
Hyperglycemia Induces Inflammatory Response of Human Macrophages to CD163-Mediated Scavenging of Hemoglobin-Haptoglobin Complexes


Abstract
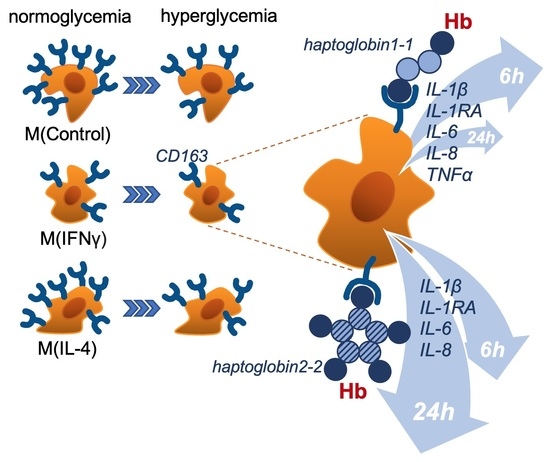
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. IFNγ and IL-4 Suppress CD163 Expression in Macrophages, and Hyperglycemia Enhances the IFNγ Effect
2.2. CD163-Mediated Internalization of Hb-Hp Complexes Is Efficient in NG and HG Conditions in M0 and M(IL-4) Macrophages
2.3. Hyperglycemia Enhances the Release of Inflammatory Cytokines in Response to Scavenging of Hemoglobin-Haptoglobin Complexes via CD163 in M(IFNγ)
3. Discussion
4. Materials and Methods
4.1. Isolation of Monocytes and Cultivation of Macrophages
4.2. RNA Samples, cDNA Synthesis, and Real-Time PCR Analysis
4.3. Flow Cytometry
4.4. ELISA
4.5. Endocytosis of Hp-Hb Complexes
4.6. Immunofluorescent Staining and Confocal Microscopy
4.7. Inflammatory Response Assay
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CD | cluster of differentiation |
| DCCT | Diabetes Control and Complications Trial |
| HG | high glucose conditions, hyperglycemia (25mM) |
| Hb | hemoglobin |
| Hp | haptoglobin |
| M0, M(Control) | non-stimulated macrophages |
| M(IFNγ) | macrophages stimulated with IFNγ |
| M(IL-4) | macrophages stimulated with IL-4 |
| M1 | classically activated macrophages |
| M2 | alternatively activated macrophages |
| MFI | mean fluorescence intensity |
| NG | normal glucose conditions, normoglycemia (5mM) |
| no compl | ‘no complexes’ |
| PBMC | peripheral blood mononuclear cells |
| PFA | paraformaldehyde |
| RAGE | receptor for advanced glycation endproducts |
| UKPDS | United Kingdom Prospective Diabetes Study |
References
- Alberti, K.G.; Zimmet, P.Z.; Aschner, P.; Assal, P.-J.; Bennet, P.H.; Groop, L.; Jervell, J.; Kanazawa, Y.; Keen, H.; Klein, R.; et al. Diagnosis and Classification of Diabetes Mellitus; Report of a WHO Consultation; WHO, Department of Noncommunicable Disease Surveillance: Geneva, Switzerland, 1999. [Google Scholar]
- Powers, A.; Niswender, K.; Evans-Molina, C. Diabetes: Diagnosis, Classification, and Pathophysiology. In Harrison’s Principles of Internal Medicine, 20th ed.; Jameson, F., Kasper, H., Longo, L., Eds.; McGraw-Hill Education: New York, NY, USA, 2018; Volume I, pp. 2850–2859. [Google Scholar]
- DCCT. The Diabetes Control and Complications Trial (DCCT). Design and methodologic considerations for the feasibility phase. Diabetes 1986, 35, 530–545. [Google Scholar] [CrossRef]
- Stevens, R.J.; Kothari, V.; Adler, A.I.; Stratton, I.M.; United Kingdom Prospective Diabetes Study (UKPDS) Group. The UKPDS risk engine: A model for the risk of coronary heart disease in Type II diabetes (UKPDS 56). Clin. Sci. 2001, 101, 671–679. [Google Scholar] [CrossRef]
- Schaper, N.C.; Havekes, B. Diabetes: Impaired damage control. Diabetologia 2012, 55, 18–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moganti, K.; Li, F.; Schmuttermaier, C.; Riemann, S.; Kluter, H.; Gratchev, A.; Harmsen, M.C.; Kzhyshkowska, J. Hyperglycemia induces mixed M1/M2 cytokine profile in primary human monocyte-derived macrophages. Immunobiology 2017, 222, 952–959. [Google Scholar] [CrossRef]
- Mossel, D.M.; Moganti, K.; Riabov, V.; Weiss, C.; Kopf, S.; Cordero, J.; Dobreva, G.; Rots, M.G.; Kluter, H.; Harmsen, M.C.; et al. Epigenetic Regulation of S100A9 and S100A12 Expression in Monocyte-Macrophage System in Hyperglycemic Conditions. Front. Immunol. 2020, 11, 1071. [Google Scholar] [CrossRef]
- Dasu, M.R.; Devaraj, S.; Park, S.; Jialal, I. Increased toll-like receptor (TLR) activation and TLR ligands in recently diagnosed type 2 diabetic subjects. Diabetes Care 2010, 33, 861–868. [Google Scholar] [CrossRef] [Green Version]
- Lorenzi, M. The polyol pathway as a mechanism for diabetic retinopathy: Attractive, elusive, and resilient. Exp. Diabetes Res. 2007, 2007, 61038. [Google Scholar] [CrossRef]
- Ishibashi, Y.; Matsui, T.; Maeda, S.; Higashimoto, Y.; Yamagishi, S. Advanced glycation end products evoke endothelial cell damage by stimulating soluble dipeptidyl peptidase-4 production and its interaction with mannose 6-phosphate/insulin-like growth factor II receptor. Cardiovasc. Diabetol. 2013, 12, 125. [Google Scholar] [CrossRef] [Green Version]
- Wendt, T.M.; Tanji, N.; Guo, J.; Kislinger, T.R.; Qu, W.; Lu, Y.; Bucciarelli, L.G.; Rong, L.L.; Moser, B.; Markowitz, G.S.; et al. RAGE drives the development of glomerulosclerosis and implicates podocyte activation in the pathogenesis of diabetic nephropathy. Am. J. Pathol. 2003, 162, 1123–1137. [Google Scholar] [CrossRef] [Green Version]
- Mathur, N.; Pedersen, B.K. Exercise as a mean to control low-grade systemic inflammation. Mediat. Inflamm 2008, 2008, 109502. [Google Scholar] [CrossRef] [Green Version]
- Torres-Castro, I.; Arroyo-Camarena, U.D.; Martinez-Reyes, C.P.; Gomez-Arauz, A.Y.; Duenas-Andrade, Y.; Hernandez-Ruiz, J.; Bejar, Y.L.; Zaga-Clavellina, V.; Morales-Montor, J.; Terrazas, L.I.; et al. Human monocytes and macrophages undergo M1-type inflammatory polarization in response to high levels of glucose. Immunol. Lett. 2016, 176, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Heizmann, C.W.; Fritz, G.; Schafer, B.W. S100 proteins: Structure, functions and pathology. Front. Biosci. 2002, 7, d1356–d1368. [Google Scholar] [PubMed]
- Chellan, B.; Sutton, N.R.; Bowman, M.A.H. S100/RAGE-Mediated Inflammation and Modified Cholesterol Lipoproteins as Mediators of Osteoblastic Differentiation of Vascular Smooth Muscle Cells. Front. Cardiovasc. Med. 2018, 5, 163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, X.; Yang, C.; Qu, S.L.; Shao, Y.D.; Zhou, C.Y.; Chao, R.; Huang, L.; Zhang, C. S100 proteins in atherosclerosis. Clin. Chim. Acta 2020, 502, 293–304. [Google Scholar] [CrossRef]
- Buechler, C.; Eisinger, K.; Krautbauer, S. Diagnostic and prognostic potential of the macrophage specific receptor CD163 in inflammatory diseases. Inflamm. Allergy Drug Targets 2013, 12, 391–402. [Google Scholar] [CrossRef]
- Fuentes-Duculan, J.; Suarez-Farinas, M.; Zaba, L.C.; Nograles, K.E.; Pierson, K.C.; Mitsui, H.; Pensabene, C.A.; Kzhyshkowska, J.; Krueger, J.G.; Lowes, M.A. A subpopulation of CD163-positive macrophages is classically activated in psoriasis. J. Investig. Dermatol. 2010, 130, 2412–2422. [Google Scholar] [CrossRef] [Green Version]
- Larionova, I.; Tuguzbaeva, G.; Ponomaryova, A.; Stakheyeva, M.; Cherdyntseva, N.; Pavlov, V.; Choinzonov, E.; Kzhyshkowska, J. Tumor-Associated Macrophages in Human Breast, Colorectal, Lung, Ovarian and Prostate Cancers. Front. Oncol. 2020, 10, 566511. [Google Scholar] [CrossRef]
- Bengtsson, E.; Hultman, K.; Edsfeldt, A.; Persson, A.; Nitulescu, M.; Nilsson, J.; Goncalves, I.; Bjorkbacka, H. CD163+ macrophages are associated with a vulnerable plaque phenotype in human carotid plaques. Sci. Rep. 2020, 10, 14362. [Google Scholar] [CrossRef]
- Kristiansen, M.; Graversen, J.; Jacobsen, C.; Sonne, O.; Hoffman, H.J.; Lawk, A.; Moestrup, S. Identifcation of the haemoglobin scavenger receptor. Nature 2001, 409, 198–201. [Google Scholar] [CrossRef]
- Buechler, C.; Ritter, M.; Orso, E.; Langmann, T.; Klucken, J.; Schmitz, G. Regulation of scavenger receptor CD163 expression in human monocytes and macrophages by pro- and antiinflammatory stimuli. J. Leukoc. Biol. 2000, 67, 97–103. [Google Scholar] [CrossRef]
- Van den Heuvel, M.M.; Tensen, C.P.; van As, J.H.; Van den Berg, T.K.; Fluitsma, D.M.; Dijkstra, C.D.; Dopp, E.A.; Droste, A.; Van Gaalen, F.A.; Sorg, C.; et al. Regulation of CD 163 on human macrophages: Cross-linking of CD163 induces signaling and activation. J. Leukoc. Biol. 1999, 66, 858–866. [Google Scholar] [CrossRef] [PubMed]
- Hogger, P.; Dreier, J.; Droste, A.; Buck, F.; Sorg, C. Identification of the integral membrane protein RM3/1 on human monocytes as a glucocorticoid-inducible member of the scavenger receptor cysteine-rich family (CD163). J. Immunol. 1998, 161, 1883–1890. [Google Scholar] [PubMed]
- Philippidis, P.; Mason, J.C.; Evans, B.J.; Nadra, I.; Taylor, K.M.; Haskard, D.O.; Landis, R.C. Hemoglobin scavenger receptor CD163 mediates interleukin-10 release and heme oxygenase-1 synthesis: Antiinflammatory monocyte-macrophage responses in vitro, in resolving skin blisters in vivo, and after cardiopulmonary bypass surgery. Circ. Res. 2004, 94, 119–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zwadlo, G.; Voegeli, R.; Schulze Osthoff, K.; Sorg, C. A monoclonal antibody to a novel differentiation antigen on human macrophages associated with the down-regulatory phase of the inflammatory process. Pathobiology 1987, 55, 295–304. [Google Scholar] [CrossRef]
- Sulahian, T.H.; Hogger, P.; Wahner, A.E.; Wardwell, K.; Goulding, N.J.; Sorg, C.; Droste, A.; Stehling, M.; Wallace, P.K.; Morganelli, P.M.; et al. Human monocytes express CD163, which is upregulated by IL-10 and identical to p155. Cytokine 2000, 12, 1312–1321. [Google Scholar] [CrossRef]
- Asleh, R.; Levy, A.P. In vivo and in vitro studies establishing haptoglobin as a major susceptibility gene for diabetic vascular disease. Vasc. Health Risk Manag. 2005, 1, 19–28. [Google Scholar] [CrossRef] [Green Version]
- Everse, J.; Hsia, N. The toxicities of native and modified hemoglobins. Free Radic. Biol. Med. 1997, 22, 1075–1099. [Google Scholar] [CrossRef]
- Schaer, C.A.; Vallelian, F.; Imhof, A.; Schoedon, G.; Schaer, D.J. CD163-expressing monocytes constitute an endotoxin-sensitive Hb clearance compartment within the vascular system. J. Leukoc. Biol. 2007, 82, 106–110. [Google Scholar] [CrossRef]
- Nielsen, M.J.; Andersen, C.B.; Moestrup, S.K. CD163 binding to haptoglobin-hemoglobin complexes involves a dual-point electrostatic receptor-ligand pairing. J. Biol. Chem. 2013, 288, 18834–18841. [Google Scholar] [CrossRef] [Green Version]
- Dubayee, M.S.A.; Alayed, H.; Almansour, R.; Alqaoud, N.; Alnamlah, R.; Obeid, D.; Alshahrani, A.; Zahra, M.M.; Nasr, A.; Al-Bawab, A.; et al. Differential Expression of Human Peripheral Mononuclear Cells Phenotype Markers in Type 2 Diabetic Patients and Type 2 Diabetic Patients on Metformin. Front. Endocrinol. 2018, 9, 537. [Google Scholar] [CrossRef] [Green Version]
- Levy, A.; Purushothaman, K.R.; Levy, N.S.; Purushothaman, M.; Strauss, M.; Asleh, R.; Marsh, S.; Cohen, O.; Moestrup, S.K.; Moller, H.J.; et al. Downregulation of the hemoglobin scavenger receptor in individuals with diabetes and the Hp 2-2 genotype: Implications for the response to intraplaque hemorrhage and plaque vulnerability. Circ. Res. 2007, 101, 106–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, A.; Asleh, R.; Blum, S.; Levy, N.S.; Miller-Lotan, R.; Kalet-Litman, S.; Anbinder, Y.; Lache, O.; Nakhoul, F.M.; Asaf, R.; et al. Haptoglobin: Basic and clinical aspects. Antioxid. Redox Signal. 2010, 12, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Langlois, M.R.; Delanghe, J.R. Biological and clinical significance of haptoglobin polymorphism in humans. Clin. Chem. 1996, 42, 1589–1600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomsen, J.H.; Etzerodt, A.; Svendsen, P.; Moestrup, S.K. The Haptoglobin-CD163-Heme Oxygenase-1 Pathway for Hemoglobin Scavenging. Oxid. Med. Cell. Longev. 2013, 2013, 523652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabay, C.; Kushner, I. Acute-phase proteins and other systemic responses to inflammation. N. Engl. J. Med. 1999, 340, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Melamed-Frank, M.; Lache, O.; Enav, B.I.; Szafranek, T.; Levy, N.S.; Ricklis, R.M.; Levy, A.P. Structure-function analysis of the antioxidant properties of haptoglobin. Blood 2001, 98, 3693–3698. [Google Scholar] [CrossRef] [Green Version]
- Asleh, R.; Marsh, S.; Shilkrut, M.; Binah, O.; Guetta, J.; Lejbkowicz, F.; Enav, B.; Shehadeh, N.; Kanter, Y.; Lache, O.; et al. Genetically determined heterogeneity in hemoglobin scavenging and susceptibility to diabetic cardiovascular disease. Circ. Res. 2003, 92, 1193–1200. [Google Scholar] [CrossRef] [Green Version]
- Levy, A.P.; Hochberg, I.; Jablonski, K.; Resnick, H.E.; Lee, E.T.; Best, L.; Howard, B.V.; Strong Heart, S. Haptoglobin phenotype is an independent risk factor for cardiovascular disease in individuals with diabetes: The Strong Heart Study. J. Am. Coll. Cardiol. 2002, 40, 1984–1990. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Kinzie, E.; Berger, F.G.; Lim, S.K.; Baumann, H. Haptoglobin, an inflammation-inducible plasma protein. Redox Rep. 2001, 6, 379–385. [Google Scholar] [CrossRef]
- Fadini, G.P.; de Kreutzenberg, S.V.; Boscaro, E.; Albiero, M.; Cappellari, R.; Krankel, N.; Landmesser, U.; Toniolo, A.; Bolego, C.; Cignarella, A.; et al. An unbalanced monocyte polarisation in peripheral blood and bone marrow of patients with type 2 diabetes has an impact on microangiopathy. Diabetologia 2013, 56, 1856–1866. [Google Scholar] [CrossRef] [Green Version]
- Etzerodt, A.; Moestrup, S.K. CD163 and Inflammation: Biological, Diagnostic, and Therapeutic Aspects. Antioxid. Redox Signal. 2013, 18, 2352–2363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moller, H.J.; Frikke-Schmidt, R.; Moestrup, S.K.; Nordestgaard, B.; Tybjærg-Hansen, A. Serum soluble CD163 predicts risk of type 2 diabetes in the general population. Clin. Chem. 2011, 57, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Fink, L.N.; Oberbach, A.; Costford, S.R.; Chan, K.L.; Sams, A.; Bluher, M.; Klip, A. Expression of anti-inflammatory macrophage genes within skeletal muscle correlates with insulin sensitivity in human obesity and type 2 diabetes. Diabetologia 2013, 56, 1623–1628. [Google Scholar] [CrossRef] [PubMed]
- Aristoteli, L.; Møller, H.J.; Bailey, B.; Moestrup, S.K.; Kritharides, L. The monocytic lineage specific soluble CD163 is a plasma marker of coronary atherosclerosis. Atherosclerosis 2006, 184, 342–347. [Google Scholar] [CrossRef]
- Zanni, M.V.; Burdo, T.H.; Makimura, H.; Williams, K.C.; Grinspoon, S.K. Relationship between monocyte/macrophage activation marker soluble CD163 and insulin resistance in obese and normal-weight subjects. Clin. Endocrinol. 2012, 77, 385–390. [Google Scholar] [CrossRef] [Green Version]
- Staples, K.J.; Hinks, T.S.; Ward, J.A.; Gunn, V.; Smith, C.; Djukanovic, R. Phenotypic characterization of lung macrophages in asthmatic patients: Overexpression of CCL17. J. Allergy Clin. Immunol. 2012, 130, 1404–1412. [Google Scholar] [CrossRef] [Green Version]
- Fadini, G.P.; Cappellari, R.; Mazzucato, M.; Agostini, C.; Vigili de Kreutzenberg, S.; Avogaro, A. Monocyte-macrophage polarization balance in pre-diabetic individuals. Acta Diabetol. 2013, 50, 977–982. [Google Scholar] [CrossRef]
- Schaer, C.A.; Schoedon, G.; Imhof, A.; Kurrer, M.O.; Schaer, D.J. Constitutive endocytosis of CD163 mediates hemoglobin-heme uptake and determines the noninflammatory and protective transcriptional response of macrophages to hemoglobin. Circ. Res. 2006, 99, 943–950. [Google Scholar] [CrossRef] [Green Version]
- Wejman, J.C.; Hovsepian, D.; Wall, J.S.; Hainfeld, J.F.; Greer, J. Structure and assembly of haptoglobin polymers by electron microscopy. J. Mol. Biol. 1984, 174, 343–368. [Google Scholar] [CrossRef]
- Andersen, C.B.; Torvund-Jensen, M.; Nielsen, M.J.; de Oliveira, C.L.; Hersleth, H.P.; Andersen, N.H.; Pedersen, J.S.; Andersen, G.R.; Moestrup, S.K. Structure of the haptoglobin-haemoglobin complex. Nature 2012, 489, 456–459. [Google Scholar] [CrossRef]
- Schaer, D.J.; Schaer, C.A.; Buehler, P.W.; Boykins, R.A.; Schoedon, G.; Alayash, A.I.; Schaffner, A. CD163 is the macrophage scavenger receptor for native and chemically modified hemoglobins in the absence of haptoglobin. Blood 2006, 107, 373–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaer, D.J. The macrophage hemoglobin scavenger receptor (CD163) as a genetically determined disease modifying pathway in atherosclerosis. Atherosclerosis 2002, 163, 199–201. [Google Scholar] [CrossRef]
- Ritter, M.; Buechler, C.; Kapinsky, M.; Schmitz, G. Interaction of CD163 with the regulatory subunit of casein kinase II (CKII) and dependence of CD163 signaling on CKII and protein kinase C. Eur. J. Immunol. 2001, 31, 999–1009. [Google Scholar] [CrossRef]
- Guo, L.; Akahori, H.; Harari, E.; Smith, S.L.; Polavarapu, R.; Karmali, V.; Otsuka, F.; Gannon, R.L.; Braumann, R.E.; Dickinson, M.H.; et al. CD163+ macrophages promote angiogenesis and vascular permeability accompanied by inflammation in atherosclerosis. J. Clin. Investig. 2018, 128, 1106–1124. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Harari, E.; Virmani, R.; Finn, A.V. Linking Hemorrhage, Angiogenesis, Macrophages, and Iron Metabolism in Atherosclerotic Vascular Diseases. Arterioscler. Thromb. Vasc. Biol. 2017, 37, e33–e39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liberale, L.; Dallegri, F.; Montecucco, F.; Carbone, F. Pathophysiological relevance of macrophage subsets in atherogenesis. Thromb. Haemost. 2017, 117, 7–18. [Google Scholar] [CrossRef]
- Finn, A.V.; Nakano, M.; Polavarapu, R.; Karmali, V.; Saeed, O.; Zhao, X.; Yazdani, S.; Otsuka, F.; Davis, T.; Habib, A.; et al. Hemoglobin directs macrophage differentiation and prevents foam cell formation in human atherosclerotic plaques. J. Am. Coll. Cardiol. 2012, 59, 166–177. [Google Scholar] [CrossRef] [Green Version]
- Boyle, J.J.; Harrington, H.A.; Piper, E.; Elderfield, K.; Stark, J.; Landis, R.C.; Haskard, D.O. Coronary intraplaque hemorrhage evokes a novel atheroprotective macrophage phenotype. Am. J. Pathol. 2009, 174, 1097–1108. [Google Scholar] [CrossRef] [Green Version]
- Szabo, P.A.; Dogra, P.; Gray, J.I.; Wells, S.B.; Connors, T.J.; Weisberg, S.P.; Krupska, I.; Matsumoto, R.; Poon, M.M.L.; Idzikowski, E.; et al. Longitudinal profiling of respiratory and systemic immune responses reveals myeloid cell-driven lung inflammation in severe COVID-19. Immunity 2021, 54, 797–814. [Google Scholar] [CrossRef]
- Abers, M.S.; Delmonte, O.M.; Ricotta, E.E.; Fintzi, J.; Fink, D.L.; de Jesus, A.A.A.; Zarember, K.A.; Alehashemi, S.; Oikonomou, V.; Desai, J.V.; et al. An immune-based biomarker signature is associated with mortality in COVID-19 patients. JCI Insight 2021, 6, e144455. [Google Scholar] [CrossRef]
- Gomez-Rial, J.; Curras-Tuala, M.J.; Rivero-Calle, I.; Gomez-Carballa, A.; Cebey-Lopez, M.; Rodriguez-Tenreiro, C.; Dacosta-Urbieta, A.; Rivero-Velasco, C.; Rodriguez-Nunez, N.; Trastoy-Pena, R.; et al. Increased Serum Levels of sCD14 and sCD163 Indicate a Preponderant Role for Monocytes in COVID-19 Immunopathology. Front. Immunol. 2020, 11, 560381. [Google Scholar] [CrossRef] [PubMed]
- Guetta, J.; Strauss, M.; Levy, N.S.; Fahoum, L.; Levy, A.P. Haptoglobin genotype modulates the balance of Th1/Th2 cytokines produced by macrophages exposed to free hemoglobin. Atherosclerosis 2007, 191, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Guest, C.B.; Park, M.J.; Johnson, D.R.; Freund, G.G. The implication of proinflammatory cytokines in type 2 diabetes. Front. Biosci. 2008, 13, 5187–5194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collier, B.; Dossett, L.A.; May, A.K.; Diaz, J.J. Glucose control and the inflammatory response. Nutr. Clin. Pract. 2008, 23, 3–15. [Google Scholar] [CrossRef]
- Esposito, K.; Nappo, F.; Marfella, R.; Giugliano, G.; Giugliano, F.; Ciotola, M.; Quagliaro, L.; Ceriello, A.; Giugliano, D. Inflammatory cytokine concentrations are acutely increased by hyperglycemia in humans: Role of oxidative stress. Circulation 2002, 106, 2067–2072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickup, J.C. Inflammation and activated innate immunity in the pathogenesis of type 2 diabetes. Diabetes Care 2004, 27, 813–823. [Google Scholar] [CrossRef] [Green Version]
- Esser, N.; Legrand-Poels, S.; Piette, J.; Scheen, A.J.; Paquot, N. Inflammation as a link between obesity, metabolic syndrome and type 2 diabetes. Diabetes Res. Clin. Pract. 2014, 105, 141–150. [Google Scholar] [CrossRef] [Green Version]
- Gresser, I.; Delers, F.; Tran Quangs, N.; Marion, S.; Engler, R.; Maury, C.; Soria, C.; Soria, J.; Fiers, W.; Tavernier, J. Tumor necrosis factor induces acute phase proteins in rats. J. Biol. Regul. Homeost. Agents 1987, 1, 173–176. [Google Scholar]
- Stentz, F.B.; Umpierrez, G.E.; Cuervo, R.; Kitabchi, A.E. Proinflammatory cytokines, markers of cardiovascular risks, oxidative stress, and lipid peroxidation in patients with hyperglycemic crises. Diabetes 2004, 53, 2079–2086. [Google Scholar] [CrossRef] [Green Version]
- Mirza, S.; Hossain, M.; Mathews, C.; Martinez, P.; Pino, P.; Gay, J.L.; Rentfro, A.; McCormick, J.B.; Fisher-Hoch, S.P. Type 2-diabetes is associated with elevated levels of TNF-alpha, IL-6 and adiponectin and low levels of leptin in a population of Mexican Americans: A cross-sectional study. Cytokine 2012, 57, 136–142. [Google Scholar] [CrossRef] [Green Version]
- Castell, J.V.; Gomez-Lechon, M.J.; David, M.; Hirano, T.; Kishimoto, T.; Heinrich, P.C. Recombinant human interleukin-6 (IL-6/BSF-2/HSF) regulates the synthesis of acute phase proteins in human hepatocytes. FEBS Lett. 1988, 232, 347–350. [Google Scholar] [CrossRef] [Green Version]
- Senn, J.J.; Klover, P.J.; Nowak, I.A.; Mooney, R.A. Interleukin-6 induces cellular insulin resistance in hepatocytes. Diabetes 2002, 51, 3391–3399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feve, B.; Bastard, J.P. The role of interleukins in insulin resistance and type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2009, 5, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Maedler, K.; Dharmadhikari, G.; Schumann, D.M.; Storling, J. Interleukin-1 beta targeted therapy for type 2 diabetes. Expert Opin. Biol. Ther. 2009, 9, 1177–1188. [Google Scholar] [CrossRef]
- Strandberg, L.; Lorentzon, M.; Hellqvist, A.; Nilsson, S.; Wallenius, V.; Ohlsson, C.; Jansson, J.O. Interleukin-1 system gene polymorphisms are associated with fat mass in young men. J. Clin. Endocrinol. Metab. 2006, 91, 2749–2754. [Google Scholar] [CrossRef] [Green Version]
- Larsen, C.M.; Faulenbach, M.; Vaag, A.; Volund, A.; Ehses, J.A.; Seifert, B.; Mandrup-Poulsen, T.; Donath, M.Y. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N. Engl. J. Med. 2007, 356, 1517–1526. [Google Scholar] [CrossRef] [Green Version]
- Kzhyshkowska, J.; Gratchev, A.; Martens, J.H.; Pervushina, O.; Mamidi, S.; Johansson, S.; Schledzewski, K.; Hansen, B.; He, X.; Tang, J.; et al. Stabilin-1 localizes to endosomes and the trans-Golgi network in human macrophages and interacts with GGA adaptors. J. Leukoc. Biol. 2004, 76, 1151–1161. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matuschik, L.; Riabov, V.; Schmuttermaier, C.; Sevastyanova, T.; Weiss, C.; Klüter, H.; Kzhyshkowska, J. Hyperglycemia Induces Inflammatory Response of Human Macrophages to CD163-Mediated Scavenging of Hemoglobin-Haptoglobin Complexes. Int. J. Mol. Sci. 2022, 23, 1385. https://doi.org/10.3390/ijms23031385
Matuschik L, Riabov V, Schmuttermaier C, Sevastyanova T, Weiss C, Klüter H, Kzhyshkowska J. Hyperglycemia Induces Inflammatory Response of Human Macrophages to CD163-Mediated Scavenging of Hemoglobin-Haptoglobin Complexes. International Journal of Molecular Sciences. 2022; 23(3):1385. https://doi.org/10.3390/ijms23031385
Chicago/Turabian StyleMatuschik, Laura, Vladimir Riabov, Christina Schmuttermaier, Tatyana Sevastyanova, Christel Weiss, Harald Klüter, and Julia Kzhyshkowska. 2022. "Hyperglycemia Induces Inflammatory Response of Human Macrophages to CD163-Mediated Scavenging of Hemoglobin-Haptoglobin Complexes" International Journal of Molecular Sciences 23, no. 3: 1385. https://doi.org/10.3390/ijms23031385
APA StyleMatuschik, L., Riabov, V., Schmuttermaier, C., Sevastyanova, T., Weiss, C., Klüter, H., & Kzhyshkowska, J. (2022). Hyperglycemia Induces Inflammatory Response of Human Macrophages to CD163-Mediated Scavenging of Hemoglobin-Haptoglobin Complexes. International Journal of Molecular Sciences, 23(3), 1385. https://doi.org/10.3390/ijms23031385