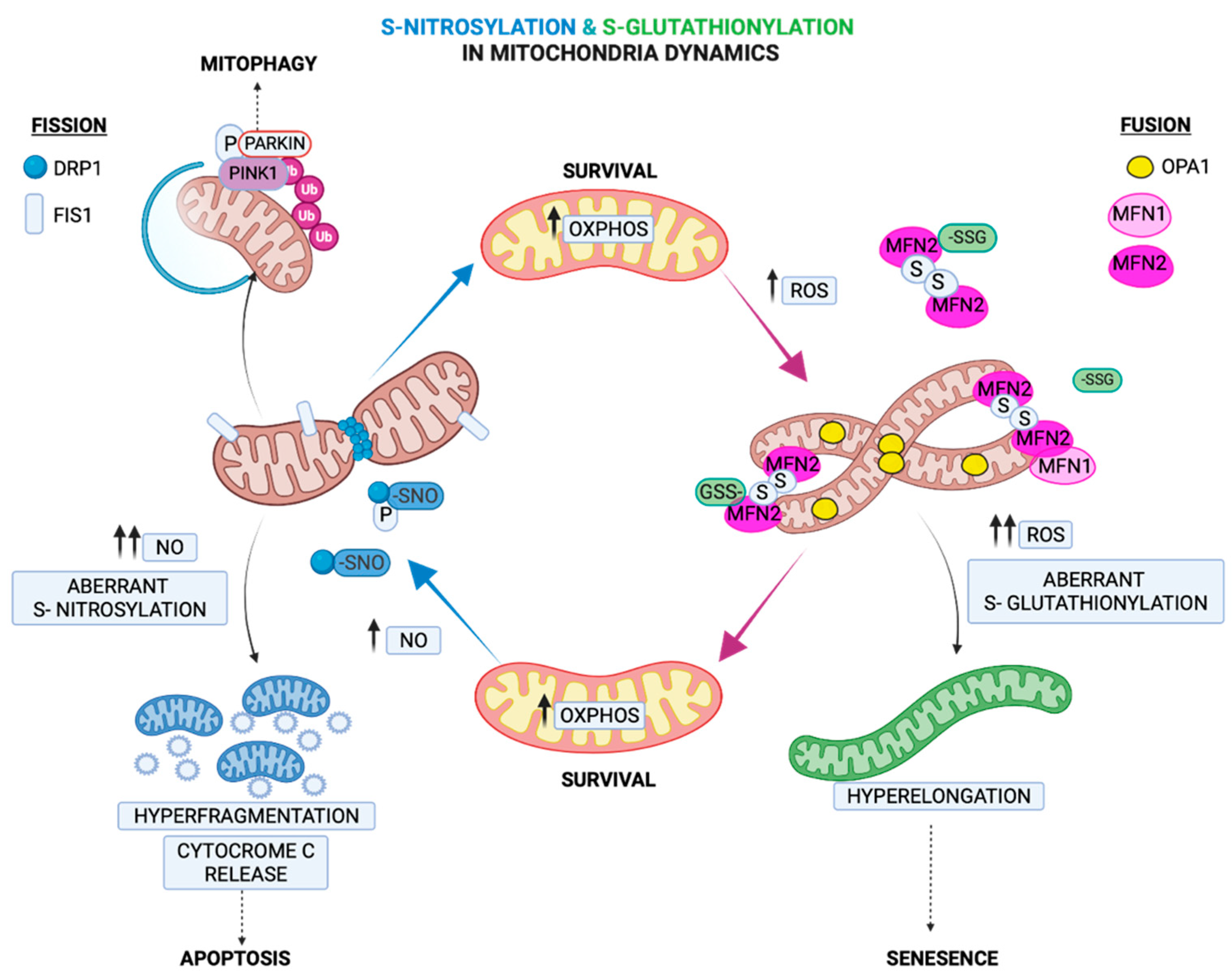{kind=link}
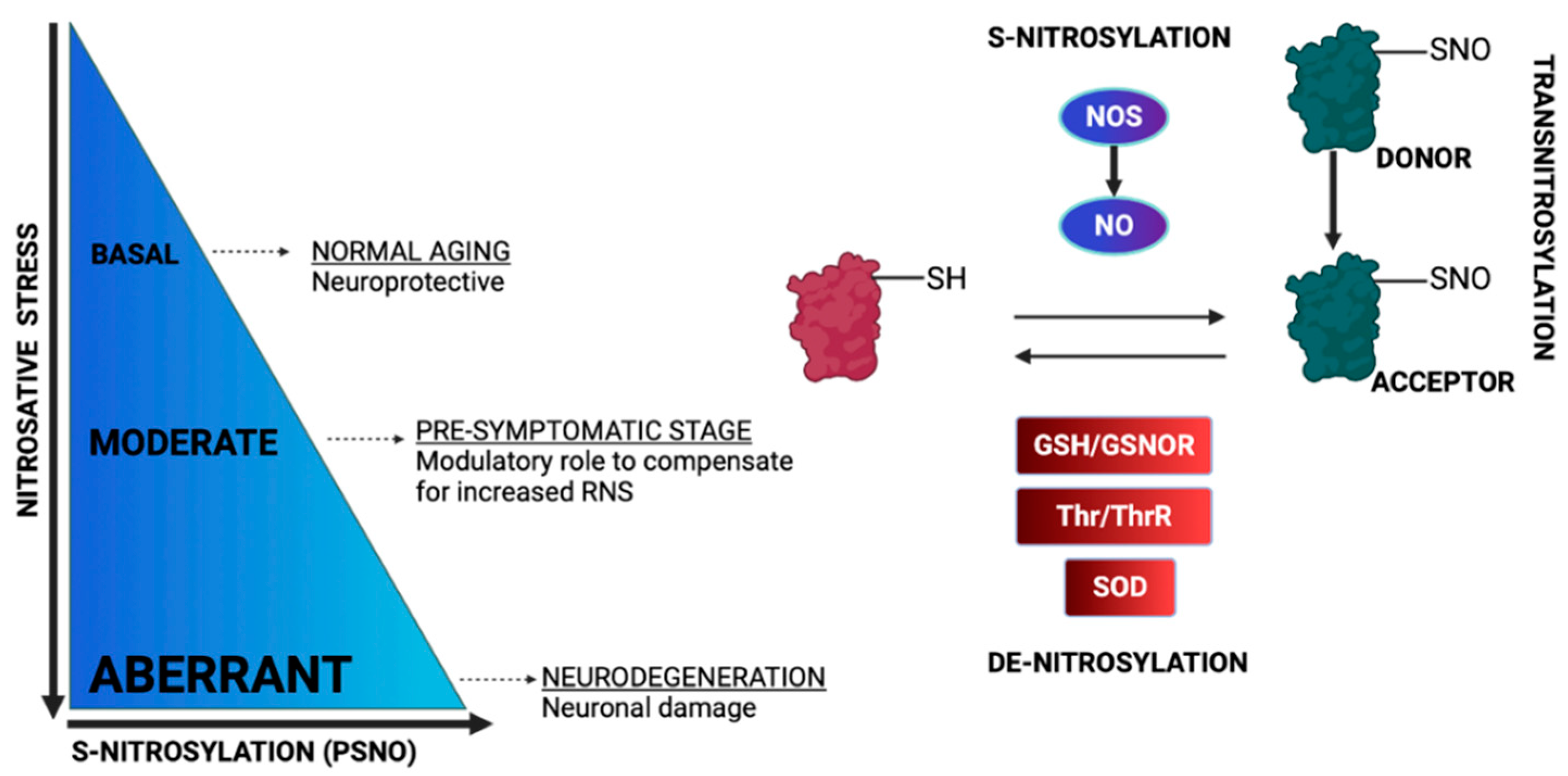{kind=link}
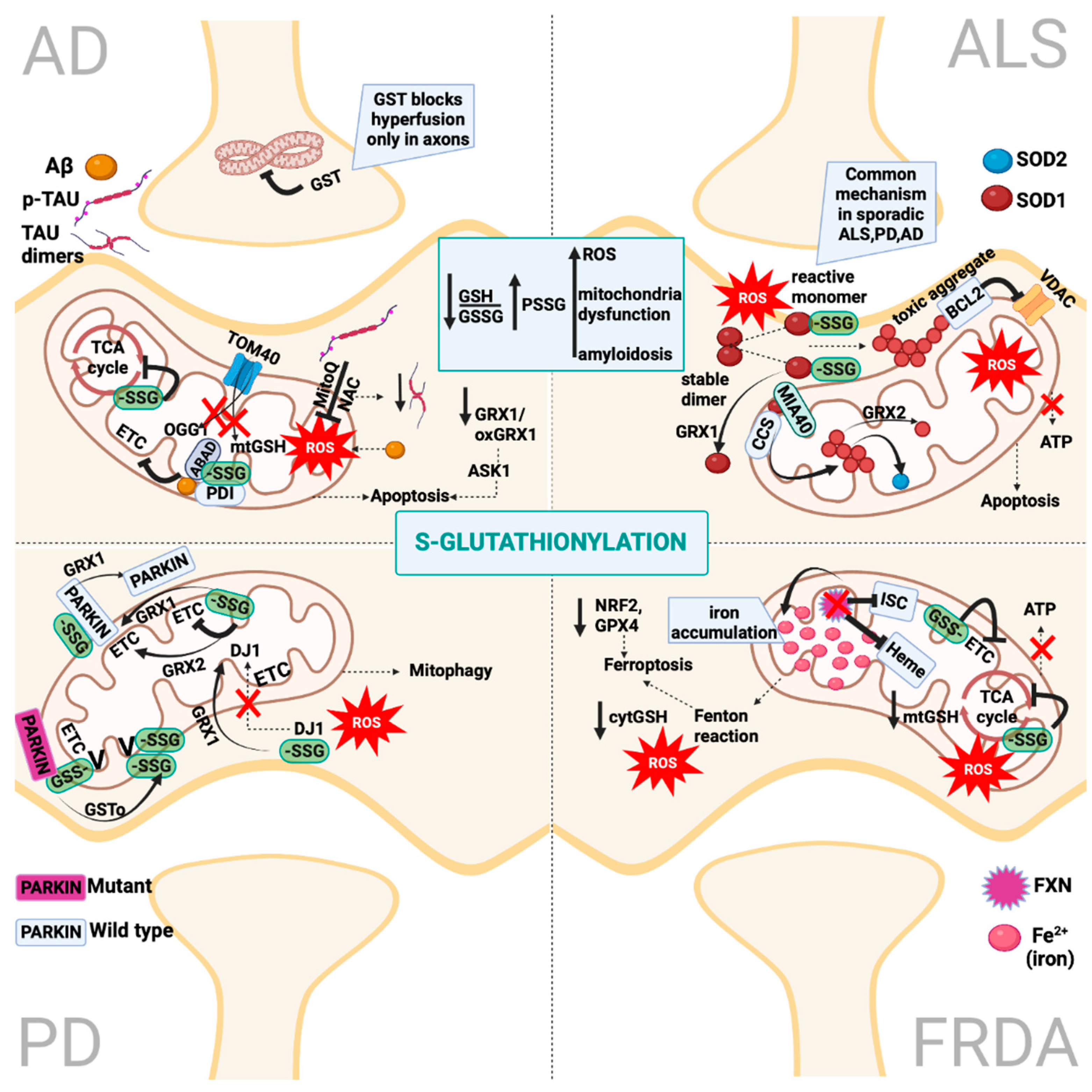{kind=link}
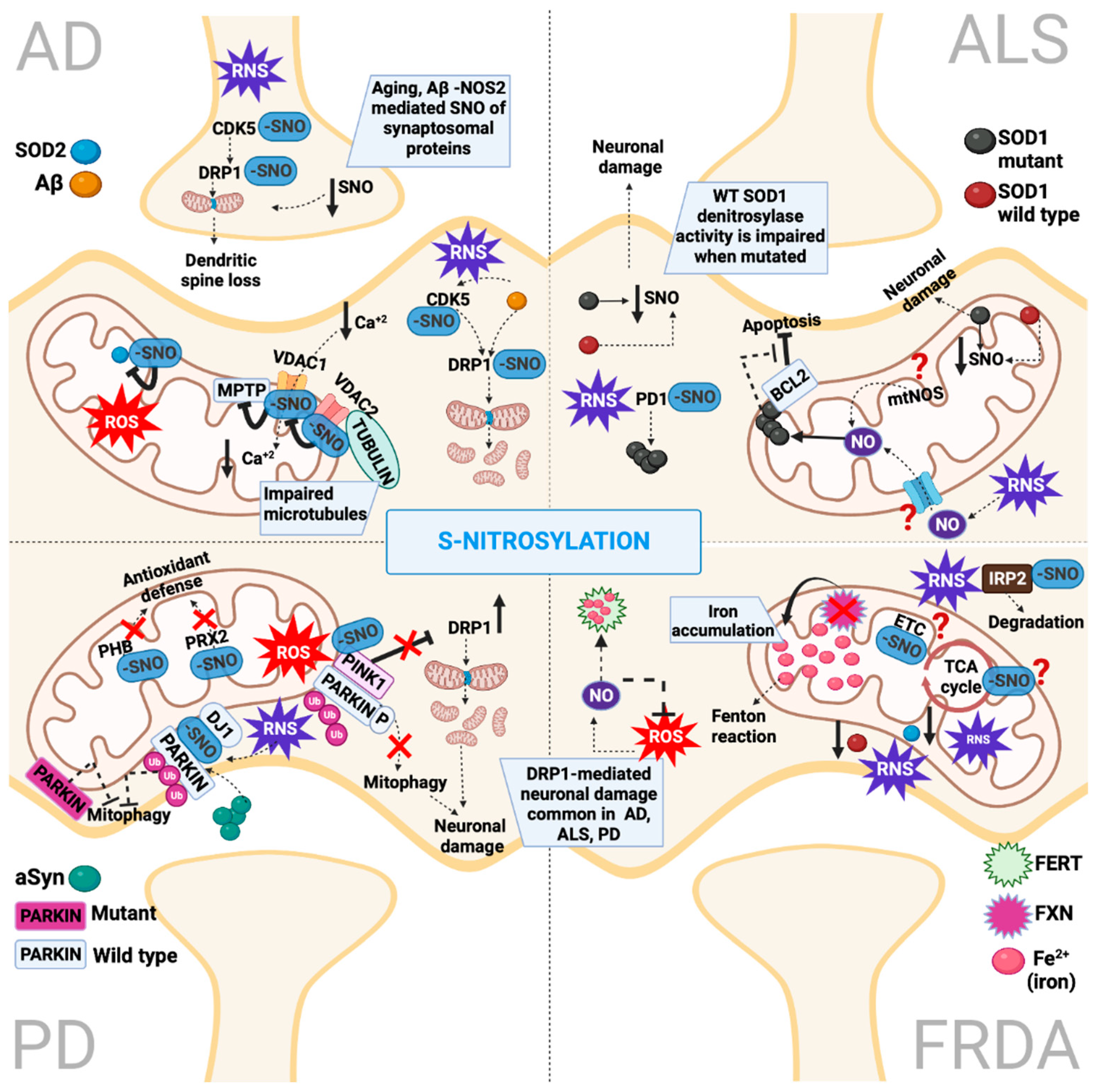{kind=link}
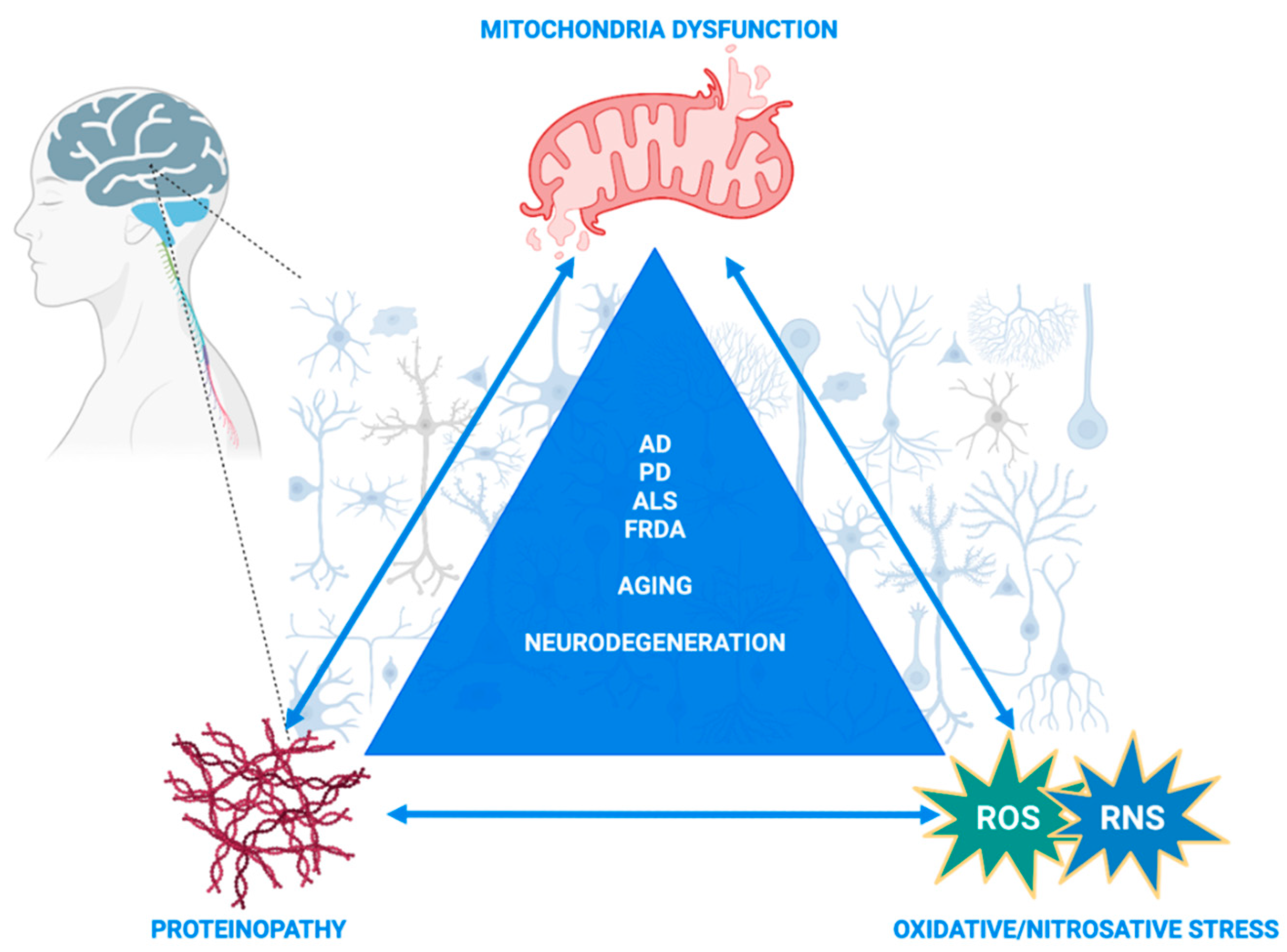{kind=link}
| Protein | Biological Function | PSSG | PSNO | Disorder | PSSG/PSNO Effect | Ref. |
|---|---|---|---|---|---|---|
| α-Ketoglutarate dehydrogenase (KGDH) (E2 subunit) | Catalyzes the conversion of α-ketoglutarate to succinyl-CoA producing NADH directly providing electrons for the respiratory chain. | + | AD | Impairs glucose utilization. Decreases ATP & ROS production. Decreases rate of mitochondria respiration. | [132,142] | |
| human Branched- chain aminotransferase protein (hBCAT) (CXXC motif) | Catalyzes reversible transamination of the α-amino group of the branched-chain amino acids to α-ketoglutarate, forming their respective branched chain α-keto acids and glutamate. | + | AD | Colocalizes with MIA40 & PDI in mitochondria. Role in Aβ misfolding. | [138,139] | |
| Aβ (Not mitochondrial) (M35 residue) | Highly oxidized residue of Aβ which affects Aβ conformation. | + | AD | Lipid peroxidation, formation of amyloid plaques & neurofibrillary tangles. Also, correlated with mitochondria dysfunction. | [136] | |
| Tau (Not mitochondrial) (C-terminal microtubule-binding region) | Microtubule-associated protein, forms insoluble filaments that accumulate as neurofibrillary tangles in AD. | + | AD | Increases tau dimerization & mitochondria dysfunction. | [140,141] | |
| Mn superoxide dismutase (SOD2) | Manganese superoxide dismutase is the essential mitochondrial antioxidant enzyme that detoxifies the free radical superoxide, the major by-product of mitochondrial respiration. | + | AD | Inhibit its detoxifying capacity leading to mitochondrial dysfunction | [143] | |
| Voltage-dependent anion-selective channel protein 1 (VDAC1) | VDACs promote mitochondrial transport of calcium ions. Part of the mitochondrial permeability transition pore (MPTP), facilitate cytochrome c release, leading to apoptosis. Interact with pro- & anti-apoptotic proteins at the outer mitochondrial membrane. | + | AD | Impaired Ca+2 transfer to mitochondria. Decreased ATP levels. | [30,144] | |
| Voltage-dependent anion-selective channel protein 2 (VDAC2) | + | AD | Interaction TUBA-1A, 1B chain and TUBB-2C leading to impaired microtubes architecture. Impaired Ca+2 transfer to mitochondria. | |||
| Dynamin-related protein 1 (DRP1) (Cys644) | Facilitates fission, promoting cytochrome c release and apoptosis. | + | AD | Increases mitochondria fragmentation leading to bioenergetics deficits & neuronal damage. | [145,146] | |
| Cyclin-dependent kinase 5 (CDK5) (Cys83, Cys157) | Proline-directed serine/threonine-protein kinase essential for neuronal cell cycle arrest and differentiation. It is involved in apoptotic cell death in neuronal diseases (AD, PD) by triggering abortive cell cycle re-entry. | + | AD | Triggers Aß-mediated dendritic spine loss and neuronal damage. Transnitrosylates Drp1 increasing mitochondria fragmentation. | [147] | |
| Succinate dehydrogenase (ubiquinone) flavoprotein subunit | Essential for assembly & activity of succinate dehydrogenase (TCA cycle). | + | AD | Unknown effect * | [148] | |
| Succinyl-CoA ligase (ADP-forming) subunit beta | Essential for assembly & activity of succinate synthase (TCA cycle). | + | AD | Unknown effect * | [148] | |
| Acyl carrier protein (ACP) | Co-factor of fatty acid biosynthesis. | + | AD | Unknown effect * | [148] | |
| Succinyl-CoA: 3-ketoacid coenzyme A transferase 1 | Transfers coenzyme A (CoA) from a donor thiol ester species (succinyl-CoA) to an acceptor carboxylate (acetoacetate), and produces acetoacetyl-CoA which is further metabolized to enter TCA cycle. | + | AD | Unknown effect * | [148] | |
| NFU1 iron–sulfur cluster scaffold homolog | Critical of iron–sulfur cluster biogenesis. | + | AD | Unknown effect * | [148] | |
| Pyruvate carboxylase (PC) | Catalyzes the conversion of pyruvate to oxaloacetate replenishing TCA cycle intermediates. Participates in gluconeogenesis, lipogenesis & neurotransmitter synthesis. | + | AD | Unknown effect * | [148] | |
| Complex I (CI) (75-kDa subunit) | Constituent of electron transport chain. First rate-limiting enzyme. | + ** | PD | Inhibits mitochondrial respiration. Induces mitochondria damage and neuronal death. | [149] | |
| PARKIN (RING & IBR domains) | Ubiquitin E3 ligase which is stratified by PINK1 in outer mitochondrial membrane to promote mitophagy. | + | PD | Mitochondrial dysfunction, protein misfolding and ubiquitin-proteasome system (UPS) impairment. SNO effect inhibits its activity to suppress DRP1-mediated fission. DJ1 activation is essential for PARKIN-SNO. | [150,151,152] | |
| ATP synthase (β subunit) | Part of membrane-bound ATP synthase complex. Role in catalytic sites. | + | PD | Impaired mitochondrial function. | [153] | |
| Protein deoxyglycase (DJ1) (Cys53, Cys106) | Multifunctional protein: chaperone, scavenger of ROS, regulator of transcription & cell signaling. Its gene PARK7 is mutated in familial PD. | + | PD | Increased proximity with CI. Increase apoptosis by impairing BCL- xL function in outer mitochondrial membrane | [154,155,156] | |
| PTEN-induced kinase 1 (PINK1) (Cys568) | Mitochondrial-targeted serine/threonine-protein kinase encoded by the PINK1 gene which is mutated in familial PD. Protects from ROS by stratifying PARKIN & triggering mitophagy. | + | PD | Inhibits its kinase activity impairing PINK1/PARKIN-mediated mitophagy leading to dopaminergic neuronal cell death | [157] | |
| Myocyte enhancer factor-2 (MEF2) (Not mitochondrial) (Cys39) | Transcriptional factor with key role in development of multiple organs. | + | PD | Inhibits MEF2-PGC1a transcriptional network, resulting in mitochondrial dysfunction and apoptosis. | [158] | |
| Peroxiredoxin 2 (PRX2) (Cys51, Cys172) | Thiol-specific peroxidase that catalyzes the reduction of hydrogen peroxide & organic hydroperoxides to water and alcohols, accordingly. | + | PD | Diminishes peroxidase activity causing hydrogen peroxide to accumulate, exacerbating oxidative stress. | [159] | |
| Prohibitin (PHB) | Mitochondrial chaperone protein | + | PD | Enhances its neuroprotective roles against ROS & glucose deprivation stress. | [160] | |
| alpha-Synuclein (α-Syn) (Tyr39) | Pre-synaptic neuronal protein implicated in familial and sporadic PD pathogenesis. | ** | PD | α-Syn nitration can potentiate a-synuclein oligomer formation. Extracellular α-Syn oligomers induce ROS/RNS-mediated nitrosylation of PARKIN leading to impaired mitophagy. | [161,162] | |
| Cu-Zn Superoxide dismutase (SOD1) (mutated & wild type) (Cys111) | Major cytosolic antioxidant enzyme (with denitrosylase activity). It has been found in mitochondrial matrix. It is genetically & neuropathologically implicated in AD, PD and ALS. | + | ALS | Triggers SOD1 dimer disassembly, aggregation, loss of activity & neuronal damage. Triggers conformational changes in BCL2 & thus cytochrome c release and eventually apoptosis. | [163] | |
| Protein disulfide isomerase (PD1) (Trx-like catalytic domain CXXC) | Multifunctional protein which associates with SOD1 misfolding. It is genetically & neuropathologically implicated in ALS. | + | + | ALS | Inhibits its activity, triggers mutant SOD1 aggregation and increases neuronal cell death | [142] |
| α-Ketoglutarate dehydrogenase (KGDH) E2 subunit | Catalyzes the conversion of α-ketoglutarate to succinyl-CoA producing NADH directly providing electrons for the respiratory chain. Constituents of electron transport chain. | + | ? *** | FRDA | Its glutathionylation limits the production of NADH and the electron flow in the respiratory chain | [55,164] |
| Complex III, Complex IV | Maintains cellular iron homeostasis. It is required for mitochondrial iron supply & function. | + | ? *** | FRDA | Impairs mitochondrial respiration. | [164] |
| Iron regulatory protein 2 (IRP2) (Cys178) | + | FRDA | Malfunction of iron homeostasis through UPS-dependent degradation of IRP2 that results in increased accumulation of iron inside the iron storage protein ferritin. | [165,166] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vrettou, S.; Wirth, B. S-Glutathionylation and S-Nitrosylation in Mitochondria: Focus on Homeostasis and Neurodegenerative Diseases. Int. J. Mol. Sci. 2022, 23, 15849. https://doi.org/10.3390/ijms232415849
Vrettou S, Wirth B. S-Glutathionylation and S-Nitrosylation in Mitochondria: Focus on Homeostasis and Neurodegenerative Diseases. International Journal of Molecular Sciences. 2022; 23(24):15849. https://doi.org/10.3390/ijms232415849
Chicago/Turabian StyleVrettou, Sofia, and Brunhilde Wirth. 2022. "S-Glutathionylation and S-Nitrosylation in Mitochondria: Focus on Homeostasis and Neurodegenerative Diseases" International Journal of Molecular Sciences 23, no. 24: 15849. https://doi.org/10.3390/ijms232415849
APA StyleVrettou, S., & Wirth, B. (2022). S-Glutathionylation and S-Nitrosylation in Mitochondria: Focus on Homeostasis and Neurodegenerative Diseases. International Journal of Molecular Sciences, 23(24), 15849. https://doi.org/10.3390/ijms232415849