
Enhanced Ca2+ Entry Sustains the Activation of Akt in Glucose Deprived SH-SY5Y Cells

 ,
,
Abstract
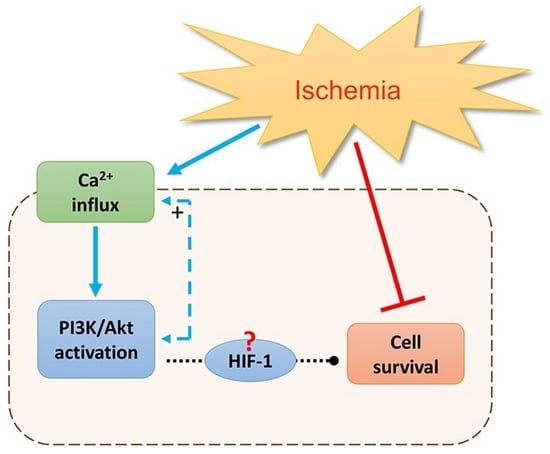
:
1. Introduction
2. Results
2.1. Effect of Glucose Deprivation on p-Akt, p-GSK3α/β and HIF-1α Protein Levels
2.2. Hypoxic Conditions Induced by CoCl2 Increases p-Akt, p-GSK3α/β and HIF-1a Protein Levels
2.3. Combined Effect of CoCl2 and Glucose Deprivation on p-Akt, and HIF-1a Protein Levels
2.4. Re-Administration of Glucose Restores p-Akt Levels
2.5. Ca2+ Measurements
2.6. Ca2+-Induced Modulation of PI3K/Akt and HIF-1α
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatments
4.2. Preparation of Total Protein Extracts and Western Blot Analysis
4.3. Cytosolic Ca2+ Measurement
4.4. Cell Viability Assay
4.5. Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Béjot, Y.; Garnier, P. Cerebral Ischemia. In Hormesis in Health and Disease; CRC Press: Boca Raton, FL, USA, 2014. [Google Scholar] [CrossRef]
- Janardhan, V.; Qureshi, A.I. Mechanisms of ischemic brain injury. Curr. Cardiol. Rep. 2004, 6, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Grabb, M.C.; Zipfel, G.J.; Choi, D.W. Brain tissue responses to ischemia. J. Clin. Investig. 2000, 106, 723–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fulda, S.; Debatin, K.M. HIF-1-regulated glucose metabolism: A key to apoptosis resistance? Cell Cycle 2007, 6, 790–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Shen, S.M.; Zhao, X.Y.; Chen, G. Q. Targeted genes and interacting proteins of hypoxia inducible factor-1. Int. J. Biochem. Mol. Biol. 2012, 3, 165–178. [Google Scholar] [PubMed]
- Li, Q.F.; Zhu, Y.S.; Jiang, H. Isoflurane preconditioning activates HIF-1α, iNOS and Erk1/2 and protects against oxygen-glucose deprivation neuronal injury. Brain Res. 2008, 1245, 26–35. [Google Scholar] [CrossRef]
- Skinner, H.D.; Zheng, J.Z.; Fang, J.; Agani, F.; Jiang, B.H. Vascular endothelial growth factor transcriptional activation is mediated by hypoxia-inducible factor 1α, HDM2, and p70S6K1 in response to phosphatidylinositol 3-kinase/AKT signaling. J. Biol. Chem. 2004, 279, 45643–45651. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Liu, A.; Lu, N.; Li, Y.; Song, Q.; Yu, H.; Li, X. Inhibitive effects of low oxygen and glucose deprivation on brain-pancreas relative protein expression via hypoxia- inducible factor-1 pathways. Cell. Physiol. Biochem. 2008, 22, 353–362. [Google Scholar] [CrossRef]
- Mishra, O.P.; Zubrow, A.B.; Ashraf, Q.M. Nitric oxide-mediated activation of extracellular signal-regulated kinase (ERK) and c-Jun N-terminal kinase (JNK) during hypoxia in cerebral cortical nuclei of newborn piglets. Neuroscience 2004, 123, 179–186. [Google Scholar] [CrossRef]
- Déry, M.A.C.; Michaud, M.D.; Richard, D.E. Hypoxia-inducible factor 1: Regulation by hypoxic and non-hypoxic activators. Int. J. Biochem. Cell Biol. 2005, 37, 535–540. [Google Scholar] [CrossRef]
- Zhong, H.; De Marzo, A.M.; Laughner, E.; Lim, M.; Hilton, D.A.; Zagzag, D.; Buechler, P.; Isaacs, W.B.; Semenza, G.L.; Simons, J.W. Overexpression of hypoxia-inducible factor 1α in common human cancers and their metastases. Cancer Res. 1999, 59, 5830–5835. [Google Scholar]
- Williams, K.J.; Telfer, B.A.; Airley, R.E.; Peters, H.P.W.; Sheridan, M.R.; Van der Kogel, A.J.; Harris, A.L.; Stratford, I.J. A protective role for HIF-1 in response to redox manipulation and glucose deprivation: Implications for tumorigenesis. Oncogene 2002, 21, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Franke, T.F.; Yang, S.I.; Chan, T.O.; Datta, K.; Kazlauskas, A.; Morrison, D.K.; Kaplan, D.R.; Tsichlis, P.N. The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell 1995, 81, 727–736. [Google Scholar] [CrossRef] [Green Version]
- Kohn, A.D.; Kovacina, K.S.; Roth, R.A. Insulin stimulates the kinase activity of RAC-PK, a pleckstrin homology domain containing ser/thr kinase. EMBO J. 1995, 14, 4288–4295. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Park, J.; Tran, H.; Hu, L.S.; Hemmings, B.A.; Greenberg, M.E. Protein Kinase SGK Mediates Survival Signals by Phosphorylating the Forkhead Transcription Factor FKHRL1 (FOXO3a). Mol. Cell. Biol. 2001, 21, 952–965. [Google Scholar] [CrossRef] [Green Version]
- Rodgers, S.J.; Ferguson, D.T.; Mitchell, C.A.; Ooms, L.M. Regulation of PI3K effector signalling in cancer by the phosphoinositide phosphatases. Biosci. Rep. 2017, 37. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.; Liang, J.; Lu, Y.; Guo, H.; German, P.; Bai, S.; Jonasch, E.; Yang, X.; Mills, G.B.; Ding, Z. Site-specific activation of AKT protects cells from death induced by glucose deprivation. Oncogene 2014, 33, 745–755. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Qu, Y.; Tang, J.; Chen, D.; Fu, X.; Mao, M.; Mu, D. PI3K/Akt signaling pathway is required for neuroprotection of thalidomide on hypoxic-ischemic cortical neurons in vitro. Brain Res. 2010, 1357, 157–165. [Google Scholar] [CrossRef]
- Sutter, C.H.; Laughner, E.; Semenza, G.L. Hypoxia-inducible factor 1α protein expression is controlled by oxygen-regulated ubiquitination that is disrupted by deletions and missense mutations. Proc. Natl. Acad. Sci. USA 2000, 97, 4748–4753. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Expression of hypoxia-inducible factor 1: Mechanisms and consequences. Biochem. Pharmacol. 2000, 59, 47–53. [Google Scholar] [CrossRef]
- Zhou, J.; Hara, K.; Inoue, M.; Hamada, S.; Yasuda, H.; Moriyama, H.; Endo, H.; Hirota, K.; Yonezawa, K.; Nagata, M.; et al. Regulation of hypoxia-inducible factor 1 by glucose availability under hypoxic conditions. Kobe J. Med. Sci. 2007, 53, 283–296. [Google Scholar]
- Serra-Pérez, A.; Planas, A.M.; Núñez-O’Mara, A.; Berra, E.; García-Villoria, J.; Ribes, A.; Santalucía, T. Extended ischemia prevents HIF1α degradation at reoxygenation by impairing prolyl-hydroxylation: Role of krebs cycle metabolites. J. Biol. Chem. 2010, 285, 18217–18224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchi, S.; Rimessi, A.; Giorgi, C.; Baldini, C.; Ferroni, L.; Rizzuto, R.; Pinton, P. Akt kinase reducing endoplasmic reticulum Ca2+ release protects cells from Ca2+-dependent apoptotic stimuli. Biochem. Biophys. Res. Commun. 2008, 375, 501–505. [Google Scholar] [CrossRef] [Green Version]
- Bhuiyan, M.I.H.; Jung, S.Y.; Kim, H.J.; Lee, Y.S.; Jin, C. Major role of the PI3K/Akt pathway in ischemic tolerance induced by sublethal oxygen-glucose deprivation in cortical neurons in vitro. Arch. Pharm. Res. 2011, 34, 1023–1034. [Google Scholar] [CrossRef]
- Brini, M.; Calì, T.; Ottolini, D.; Carafoli, E. Neuronal calcium signaling: Function and dysfunction. Cell. Mol. Life Sci. 2014, 71, 2787–2814. [Google Scholar] [CrossRef]
- Augustine, G.J.; Santamaria, F.; Tanaka, K. Local calcium signaling in neurons. Neuron 2003, 40, 331–346. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Fonseca, K.; Massieu, L. Disruption of endoplasmic reticulum calcium stores is involved in neuronal death induced by glycolysis inhibition in cultured hippocampal neurons. J. Neurosci. Res. 2005, 82, 196–205. [Google Scholar] [CrossRef]
- Larsen, G.A.; Skjellegrind, H.K.; Berg-Johnsen, J.; Moe, M.C.; Vinje, M.L. Depolarization of mitochondria in isolated CA1 neurons during hypoxia, glucose deprivation and glutamate excitotoxicity. Brain Res. 2006, 1077, 153–160. [Google Scholar] [CrossRef]
- Henrich, M.; Buckler, K.J. Effects of anoxia, aglycemia, and acidosis on cytosolic Mg2+, ATP, and pH in rat sensory neurons. Am. J. Physiol. Cell Physiol. 2008, 294, C280–C294. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, H.N.; Wang, C.; Perry, D.C. Depletion of intracellular calcium stores is toxic to SH-SY5Y neuronal cells. Brain Res. 2002, 924, 159–166. [Google Scholar] [CrossRef]
- Stegeman, H.; Span, P.N.; Peeters, W.J.M.; Verheijen, M.M.G.; Grénman, R.; Meijer, T.W.H.; Kaanders, J.H.A.M.; Bussink, J. Interaction between hypoxia, AKT and HIF-1 signaling in HNSCC and NSCLC: Implications for future treatment strategies. Future Sci. OA 2016, 2. [Google Scholar] [CrossRef] [Green Version]
- Arsham, A.M.; Plas, D.R.; Thompson, C.B.; Simon, M.C. Akt and hypoxia-inducible factor-1 independently enhance tumor growth and angiogenesis. Cancer Res. 2004, 64, 3500–3507. [Google Scholar] [CrossRef] [Green Version]
- Thastrup, O.; Cullen, P.J.; Drobak, B.K.; Hanley, M.R.; Dawson, A.P. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2+-ATPase. Proc. Natl. Acad. Sci. USA 1990, 87, 2466–2470. [Google Scholar] [CrossRef] [Green Version]
- Divolis, G.; Mavroeidi, P.; Mavrofrydi, O.; Papazafiri, P. Differential effects of calcium on PI3K-Akt and HIF-1α survival pathways. Cell Biol. Toxicol. 2016, 32, 437–449. [Google Scholar] [CrossRef]
- Du, C.; Hu, R.; Csernansky, C.A.; Hsu, C.Y.; Choi, D.W. Very delayed infarction after mild focal cerebral ischemia: A role for apoptosis? J. Cereb. Blood Flow Metab. 1996, 16, 195–201. [Google Scholar] [CrossRef]
- Dirnagl, U.; Iadecola, C.; Moskowitz, M.A. Pathobiology of ischaemic stroke: An integrated view. Trends Neurosci. 1999, 22, 391–397. [Google Scholar] [CrossRef]
- Lee, D.R.; Helps, S.C.; Gibbins, I.L.; Nilsson, M.; Sims, N.R. Losses of NG2 and NeuN immunoreactivity but not astrocytic markers during early reperfusion following severe focal cerebral ischemia. Brain Res. 2003, 989, 221–230. [Google Scholar] [CrossRef]
- Thoren, A.E.; Helps, S.C.; Nilsson, M.; Sims, N.R. Astrocytic function assessed from 1-14C-acetate metabolism affer temporary focal cerebral ischemia in rats. J. Cereb. Blood Flow Metab. 2005, 25, 440–450. [Google Scholar] [CrossRef] [Green Version]
- Thoren, A.E.; Helps, S.C.; Nilsson, M.; Sims, N.R. The metabolism of 14C-glucose by neurons and astrocytes in brain subregions following focal cerebral ischemia in rats. J. Neurochem. 2006, 97, 968–978. [Google Scholar] [CrossRef]
- Kimbro, K.S.; Simons, J.W. Hypoxia-inducible factor-1 in human breast and prostate cancer. Endocr. Relat. Cancer 2006, 13, 739–749. [Google Scholar] [CrossRef]
- Lee, M.; Spokes, K.C.; Aird, W.C.; Abid, M.R. Intracellular Ca2+ can compensate for the lack of NADPH oxidase-derived ROS in endothelial cells. FEBS Lett. 2010, 584, 3131–3136. [Google Scholar] [CrossRef] [Green Version]
- Mavrofrydi, O.; Papazafiri, P. Hypoxia-inducible factor-1α increase is an early and sensitive marker of lung cells responding to benzo[a] pyrene. J. Environ. Pathol. Toxicol. Oncol. 2012, 31, 335–347. [Google Scholar] [CrossRef] [PubMed]
- Werno, C.; Zhou, J.; Brüne, B. A23187, ionomycin and thapsigargin upregulate mRNA of HIF-1α via endoplasmic reticulum stress rather than a rise in intracellular calcium. J. Cell. Physiol. 2008, 215, 708–714. [Google Scholar] [CrossRef] [PubMed]
- Yuan, G.; Nanduri, J.; Khan, S.; Semenza, G.L.; Prabhakar, N.R. Induction of HIF-1α expression by intermittent hypoxia: Involvement of NADPH oxidase, Ca2+ signaling, prolyl hydroxylases, and mTOR. J. Cell. Physiol. 2008, 217, 674–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.; Denmeade, S.; Carducci, M. HIF-1α and Calcium Signaling as Targets for Treatment of Prostate Cancer by Cardiac Glycosides. Curr. Cancer Drug Targets 2009, 9, 881–887. [Google Scholar] [CrossRef] [Green Version]
- Berchner-Pfannschmidt, U.; Petrat, F.; Doege, K.; Trinidad, B.; Freitag, P.; Metzen, E.; De Groot, H.; Fandrey, J. Chelation of cellular calcium modulates hypoxia-inducible gene expression through activation of Hypoxia-Inducible Factor-1α. J. Biol. Chem. 2004, 279, 44976–44986. [Google Scholar] [CrossRef] [Green Version]
- Oda, S.; Oda, T.; Takabuchi, S.; Nishi, K.; Wakamatsu, T.; Tanaka, T.; Adachi, T.; Fukuda, K.; Nohara, R.; Hirota, K. The calcium channel blocker cilnidipine selectively suppresses hypoxia-inducible factor 1 activity in vascular cells. Eur. J. Pharmacol. 2009, 606, 130–136. [Google Scholar] [CrossRef]
- Parekh, A.B. Store-operated CRAC channels: Function in health and disease. Nat. Rev. Drug Discov. 2010, 9, 399–410. [Google Scholar] [CrossRef]
- Gruszczynska-Biegala, J.; Pomorski, P.; Wisniewska, M.B.; Kuznicki, J. Differential roles for STIM1 and STIM2 in store-operated calcium entry in rat neurons. PLoS ONE 2011, 6, e19285. [Google Scholar] [CrossRef] [Green Version]
- Secondo, A.; Petrozziello, T.; Tedeschi, V.; Boscia, F.; Vinciguerra, A.; Ciccone, R.; Pannaccione, A.; Molinaro, P.; Pignataro, G.; Annunziato, L. ORAI1/STIM1 Interaction Intervenes in Stroke and in Neuroprotection Induced by Ischemic Preconditioning Through Store-Operated Calcium Entry. Stroke 2019, 50, 1240–1249. [Google Scholar] [CrossRef]
- Wang, L.; Lei, Q.; Zhao, S.; Xu, W.J.; Dong, W.; Ran, J.H.; Shi, Q.H.; Fu, J.F. Ginkgolide B Maintains Calcium Homeostasis in Hypoxic Hippocampal Neurons by Inhibiting Calcium Influx and Intracellular Calcium Release. Front. Cell. Neurosci. 2021, 14, 486. [Google Scholar] [CrossRef]
- Price, J.; Zaidi, A.K.; Bohensky, J.; Srinivas, V.; Shapiro, I.M.; Ali, H. Akt-1 mediates survival of chondrocytes from endoplasmic reticulum-induced stress. J. Cell. Physiol. 2010, 222, 502–508. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Han, Z.; Couvillon, A.D.; Exton, J.H. Critical role of endogenous Akt/IAPs and MEK1/ERK pathways in counteracting endoplasmic reticulum stress-induced cell death. J. Biol. Chem. 2004, 279, 49420–49429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scherbik, S.V.; Brinton, M.A. Virus-Induced Ca 2+ Influx Extends Survival of West Nile Virus-Infected Cells. J. Virol. 2010, 84, 8721–8731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.H.; Park, E.Y.; Hwang, K.H.; Park, K.S.; Choi, S.J.; Cha, S.K. Soluble αKlotho downregulates Orai1-mediated store-operated Ca2+ entry via PI3K-dependent signaling. Pflugers Arch. Eur. J. Physiol. 2021, 473, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Fedida-Metula, S.; Feldman, B.; Koshelev, V.; Levin-Gromiko, U.; Voronov, E.; Fishman, D. Lipid rafts couple store-operated Ca2+ entry to constitutive activation of PKB/Akt in a Ca2+/calmodulin-, Src- and PP2A-mediated pathway and promote melanoma tumor growth. Carcinogenesis 2012, 33, 740–750. [Google Scholar] [CrossRef] [Green Version]
- Grynkiewicz, G.; Poenie, M.; Tsien, R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985, 260, 3440–3450. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Culture Condition | [Ca2+]i, nM | ER Ca2+ Content (% [Ca2+]i) | Ca2+ Entry (% of ER Ca2+ Content) |
|---|---|---|---|
| Control | 47.8 ± 4 | 297.9 ± 21 | 71.8 ± 8 |
| GD 4 h | 64.3 ± 8 * | 107.5 ± 14 ** | 98.3 ± 6 ** |
| GD 4 h & 1 h Gluc | 62.5 ± 8 * | 146.8 ± 12 **‡ | 82.4 ± 5 |
| GD 24 h | 83.2 ± 12 * | 23.6 ± 2 † | 482.6 ± 3 † |
| GD 24 h & 2 h Gluc | 78.9 ± 6 * | 106.4 ± 12 ‡ | 66.7 ± 9 ‡ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kourti, M.; Liaropoulou, D.; Paschou, M.; Giagklisi, I.; Paschalidi, M.; Petani, E.; Papazafiri, P. Enhanced Ca2+ Entry Sustains the Activation of Akt in Glucose Deprived SH-SY5Y Cells. Int. J. Mol. Sci. 2022, 23, 1386. https://doi.org/10.3390/ijms23031386
Kourti M, Liaropoulou D, Paschou M, Giagklisi I, Paschalidi M, Petani E, Papazafiri P. Enhanced Ca2+ Entry Sustains the Activation of Akt in Glucose Deprived SH-SY5Y Cells. International Journal of Molecular Sciences. 2022; 23(3):1386. https://doi.org/10.3390/ijms23031386
Chicago/Turabian StyleKourti, Maria, Danai Liaropoulou, Maria Paschou, Ioanna Giagklisi, Maria Paschalidi, Evangelia Petani, and Panagiota Papazafiri. 2022. "Enhanced Ca2+ Entry Sustains the Activation of Akt in Glucose Deprived SH-SY5Y Cells" International Journal of Molecular Sciences 23, no. 3: 1386. https://doi.org/10.3390/ijms23031386
APA StyleKourti, M., Liaropoulou, D., Paschou, M., Giagklisi, I., Paschalidi, M., Petani, E., & Papazafiri, P. (2022). Enhanced Ca2+ Entry Sustains the Activation of Akt in Glucose Deprived SH-SY5Y Cells. International Journal of Molecular Sciences, 23(3), 1386. https://doi.org/10.3390/ijms23031386