
Novel NK1R-Targeted 68Ga-/177Lu-Radioconjugates with Potential Application against Glioblastoma Multiforme: Preliminary Exploration of Structure–Activity Relationships

 , , , , and
, , , , and 
Abstract
:1. Introduction
2. Results and Discussion
2.1. Structure–Activity Relationships of the Extended L732,138 Analogues
2.1.1. Design Rationale
2.1.2. Synthesis
2.1.3. Binding Affinity Determination
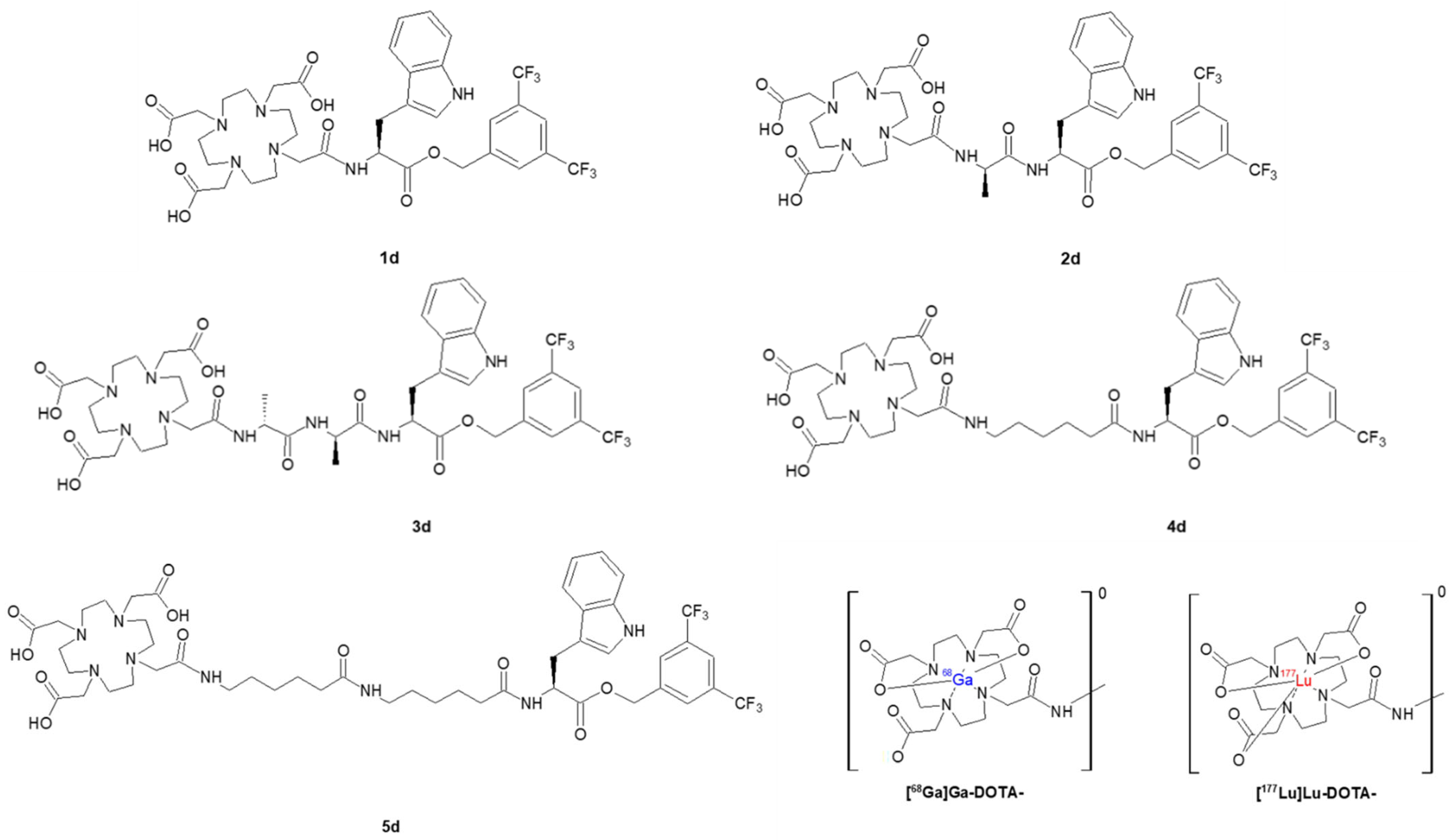
2.2. Radioconjugates
2.2.1. Synthesis of Radioconjugates
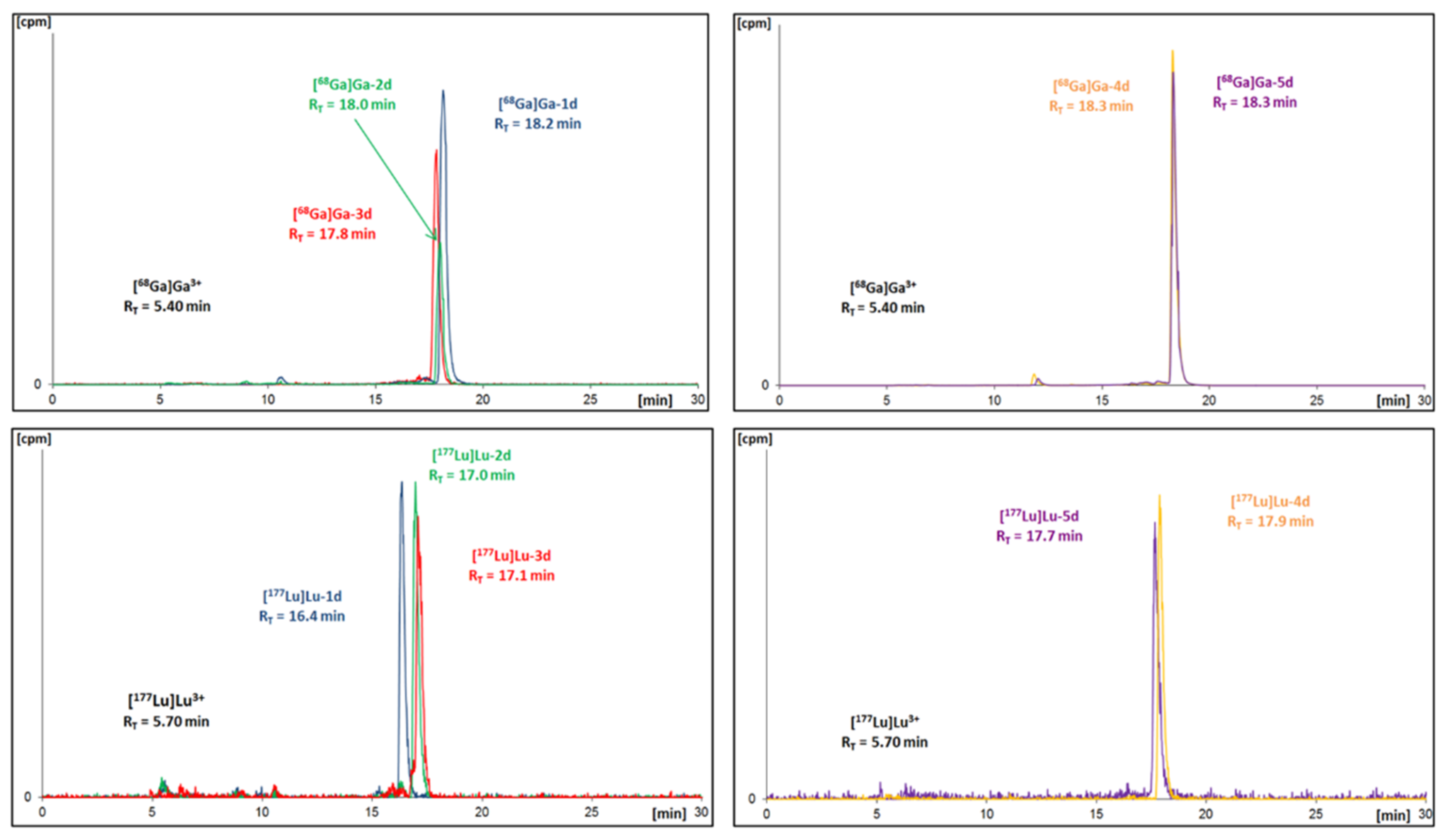
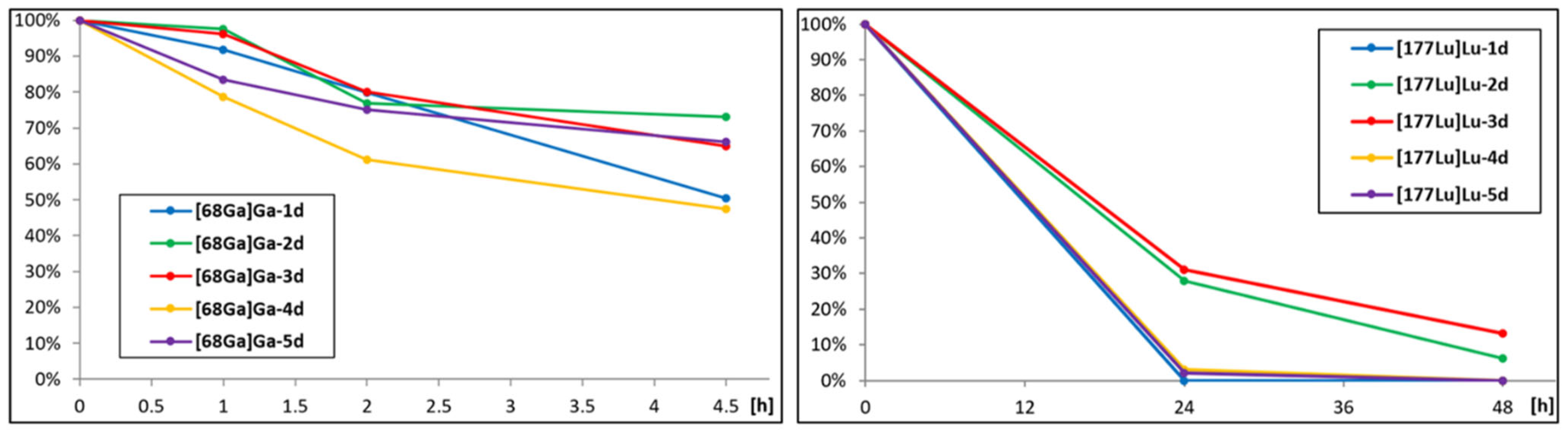
2.2.2. Plasma Stability
2.2.3. Lipophilicity Study
2.2.4. Binding Affinity (Competitive Assay)
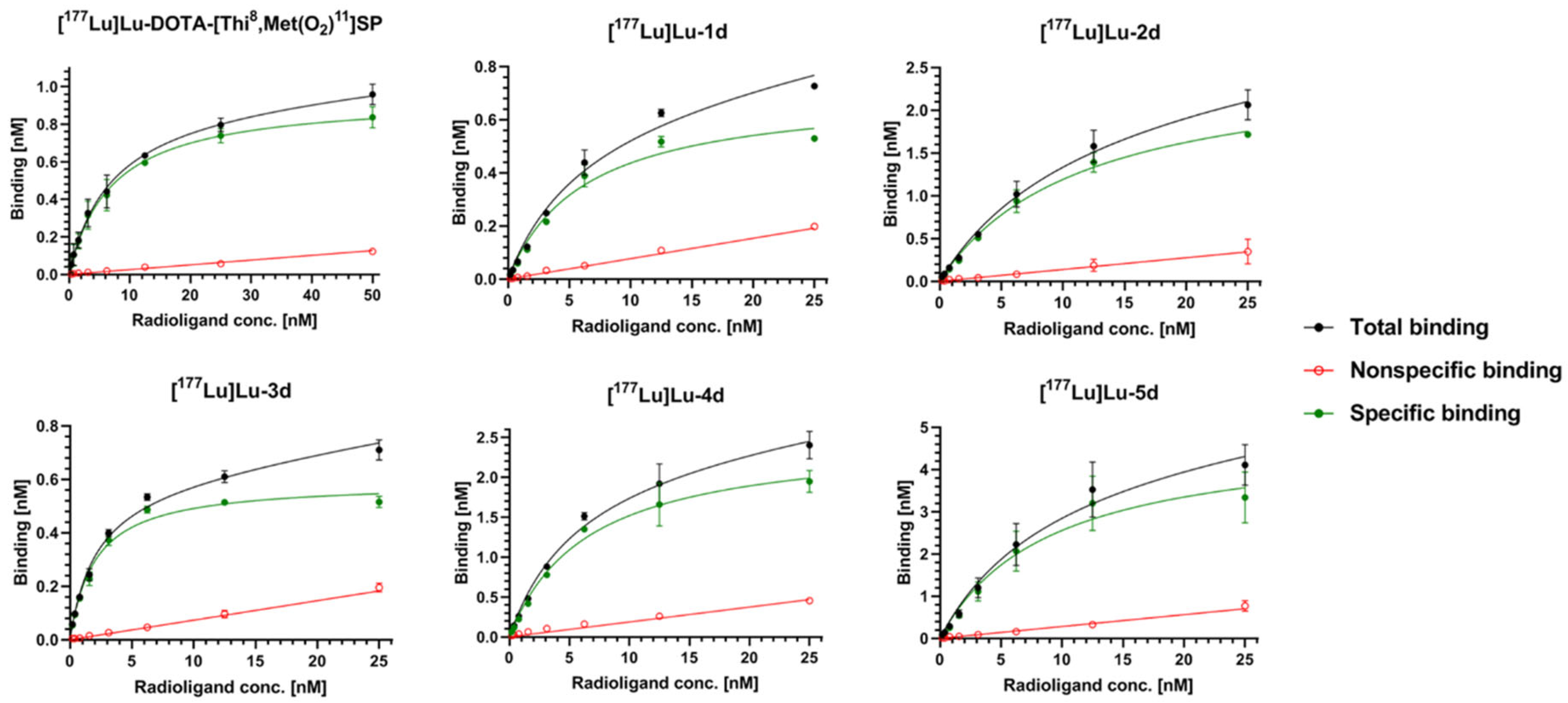
2.2.5. Binding Affinity (Saturation Assay)
2.3. Retrospective Molecular Modelling
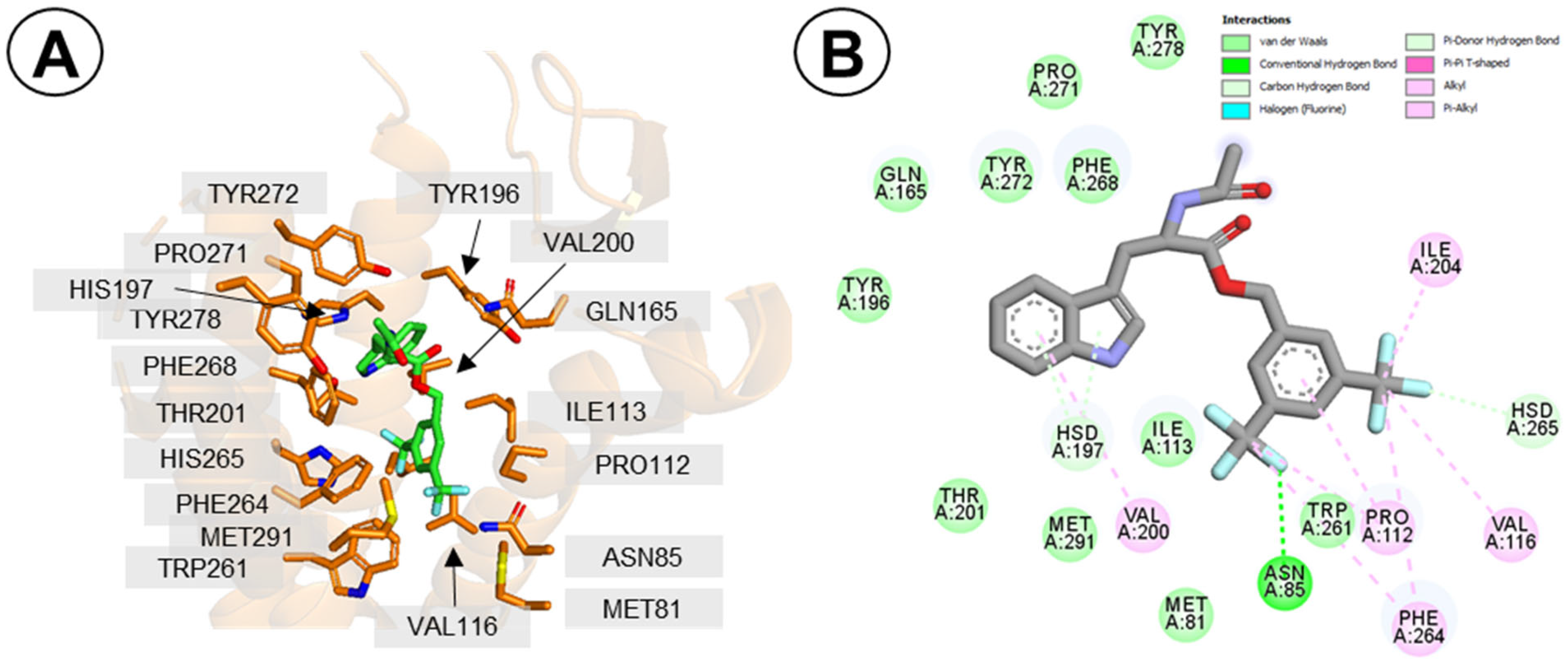
2.3.1. Binding Mode of L732,138
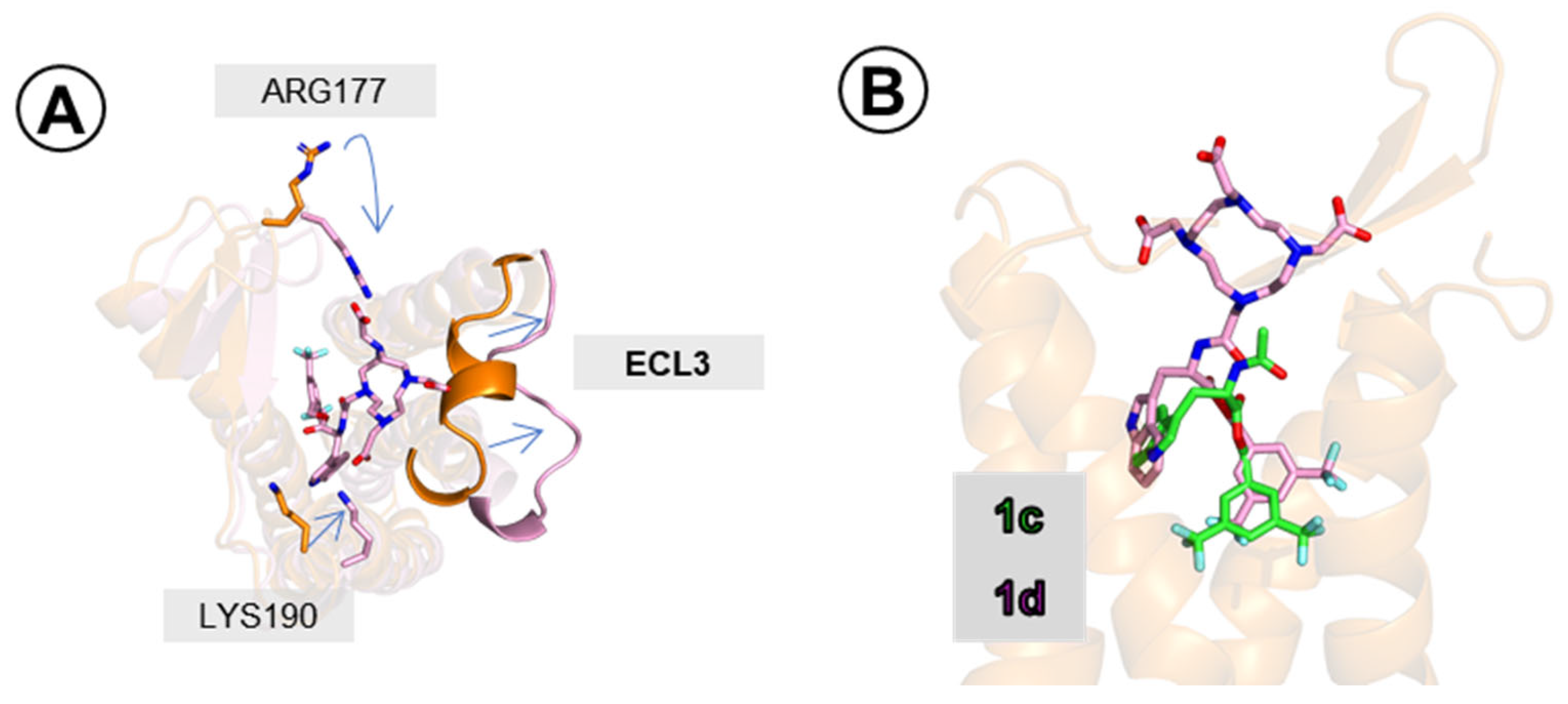
2.3.2. Binding Mode of Compound 1d
2.3.3. Binding Modes of Compounds 5a and 5b
3. Materials and Methods
3.1. Synthesis of L732,138 Analogues
3.1.1. General
3.1.2. Synthesis of Compound 1a
3.1.3. Synthesis of Compounds 2a and 3a
3.1.4. Synthesis of Compounds 4a and 5a
3.1.5. Deprotection Reaction (1b, 2b, 3b, 4b, 5b)
3.1.6. Acetylation Reaction (1c, 2c, 3c, 4c, 5c)
3.2. Synthesis of Conjugates and Radioconjugates
3.2.1. General
3.2.2. General Procedure of Syntheses of Conjugates with DOTA
- MS: Calculated monoisotopic mass for 1d, C36H42F6N6O9: 816.74; found: 817.38 m/z [M + H]+
- MS: Calculated monoisotopic mass for 2d, C39H47F6N7O10: 887.82; found: 888.35 m/z [M + H]+
- MS: Calculated monoisotopic mass for 3d, C42H52F8N8O11: 958.90; found: 959.40 m/z [M + H]+
- MS: Calculated monoisotopic mass for 4d, C42H53F6N7O10: 929.90; found: 930.38 m/z [M + H]+
- MS: Calculated monoisotopic mass for 5d, C48H64F6N8O11: 1043.06; found: 1043.58 m/z [M + H]+
3.2.3. 68Ga Radiolabelling
3.2.4. 177Lu Radiolabelling
3.2.5. Preparation of Non-Radioactive References
- MS: Calculated for monoisotopic mass Ga-1d, C41H50F7N9O10Ga: 883.20 and 885.20; found: 883.31 and 885.28 m/z [M]+
- MS: Calculated for monoisotopic mass Ga-2d, C41H49F7N10O11Ga: 954.24 and 956.24; found: 954.33 and 956.65 m/z [M]+
- MS: Calculated for monoisotopic mass Ga-3d, C43H53F7N10O11Ga: 1025.28 and 1027.27; found: 1025.37 and 1027.38 m/z [M+]
- MS: Calculated for monoisotopic mass Ga-4d, C42H52F7N9O10Ga: 996.29 and 998.28; found: 996.39 and 998.38 m/z [M]+
- MS: Calculated for monoisotopic mass Ga-5d, C43H54F7N9O10Ga: 1109.37 and 1111.37; found: 1109.09 and 1111.20 m/z [M]+
3.3. Plasma Stability Study
3.4. Lipophilicity Determination
3.5. Cell Culture
3.6. hNK1-CHO Membrane Preparation
3.7. Competitive Binding Assays
3.8. Saturation Binding Assays
3.9. Molecular Docking
3.10. Molecular Dynamics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Holland, E.C. Glioblastoma multiforme: The terminator. Proc. Natl. Acad. Sci. USA 2000, 97, 6242–6244. [Google Scholar] [CrossRef] [Green Version]
- Lapointe, S.; Perry, A.; Butowski, N.A. Primary brain tumours in adults. Lancet 2018, 392, 432–446. [Google Scholar] [CrossRef]
- Reulen, H.-J.; Suero Molina, E.; Zeidler, R.; Gildehaus, F.J.; Böning, G.; Gosewisch, A.; Stummer, W. Intracavitary radioimmunotherapy of high-grade gliomas: Present status and future developments. Acta Neurochir. 2019, 161, 1109–1124. [Google Scholar] [CrossRef]
- Majkowska-Pilip, A.; Halik, P.K.; Gniazdowska, E. The Significance of NK1 Receptor Ligands and Their Application in Targeted Radionuclide Tumour Therapy. Pharmaceutics 2019, 11, 443. [Google Scholar] [CrossRef] [Green Version]
- Kneifel, S.; Cordier, D.; Good, S.; Ionescu, M.C.S.; Ghaffari, A.; Hofer, S.; Kretzschmar, M.; Tolnay, M.; Apostolidis, C.; Waser, B.; et al. Local Targeting of Malignant Gliomas by the Diffusible Peptidic Vector 1,4,7,10-Tetraazacyclododecane-1-Glutaric Acid-4,7,10-Triacetic Acid-Substance P. Clin. Cancer Res. 2006, 12, 3843–3850. [Google Scholar] [CrossRef] [Green Version]
- Cordier, D.; Forrer, F.; Bruchertseifer, F.; Morgenstern, A.; Apostolidis, C.; Good, S.; Müller-Brand, J.; Mäcke, H.; Reubi, J.C.; Merlo, A. Targeted alpha-radionuclide therapy of functionally critically located gliomas with 213Bi-DOTA-[Thi8,Met(O2)11]- substance P: A pilot trial. Eur. J. Nucl. Med. Mol. Imaging 2010, 37, 1335–1344. [Google Scholar] [CrossRef] [Green Version]
- Cordier, D.; Forrer, F.; Kneifel, S.; Sailer, M.; Mariani, L.; Mäcke, H.; Müller-Brand, J.; Merlo, A. Neoadjuvant targeting of glioblastoma multiforme with radiolabeled DOTAGA–substance P—results from a phase I study. J. Neurooncol. 2010, 100, 129–136. [Google Scholar] [CrossRef]
- Królicki, L.; Kunikowska, J.; Bruchertseifer, F.; Koziara, H.; Królicki, B.; Jakuciński, M.; Pawlak, D.; Rola, R.; Morgenstern, A.; Rosiak, E.; et al. 225Ac- and 213Bi-Substance P Analogues for Glioma Therapy. Semin. Nucl. Med. 2020, 50, 141–151. [Google Scholar] [CrossRef]
- Królicki, L.; Bruchertseifer, F.; Kunikowska, J.; Koziara, H.; Królicki, B.; Jakuciński, M.; Pawlak, D.; Apostolidis, C.; Mirzadeh, S.; Rola, R.; et al. Safety and efficacy of targeted alpha therapy with 213Bi-DOTA-substance P in recurrent glioblastoma. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 614–622. [Google Scholar] [CrossRef]
- Merlo, A.; Mäcke, H.; Reubi, J.C.; Good, S. Radiolabeled Conjugates Based on Substance P and the Uses. Thereof. Patent Application No. WO 2004/082722, 30 September 2004. [Google Scholar]
- Majkowska-Pilip, A.; Rius, M.; Bruchertseifer, F.; Apostolidis, C.; Weis, M.; Bonelli, M.; Laurenza, M.; Królicki, L.; Morgenstern, A. In vitro evaluation of 225 Ac-DOTA-substance P for targeted alpha therapy of glioblastoma multiforme. Chem. Biol. Drug Des. 2018, 92, 1344–1356. [Google Scholar] [CrossRef]
- Królicki, L.; Bruchertseifer, F.; Kunikowska, J.; Koziara, H.; Pawlak, D.; Kuliński, R.; Rola, R.; Merlo, A.; Morgenstern, A. Dose escalation study of targeted alpha therapy with [225Ac]Ac-DOTA-substance P in recurrence glioblastoma–safety and efficacy. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 3595–3605. [Google Scholar] [CrossRef] [PubMed]
- Majkowska-Pilip, A.; Koźmiński, P.; Wawrzynowska, A.; Budlewski, T.; Kostkiewicz, B.; Gniazdowska, E. Application of Neurokinin-1 Receptor in Targeted Strategies for Glioma Treatment. Part I: Synthesis and Evaluation of Substance P Fragments Labeled with 99mTc and 177Lu as Potential Receptor Radiopharmaceuticals. Molecules 2018, 23, 2542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyczko, M.; Pruszynski, M.; Majkowska-Pilip, A.; Lyczko, K.; Was, B.; Meczynska-Wielgosz, S.; Kruszewski, M.; Szkliniarz, K.; Jastrzebski, J.; Stolarz, A.; et al. 211 At labeled substance P (5–11) as potential radiopharmaceutical for glioma treatment. Nucl. Med. Biol. 2017, 53, 1–8. [Google Scholar] [CrossRef]
- Halik, P.K.; Lipiński, P.F.J.; Matalińska, J.; Koźmiński, P.; Misicka, A.; Gniazdowska, E. Radiochemical Synthesis and Evaluation of Novel Radioconjugates of Neurokinin 1 Receptor Antagonist Aprepitant Dedicated for NK1R-Positive Tumors. Molecules 2020, 25, 3756. [Google Scholar] [CrossRef] [PubMed]
- MacLeod, A.M.; Merchant, K.J.; Cascieri, M.A.; Sadowski, S.; Ber, E.; Swain, C.J.; Baker, R. N-Acyl-L-tryptophan benzyl esters: Potent substance P receptor antagonists. J. Med. Chem. 1993, 36, 2044–2045. [Google Scholar] [CrossRef]
- Spector, R.; Robert Snodgrass, S.; Johanson, C.E. A balanced view of the cerebrospinal fluid composition and functions: Focus on adult humans. Exp. Neurol. 2015, 273, 57–68. [Google Scholar] [CrossRef] [Green Version]
- Arnott, J.A.; Planey, S.L. The influence of lipophilicity in drug discovery and design. Expert Opin. Drug Discov. 2012, 7, 863–875. [Google Scholar] [CrossRef]
- Waring, M.J. Lipophilicity in drug discovery. Expert Opin. Drug Discov. 2010, 5, 235–248. [Google Scholar] [CrossRef]
- Leffler, A.; Ahlstedt, I.; Engberg, S.; Svensson, A.; Billger, M.; Öberg, L.; Bjursell, M.K.; Lindström, E.; von Mentzer, B. Characterization of species-related differences in the pharmacology of tachykinin NK receptors 1, 2 and 3. Biochem. Pharmacol. 2009, 77, 1522–1530. [Google Scholar] [CrossRef] [Green Version]
- MacLeod, A.M.; Merchant, K.J.; Brookfield, F.; Kelleher, F.; Stevenson, G.; Owens, A.P.; Swain, C.J.; Cascieri, M.A.; Sadowski, S. Identification of L-Tryptophan Derivatives with Potent and Selective Antagonist Activity at the NK1 Receptor. J. Med. Chem. 1994, 37, 1269–1274. [Google Scholar] [CrossRef]
- Vardanyan, R.; Kumirov, V.K.; Nichol, G.S.; Davis, P.; Liktor-Busa, E.; Rankin, D.; Varga, E.; Vanderah, T.; Porreca, F.; Lai, J.; et al. Synthesis and biological evaluation of new opioid agonist and neurokinin-1 antagonist bivalent ligands. Bioorg. Med. Chem. 2011, 19, 6135–6142. [Google Scholar] [CrossRef] [Green Version]
- Cascieri, M.A.; Macleod, A.M.; Underwood, D.; Shiao, L.L.; Ber, E.; Sadowski, S.; Yu, H.; Merchant, K.J.; Swain, C.J.; Strader, C.D. Characterization of the interaction of N-acyl-L-tryptophan benzyl ester neurokinin antagonists with the human neurokinin-1 receptor. J. Biol. Chem. 1994, 269, 6587–6591. [Google Scholar] [CrossRef]
- Morgenstern, A.; Apostolidis, C.; Kratochwil, C.; Sathekge, M.; Krolicki, L.; Bruchertseifer, F. An Overview of Targeted Alpha Therapy with 225 Actinium and 213 Bismuth. Curr. Radiopharm. 2018, 11, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Schöppe, J.; Ehrenmann, J.; Klenk, C.; Rucktooa, P.; Schütz, M.; Doré, A.S.; Plückthun, A. Crystal structures of the human neurokinin 1 receptor in complex with clinically used antagonists. Nat. Commun. 2019, 10, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacLeod, A.M.; Cascieri, M.A.; Merchant, K.J.; Sadowski, S.; Hardwicke, S.; Lewis, R.T.; MacIntyre, D.E.; Metzger, J.M.; Fong, T.M. Synthesis and Biological Evaluation of NK1 Antagonists Derived from L-Tryptophan. J. Med. Chem. 1995, 38, 934–941. [Google Scholar] [CrossRef]
- Lewis, R.T.; Macleod, A.M.; Merchant, K.J.; Kelleher, F.; Sanderson, I.; Herbert, R.H.; Cascieri, M.A.; Sadowski, S.; Ball, R.G.; Hoogsteen, K. Tryptophan-Derived NK1 Antagonists: Conformationally Constrained Heterocyclic Bioisosteres of the Ester Linkage. J. Med. Chem. 1995, 38, 923–933. [Google Scholar] [CrossRef]
- Branik, M.; Kessler, H. Zur Konformation geschützter Aminosäuren, III. NMR- und IR-Untersuchungen von Boc-L-α-Aminosäuren. Chem. Ber. 1975, 108, 2176–2188. [Google Scholar] [CrossRef]
- Starnowska, J.; Costante, R.; Guillemyn, K.; Popiolek-Barczyk, K.; Chung, N.N.; Lemieux, C.; Keresztes, A.; Van Duppen, J.; Mollica, A.; Streicher, J.M.; et al. Analgesic properties of opioid/NK1 multitarget ligands with distinct in vitro profiles in naive and chronic constriction injury (CCI)-mice. ACS Chem. Neurosci. 2017, 8, 2315–2324. [Google Scholar] [CrossRef]
- Matalińska, J.; Lipiński, P.F.J.; Kotlarz, A.; Kosson, P.; Muchowska, A.; Dyniewicz, J. Evaluation of Receptor Affinity, Analgesic Activity and Cytotoxicity of a Hybrid Peptide, AWL3020. Int. J. Pept. Res. Ther. 2020, 26, 2603–2617. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Pándy-Szekeres, G.; Munk, C.; Tsonkov, T.M.; Mordalski, S.; Harpsøe, K.; Hauser, A.S.; Bojarski, A.J.; Gloriam, D.E. GPCRdb in 2018: Adding GPCR structure models and ligands. Nucleic Acids Res. 2018, 46, D440–D446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lomize, M.A.; Lomize, A.L.; Pogozheva, I.D.; Mosberg, H.I. OPM: Orientations of proteins in membranes database. Bioinformatics 2006, 22, 623–625. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2009, 31, 671–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Schrödinger LLC. The PyMOL Molecular Graphics System. 2018. Available online: https://sourceforge.net/p/pymol/code/HEAD/tree/trunk/pymol/ (accessed on 1 November 2020).
- Biovia Discovery Studio Visualizer v.19; Dassault Systèmes: San Diego, CA, USA, 2018.
- Matalińska, J.; Lipiński, P.F.J.; Kosson, P.; Kosińska, K.; Misicka, A. In Vivo, In Vitro and In Silico Studies of the Hybrid Compound AA3266, an Opioid Agonist/NK1R Antagonist with Selective Cytotoxicity. Int. J. Mol. Sci. 2020, 21, 7738. [Google Scholar] [CrossRef]
- Dyniewicz, J.; Lipiński, P.F.J.; Kosson, P.; Leśniak, A.; Bochyńska-Czyż, M.; Muchowska, A.; Tourwé, D.; Ballet, S.; Misicka, A.; Lipkowski, A.W. Hydrazone Linker as a Useful Tool for Preparing Chimeric Peptide/Nonpeptide Bifunctional Compounds. ACS Med. Chem. Lett. 2017, 8, 73–77. [Google Scholar] [CrossRef] [Green Version]
- Kleczkowska, P.; Nowicka, K.; Bujalska-Zadrozny, M.; Hermans, E. Neurokinin-1 receptor-based bivalent drugs in pain management: The journey to nowhere? Pharmacol. Ther. 2019, 196, 44–58. [Google Scholar] [CrossRef]
- Rizk, S.S.; Misiura, A.; Paduch, M.; Kossiakoff, A.A. Substance P Derivatives as Versatile Tools for Specific Delivery of Various Types of Biomolecular Cargo. Bioconjug. Chem. 2012, 23, 42–46. [Google Scholar] [CrossRef] [Green Version]
- Ding, G.; Wang, T.; Han, Z.; Tian, L.; Cheng, Q.; Luo, L.; Zhao, B.; Wang, C.; Feng, S.; Wang, L.; et al. Substance P containing peptide gene delivery vectors for specifically transfecting glioma cells mediated by a neurokinin-1 receptor. J. Mater. Chem. B 2021, 9, 6347–6356. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Linker Length | Linker | N-Terminus | IC50 (Mean ± SD) 1 | |
|---|---|---|---|---|---|
| rNK1R | hNK1R | ||||
| (n Atoms) | (nM) | (nM) | |||
| 1a | 0 | none | Boc | 2928 ± 1518 | 180.4 ± 112.4 |
| 1b | 0 | none | H | >10000 | 358.9 ± 132.0 |
| 1c L732,138 2 (parent) | 0 | none | Ac | 2879 ± 999 | 25.6 ± 15.9 |
| 2a | 3 | -DAla- | Boc | 1532 ± 279 | 135.0 ± 108.3 |
| 2b | 3 | -DAla- | H | 4681 ± 1322 | 25.4 ± 12.5 |
| 2c | 3 | -DAla- | Ac | 2280 ± 703 | 25.3 ± 0.4 |
| 3a | 6 | -DAla-DAla- | Boc | 745 ± 311 | 13.9 ± 11.2 |
| 3b | 6 | -DAla-DAla- | H | 4007 ± 1373 | 80.8 ± 59.7 |
| 3c | 6 | -DAla-DAla- | Ac | 1141 ± 289 | 13.5 ± 12.1 |
| 4a | 7 | -Ahx- | Boc | 1658 ± 1051 | 60.7 ± 31.9 |
| 4b | 7 | -Ahx- | H | 1399 ± 319 | 35.4 ± 23.6 |
| 4c | 7 | -Ahx- | Ac | 1951 ± 641 | 83.1 ± 69.3 |
| 5a | 14 | -Ahx-Ahx- | Boc | 762 ± 295 | 4.5 ± 2.8 |
| 5b | 14 | -Ahx-Ahx- | H | 572 ± 41 | 20.4 ± 10.2 |
| 5c | 14 | -Ahx-Ahx- | Ac | 655 ± 120 | 14.3 ± 5.2 |
| Reference compounds | |||||
| Substance P | - | - | - | 3.6 (n = 1) | 30.7 ± 4.2 (n = 2) |
| Aprepitant | - | - | - | 130 ± 60 [15] | 27.7 (n = 1) |
| Stable Ga-Conjugate | tR 1 | 68Ga-Radioconjugate | tR |
|---|---|---|---|
| Ga-1d | 17.9 min | [68Ga]Ga-1d | 18.2 min |
| Ga-2d | 17.7 min | [68Ga]Ga-2d | 18.0 min |
| Ga-3d | 17.6 min | [68Ga]Ga-3d | 17.8 min |
| Ga-4d | 18.1 min | [68Ga]Ga-4d | 18.3 min |
| Ga-5d | 18.2 min | [68Ga]Ga-5d | 18.3 min |
| Radioconjugate | logD7.4 (Mean ± SD) 1 | |
|---|---|---|
| 68Ga- | 177Lu- | |
| 1d- | −0.085 ± 0.015 | 0.400 ± 0.004 |
| 2d- | −0.06 ± 0.02 | 0.704 ± 0.005 |
| 3d- | −0.259 ± 0.017 | 0.619 ± 0.007 |
| 4d- | 0.071 ± 0.023 | 0.866 ± 0.019 |
| 5d- | −0.466 ± 0.016 | 0.294 ± 0.013 |
| Compound | Linker Length | Linker | N-Terminus | IC50 (Mean ± SD) 1 | |
|---|---|---|---|---|---|
| rNK1R | hNK1R | ||||
| (nM) | (nM) | ||||
| 1d | 0 | none | DOTA | n/d 2 | 7.2 ± 1.4 |
| Ga-1d | 0 | none | Ga-DOTA | 112 ± 3 | 5.1 ± 2.0 |
| 2d | 3 | -DAla- | DOTA | 1536 ± 231 | 21.4 ± 5.7 |
| Ga-2d | 3 | -DAla- | Ga-DOTA | 747 ± 348 | 38.7 ± 22.9 |
| 3d | 6 | -DAla-DAla- | DOTA | 1270 ± 415 | 27.1 ± 1.1 |
| Ga-3d | 6 | -DAla-DAla- | Ga-DOTA | 2393 ± 1114 | 14.6 ± 2.3 |
| 4d | 7 | -Ahx- | DOTA | n/d 2 | 8.7 ± 3.2 |
| Ga-4d | 7 | -Ahx- | Ga-DOTA | 1000 ± 71 | 15.5 ± 2.0 |
| 5d | 14 | -Ahx-Ahx- | DOTA | 830 ± 236 | 4.2 ± 0.8 |
| Ga-5d | 14 | -Ahx-Ahx- | Ga-DOTA | 411 ± 183 | 8.7 ± 2.4 |
| Radioconjugate | Kd ± SD 1 (nM) | Ratio to SP | BMAX ± SD 1 (nM) | Ratio to SP |
|---|---|---|---|---|
| [177Lu]Lu-DOTA-[Thi8,Met(O2)11]SP (reference) | 6.15 ± 0.64 | 1.00 | 0.929 ± 0.027 | 1.00 |
| [177Lu]Lu-1d | 6.98 ± 0.69 | 1.13 | 0.762 ± 0.046 | 0.82 |
| [177Lu]Lu-2d | 10.52 ± 0.77 | 1.71 | 2.497 ± 0.046 | 2.69 |
| [177Lu]Lu-3d | 2.002 ± 0.051 | 0.33 | 0.5939 ± 0.0063 | 0.64 |
| [177Lu]Lu-4d | 5.40 ± 0.30 | 0.88 | 2.363 ± 0.060 | 2.54 |
| [177Lu]Lu-5d | 8.70 ± 0.46 | 1.42 | 4.93 ± 0.26 | 5.31 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matalińska, J.; Kosińska, K.; Halik, P.K.; Koźmiński, P.; Lipiński, P.F.J.; Gniazdowska, E.; Misicka, A. Novel NK1R-Targeted 68Ga-/177Lu-Radioconjugates with Potential Application against Glioblastoma Multiforme: Preliminary Exploration of Structure–Activity Relationships. Int. J. Mol. Sci. 2022, 23, 1214. https://doi.org/10.3390/ijms23031214
Matalińska J, Kosińska K, Halik PK, Koźmiński P, Lipiński PFJ, Gniazdowska E, Misicka A. Novel NK1R-Targeted 68Ga-/177Lu-Radioconjugates with Potential Application against Glioblastoma Multiforme: Preliminary Exploration of Structure–Activity Relationships. International Journal of Molecular Sciences. 2022; 23(3):1214. https://doi.org/10.3390/ijms23031214
Chicago/Turabian StyleMatalińska, Joanna, Katarzyna Kosińska, Paweł K. Halik, Przemysław Koźmiński, Piotr F. J. Lipiński, Ewa Gniazdowska, and Aleksandra Misicka. 2022. "Novel NK1R-Targeted 68Ga-/177Lu-Radioconjugates with Potential Application against Glioblastoma Multiforme: Preliminary Exploration of Structure–Activity Relationships" International Journal of Molecular Sciences 23, no. 3: 1214. https://doi.org/10.3390/ijms23031214
APA StyleMatalińska, J., Kosińska, K., Halik, P. K., Koźmiński, P., Lipiński, P. F. J., Gniazdowska, E., & Misicka, A. (2022). Novel NK1R-Targeted 68Ga-/177Lu-Radioconjugates with Potential Application against Glioblastoma Multiforme: Preliminary Exploration of Structure–Activity Relationships. International Journal of Molecular Sciences, 23(3), 1214. https://doi.org/10.3390/ijms23031214