
An Overview of Epigenetics in Obesity: The Role of Lifestyle and Therapeutic Interventions


{kind=link}
Abstract
:1. Introduction
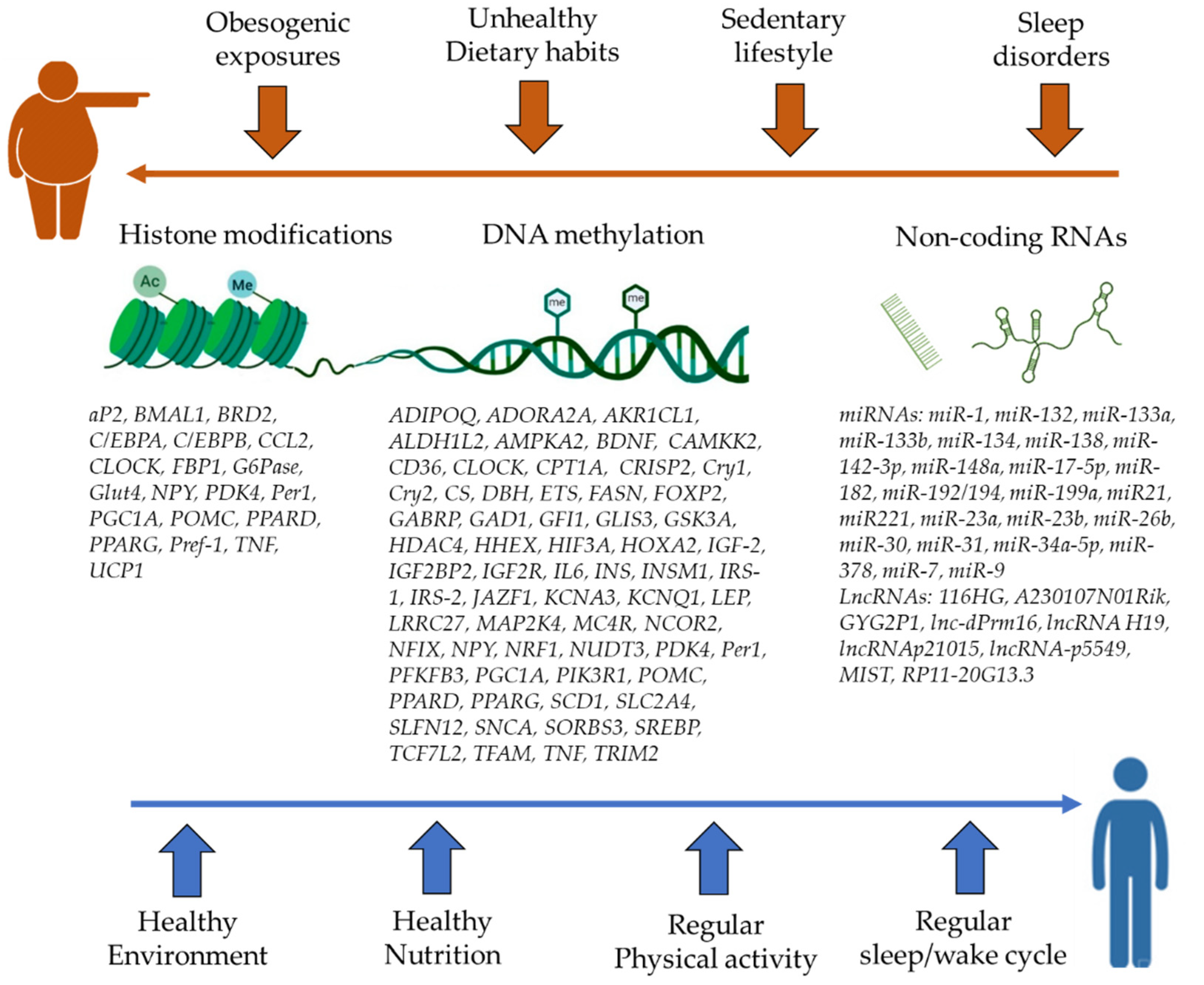
2. The Role of Epigenetics in Obesity
2.1. DNA Methylation
2.2. Histone Modifications
2.3. Non-Coding RNAs
3. Epigenetic Effects of Environmental Factors and Lifestyle
3.1. Obesogenic Exposures
3.2. Dietary Factors
3.3. Physical Activity
3.4. Sleep Deprivation
3.5. Alcohol Intake
3.6. Weight Loss Interventions
3.7. Epigenetic Drugs
4. Conclusions and Future Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Peeters, A.; Barendregt, J.J.; Willekens, F.; Mackenbach, J.P.; Al Mamun, A.; Bonneux, L. The Netherlands Epidemiology and Demography Compression of Morbidity Research Group. Obesity in adulthood and its consequences for life expectancy: A life-table analysis. Ann. Intern. Med. 2003, 138, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Pantalone, K.M.; Hobbs, T.M.; Chagin, K.M.; Kong, S.X.; Wells, B.J.; Kattan, M.W.; Bouchard, J.; Sakurada, B.; Milinovich, A.; Weng, W.; et al. Prevalence and recognition of obesity and its associated comorbidities: Cross-sectional analysis of electronic health record data from a large US integrated health system. BMJ Open 2017, 7, e017583. [Google Scholar] [CrossRef] [PubMed]
- Gammone, M.A.; D’Orazio, N. COVID-19 and Obesity: Overlapping of Two Pandemics. Obes. Facts 2021, 14, 579–585. [Google Scholar] [CrossRef] [PubMed]
- Yengo, L.; Sidorenko, J.; Kemper, K.E.; Zheng, Z.; Wood, A.R.; Weedon, M.N.; Frayling, T.M.; Hirschhorn, J.; Yang, J.; Visscher, P.M.; et al. Meta-analysis of genome-wide association studies for height and body mass index in approximately 700,000 individuals of European ancestry. Hum. Mol. Genet. 2018, 27, 3641–3649. [Google Scholar] [CrossRef]
- Meldrum, D.R.; Morris, M.A.; Gambone, J.C. Obesity pandemic: Causes, consequences, and solutions-but do we have the will? Fertil. Steril. 2017, 107, 833–839. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.J.; Han, S.M.; Huh, J.Y.; Kim, J.B. Emerging roles of epigenetic regulation in obesity and metabolic disease. J. Biol. Chem. 2021, 297, 101296. [Google Scholar] [CrossRef]
- Weinhold, B. Epigenetics: The science of change. Environ. Health Perspect. 2006, 114, A160–A167. [Google Scholar] [CrossRef] [Green Version]
- Dolinoy, D.C. The agouti mouse model: An epigenetic biosensor for nutritional and environmental alterations on the fetal epigenome. Nutr. Rev. 2008, 66 (Suppl. 1), S7–S11. [Google Scholar] [CrossRef] [Green Version]
- Turcot, V.; Tchernof, A.; Deshaies, Y.; Perusse, L.; Belisle, A.; Marceau, S.; Biron, S.; Lescelleur, O.; Biertho, L.; Vohl, M.C. LINE-1 methylation in visceral adipose tissue of severely obese individuals is associated with metabolic syndrome status and related phenotypes. Clin. Epigenet. 2012, 4, 10. [Google Scholar] [CrossRef] [Green Version]
- Petrus, P.; Bialesova, L.; Checa, A.; Kerr, A.; Naz, S.; Backdahl, J.; Gracia, A.; Toft, S.; Dahlman-Wright, K.; Heden, P.; et al. Adipocyte Expression of SLC19A1 Links DNA Hypermethylation to Adipose Tissue Inflammation and Insulin Resistance. J. Clin. Endocrinol. Metab. 2018, 103, 710–721. [Google Scholar] [CrossRef]
- Sadashiv; Modi, A.; Khokhar, M.; Sharma, P.; Joshi, R.; Mishra, S.S.; Bharshankar, R.N.; Tiwari, S.; Singh, P.K.; Bhosale, V.V.; et al. Leptin DNA Methylation and Its Association with Metabolic Risk Factors in a Northwest Indian Obese Population. J. Obes. Metab. Syndr. 2021, 30, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Houde, A.A.; Legare, C.; Biron, S.; Lescelleur, O.; Biertho, L.; Marceau, S.; Tchernof, A.; Vohl, M.C.; Hivert, M.F.; Bouchard, L. Leptin and adiponectin DNA methylation levels in adipose tissues and blood cells are associated with BMI, waist girth and LDL-cholesterol levels in severely obese men and women. BMC Med. Genet. 2015, 16, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houshmand-Oeregaard, A.; Hansen, N.S.; Hjort, L.; Kelstrup, L.; Broholm, C.; Mathiesen, E.R.; Clausen, T.D.; Damm, P.; Vaag, A. Differential adipokine DNA methylation and gene expression in subcutaneous adipose tissue from adult offspring of women with diabetes in pregnancy. Clin. Epigenet. 2017, 9, 37. [Google Scholar] [CrossRef] [PubMed]
- Ott, R.; Stupin, J.H.; Melchior, K.; Schellong, K.; Ziska, T.; Dudenhausen, J.W.; Henrich, W.; Rancourt, R.C.; Plagemann, A. Alterations of adiponectin gene expression and DNA methylation in adipose tissues and blood cells are associated with gestational diabetes and neonatal outcome. Clin. Epigenet. 2018, 10, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, A.Y.; Park, Y.J.; Pan, X.; Shin, K.C.; Kwak, S.H.; Bassas, A.F.; Sallam, R.M.; Park, K.S.; Alfadda, A.A.; Xu, A.; et al. Obesity-induced DNA hypermethylation of the adiponectin gene mediates insulin resistance. Nat. Commun. 2015, 6, 7585. [Google Scholar] [CrossRef] [Green Version]
- Kuroda, A.; Rauch, T.A.; Todorov, I.; Ku, H.T.; Al-Abdullah, I.H.; Kandeel, F.; Mullen, Y.; Pfeifer, G.P.; Ferreri, K. Insulin gene expression is regulated by DNA methylation. PLoS ONE 2009, 4, e6953. [Google Scholar] [CrossRef]
- Nilsson, E.; Jansson, P.A.; Perfilyev, A.; Volkov, P.; Pedersen, M.; Svensson, M.K.; Poulsen, P.; Ribel-Madsen, R.; Pedersen, N.L.; Almgren, P.; et al. Altered DNA methylation and differential expression of genes influencing metabolism and inflammation in adipose tissue from subjects with type 2 diabetes. Diabetes 2014, 63, 2962–2976. [Google Scholar] [CrossRef] [Green Version]
- Pinhel, M.A.S.; Noronha, N.Y.; Nicoletti, C.F.; Pereira, V.A.; de Oliveira, B.A.; Cortes-Oliveira, C.; Salgado, W., Jr.; Barbosa, F., Jr.; Marchini, J.S.; Souza, D.R.; et al. Changes in DNA Methylation and Gene Expression of Insulin and Obesity-Related Gene PIK3R1 after Roux-en-Y Gastric Bypass. Int. J. Mol. Sci. 2020, 21, 4476. [Google Scholar] [CrossRef]
- Rohde, K.; Klos, M.; Hopp, L.; Liu, X.; Keller, M.; Stumvoll, M.; Dietrich, A.; Schon, M.R.; Gartner, D.; Lohmann, T.; et al. IRS1 DNA promoter methylation and expression in human adipose tissue are related to fat distribution and metabolic traits. Sci. Rep. 2017, 7, 12369. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, S.A.H.; Ansari, S.A.; Mensah-Brown, E.P.K.; Emerald, B.S. The role of DNA methylation in the pathogenesis of type 2 diabetes mellitus. Clin. Epigenet. 2020, 12, 104. [Google Scholar] [CrossRef]
- Gemma, C.; Sookoian, S.; Alvarinas, J.; Garcia, S.I.; Quintana, L.; Kanevsky, D.; Gonzalez, C.D.; Pirola, C.J. Maternal pregestational BMI is associated with methylation of the PPARGC1A promoter in newborns. Obesity 2009, 17, 1032–1039. [Google Scholar] [CrossRef] [PubMed]
- Brons, C.; Jacobsen, S.; Nilsson, E.; Ronn, T.; Jensen, C.B.; Storgaard, H.; Poulsen, P.; Groop, L.; Ling, C.; Astrup, A.; et al. Deoxyribonucleic acid methylation and gene expression of PPARGC1A in human muscle is influenced by high-fat overfeeding in a birth-weight-dependent manner. J. Clin. Endocrinol. Metab. 2010, 95, 3048–3056. [Google Scholar] [CrossRef] [PubMed]
- Perkins, E.; Murphy, S.K.; Murtha, A.P.; Schildkraut, J.; Jirtle, R.L.; Demark-Wahnefried, W.; Forman, M.R.; Kurtzberg, J.; Overcash, F.; Huang, Z.; et al. Insulin-like growth factor 2/H19 methylation at birth and risk of overweight and obesity in children. J. Pediatr. 2012, 161, 31–39. [Google Scholar] [CrossRef] [Green Version]
- Crujeiras, A.B.; Campion, J.; Diaz-Lagares, A.; Milagro, F.I.; Goyenechea, E.; Abete, I.; Casanueva, F.F.; Martinez, J.A. Association of weight regain with specific methylation levels in the NPY and POMC promoters in leukocytes of obese men: A translational study. Regul. Pept. 2013, 186, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Dick, K.J.; Nelson, C.P.; Tsaprouni, L.; Sandling, J.K.; Aissi, D.; Wahl, S.; Meduri, E.; Morange, P.E.; Gagnon, F.; Grallert, H.; et al. DNA methylation and body-mass index: A genome-wide analysis. Lancet 2014, 383, 1990–1998. [Google Scholar] [CrossRef] [Green Version]
- Pan, H.; Lin, X.; Wu, Y.; Chen, L.; Teh, A.L.; Soh, S.E.; Lee, Y.S.; Tint, M.T.; MacIsaac, J.L.; Morin, A.M.; et al. HIF3A association with adiposity: The story begins before birth. Epigenomics 2015, 7, 937–950. [Google Scholar] [CrossRef] [Green Version]
- Na, Y.K.; Hong, H.S.; Lee, W.K.; Kim, Y.H.; Kim, D.S. Increased methylation of interleukin 6 gene is associated with obesity in Korean women. Mol. Cells 2015, 38, 452–456. [Google Scholar] [CrossRef] [Green Version]
- Ali, M.M.; Naquiallah, D.; Qureshi, M.; Mirza, M.I.; Hassan, C.; Masrur, M.; Bianco, F.M.; Frederick, P.; Cristoforo, G.P.; Gangemi, A.; et al. DNA methylation profile of genes involved in inflammation and autoimmunity correlates with vascular function in morbidly obese adults. Epigenetics 2021, 25, 1–17. [Google Scholar] [CrossRef]
- Ali, M.M.; Hassan, C.; Masrur, M.; Bianco, F.M.; Naquiallah, D.; Mirza, I.; Frederick, P.; Fernandes, E.T.; Giulianotti, C.P.; Gangemi, A.; et al. Adipose Tissue Hypoxia Correlates with Adipokine Hypomethylation and Vascular Dysfunction. Biomedicines 2021, 9, 1034. [Google Scholar] [CrossRef]
- You, D.; Nilsson, E.; Tenen, D.E.; Lyubetskaya, A.; Lo, J.C.; Jiang, R.; Deng, J.; Dawes, B.A.; Vaag, A.; Ling, C.; et al. Dnmt3a is an epigenetic mediator of adipose insulin resistance. Elife 2017, 6, e30766. [Google Scholar] [CrossRef]
- Ma, X.; Kang, S. Functional Implications of DNA Methylation in Adipose Biology. Diabetes 2019, 68, 871–878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castillo, J.; Lopez-Rodas, G.; Franco, L. Histone Post-Translational Modifications and Nucleosome Organisation in Transcriptional Regulation: Some Open Questions. Adv. Exp. Med. Biol. 2017, 966, 65–92. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Yang, K.; Gu, H.; Sun, C. Dynamic post-translational modifications in obesity. J. Cell Mol. Med. 2020, 24, 2384–2387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funato, H.; Oda, S.; Yokofujita, J.; Igarashi, H.; Kuroda, M. Fasting and high-fat diet alter histone deacetylase expression in the medial hypothalamus. PLoS ONE 2011, 6, e18950. [Google Scholar] [CrossRef] [PubMed]
- Tateishi, K.; Okada, Y.; Kallin, E.M.; Zhang, Y. Role of Jhdm2a in regulating metabolic gene expression and obesity resistance. Nature 2009, 458, 757–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Ramlee, M.K.; Brunmeir, R.; Villanueva, C.J.; Halperin, D.; Xu, F. Dynamic and distinct histone modifications modulate the expression of key adipogenesis regulatory genes. Cell Cycle 2012, 11, 4310–4322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikula, M.; Majewska, A.; Ledwon, J.K.; Dzwonek, A.; Ostrowski, J. Obesity increases histone H3 lysine 9 and 18 acetylation at Tnfa and Ccl2 genes in mouse liver. Int. J. Mol. Med. 2014, 34, 1647–1654. [Google Scholar] [CrossRef] [Green Version]
- Wheatley, K.E.; Nogueira, L.M.; Perkins, S.N.; Hursting, S.D. Differential effects of calorie restriction and exercise on the adipose transcriptome in diet-induced obese mice. J. Obes. 2011, 2011, 265417. [Google Scholar] [CrossRef] [Green Version]
- Landrier, J.F.; Derghal, A.; Mounien, L. MicroRNAs in Obesity and Related Metabolic Disorders. Cells 2019, 8, 859. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Ji, Y.; Li, M.; Wang, M.; Yi, X.; Yin, C.; Wang, S.; Zhang, M.; Zhao, Z.; Xiao, Y. Integrated analysis of long noncoding RNA and mRNA expression profile in children with obesity by microarray analysis. Sci. Rep. 2018, 8, 8750. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Ruan, Y.; Wang, M.; Chen, R.; Yu, N.; Sun, L.; Liu, T.; Chen, H. Differentially expressed circulating LncRNAs and mRNA identified by microarray analysis in obese patients. Sci. Rep. 2016, 6, 35421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, C.; Lim, Y.C.; Chia, S.Y.; Walet, A.C.E.; Xu, S.; Lo, K.A.; Zhao, Y.; Zhu, D.; Shan, Z.; Chen, Q.; et al. De novo reconstruction of human adipose transcriptome reveals conserved lncRNAs as regulators of brown adipogenesis. Nat. Commun. 2018, 9, 1329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stapleton, K.; Das, S.; Reddy, M.A.; Leung, A.; Amaram, V.; Lanting, L.; Chen, Z.; Zhang, L.; Palanivel, R.; Deiuliis, J.A.; et al. Novel Long Noncoding RNA, Macrophage Inflammation-Suppressing Transcript (MIST), Regulates Macrophage Activation During Obesity. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 914–928. [Google Scholar] [CrossRef] [PubMed]
- Squillaro, T.; Peluso, G.; Galderisi, U.; Di Bernardo, G. Long non-coding RNAs in regulation of adipogenesis and adipose tissue function. Elife 2020, 9, e59053. [Google Scholar] [CrossRef]
- Baillie-Hamilton, P.F. Chemical toxins: A hypothesis to explain the global obesity epidemic. J. Altern. Complement. Med. 2002, 8, 185–192. [Google Scholar] [CrossRef]
- Grun, F.; Blumberg, B. Environmental obesogens: Organotins and endocrine disruption via nuclear receptor signaling. Endocrinology 2006, 147, S50–S55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savastano, S.; Tarantino, G.; D’Esposito, V.; Passaretti, F.; Cabaro, S.; Liotti, A.; Liguoro, D.; Perruolo, G.; Ariemma, F.; Finelli, C.; et al. Bisphenol-A plasma levels are related to inflammatory markers, visceral obesity and insulin-resistance: A cross-sectional study on adult male population. J. Transl. Med. 2015, 13, 169. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.J.; Lee, Y.A.; Hong, Y.C.; Cho, J.; Lee, K.S.; Shin, C.H.; Kim, B.N.; Kim, J.I.; Park, S.J.; Bisgaard, H.; et al. Effect of prenatal bisphenol A exposure on early childhood body mass index through epigenetic influence on the insulin-like growth factor 2 receptor (IGF2R) gene. Environ. Int. 2020, 143, 105929. [Google Scholar] [CrossRef]
- Bastos Sales, L.; Kamstra, J.H.; Cenijn, P.H.; van Rijt, L.S.; Hamers, T.; Legler, J. Effects of endocrine disrupting chemicals on in vitro global DNA methylation and adipocyte differentiation. Toxicol. Vitr. 2013, 27, 1634–1643. [Google Scholar] [CrossRef]
- Chevalier, N.; Fenichel, P. Endocrine disruptors: New players in the pathophysiology of type 2 diabetes? Diabetes Metab. 2015, 41, 107–115. [Google Scholar] [CrossRef]
- Strakovsky, R.S.; Wang, H.; Engeseth, N.J.; Flaws, J.A.; Helferich, W.G.; Pan, Y.X.; Lezmi, S. Developmental bisphenol A (BPA) exposure leads to sex-specific modification of hepatic gene expression and epigenome at birth that may exacerbate high-fat diet-induced hepatic steatosis. Toxicol. Appl. Pharmacol. 2015, 284, 101–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutta, S.; Haggerty, D.K.; Rappolee, D.A.; Ruden, D.M. Phthalate Exposure and Long-Term Epigenomic Consequences: A Review. Front. Genet. 2020, 11, 405. [Google Scholar] [CrossRef]
- Meruvu, S.; Zhang, J.; Choudhury, M. Butyl Benzyl Phthalate Promotes Adipogenesis in 3T3-L1 Cells via the miRNA-34a-5p Signaling Pathway in the Absence of Exogenous Adipogenic Stimuli. Chem. Res. Toxicol. 2021, 34, 2251–2260. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Choudhury, M. Benzyl Butyl Phthalate Induced Early lncRNA H19 Regulation in C3H10T1/2 Stem Cell Line. Chem. Res. Toxicol. 2021, 34, 54–62. [Google Scholar] [CrossRef]
- Janesick, A.; Blumberg, B. Minireview: PPARgamma as the target of obesogens. J. Steroid Biochem. Mol. Biol. 2011, 127, 4–8. [Google Scholar] [CrossRef] [Green Version]
- Slotkin, T.A. Does early-life exposure to organophosphate insecticides lead to prediabetes and obesity? Reprod. Toxicol. 2011, 31, 297–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maugeri, A.; Barchitta, M. How Dietary Factors Affect DNA Methylation: Lesson from Epidemiological Studies. Medicina 2020, 56, 374. [Google Scholar] [CrossRef]
- Piyathilake, C.J.; Badiga, S.; Kabagambe, E.K.; Azuero, A.; Alvarez, R.D.; Johanning, G.L.; Partridge, E.E. A dietary pattern associated with LINE-1 methylation alters the risk of developing cervical intraepithelial neoplasia. Cancer Prev. Res. 2012, 5, 385–392. [Google Scholar] [CrossRef] [Green Version]
- Jacobsen, S.C.; Brons, C.; Bork-Jensen, J.; Ribel-Madsen, R.; Yang, B.; Lara, E.; Hall, E.; Calvanese, V.; Nilsson, E.; Jorgensen, S.W.; et al. Effects of short-term high-fat overfeeding on genome-wide DNA methylation in the skeletal muscle of healthy young men. Diabetologia 2012, 55, 3341–3349. [Google Scholar] [CrossRef] [Green Version]
- Widiker, S.; Karst, S.; Wagener, A.; Brockmann, G.A. High-fat diet leads to a decreased methylation of the Mc4r gene in the obese BFMI and the lean B6 mouse lines. J. Appl. Genet. 2010, 51, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, T.K.; Basford, J.E.; Yiew, K.H.; Stepp, D.W.; Hui, D.Y.; Weintraub, N.L. Role of histone deacetylase 9 in regulating adipogenic differentiation and high fat diet-induced metabolic disease. Adipocyte 2014, 3, 333–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagchi, R.A.; Ferguson, B.S.; Stratton, M.S.; Hu, T.; Cavasin, M.A.; Sun, L.; Lin, Y.H.; Liu, D.; Londono, P.; Song, K.; et al. HDAC11 suppresses the thermogenic program of adipose tissue via BRD2. JCI Insight 2018, 3, e120159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couvreur, O.; Ferezou, J.; Gripois, D.; Serougne, C.; Crepin, D.; Aubourg, A.; Gertler, A.; Vacher, C.M.; Taouis, M. Unexpected long-term protection of adult offspring born to high-fat fed dams against obesity induced by a sucrose-rich diet. PLoS ONE 2011, 6, e18043. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Xiao, X.; Zheng, J.; Li, M.; Yu, M.; Ping, F.; Wang, T.; Wang, X. A Maternal High-Fat Diet Induces DNA Methylation Changes That Contribute to Glucose Intolerance in Offspring. Front. Endocrinol. 2019, 10, 871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.F.; Morabia, A.; Carroll, J.; Gonzalez, K.; Fulda, K.; Kaur, M.; Vishwanatha, J.K.; Santella, R.M.; Cardarelli, R. Dietary patterns are associated with levels of global genomic DNA methylation in a cancer-free population. J. Nutr. 2011, 141, 1165–1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Dam, R.M.; Seidell, J.C. Carbohydrate intake and obesity. Eur. J. Clin. Nutr. 2007, 61 (Suppl. 1), S75–S99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, C.Q.; Parnell, L.D.; Smith, C.E.; Guo, T.; Sayols-Baixeras, S.; Aslibekyan, S.; Tiwari, H.K.; Irvin, M.R.; Bender, C.; Fei, D.; et al. Carbohydrate and fat intake associated with risk of metabolic diseases through epigenetics of CPT1A. Am. J. Clin. Nutr. 2020, 112, 1200–1211. [Google Scholar] [CrossRef]
- Moody, L.; Xu, G.B.; Chen, H.; Pan, Y.X. Epigenetic regulation of carnitine palmitoyltransferase 1 (Cpt1a) by high fat diet. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862, 141–152. [Google Scholar] [CrossRef]
- Ohashi, K.; Munetsuna, E.; Yamada, H.; Ando, Y.; Yamazaki, M.; Taromaru, N.; Nagura, A.; Ishikawa, H.; Suzuki, K.; Teradaira, R.; et al. High fructose consumption induces DNA methylation at PPARalpha and CPT1A promoter regions in the rat liver. Biochem. Biophys. Res. Commun. 2015, 468, 185–189. [Google Scholar] [CrossRef]
- Ramos-Lopez, O.; Riezu-Boj, J.I.; Milagro, F.I.; Martinez, J.A.; Project, M. Dopamine gene methylation patterns are associated with obesity markers and carbohydrate intake. Brain Behav. 2018, 8, e01017. [Google Scholar] [CrossRef] [Green Version]
- Samblas, M.; Milagro, F.I.; Gomez-Abellan, P.; Martinez, J.A.; Garaulet, M. Methylation on the Circadian Gene BMAL1 Is Associated with the Effects of a Weight Loss Intervention on Serum Lipid Levels. J. Biol. Rhythms. 2016, 31, 308–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos-Lopez, O.; Samblas, M.; Milagro, F.I.; Riezu-Boj, J.I.; Crujeiras, A.B.; Martinez, J.A.; Project, M. Circadian gene methylation profiles are associated with obesity, metabolic disturbances and carbohydrate intake. Chronobiol. Int. 2018, 35, 969–981. [Google Scholar] [CrossRef] [PubMed]
- Martin-Gronert, M.S.; Ozanne, S.E. Mechanisms linking suboptimal early nutrition and increased risk of type 2 diabetes and obesity. J. Nutr. 2010, 140, 662–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heijmans, B.T.; Tobi, E.W.; Stein, A.D.; Putter, H.; Blauw, G.J.; Susser, E.S.; Slagboom, P.E.; Lumey, L.H. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc. Natl. Acad. Sci. USA 2008, 105, 17046–17049. [Google Scholar] [CrossRef] [Green Version]
- Zheng, S.; Rollet, M.; Pan, Y.X. Maternal protein restriction during pregnancy induces CCAAT/enhancer-binding protein (C/EBPbeta) expression through the regulation of histone modification at its promoter region in female offspring rat skeletal muscle. Epigenetics 2011, 6, 161–170. [Google Scholar] [CrossRef] [Green Version]
- Sohi, G.; Marchand, K.; Revesz, A.; Arany, E.; Hardy, D.B. Maternal protein restriction elevates cholesterol in adult rat offspring due to repressive changes in histone modifications at the cholesterol 7alpha-hydroxylase promoter. Mol. Endocrinol. 2011, 25, 785–798. [Google Scholar] [CrossRef] [Green Version]
- Calder, P.C.; Ahluwalia, N.; Brouns, F.; Buetler, T.; Clement, K.; Cunningham, K.; Esposito, K.; Jonsson, L.S.; Kolb, H.; Lansink, M.; et al. Dietary factors and low-grade inflammation in relation to overweight and obesity. Br. J. Nutr. 2011, 106 (Suppl. 3), S5–S78. [Google Scholar] [CrossRef]
- Zeisel, S.H. Epigenetic mechanisms for nutrition determinants of later health outcomes. Am. J. Clin. Nutr. 2009, 89, 1488S–1493S. [Google Scholar] [CrossRef]
- Ramos-Lopez, O.; Samblas, M.; Milagro, F.I.; Zulet, M.A.; Mansego, M.L.; Riezu-Boj, J.I.; Martinez, J.A. Association of low dietary folate intake with lower CAMKK2 gene methylation, adiposity, and insulin resistance in obese subjects. Nutr. Res. 2018, 50, 53–62. [Google Scholar] [CrossRef]
- Ono, H.; Iwasaki, M.; Kuchiba, A.; Kasuga, Y.; Yokoyama, S.; Onuma, H.; Nishimura, H.; Kusama, R.; Ohnami, S.; Sakamoto, H.; et al. Association of dietary and genetic factors related to one-carbon metabolism with global methylation level of leukocyte DNA. Cancer Sci. 2012, 103, 2159–2164. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.F.; Cardarelli, R.; Carroll, J.; Fulda, K.G.; Kaur, M.; Gonzalez, K.; Vishwanatha, J.K.; Santella, R.M.; Morabia, A. Significant differences in global genomic DNA methylation by gender and race/ethnicity in peripheral blood. Epigenetics 2011, 6, 623–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milagro, F.I.; Mansego, M.L.; De Miguel, C.; Martinez, J.A. Dietary factors, epigenetic modifications and obesity outcomes: Progresses and perspectives. Mol. Aspects Med. 2013, 34, 782–812. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.K.; Cheung, C.; Reuhl, K.R.; Liu, A.B.; Lee, M.J.; Lu, Y.P.; Yang, C.S. Effects of green tea polyphenol (-)-epigallocatechin-3-gallate on newly developed high-fat/Western-style diet-induced obesity and metabolic syndrome in mice. J. Agric. Food Chem. 2011, 59, 11862–11871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, W.; Yu, Z.; Chiang, Y.; Yang, Y.; Chai, T.; Foltz, W.; Lu, H.; Fantus, I.G.; Jin, T. Curcumin prevents high fat diet induced insulin resistance and obesity via attenuating lipogenesis in liver and inflammatory pathway in adipocytes. PLoS ONE 2012, 7, e28784. [Google Scholar] [CrossRef]
- Camins, A.; Sureda, F.X.; Junyent, F.; Verdaguer, E.; Folch, J.; Pelegri, C.; Vilaplana, J.; Beas-Zarate, C.; Pallas, M. Sirtuin activators: Designing molecules to extend life span. Biochim. Biophys. Acta 2010, 1799, 740–749. [Google Scholar] [CrossRef]
- Choi, S.W.; Friso, S. Epigenetics: A New Bridge between Nutrition and Health. Adv. Nutr. 2010, 1, 8–16. [Google Scholar] [CrossRef]
- Bajpeyi, S.; Covington, J.D.; Taylor, E.M.; Stewart, L.K.; Galgani, J.E.; Henagan, T.M. Skeletal Muscle PGC1alpha -1 Nucleosome Position and -260 nt DNA Methylation Determine Exercise Response and Prevent Ectopic Lipid Accumulation in Men. Endocrinology 2017, 158, 2190–2199. [Google Scholar] [CrossRef]
- Ronn, T.; Volkov, P.; Davegardh, C.; Dayeh, T.; Hall, E.; Olsson, A.H.; Nilsson, E.; Tornberg, A.; Dekker Nitert, M.; Eriksson, K.F.; et al. A six months exercise intervention influences the genome-wide DNA methylation pattern in human adipose tissue. PLoS Genet. 2013, 9, e1003572. [Google Scholar] [CrossRef]
- Rowlands, D.S.; Page, R.A.; Sukala, W.R.; Giri, M.; Ghimbovschi, S.D.; Hayat, I.; Cheema, B.S.; Lys, I.; Leikis, M.; Sheard, P.W.; et al. Multi-omic integrated networks connect DNA methylation and miRNA with skeletal muscle plasticity to chronic exercise in Type 2 diabetic obesity. Physiol. Genomics 2014, 46, 747–765. [Google Scholar] [CrossRef]
- Nielsen, S.; Scheele, C.; Yfanti, C.; Akerstrom, T.; Nielsen, A.R.; Pedersen, B.K.; Laye, M.J. Muscle specific microRNAs are regulated by endurance exercise in human skeletal muscle. J. Physiol. 2010, 588, 4029–4037. [Google Scholar] [CrossRef]
- McLean, C.S.; Mielke, C.; Cordova, J.M.; Langlais, P.R.; Bowen, B.; Miranda, D.; Coletta, D.K.; Mandarino, L.J. Gene and MicroRNA Expression Responses to Exercise; Relationship with Insulin Sensitivity. PLoS ONE 2015, 10, e0127089. [Google Scholar] [CrossRef] [PubMed]
- Barres, R.; Yan, J.; Egan, B.; Treebak, J.T.; Rasmussen, M.; Fritz, T.; Caidahl, K.; Krook, A.; O’Gorman, D.J.; Zierath, J.R. Acute exercise remodels promoter methylation in human skeletal muscle. Cell Metab. 2012, 15, 405–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King-Himmelreich, T.S.; Schramm, S.; Wolters, M.C.; Schmetzer, J.; Moser, C.V.; Knothe, C.; Resch, E.; Peil, J.; Geisslinger, G.; Niederberger, E. The impact of endurance exercise on global and AMPK gene-specific DNA methylation. Biochem. Biophys. Res. Commun. 2016, 474, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Russell, A.P.; Lamon, S.; Boon, H.; Wada, S.; Guller, I.; Brown, E.L.; Chibalin, A.V.; Zierath, J.R.; Snow, R.J.; Stepto, N.; et al. Regulation of miRNAs in human skeletal muscle following acute endurance exercise and short-term endurance training. J. Physiol. 2013, 591, 4637–4653. [Google Scholar] [CrossRef]
- Nitert, M.D.; Dayeh, T.; Volkov, P.; Elgzyri, T.; Hall, E.; Nilsson, E.; Yang, B.T.; Lang, S.; Parikh, H.; Wessman, Y.; et al. Impact of an exercise intervention on DNA methylation in skeletal muscle from first-degree relatives of patients with type 2 diabetes. Diabetes 2012, 61, 3322–3332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denham, J.; Marques, F.Z.; Bruns, E.L.; O’Brien, B.J.; Charchar, F.J. Epigenetic changes in leukocytes after 8 weeks of resistance exercise training. Eur. J. Appl. Physiol. 2016, 116, 1245–1253. [Google Scholar] [CrossRef] [PubMed]
- Barron-Cabrera, E.; Ramos-Lopez, O.; Gonzalez-Becerra, K.; Riezu-Boj, J.I.; Milagro, F.I.; Martinez-Lopez, E.; Martinez, J.A. Epigenetic Modifications as Outcomes of Exercise Interventions Related to Specific Metabolic Alterations: A Systematic Review. Lifestyle Genom. 2019, 12, 25–44. [Google Scholar] [CrossRef]
- Calhoun, D.A.; Harding, S.M. Sleep and hypertension. Chest 2010, 138, 434–443. [Google Scholar] [CrossRef]
- Al-Abri, M.A.; Jaju, D.; Al-Sinani, S.; Al-Mamari, A.; Albarwani, S.; Al-Resadi, K.; Bayoumi, R.; Hassan, M.; Al-Hashmi, K. Habitual Sleep Deprivation is Associated with Type 2 Diabetes: A Case-Control Study. Oman Med. J. 2016, 31, 399–403. [Google Scholar] [CrossRef]
- Cooper, C.B.; Neufeld, E.V.; Dolezal, B.A.; Martin, J.L. Sleep deprivation and obesity in adults: A brief narrative review. BMJ Open Sport Exerc. Med. 2018, 4, e000392. [Google Scholar] [CrossRef] [Green Version]
- Gaine, M.E.; Chatterjee, S.; Abel, T. Sleep Deprivation and the Epigenome. Front. Neural Circuits 2018, 12, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortese, R. Epigenetics of Sleep Disorders: An Emerging Field in Diagnosis and Therapeutics. Diagnostics 2021, 11, 851. [Google Scholar] [CrossRef] [PubMed]
- Cedernaes, J.; Osler, M.E.; Voisin, S.; Broman, J.E.; Vogel, H.; Dickson, S.L.; Zierath, J.R.; Schioth, H.B.; Benedict, C. Acute Sleep Loss Induces Tissue-Specific Epigenetic and Transcriptional Alterations to Circadian Clock Genes in Men. J. Clin. Endocrinol. Metab. 2015, 100, E1255–E1261. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Stevens, R.G.; Hoffman, A.E.; Tjonneland, A.; Vogel, U.B.; Zheng, T.; Hansen, J. Epigenetic impact of long-term shiftwork: Pilot evidence from circadian genes and whole-genome methylation analysis. Chronobiol. Int. 2011, 28, 852–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skuladottir, G.V.; Nilsson, E.K.; Mwinyi, J.; Schioth, H.B. One-night sleep deprivation induces changes in the DNA methylation and serum activity indices of stearoyl-CoA desaturase in young healthy men. Lipids Health Dis. 2016, 15, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, C.C.; Parsons, M.J.; Lester, K.J.; Burrage, J.; Eley, T.C.; Mill, J.; Dempster, E.L.; Gregory, A.M. Epigenome-Wide DNA Methylation Analysis of Monozygotic Twins Discordant for Diurnal Preference. Twin Res. Hum. Genet. 2015, 18, 662–669. [Google Scholar] [CrossRef] [Green Version]
- Massart, R.; Freyburger, M.; Suderman, M.; Paquet, J.; El Helou, J.; Belanger-Nelson, E.; Rachalski, A.; Koumar, O.C.; Carrier, J.; Szyf, M.; et al. The genome-wide landscape of DNA methylation and hydroxymethylation in response to sleep deprivation impacts on synaptic plasticity genes. Transl. Psychiatry 2014, 4, e347. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Zhu, Y.; Eliot, M.N.; Knopik, V.S.; McGeary, J.E.; Carskadon, M.A.; Hart, A.C. Combining Human Epigenetics and Sleep Studies in Caenorhabditis elegans: A Cross-Species Approach for Finding Conserved Genes Regulating Sleep. Sleep 2017, 40, zsx063. [Google Scholar] [CrossRef]
- Cedernaes, J.; Schonke, M.; Westholm, J.O.; Mi, J.; Chibalin, A.; Voisin, S.; Osler, M.; Vogel, H.; Hornaeus, K.; Dickson, S.L.; et al. Acute sleep loss results in tissue-specific alterations in genome-wide DNA methylation state and metabolic fuel utilization in humans. Sci. Adv. 2018, 4, eaar8590. [Google Scholar] [CrossRef] [Green Version]
- Duan, R.; Liu, X.; Wang, T.; Wu, L.; Gao, X.; Zhang, Z. Histone Acetylation Regulation in Sleep Deprivation-Induced Spatial Memory Impairment. Neurochem. Res. 2016, 41, 2223–2232. [Google Scholar] [CrossRef]
- Naruse, Y.; Oh-hashi, K.; Iijima, N.; Naruse, M.; Yoshioka, H.; Tanaka, M. Circadian and light-induced transcription of clock gene Per1 depends on histone acetylation and deacetylation. Mol. Cell Biol. 2004, 24, 6278–6287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, C.J.; Taishi, P.; Honn, K.A.; Koberstein, J.N.; Krueger, J.M. P2X7 receptors in body temperature, locomotor activity, and brain mRNA and lncRNA responses to sleep deprivation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 311, R1004–R1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shende, V.R.; Neuendorff, N.; Earnest, D.J. Role of miR-142-3p in the post-transcriptional regulation of the clock gene Bmal1 in the mouse SCN. PLoS ONE 2013, 8, e65300. [Google Scholar] [CrossRef] [PubMed]
- Nagel, R.; Clijsters, L.; Agami, R. The miRNA-192/194 cluster regulates the Period gene family and the circadian clock. FEBS J. 2009, 276, 5447–5455. [Google Scholar] [CrossRef] [PubMed]
- Saus, E.; Soria, V.; Escaramis, G.; Vivarelli, F.; Crespo, J.M.; Kagerbauer, B.; Menchon, J.M.; Urretavizcaya, M.; Gratacos, M.; Estivill, X. Genetic variants and abnormal processing of pre-miR-182, a circadian clock modulator, in major depression patients with late insomnia. Hum. Mol. Genet. 2010, 19, 4017–4025. [Google Scholar] [CrossRef] [Green Version]
- Traversy, G.; Chaput, J.P. Alcohol Consumption and Obesity: An Update. Curr. Obes. Rep. 2015, 4, 122–130. [Google Scholar] [CrossRef] [Green Version]
- Halsted, C.H.; Villanueva, J.A.; Devlin, A.M.; Chandler, C.J. Metabolic interactions of alcohol and folate. J. Nutr. 2002, 132, 2367S–2372S. [Google Scholar] [CrossRef] [Green Version]
- Haloul, M.; Vinjamuri, S.J.; Naquiallah, D.; Mirza, M.I.; Qureshi, M.; Hassan, C.; Masrur, M.; Bianco, F.M.; Frederick, P.; Cristoforo, G.P.; et al. Hyperhomocysteinemia and Low Folate and Vitamin B12 Are Associated with Vascular Dysfunction and Impaired Nitric Oxide Sensitivity in Morbidly Obese Patients. Nutrients 2020, 12, 2014. [Google Scholar] [CrossRef]
- Zhang, X.; Kusumo, H.; Sakharkar, A.J.; Pandey, S.C.; Guizzetti, M. Regulation of DNA methylation by ethanol induces tissue plasminogen activator expression in astrocytes. J. Neurochem. 2014, 128, 344–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warnault, V.; Darcq, E.; Levine, A.; Barak, S.; Ron, D. Chromatin remodeling--a novel strategy to control excessive alcohol drinking. Transl. Psychiatry 2013, 3, e231. [Google Scholar] [CrossRef]
- Rodriguez-Araujo, G.; Nakagami, H.; Takami, Y.; Katsuya, T.; Akasaka, H.; Saitoh, S.; Shimamoto, K.; Morishita, R.; Rakugi, H.; Kaneda, Y. Low alpha-synuclein levels in the blood are associated with insulin resistance. Sci. Rep. 2015, 5, 12081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciafre, S.; Carito, V.; Ferraguti, G.; Greco, A.; Chaldakov, G.N.; Fiore, M.; Ceccanti, M. How alcohol drinking affects our genes: An epigenetic point of view. Biochem. Cell Biol. 2019, 97, 345–356. [Google Scholar] [CrossRef] [Green Version]
- Ponomarev, I.; Wang, S.; Zhang, L.; Harris, R.A.; Mayfield, R.D. Gene coexpression networks in human brain identify epigenetic modifications in alcohol dependence. J. Neurosci. 2012, 32, 1884–1897. [Google Scholar] [CrossRef] [PubMed]
- Qiang, M.; Denny, A.; Lieu, M.; Carreon, S.; Li, J. Histone H3K9 modifications are a local chromatin event involved in ethanol-induced neuroadaptation of the NR2B gene. Epigenetics 2011, 6, 1095–1104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campion, J.; Milagro, F.I.; Goyenechea, E.; Martinez, J.A. TNF-alpha promoter methylation as a predictive biomarker for weight-loss response. Obesity 2009, 17, 1293–1297. [Google Scholar] [CrossRef] [PubMed]
- Cordero, P.; Campion, J.; Milagro, F.I.; Goyenechea, E.; Steemburgo, T.; Javierre, B.M.; Martinez, J.A. Leptin and TNF-alpha promoter methylation levels measured by MSP could predict the response to a low-calorie diet. J. Physiol. Biochem. 2011, 67, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, L.; Rabasa-Lhoret, R.; Faraj, M.; Lavoie, M.E.; Mill, J.; Perusse, L.; Vohl, M.C. Differential epigenomic and transcriptomic responses in subcutaneous adipose tissue between low and high responders to caloric restriction. Am. J. Clin. Nutr. 2010, 91, 309–320. [Google Scholar] [CrossRef] [Green Version]
- Keller, M.; Yaskolka Meir, A.; Bernhart, S.H.; Gepner, Y.; Shelef, I.; Schwarzfuchs, D.; Tsaban, G.; Zelicha, H.; Hopp, L.; Muller, L.; et al. DNA methylation signature in blood mirrors successful weight-loss during lifestyle interventions: The CENTRAL trial. Genome Med. 2020, 12, 97. [Google Scholar] [CrossRef]
- Milagro, F.I.; Gomez-Abellan, P.; Campion, J.; Martinez, J.A.; Ordovas, J.M.; Garaulet, M. CLOCK, PER2 and BMAL1 DNA methylation: Association with obesity and metabolic syndrome characteristics and monounsaturated fat intake. Chronobiol. Int. 2012, 29, 1180–1194. [Google Scholar] [CrossRef]
- Uriarte, G.; Paternain, L.; Milagro, F.I.; Martinez, J.A.; Campion, J. Shifting to a control diet after a high-fat, high-sucrose diet intake induces epigenetic changes in retroperitoneal adipocytes of Wistar rats. J. Physiol. Biochem. 2013, 69, 601–611. [Google Scholar] [CrossRef]
- Day, S.E.; Garcia, L.A.; Coletta, R.L.; Campbell, L.E.; Benjamin, T.R.; De Filippis, E.A.; Madura, J.A., II; Mandarino, L.J.; Roust, L.R.; Coletta, D.K. Alterations of sorbin and SH3 domain containing 3 (SORBS3) in human skeletal muscle following Roux-en-Y gastric bypass surgery. Clin. Epigenet. 2017, 9, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barres, R.; Kirchner, H.; Rasmussen, M.; Yan, J.; Kantor, F.R.; Krook, A.; Naslund, E.; Zierath, J.R. Weight loss after gastric bypass surgery in human obesity remodels promoter methylation. Cell Rep. 2013, 3, 1020–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benton, M.C.; Johnstone, A.; Eccles, D.; Harmon, B.; Hayes, M.T.; Lea, R.A.; Griffiths, L.; Hoffman, E.P.; Stubbs, R.S.; Macartney-Coxson, D. An analysis of DNA methylation in human adipose tissue reveals differential modification of obesity genes before and after gastric bypass and weight loss. Genome Biol. 2015, 16, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahrens, M.; Ammerpohl, O.; von Schonfels, W.; Kolarova, J.; Bens, S.; Itzel, T.; Teufel, A.; Herrmann, A.; Brosch, M.; Hinrichsen, H.; et al. DNA methylation analysis in nonalcoholic fatty liver disease suggests distinct disease-specific and remodeling signatures after bariatric surgery. Cell Metab. 2013, 18, 296–302. [Google Scholar] [CrossRef] [Green Version]
- Morcillo, S.; Martin-Nunez, G.M.; Garcia-Serrano, S.; Gutierrez-Repiso, C.; Rodriguez-Pacheco, F.; Valdes, S.; Gonzalo, M.; Rojo-Martinez, G.; Moreno-Ruiz, F.J.; Rodriguez-Canete, A.; et al. Changes in SCD gene DNA methylation after bariatric surgery in morbidly obese patients are associated with free fatty acids. Sci. Rep. 2017, 7, 46292. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, E.K.; Ernst, B.; Voisin, S.; Almen, M.S.; Benedict, C.; Mwinyi, J.; Fredriksson, R.; Schultes, B.; Schioth, H.B. Roux-en Y gastric bypass surgery induces genome-wide promoter-specific changes in DNA methylation in whole blood of obese patients. PLoS ONE 2015, 10, e0115186. [Google Scholar] [CrossRef]
- Liang, Y.; Yu, B.; Wang, Y.; Qiao, Z.; Cao, T.; Zhang, P. Duodenal long noncoding RNAs are associated with glycemic control after bariatric surgery in high-fat diet-induced diabetic mice. Surg. Obes. Relat. Dis. 2017, 13, 1212–1226. [Google Scholar] [CrossRef]
- Alkandari, A.; Ashrafian, H.; Sathyapalan, T.; Sedman, P.; Darzi, A.; Holmes, E.; Athanasiou, T.; Atkin, S.L.; Gooderham, N.J. Improved physiology and metabolic flux after Roux-en-Y gastric bypass is associated with temporal changes in the circulating microRNAome: A longitudinal study in humans. BMC Obes. 2018, 5, 20. [Google Scholar] [CrossRef] [Green Version]
- Ortega, F.J.; Mercader, J.M.; Moreno-Navarrete, J.M.; Nonell, L.; Puigdecanet, E.; Rodriquez-Hermosa, J.I.; Rovira, O.; Xifra, G.; Guerra, E.; Moreno, M.; et al. Surgery-Induced Weight Loss Is Associated With the Downregulation of Genes Targeted by MicroRNAs in Adipose Tissue. J. Clin. Endocrinol. Metab. 2015, 100, E1467–E1476. [Google Scholar] [CrossRef] [Green Version]
- Avery, L.B.; Bumpus, N.N. Valproic acid is a novel activator of AMP-activated protein kinase and decreases liver mass, hepatic fat accumulation, and serum glucose in obese mice. Mol. Pharmacol. 2014, 85, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Ozcan, U.; Yilmaz, E.; Ozcan, L.; Furuhashi, M.; Vaillancourt, E.; Smith, R.O.; Gorgun, C.Z.; Hotamisligil, G.S. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 2006, 313, 1137–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahllof, M.S.; Christensen, D.P.; Lundh, M.; Dinarello, C.A.; Mascagni, P.; Grunnet, L.G.; Mandrup-Poulsen, T. The lysine deacetylase inhibitor Givinostat inhibits beta-cell IL-1beta induced IL-1beta transcription and processing. Islets 2012, 4, 417–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leoni, F.; Fossati, G.; Lewis, E.C.; Lee, J.K.; Porro, G.; Pagani, P.; Modena, D.; Moras, M.L.; Pozzi, P.; Reznikov, L.L.; et al. The histone deacetylase inhibitor ITF2357 reduces production of pro-inflammatory cytokines in vitro and systemic inflammation in vivo. Mol. Med. 2005, 11, 1–15. [Google Scholar] [CrossRef]
- Cabrera, S.M.; Colvin, S.C.; Tersey, S.A.; Maier, B.; Nadler, J.L.; Mirmira, R.G. Effects of combination therapy with dipeptidyl peptidase-IV and histone deacetylase inhibitors in the non-obese diabetic mouse model of type 1 diabetes. Clin. Exp. Immunol. 2013, 172, 375–382. [Google Scholar] [CrossRef]
- Lewis, E.C.; Blaabjerg, L.; Storling, J.; Ronn, S.G.; Mascagni, P.; Dinarello, C.A.; Mandrup-Poulsen, T. The oral histone deacetylase inhibitor ITF2357 reduces cytokines and protects islet beta cells in vivo and in vitro. Mol. Med. 2011, 17, 369–377. [Google Scholar] [CrossRef]
- Xiao, C.; Giacca, A.; Lewis, G.F. Sodium phenylbutyrate, a drug with known capacity to reduce endoplasmic reticulum stress, partially alleviates lipid-induced insulin resistance and beta-cell dysfunction in humans. Diabetes 2011, 60, 918–924. [Google Scholar] [CrossRef] [Green Version]
- Arguelles, A.O.; Meruvu, S.; Bowman, J.D.; Choudhury, M. Are epigenetic drugs for diabetes and obesity at our door step? Drug Discov. Today 2016, 21, 499–509. [Google Scholar] [CrossRef]
- Han, H.S.; Choi, D.; Choi, S.; Koo, S.H. Roles of protein arginine methyltransferases in the control of glucose metabolism. Endocrinol. Metab. 2014, 29, 435–440. [Google Scholar] [CrossRef] [Green Version]
- vanLieshout, T.L.; Ljubicic, V. The emergence of protein arginine methyltransferases in skeletal muscle and metabolic disease. Am. J. Physiol. Endocrinol. Metab. 2019, 317, E1070–E1080. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.; Oh, K.J.; Han, H.S.; Yoon, Y.S.; Jung, C.Y.; Kim, S.T.; Koo, S.H. Protein arginine methyltransferase 1 regulates hepatic glucose production in a FoxO1-dependent manner. Hepatology 2012, 56, 1546–1556. [Google Scholar] [CrossRef]
- Wang, J.; Chen, L.; Sinha, S.H.; Liang, Z.; Chai, H.; Muniyan, S.; Chou, Y.W.; Yang, C.; Yan, L.; Feng, Y.; et al. Pharmacophore-based virtual screening and biological evaluation of small molecule inhibitors for protein arginine methylation. J. Med. Chem. 2012, 55, 7978–7987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hino, S.; Sakamoto, A.; Nagaoka, K.; Anan, K.; Wang, Y.; Mimasu, S.; Umehara, T.; Yokoyama, S.; Kosai, K.; Nakao, M. FAD-dependent lysine-specific demethylase-1 regulates cellular energy expenditure. Nat. Commun. 2012, 3, 758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, D.; Mao, C.; Wang, Y.X. Suppression of gluconeogenic gene expression by LSD1-mediated histone demethylation. PLoS ONE 2013, 8, e66294. [Google Scholar] [CrossRef] [PubMed]
- Chase, K.; Sharma, R.P. Epigenetic developmental programs and adipogenesis: Implications for psychotropic induced obesity. Epigenetics 2013, 8, 1133–1140. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.T.; Dayeh, T.A.; Volkov, P.A.; Kirkpatrick, C.L.; Malmgren, S.; Jing, X.; Renstrom, E.; Wollheim, C.B.; Nitert, M.D.; Ling, C. Increased DNA methylation and decreased expression of PDX-1 in pancreatic islets from patients with type 2 diabetes. Mol. Endocrinol. 2012, 26, 1203–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halby, L.; Champion, C.; Senamaud-Beaufort, C.; Ajjan, S.; Drujon, T.; Rajavelu, A.; Ceccaldi, A.; Jurkowska, R.; Lequin, O.; Nelson, W.G.; et al. Rapid synthesis of new DNMT inhibitors derivatives of procainamide. Chembiochem 2012, 13, 157–165. [Google Scholar] [CrossRef]
- Lee, B.H.; Yegnasubramanian, S.; Lin, X.; Nelson, W.G. Procainamide is a specific inhibitor of DNA methyltransferase 1. J. Biol. Chem. 2005, 280, 40749–40756. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahmoud, A.M. An Overview of Epigenetics in Obesity: The Role of Lifestyle and Therapeutic Interventions. Int. J. Mol. Sci. 2022, 23, 1341. https://doi.org/10.3390/ijms23031341
Mahmoud AM. An Overview of Epigenetics in Obesity: The Role of Lifestyle and Therapeutic Interventions. International Journal of Molecular Sciences. 2022; 23(3):1341. https://doi.org/10.3390/ijms23031341
Chicago/Turabian StyleMahmoud, Abeer M. 2022. "An Overview of Epigenetics in Obesity: The Role of Lifestyle and Therapeutic Interventions" International Journal of Molecular Sciences 23, no. 3: 1341. https://doi.org/10.3390/ijms23031341
APA StyleMahmoud, A. M. (2022). An Overview of Epigenetics in Obesity: The Role of Lifestyle and Therapeutic Interventions. International Journal of Molecular Sciences, 23(3), 1341. https://doi.org/10.3390/ijms23031341