
Inflammatory Arthritis and Bone Metabolism Regulated by Type 2 Innate and Adaptive Immunity

 , ,
, ,  and
and 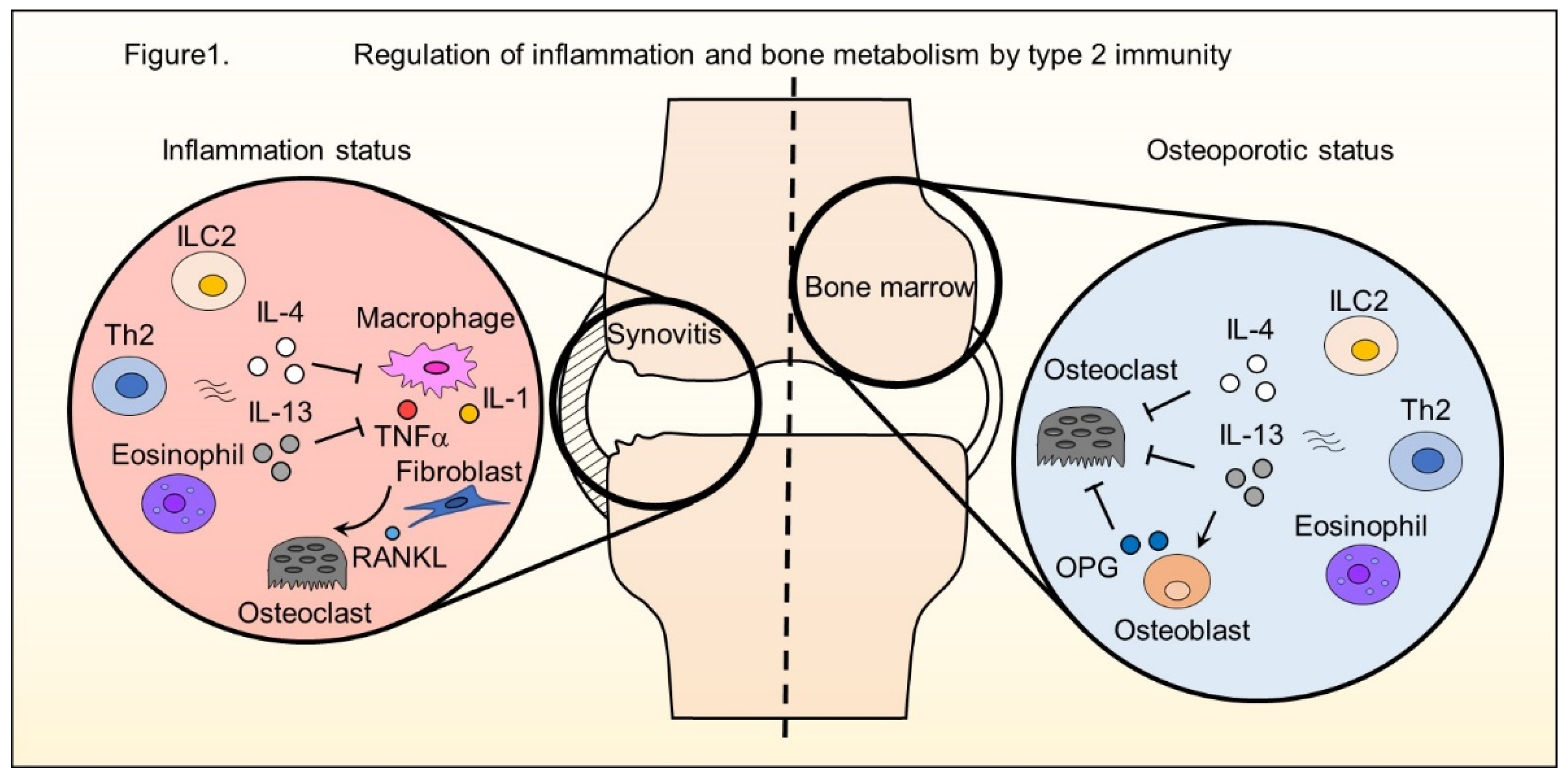{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Type 2 Immune Responses in Arthritis
3. Type 2 Innate Lymphoid Cells in Arthritis
4. Type 2 Immunity and Bone Metabolism
5. ILC2 and Bone Homeostasis
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wynn, T.A. Type 2 cytokines: Mechanisms and therapeutic strategies. Nat. Rev. Immunol. 2015, 15, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, N.; Takayanagi, H. Inflammation and bone destruction in arthritis: Synergistic activity of immune and mesenchymal cells in joints. Front. Immunol. 2012, 3, 77. [Google Scholar] [CrossRef] [Green Version]
- Takayanagi, H.; Iizuka, H.; Juji, T.; Nakagawa, T.; Yamamoto, A.; Miyazaki, T.; Koshihara, Y.; Oda, H.; Nakamura, K.; Tanaka, S. Involvement of receptor activator of nuclear factor kappaB ligand/osteoclast differentiation factor in osteoclastogenesis from synoviocytes in rheumatoid arthritis. Arthritis Rheumatol. 2000, 43, 259–269. [Google Scholar] [CrossRef]
- Moser, M.; Murphy, K.M. Dendritic cell regulation of TH1-TH2 development. Nat. Immunol. 2000, 1, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Amsen, D.; Blander, J.M.; Lee, G.R.; Tanigaki, K.; Honjo, T.; Flavell, R.A. Instruction of distinct CD4 T helper cell fates by different notch ligands on antigen-presenting cells. Cell 2004, 117, 515–526. [Google Scholar] [CrossRef] [Green Version]
- Urban, J.F., Jr.; Noben-Trauth, N.; Donaldson, D.D.; Madden, K.B.; Morris, S.C.; Collins, M.; Finkelman, F.D. IL-13, IL-4Ralpha, and Stat6 are required for the expulsion of the gastrointestinal nematode parasite Nippostrongylus brasiliensis. Immunity 1998, 8, 255–264. [Google Scholar] [CrossRef] [Green Version]
- West, G.A.; Matsuura, T.; Levine, A.D.; Klein, J.S.; Fiocchi, C. Interleukin 4 in inflammatory bowel disease and mucosal immune reactivity. Gastroenterology 1996, 110, 1683–1695. [Google Scholar] [CrossRef]
- Omata, Y.; Frech, M.; Primbs, T.; Lucas, S.; Andreev, D.; Scholtysek, C.; Sarter, K.; Kindermann, M.; Yeremenko, N.; Baeten, D.L.; et al. Group 2 Innate Lymphoid Cells Attenuate Inflammatory Arthritis and Protect from Bone Destruction in Mice. Cell Rep. 2018, 24, 169–180. [Google Scholar] [CrossRef] [Green Version]
- Palm, N.W.; Rosenstein, R.K.; Medzhitov, R. Allergic host defences. Nature 2012, 484, 465–472. [Google Scholar] [CrossRef] [PubMed]
- King, I.L.; Mohrs, M. IL-4-producing CD4+ T cells in reactive lymph nodes during helminth infection are T follicular helper cells. J. Exp. Med. 2009, 206, 1001–1007. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, C.M.; Snelgrove, R.J. Type 2 immunity: Expanding our view. Sci. Immunol. 2018, 3, eaat1604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fort, M.M.; Cheung, J.; Yen, D.; Li, J.; Zurawski, S.M.; Lo, S.; Menon, S.; Clifford, T.; Hunte, B.; Lesley, R.; et al. IL-25 induces IL-4, IL-5, and IL-13 and Th2-associated pathologies in vivo. Immunity 2001, 15, 985–995. [Google Scholar] [CrossRef] [Green Version]
- Moro, K.; Yamada, T.; Tanabe, M.; Takeuchi, T.; Ikawa, T.; Kawamoto, H.; Furusawa, J.; Ohtani, M.; Fujii, H.; Koyasu, S. Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature 2010, 463, 540–544. [Google Scholar] [CrossRef]
- Neill, D.R.; Wong, S.H.; Bellosi, A.; Flynn, R.J.; Daly, M.; Langford, T.K.; Bucks, C.; Kane, C.M.; Fallon, P.G.; Pannell, R.; et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature 2010, 464, 1367–1370. [Google Scholar] [CrossRef] [Green Version]
- Price, A.E.; Liang, H.E.; Sullivan, B.M.; Reinhardt, R.L.; Eisley, C.J.; Erle, D.J.; Locksley, R.M. Systemically dispersed innate IL-13-expressing cells in type 2 immunity. Proc. Natl. Acad. Sci. USA 2010, 107, 11489–11494. [Google Scholar] [CrossRef] [Green Version]
- Saenz, S.A.; Siracusa, M.C.; Perrigoue, J.G.; Spencer, S.P.; Urban, J.F., Jr.; Tocker, J.E.; Budelsky, A.L.; Kleinschek, M.A.; Kastelein, R.A.; Kambayashi, T.; et al. IL25 elicits a multipotent progenitor cell population that promotes T(H)2 cytokine responses. Nature 2010, 464, 1362–1366. [Google Scholar] [CrossRef]
- Vivier, E.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.J.; Mebius, R.E.; et al. Innate Lymphoid Cells: 10 Years On. Cell 2018, 174, 1054–1066. [Google Scholar] [CrossRef] [Green Version]
- Hewagama, A.; Richardson, B. The genetics and epigenetics of autoimmune diseases. J. Autoimmun. 2009, 33, 3–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Stefano, L.; D’Onofrio, B.; Manzo, A.; Montecucco, C.; Bugatti, S. The Genetic, Environmental, and Immunopathological Complexity of Autoantibody-Negative Rheumatoid Arthritis. Int. J. Mol. Sci. 2021, 22, 12386. [Google Scholar] [CrossRef]
- Petrovic-Rackov, L.; Pejnovic, N. Clinical significance of IL-18, IL-15, IL-12 and TNF-alpha measurement in rheumatoid arthritis. Clin. Rheumatol. 2006, 25, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Cortez, V.S.; Colonna, M. Diversity and function of group 1 innate lymphoid cells. Immunol. Lett. 2016, 179, 19–24. [Google Scholar] [CrossRef] [Green Version]
- Akatsu, T.; Takahashi, N.; Udagawa, N.; Imamura, K.; Yamaguchi, A.; Sato, K.; Nagata, N.; Suda, T. Role of prostaglandins in interleukin-1-induced bone resorption in mice in vitro. J. Bone Miner. Res. 1991, 6, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Kay, J.; Calabrese, L. The role of interleukin-1 in the pathogenesis of rheumatoid arthritis. Rheumatology (Oxf.) 2004, 43 (Suppl. S3), iii2–iii9. [Google Scholar] [CrossRef] [Green Version]
- Infante-Duarte, C.; Horton, H.F.; Byrne, M.C.; Kamradt, T. Microbial lipopeptides induce the production of IL-17 in Th cells. J. Immunol. 2000, 165, 6107–6115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrington, L.E.; Hatton, R.D.; Mangan, P.R.; Turner, H.; Murphy, T.L.; Murphy, K.M.; Weaver, C.T. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat. Immunol. 2005, 6, 1123–1132. [Google Scholar] [CrossRef]
- Nakae, S.; Nambu, A.; Sudo, K.; Iwakura, Y. Suppression of immune induction of collagen-induced arthritis in IL-17-deficient mice. J. Immunol. 2003, 171, 6173–6177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hart, P.H.; Vitti, G.F.; Burgess, D.R.; Whitty, G.A.; Piccoli, D.S.; Hamilton, J.A. Potential antiinflammatory effects of interleukin 4: Suppression of human monocyte tumor necrosis factor alpha, interleukin 1, and prostaglandin E2. Proc. Natl. Acad. Sci. USA 1989, 86, 3803–3807. [Google Scholar] [CrossRef] [Green Version]
- Gautam, S.; Tebo, J.M.; Hamilton, T.A. IL-4 suppresses cytokine gene expression induced by IFN-gamma and/or IL-2 in murine peritoneal macrophages. J. Immunol. 1992, 148, 1725–1730. [Google Scholar]
- Cantagrel, A.; Navaux, F.; Loubet-Lescoulie, P.; Nourhashemi, F.; Enault, G.; Abbal, M.; Constantin, A.; Laroche, M.; Mazieres, B. Interleukin-1beta, interleukin-1 receptor antagonist, interleukin-4, and interleukin-10 gene polymorphisms: Relationship to occurrence and severity of rheumatoid arthritis. Arthritis Rheumatol. 1999, 42, 1093–1100. [Google Scholar] [CrossRef]
- Pawlik, A.; Wrzesniewska, J.; Florczak, M.; Gawronska-Szklarz, B.; Herczynska, M. The -590 IL-4 promoter polymorphism in patients with rheumatoid arthritis. Rheumatol. Int. 2005, 26, 48–51. [Google Scholar] [CrossRef]
- Moreno, O.; Gonzalez, C.I.; Saaibi, D.L.; Otero, W.; Badillo, R.; Martin, J.; Ramirez, G. Polymorphisms in the IL4 and IL4RA genes in Colombian patients with rheumatoid arthritis. J. Rheumatol. 2007, 34, 36–42. [Google Scholar]
- Sun, Y.H.; Wei, S.T.; Zong, S.H. Correlation between IL-4 gene polymorphismas well as its mRNA expressionand rheumatoid arthritis. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 3879–3885. [Google Scholar] [PubMed]
- Yucel, B.; Sumer, C.; Gok, I.; Karkucak, M.; Alemdaroglu, E.; Ucar, F. Associations between cytokine gene polymorphisms and rheumatoid arthritis in Turkish population. North Clin. Istanb. 2020, 7, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Skapenko, A.; Smolen, J.S.; Kavanaugh, A.; Arora, V.; Kupper, H.; Schulze-Koops, H. Genetic markers associated with clinical and radiographic response in adalimumab plus methotrexate- or methotrexate-treated rheumatoid arthritis patients in OPTIMA. Clin. Exp. Rheumatol. 2019, 37, 783–790. [Google Scholar]
- Horsfall, A.C.; Butler, D.M.; Marinova, L.; Warden, P.J.; Williams, R.O.; Maini, R.N.; Feldmann, M. Suppression of collagen-induced arthritis by continuous administration of IL-4. J. Immunol. 1997, 159, 5687–5696. [Google Scholar] [PubMed]
- Finnegan, A.; Mikecz, K.; Tao, P.; Glant, T.T. Proteoglycan (aggrecan)-induced arthritis in BALB/c mice is a Th1-type disease regulated by Th2 cytokines. J. Immunol. 1999, 163, 5383–5390. [Google Scholar]
- Lawlor, K.E.; Wong, P.K.; Campbell, I.K.; van Rooijen, N.; Wicks, I.P. Acute CD4+ T lymphocyte-dependent interleukin-1-driven arthritis selectively requires interleukin-2 and interleukin-4, joint macrophages, granulocyte-macrophage colony-stimulating factor, interleukin-6, and leukemia inhibitory factor. Arthritis Rheumatol. 2005, 52, 3749–3754. [Google Scholar] [CrossRef]
- Cao, Y.; Brombacher, F.; Tunyogi-Csapo, M.; Glant, T.T.; Finnegan, A. Interleukin-4 regulates proteoglycan-induced arthritis by specifically suppressing the innate immune response. Arthritis Rheumatol. 2007, 56, 861–870. [Google Scholar] [CrossRef]
- Hong, K.H.; Cho, M.L.; Min, S.Y.; Shin, Y.J.; Yoo, S.A.; Choi, J.J.; Kim, W.U.; Song, S.W.; Cho, C.S. Effect of interleukin-4 on vascular endothelial growth factor production in rheumatoid synovial fibroblasts. Clin. Exp. Immunol. 2007, 147, 573–579. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Andreev, D.; Oeser, K.; Krljanac, B.; Hueber, A.; Kleyer, A.; Voehringer, D.; Schett, G.; Bozec, A. Th2 and eosinophil responses suppress inflammatory arthritis. Nat. Commun. 2016, 7, 11596. [Google Scholar] [CrossRef] [Green Version]
- Gately, M.K.; Renzetti, L.M.; Magram, J.; Stern, A.S.; Adorini, L.; Gubler, U.; Presky, D.H. The interleukin-12/interleukin-12-receptor system: Role in normal and pathologic immune responses. Annu. Rev. Immunol. 1998, 16, 495–521. [Google Scholar] [CrossRef]
- Lavocat, F.; Ndongo-Thiam, N.; Miossec, P. Interleukin-25 Produced by Synoviocytes Has Anti-inflammatory Effects by Acting As a Receptor Antagonist for Interleukin-17A Function. Front. Immunol. 2017, 8, 647. [Google Scholar] [CrossRef] [Green Version]
- Kaiwen, W.; Zhaoliang, S.; Yinxia, Z.; Siamak, S.S.; Zhijun, J.; Yuan, X.; Heng, Y.; Dong, Z.; Yanfang, L.; Pei, S.; et al. Changes and significance of IL-25 in chicken collagen II-induced experimental arthritis (CIA). Rheumatol. Int. 2012, 32, 2331–2338. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.H.; Angkasekwinai, P.; Lu, N.; Voo, K.S.; Arima, K.; Hanabuchi, S.; Hippe, A.; Corrigan, C.J.; Dong, C.; Homey, B.; et al. IL-25 augments type 2 immune responses by enhancing the expansion and functions of TSLP-DC-activated Th2 memory cells. J. Exp. Med. 2007, 204, 1837–1847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, D.; Jiang, H.R.; Kewin, P.; Li, Y.; Mu, R.; Fraser, A.R.; Pitman, N.; Kurowska-Stolarska, M.; McKenzie, A.N.; McInnes, I.B.; et al. IL-33 exacerbates antigen-induced arthritis by activating mast cells. Proc. Natl. Acad. Sci. USA 2008, 105, 10913–10918. [Google Scholar] [CrossRef] [Green Version]
- Matsuyama, Y.; Okazaki, H.; Tamemoto, H.; Kimura, H.; Kamata, Y.; Nagatani, K.; Nagashima, T.; Hayakawa, M.; Iwamoto, M.; Yoshio, T.; et al. Increased levels of interleukin 33 in sera and synovial fluid from patients with active rheumatoid arthritis. J. Rheumatol. 2010, 37, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Athari, S.K.; Poirier, E.; Biton, J.; Semerano, L.; Herve, R.; Raffaillac, A.; Lemeiter, D.; Herbelin, A.; Girard, J.P.; Caux, F.; et al. Collagen-induced arthritis and imiquimod-induced psoriasis develop independently of interleukin-33. Arthritis Res. Ther. 2016, 18, 143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaiss, M.M.; Kurowska-Stolarska, M.; Bohm, C.; Gary, R.; Scholtysek, C.; Stolarski, B.; Reilly, J.; Kerr, S.; Millar, N.L.; Kamradt, T.; et al. IL-33 shifts the balance from osteoclast to alternatively activated macrophage differentiation and protects from TNF-alpha-mediated bone loss. J. Immunol. 2011, 186, 6097–6105. [Google Scholar] [CrossRef]
- Doran, E.; Cai, F.; Holweg, C.T.J.; Wong, K.; Brumm, J.; Arron, J.R. Interleukin-13 in Asthma and Other Eosinophilic Disorders. Front. Med. (Lausanne) 2017, 4, 139. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.B.; Chen, C.Y.; Liu, B.; Mugge, L.; Angkasekwinai, P.; Facchinetti, V.; Dong, C.; Liu, Y.J.; Rothenberg, M.E.; Hogan, S.P.; et al. IL-25 and CD4(+) TH2 cells enhance type 2 innate lymphoid cell-derived IL-13 production, which promotes IgE-mediated experimental food allergy. J. Allergy Clin. Immunol. 2016, 137, 1216–1225.e5. [Google Scholar] [CrossRef] [Green Version]
- Mao, Y.M.; Zhao, C.N.; Leng, J.; Leng, R.X.; Ye, D.Q.; Zheng, S.G.; Pan, H.F. Interleukin-13: A promising therapeutic target for autoimmune disease. Cytokine Growth Factor Rev. 2019, 45, 9–23. [Google Scholar] [CrossRef]
- Hart, P.H.; Ahern, M.J.; Smith, M.D.; Finlay-Jones, J.J. Regulatory effects of IL-13 on synovial fluid macrophages and blood monocytes from patients with inflammatory arthritis. Clin. Exp. Immunol. 1995, 99, 331–337. [Google Scholar] [CrossRef]
- Bessis, N.; Boissier, M.C.; Ferrara, P.; Blankenstein, T.; Fradelizi, D.; Fournier, C. Attenuation of collagen-induced arthritis in mice by treatment with vector cells engineered to secrete interleukin-13. Eur. J. Immunol. 1996, 26, 2399–2403. [Google Scholar] [CrossRef] [PubMed]
- Marinou, I.; Till, S.H.; Moore, D.J.; Wilson, A.G. Lack of association or interactions between the IL-4, IL-4Ralpha and IL-13 genes, and rheumatoid arthritis. Arthritis Res. Ther. 2008, 10, R80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavkova Goldbergova, M.; Nemec, P.; Lipkova, J.; Jarkovsky, J.; Gatterova, J.; Ambrozkova, D.; Vasku, A.; Soucek, M.; Pavek, N. Relation of IL-6, IL-13 and IL-15 gene polymorphisms to the rheumatoid factors, anti-CCP and other measures of rheumatoid arthritis activity. Int. J. Immunogenet. 2014, 41, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Bakakos, A.; Rovina, N.; Bakakos, P. Treatment Challenges in Severe Eosinophilic Asthma: Differential Response to Anti-IL-5 and Anti-IL-5R Therapy. Int. J. Mol. Sci. 2021, 22, 3969. [Google Scholar] [CrossRef]
- Ortega, H.G.; Liu, M.C.; Pavord, I.D.; Brusselle, G.G.; FitzGerald, J.M.; Chetta, A.; Humbert, M.; Katz, L.E.; Keene, O.N.; Yancey, S.W.; et al. Mepolizumab treatment in patients with severe eosinophilic asthma. N. Engl. J. Med. 2014, 371, 1198–1207. [Google Scholar] [CrossRef] [Green Version]
- Castro, M.; Zangrilli, J.; Wechsler, M.E.; Bateman, E.D.; Brusselle, G.G.; Bardin, P.; Murphy, K.; Maspero, J.F.; O’Brien, C.; Korn, S. Reslizumab for inadequately controlled asthma with elevated blood eosinophil counts: Results from two multicentre, parallel, double-blind, randomised, placebo-controlled, phase 3 trials. Lancet Respir. Med. 2015, 3, 355–366. [Google Scholar] [CrossRef]
- Bleecker, E.R.; FitzGerald, J.M.; Chanez, P.; Papi, A.; Weinstein, S.F.; Barker, P.; Sproule, S.; Gilmartin, G.; Aurivillius, M.; Werkstrom, V.; et al. Efficacy and safety of benralizumab for patients with severe asthma uncontrolled with high-dosage inhaled corticosteroids and long-acting beta2-agonists (SIROCCO): A randomised, multicentre, placebo-controlled phase 3 trial. Lancet 2016, 388, 2115–2127. [Google Scholar] [CrossRef]
- Barra, L.; Summers, K.; Bell, D.; Cairns, E. Serum cytokine profile of unaffected first-degree relatives of patients with rheumatoid arthritis. J. Rheumatol. 2014, 41, 280–285. [Google Scholar] [CrossRef]
- Raza, K.; Falciani, F.; Curnow, S.J.; Ross, E.J.; Lee, C.Y.; Akbar, A.N.; Lord, J.M.; Gordon, C.; Buckley, C.D.; Salmon, M. Early rheumatoid arthritis is characterized by a distinct and transient synovial fluid cytokine profile of T cell and stromal cell origin. Arthritis Res. Ther. 2005, 7, R784–R795. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Bozec, A.; Ramming, A.; Schett, G. Anti-inflammatory and immune-regulatory cytokines in rheumatoid arthritis. Nat. Rev. Rheumatol. 2019, 15, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Do-Thi, V.A.; Lee, J.O.; Lee, H.; Kim, Y.S. Crosstalk between the Producers and Immune Targets of IL-9. Immune Netw. 2020, 20, e45. [Google Scholar] [CrossRef]
- Dardalhon, V.; Awasthi, A.; Kwon, H.; Galileos, G.; Gao, W.; Sobel, R.A.; Mitsdoerffer, M.; Strom, T.B.; Elyaman, W.; Ho, I.C.; et al. IL-4 inhibits TGF-beta-induced Foxp3+ T cells and, together with TGF-beta, generates IL-9+ IL-10+ Foxp3(-) effector T cells. Nat. Immunol. 2008, 9, 1347–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veldhoen, M.; Uyttenhove, C.; van Snick, J.; Helmby, H.; Westendorf, A.; Buer, J.; Martin, B.; Wilhelm, C.; Stockinger, B. Transforming growth factor-beta ‘reprograms’ the differentiation of T helper 2 cells and promotes an interleukin 9-producing subset. Nat. Immunol. 2008, 9, 1341–1346. [Google Scholar] [CrossRef] [PubMed]
- Yao, W.; Zhang, Y.; Jabeen, R.; Nguyen, E.T.; Wilkes, D.S.; Tepper, R.S.; Kaplan, M.H.; Zhou, B. Interleukin-9 is required for allergic airway inflammation mediated by the cytokine TSLP. Immunity 2013, 38, 360–372. [Google Scholar] [CrossRef] [Green Version]
- Ciccia, F.; Guggino, G.; Rizzo, A.; Manzo, A.; Vitolo, B.; La Manna, M.P.; Giardina, G.; Sireci, G.; Dieli, F.; Montecucco, C.M.; et al. Potential involvement of IL-9 and Th9 cells in the pathogenesis of rheumatoid arthritis. Rheumatology 2015, 54, 2264–2272. [Google Scholar] [CrossRef] [Green Version]
- Erpenbeck, V.J.; Hohlfeld, J.M.; Volkmann, B.; Hagenberg, A.; Geldmacher, H.; Braun, A.; Krug, N. Segmental allergen challenge in patients with atopic asthma leads to increased IL-9 expression in bronchoalveolar lavage fluid lymphocytes. J. Allergy Clin. Immunol. 2003, 111, 1319–1327. [Google Scholar] [CrossRef] [PubMed]
- Rauber, S.; Luber, M.; Weber, S.; Maul, L.; Soare, A.; Wohlfahrt, T.; Lin, N.Y.; Dietel, K.; Bozec, A.; Herrmann, M.; et al. Resolution of inflammation by interleukin-9-producing type 2 innate lymphoid cells. Nat. Med. 2017, 23, 938–944. [Google Scholar] [CrossRef] [Green Version]
- Kundu-Raychaudhuri, S.; Abria, C.; Raychaudhuri, S.P. IL-9, a local growth factor for synovial T cells in inflammatory arthritis. Cytokine 2016, 79, 45–51. [Google Scholar] [CrossRef]
- Dantas, A.T.; Marques, C.D.; da Rocha Junior, L.F.; Cavalcanti, M.B.; Goncalves, S.M.; Cardoso, P.R.; Mariz Hde, A.; Rego, M.J.; Duarte, A.L.; Pitta Ida, R.; et al. Increased Serum Interleukin-9 Levels in Rheumatoid Arthritis and Systemic Lupus Erythematosus: Pathogenic Role or Just an Epiphenomenon? Dis. Markers 2015, 2015, 519638. [Google Scholar] [CrossRef]
- Bal, S.M.; Golebski, K.; Spits, H. Plasticity of innate lymphoid cell subsets. Nat. Rev. Immunol. 2020, 20, 552–565. [Google Scholar] [CrossRef]
- Asghar Pasha, M.; Yang, Q. Innate Lymphoid Cells in Airway Inflammation. Adv. Exp. Med. Biol. 2021, 1303, 183–191. [Google Scholar]
- Stier, M.T.; Zhang, J.; Goleniewska, K.; Cephus, J.Y.; Rusznak, M.; Wu, L.; Van Kaer, L.; Zhou, B.; Newcomb, D.C.; Peebles, R.S., Jr. IL-33 promotes the egress of group 2 innate lymphoid cells from the bone marrow. J. Exp. Med. 2018, 215, 263–281. [Google Scholar] [CrossRef]
- Omata, Y.; Frech, M.; Lucas, S.; Primbs, T.; Knipfer, L.; Wirtz, S.; Kadono, Y.; Saito, T.; Tanaka, S.; Sarter, K.; et al. Type 2 innate lymphoid cells inhibit the differentiation of osteoclasts and protect from ovariectomy-induced bone loss. Bone 2020, 136, 115335. [Google Scholar] [CrossRef]
- McHedlidze, T.; Waldner, M.; Zopf, S.; Walker, J.; Rankin, A.L.; Schuchmann, M.; Voehringer, D.; McKenzie, A.N.; Neurath, M.F.; Pflanz, S.; et al. Interleukin-33-dependent innate lymphoid cells mediate hepatic fibrosis. Immunity 2013, 39, 357–371. [Google Scholar] [CrossRef] [Green Version]
- Frech, M.; Knipfer, L.; Wirtz, S.; Zaiss, M.M. An in vivo gene delivery approach for the isolation of reasonable numbers of type 2 innate lymphoid cells. MethodsX 2020, 7, 101054. [Google Scholar] [CrossRef] [PubMed]
- Frisbee, A.L.; Saleh, M.M.; Young, M.K.; Leslie, J.L.; Simpson, M.E.; Abhyankar, M.M.; Cowardin, C.A.; Ma, J.Z.; Pramoonjago, P.; Turner, S.D.; et al. IL-33 drives group 2 innate lymphoid cell-mediated protection during Clostridium difficile infection. Nat. Commun. 2019, 10, 2712. [Google Scholar] [CrossRef]
- Ngo Thi Phuong, N.; Palmieri, V.; Adamczyk, A.; Klopfleisch, R.; Langhorst, J.; Hansen, W.; Westendorf, A.M.; Pastille, E. IL-33 Drives Expansion of Type 2 Innate Lymphoid Cells and Regulatory T Cells and Protects Mice From Severe, Acute Colitis. Front. Immunol. 2021, 12, 669787. [Google Scholar] [CrossRef] [PubMed]
- You, Y.; Zhang, X.; Wang, X.; Yue, D.; Meng, F.; Zhu, J.; Wang, Y.; Sun, X. ILC2 Proliferated by IL-33 Stimulation Alleviates Acute Colitis in Rag1(-/-) Mouse through Promoting M2 Macrophage Polarization. J. Immunol. Res. 2020, 2020, 5018975. [Google Scholar] [CrossRef] [PubMed]
- Soare, A.; Weber, S.; Maul, L.; Rauber, S.; Gheorghiu, A.M.; Luber, M.; Houssni, I.; Kleyer, A.; von Pickardt, G.; Gado, M.; et al. Cutting Edge: Homeostasis of Innate Lymphoid Cells Is Imbalanced in Psoriatic Arthritis. J. Immunol. 2018, 200, 1249–1254. [Google Scholar] [CrossRef] [Green Version]
- Bridgewood, C.; Sharif, K.; Freeston, J.; Saleem, B.; Russell, T.; Watad, A.; Khan, A.; Loughenbury, P.; Rao, A.; Wittmann, M.; et al. Regulation of entheseal IL-23 expression by IL-4 and IL-13 as an explanation for arthropathy development under dupilumab therapy. Rheumatology (Oxf.) 2021, 60, 2461–2466. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S. Signaling axis in osteoclast biology and therapeutic targeting in the RANKL/RANK/OPG system. Am. J. Nephrol. 2007, 27, 466–478. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Ando, F.; Niino, N.; Shimokata, H. Association of polymorphisms of the osteoprotegerin gene with bone mineral density in Japanese women but not men. Mol. Genet. Metab. 2003, 80, 344–349. [Google Scholar] [CrossRef]
- Arko, B.; Prezelj, J.; Kocijancic, A.; Komel, R.; Marc, J. Association of the osteoprotegerin gene polymorphisms with bone mineral density in postmenopausal women. Maturitas 2005, 51, 270–279. [Google Scholar] [CrossRef]
- Garcia-Unzueta, M.T.; Riancho, J.A.; Zarrabeitia, M.T.; Sanudo, C.; Berja, A.; Valero, C.; Pesquera, C.; Paule, B.; Gonzalez-Macias, J.; Amado, J.A. Association of the 163A/G and 1181G/C osteoprotegerin polymorphism with bone mineral density. Horm. Metab. Res. 2008, 40, 219–224. [Google Scholar] [CrossRef]
- Kim, Y.G.; Lee, C.K.; Nah, S.S.; Mun, S.H.; Yoo, B.; Moon, H.B. Human CD4+CD25+ regulatory T cells inhibit the differentiation of osteoclasts from peripheral blood mononuclear cells. Biochem. Biophys. Res. Commun. 2007, 357, 1046–1052. [Google Scholar] [CrossRef]
- Zaiss, M.M.; Axmann, R.; Zwerina, J.; Polzer, K.; Guckel, E.; Skapenko, A.; Schulze-Koops, H.; Horwood, N.; Cope, A.; Schett, G. Treg cells suppress osteoclast formation: A new link between the immune system and bone. Arthritis Rheumatol. 2007, 56, 4104–4112. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Tanaka, Y.; Morimoto, I.; Yahata, K.; Zeki, K.; Fujihira, T.; Yamashita, U.; Eto, S. Interleukin-4 as a potent inhibitor of bone resorption. Biochem. Biophys. Res. Commun. 1990, 172, 1035–1041. [Google Scholar] [CrossRef]
- Shioi, A.; Teitelbaum, S.L.; Ross, F.P.; Welgus, H.G.; Suzuki, H.; Ohara, J.; Lacey, D.L. Interleukin 4 inhibits murine osteoclast formation in vitro. J. Cell. Biochem. 1991, 47, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Kasono, K.; Sato, K.; Sato, Y.; Tsushima, T.; Shizume, K.; Demura, H. Inhibitory effect of interleukin-4 on osteoclast-like cell formation in mouse bone marrow culture. Bone Miner. 1993, 21, 179–188. [Google Scholar] [CrossRef]
- Riancho, J.A.; Zarrabeitia, M.T.; Mundy, G.R.; Yoneda, T.; Gonzalez-Macias, J. Effects of interleukin-4 on the formation of macrophages and osteoclast-like cells. J. Bone Miner. Res. 1993, 8, 1337–1344. [Google Scholar] [CrossRef]
- Wei, S.; Wang, M.W.; Teitelbaum, S.L.; Ross, F.P. Interleukin-4 reversibly inhibits osteoclastogenesis via inhibition of NF-kappa B and mitogen-activated protein kinase signaling. J. Biol. Chem. 2002, 277, 6622–6630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitaura, H.; Nagata, N.; Fujimura, Y.; Hotokezaka, H.; Tatamiya, M.; Nakao, N.; Yoshida, N.; Nakayama, K. Interleukin-4 directly inhibits tumor necrosis factor-alpha-mediated osteoclast formation in mouse bone marrow macrophages. Immunol. Lett. 2003, 88, 193–198. [Google Scholar] [CrossRef]
- Mirosavljevic, D.; Quinn, J.M.; Elliott, J.; Horwood, N.J.; Martin, T.J.; Gillespie, M.T. T-cells mediate an inhibitory effect of interleukin-4 on osteoclastogenesis. J. Bone Miner. Res. 2003, 18, 984–993. [Google Scholar] [CrossRef]
- Abu-Amer, Y. IL-4 abrogates osteoclastogenesis through STAT6-dependent inhibition of NF-kappaB. J. Clin. Investig. 2001, 107, 1375–1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, D.B.; Liggitt, H.D.; Effmann, E.L.; Motley, S.T.; Teitelbaum, S.L.; Jepsen, K.J.; Goldstein, S.A.; Bonadio, J.; Carpenter, J.; Perlmutter, R.M. Osteoporosis induced in mice by overproduction of interleukin 4. Proc. Natl. Acad. Sci. USA 1993, 90, 11618–11622. [Google Scholar] [CrossRef] [Green Version]
- Ahn, J.S.; Agrawal, B. IL-4 is more effective than IL-13 for in vitro differentiation of dendritic cells from peripheral blood mononuclear cells. Int. Immunol. 2005, 17, 1337–1346. [Google Scholar] [CrossRef] [PubMed]
- Yamada, A.; Takami, M.; Kawawa, T.; Yasuhara, R.; Zhao, B.; Mochizuki, A.; Miyamoto, Y.; Eto, T.; Yasuda, H.; Nakamichi, Y.; et al. Interleukin-4 inhibition of osteoclast differentiation is stronger than that of interleukin-13 and they are equivalent for induction of osteoprotegerin production from osteoblasts. Immunology 2007, 120, 573–579. [Google Scholar] [CrossRef] [PubMed]
- Min, H.K.; Won, J.Y.; Kim, B.M.; Lee, K.A.; Lee, S.J.; Lee, S.H.; Kim, H.R.; Kim, K.W. Interleukin (IL)-25 suppresses IL-22-induced osteoclastogenesis in rheumatoid arthritis via STAT3 and p38 MAPK/IkappaBalpha pathway. Arthritis Res. Ther. 2020, 22, 222. [Google Scholar] [CrossRef] [PubMed]
- Ge, S.; Hertel, B.; Susnik, N.; Rong, S.; Dittrich, A.M.; Schmitt, R.; Haller, H.; von Vietinghoff, S. Interleukin 17 receptor A modulates monocyte subsets and macrophage generation in vivo. PLoS ONE 2014, 9, e85461. [Google Scholar] [CrossRef]
- Schulze, J.; Bickert, T.; Beil, F.T.; Zaiss, M.M.; Albers, J.; Wintges, K.; Streichert, T.; Klaetschke, K.; Keller, J.; Hissnauer, T.N.; et al. Interleukin-33 is expressed in differentiated osteoblasts and blocks osteoclast formation from bone marrow precursor cells. J. Bone Miner. Res. 2011, 26, 704–717. [Google Scholar] [CrossRef] [PubMed]
- Lima, I.L.; Macari, S.; Madeira, M.F.; Rodrigues, L.F.; Colavite, P.M.; Garlet, G.P.; Soriani, F.M.; Teixeira, M.M.; Fukada, S.Y.; Silva, T.A. Osteoprotective Effects of IL-33/ST2 Link to Osteoclast Apoptosis. Am. J. Pathol. 2015, 185, 3338–3348. [Google Scholar] [CrossRef] [Green Version]
- Vladich, F.D.; Brazille, S.M.; Stern, D.; Peck, M.L.; Ghittoni, R.; Vercelli, D. IL-13 R130Q, a common variant associated with allergy and asthma, enhances effector mechanisms essential for human allergic inflammation. J. Clin. Investig. 2005, 115, 747–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frost, A.; Jonsson, K.B.; Brandstrom, H.; Ljunghall, S.; Nilsson, O.; Ljunggren, O. Interleukin (IL)-13 and IL-4 inhibit proliferation and stimulate IL-6 formation in human osteoblasts: Evidence for involvement of receptor subunits IL-13R, IL-13Ralpha, and IL-4Ralpha. Bone 2001, 28, 268–274. [Google Scholar] [CrossRef]
- Silfversward, C.J.; Frost, A.; Brandstrom, H.; Nilsson, O.; Ljunggren, O. Interleukin-4 and interleukin-13 potentiate interleukin-1 induced secretion of interleukin-6 in human osteoblast-like cells. J. Orthop. Res. 2004, 22, 1058–1062. [Google Scholar] [CrossRef] [PubMed]
- Stein, N.C.; Kreutzmann, C.; Zimmermann, S.P.; Niebergall, U.; Hellmeyer, L.; Goettsch, C.; Schoppet, M.; Hofbauer, L.C. Interleukin-4 and interleukin-13 stimulate the osteoclast inhibitor osteoprotegerin by human endothelial cells through the STAT6 pathway. J. Bone Miner. Res. 2008, 23, 750–758. [Google Scholar] [CrossRef] [PubMed]
- Clutterbuck, E.J.; Hirst, E.M.; Sanderson, C.J. Human interleukin-5 (IL-5) regulates the production of eosinophils in human bone marrow cultures: Comparison and interaction with IL-1, IL-3, IL-6, and GMCSF. Blood 1989, 73, 1504–1512. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, Y.; Suda, T.; Suda, J.; Eguchi, M.; Miura, Y.; Harada, N.; Tominaga, A.; Takatsu, K. Purified interleukin 5 supports the terminal differentiation and proliferation of murine eosinophilic precursors. J. Exp. Med. 1988, 167, 43–56. [Google Scholar] [CrossRef] [Green Version]
- Bjerke, T.; Gaustadnes, M.; Nielsen, S.; Nielsen, L.P.; Schiotz, P.O.; Rudiger, N.; Reimert, C.M.; Dahl, R.; Christensen, I.; Poulsen, L.K. Human blood eosinophils produce and secrete interleukin 4. Respir. Med. 1996, 90, 271–277. [Google Scholar] [CrossRef] [Green Version]
- Dent, L.A.; Strath, M.; Mellor, A.L.; Sanderson, C.J. Eosinophilia in transgenic mice expressing interleukin 5. J. Exp. Med. 1990, 172, 1425–1431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macias, M.P.; Fitzpatrick, L.A.; Brenneise, I.; McGarry, M.P.; Lee, J.J.; Lee, N.A. Expression of IL-5 alters bone metabolism and induces ossification of the spleen in transgenic mice. J. Clin. Investig. 2001, 107, 949–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirchandani, A.S.; Besnard, A.G.; Yip, E.; Scott, C.; Bain, C.C.; Cerovic, V.; Salmond, R.J.; Liew, F.Y. Type 2 innate lymphoid cells drive CD4+ Th2 cell responses. J. Immunol. 2014, 192, 2442–2448. [Google Scholar] [CrossRef] [Green Version]
- Zaiss, M.M.; Frey, B.; Hess, A.; Zwerina, J.; Luther, J.; Nimmerjahn, F.; Engelke, K.; Kollias, G.; Hunig, T.; Schett, G.; et al. Regulatory T cells protect from local and systemic bone destruction in arthritis. J. Immunol. 2010, 184, 7238–7246. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Omata, Y.; Frech, M.; Saito, T.; Schett, G.; Zaiss, M.M.; Tanaka, S. Inflammatory Arthritis and Bone Metabolism Regulated by Type 2 Innate and Adaptive Immunity. Int. J. Mol. Sci. 2022, 23, 1104. https://doi.org/10.3390/ijms23031104
Omata Y, Frech M, Saito T, Schett G, Zaiss MM, Tanaka S. Inflammatory Arthritis and Bone Metabolism Regulated by Type 2 Innate and Adaptive Immunity. International Journal of Molecular Sciences. 2022; 23(3):1104. https://doi.org/10.3390/ijms23031104
Chicago/Turabian StyleOmata, Yasunori, Michael Frech, Taku Saito, Georg Schett, Mario M. Zaiss, and Sakae Tanaka. 2022. "Inflammatory Arthritis and Bone Metabolism Regulated by Type 2 Innate and Adaptive Immunity" International Journal of Molecular Sciences 23, no. 3: 1104. https://doi.org/10.3390/ijms23031104
APA StyleOmata, Y., Frech, M., Saito, T., Schett, G., Zaiss, M. M., & Tanaka, S. (2022). Inflammatory Arthritis and Bone Metabolism Regulated by Type 2 Innate and Adaptive Immunity. International Journal of Molecular Sciences, 23(3), 1104. https://doi.org/10.3390/ijms23031104