
NSAIDs and Cancer Resolution: New Paradigms beyond Cyclooxygenase



Abstract
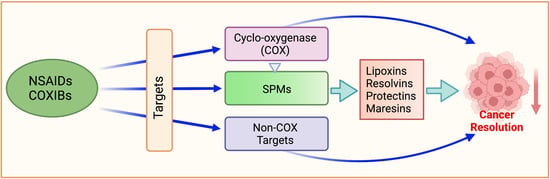
:
1. Introduction
2. NSAIDs Protect against Cancer: Proof of Principle
3. Molecular Targets of NSAIDs
3.1. COX Cascade
COX-2 Inhibition
4. NSAID Targets beyond COX
4.1. NF-κB
4.2. PDK-1/Akt Pathway
4.3. PPARs
4.4. Mitogen-Activated Protein Kinases (MAPKs)
4.5. Wnt/β-Catenin Pathway
4.6. Phosphodiesterases (PDEs)
4.7. NSAIDs and the mTOR Pathway
4.8. Autophagy
4.9. Cell Kinetics
4.10. Cytochrome C Release
4.11. NSAID-Activated Gene (NAG-1)
4.12. Ca2+ Mobilization
4.13. Inhibition of Angiogenesis
4.14. Carbonic Anhydrase (CA) Inhibition
5. Specialized Pro-Resolving Mediators (SPMs)
5.1. Lipid Class Switching
5.1.1. Lipoxins
5.1.2. Resolvins
5.1.3. Protectins and Maresins
6. Summary and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 13-HDPA | 13-hydroxyDPA |
| 13-HODE | 13-Hydroxyoctadecadienoic acid |
| 13-HpDPA | 13-hydroperoxyDPA |
| 15-HEPE | 15-hydroxyeicosapentaenoic acid |
| 15-HETE | 15-Hydroxyeicosatetraenoic acid |
| 17-R-HDHA | 17-R-hydroperoxyDHA |
| 18-HEPE | 18-hydroxyeicosapentaenoic acid |
| AA | Arachidonic acid; (20:4, n-6) |
| Akt | Protein kinase B (PKB) |
| Apaf-1 | Apoptotic protease activating factor 1 |
| APC | Adenomatous polyposis coli |
| APPROVe | Adenomatous Polyp Prevention on Vioxx |
| AT-LXA4 | Aspirin-triggered lipoxin A4 |
| ATL | Aspirin-triggered lipoxin |
| AT-PD1/AT-NPD1 | Aspirin-triggered protectin D1/Aspirin-triggered neuroprotectin D1 |
| AT-RvD | Aspirin-triggered D-series resolvin |
| AT-RvE | Aspirin-triggered E-series resolvin |
| Bad | Bcl-2-agonist of death |
| Bak | Bcl-2 homologous antagonist/killer |
| Bax | Bcl-2-like protein 4 |
| Bcl-2 | B-cell lymphoma 2 |
| BH3 | Bcl-2 homology 3 |
| Bid | BH3-interacting domain death agonist |
| Bim | Bcl-2-like protein 11 |
| CA | Carbonic anhydrase |
| CAI | Carbonic anhydrase inhibitor |
| cAMP | Cyclic adenosine monophosphate |
| cdk4 | Cyclin-dependent kinase 4 |
| cGMP | Cyclic guanosine monophosphate |
| CLASS | Celecoxib Long-Term Arthritis Safety Study |
| CO2 | Carbon dioxide |
| COX | Cyclooxygenase |
| COXIBs | COX-2 selective NSAIDs |
| DCP | Dichlorophenamide |
| DHA | Docosahexaenoic acid; (22:6, n-3) |
| DMC | 2,5-Dimethyl-celecoxib |
| DPA | Docosapentaenoic acid (22:5, n-3) |
| EGR-1 | Early growth response factor 1 |
| EPA | Eicosapentaenoic acid; (20:5, n-3) |
| ER | Endoplasmic reticulum |
| ERK | Extracellular-signal-regulated kinase |
| FAP | Familial adenomatous polyposis |
| GDP | Guanosine diphosphate |
| GI | Gastrointestinal |
| GSK-3β | Glycogen synthase kinase-3beta |
| GTP | Guanosine triphosphate |
| IL | Interleukin |
| IP3 | Inositol trisphosphate |
| IκB | I-kappa-B |
| JNK | c-Jun N-terminal kinase |
| LA | Linoleic acid; (18:2, n-6) |
| LOX | Lipoxygenase |
| LT | Leukotriene |
| LTA4 | Leukotriene A4 |
| LX | Lipoxin |
| MAPK | Mitogen-activated protein kinase |
| MaR | Maresin |
| MEK | MAPK/ERK kinase |
| MIF | migration inhibitory factor |
| mTOR | Mammalian target of rapamycin |
| mTORC1 | Mammalian target of rapamycin complex 1 |
| mTORC2 | Mammalian target of rapamycin complex 2 |
| NAG-1 | NSAID-activated gene |
| NF-κB | Nuclear factor kappa light-chain enhancer of activated B cells |
| NK cells | Natural killer cells |
| NSAIDs | Nonsteroidal anti-inflammatory drugs |
| PD1/NPD1 | Protectin D1/Neuroprotectin D1 |
| PDE | Phosphodiesterase |
| PDK-1 | phosphoinositide-dependent kinase-1 |
| PDs | Protectins |
| PG | Prostaglandin |
| PGI2 | Prostacyclin |
| PI3K | Phosphoinositide 3-kinase |
| PIP2 | Phosphatidylinositol (4,5)- bisphosphate |
| PIP3 | Phosphatidylinositol (3,4,5)- trisphosphate |
| PKG | Protein kinase G |
| PMNs | Polymorphonuclear leukocytes |
| PPAR | Peroxisome-proliferator-activated receptor |
| PUFAs | Polyunsaturated fatty acids |
| Puma | p53 upregulated modulator of apoptosis; Bcl-2-binding component 3 |
| RTX | retinoid X receptor |
| Rv | Resolvin |
| RvD | D-series resolvin |
| RvE | E-series resolvin |
| RvT | T-series resolvin |
| SPMs | Specialized pro-resolving mediators |
| TAM | Tumor-associated macrophages |
| Tcf-4 | Transcription factor 4 |
| TGF-β | Transforming growth factor-beta |
| TNF-α | Tumor necrosis factor-alpha |
| TXA2 | Thromboxane A2 |
| VEGF | Vascular endothelial growth factor |
| VIGOR | Vioxx Gastrointestinal Outcomes Research |
| Wnt/β-catenin | Wingless-related integration site/beta-catenin |
References
- Ley, K. Physiology of Inflammation; Methods in Physiology Series; Springer: Berlin/Heidelberg, Germany, 2001; ISBN 9781461475125. [Google Scholar]
- Kashfi, K. Anti-inflammatory agents as cancer therapeutics. Adv. Pharm. 2009, 57, 31–89. [Google Scholar]
- Balkwill, F.A. Mantovani Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef]
- Kumar, R.; Clermont, G.; Vodovotz, Y.; Chow, C.C. The dynamics of acute inflammation. J. Theor. Biol. 2004, 230, 145–155. [Google Scholar] [CrossRef] [Green Version]
- Freire, O.M.; van Dyke, T.E. Natural resolution of inflammation. Periodontol. 2000 2013, 63, 149–164. [Google Scholar] [CrossRef] [Green Version]
- Nathan, C.; Ding, A. Nonresolving Inflammation. Cell 2010, 140, 871–882. [Google Scholar] [CrossRef] [Green Version]
- Thun, M.J.; Henley, S.J.; Patrono, C. Nonsteroidal anti-inflammatory drugs as anticancer agents: Mechanistic pharmacologic and clinical issues. J. Natl. Cancer Inst. 2002, 94, 252–266. [Google Scholar] [CrossRef] [Green Version]
- Sandler, R.S.; Halabi, S.; Baron, J.A.; Budinger, S.; Paskett, E.; Keresztes, R.; Petrelli, N.; Pipas, J.M.; Karp, D.D.; Loprinzi, C.L.; et al. A randomized trial of aspirin to prevent colorectal adenomas in patients with previous colorectal cancer. N. Engl. J. Med. 2003, 348, 883–890. [Google Scholar] [CrossRef]
- Baron, J.A.; Cole, B.F.; Sandler, R.S.; Haile, R.W.; Ahnen, D.; Bresalier, R.; Mckeown-Eyssen, G.; Summers, R.W.; Rothstein, R.; Burke, C.A.; et al. A randomized trial of aspirin to prevent colorectal adenomas. N. Engl. J. Med. 2003, 348, 891–899. [Google Scholar] [CrossRef]
- Baandrup, L.; Kjaer, S.K.; Olsen, J.H.; Dehlendorff, C.; Friis, S. Low-dose aspirin use and the risk of ovarian cancer in Denmark. Ann. Oncol. 2015, 26, 787–792. [Google Scholar] [CrossRef] [PubMed]
- Trabert, B.; Ness, R.B.; Lo-Ciganic, W.-H.; Murphy, M.A.; Goode, E.L.; Poole, E.M.; Brinton, L.A.; Webb, P.M.; Nagle, C.M.; Jordan, S.J.; et al. Aspirin nonaspirin nonsteroidal anti-inflammatory drug and acetaminophen use and risk of invasive epithelial ovarian cancer: A pooled analysis in the Ovarian Cancer Association Consortium. J. Natl. Cancer Inst. 2014, 106, djt431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shebl, F.M.; Sakoda, L.C.; Black, A.; Koshiol, J.; Andriole, G.L.; Grubb, R.; Church, T.R.; Chia, D.; Zhou, C.K.; Chu, L.W.; et al. Aspirin but not ibuprofen use is associated with reduced risk of Prostate cancer: A PLCO study. Br. J. Cancer 2012, 107, 207–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahasrabuddhe, V.V.; Sahasrabuddhe, V.V.; Chan, A.T.; Alavanja, M.C.; Beane-Freeman, L.E.; Buring, J.E.; Chen, J.; Chong, D.Q.; Freedman, N.D.; Fuchs, C.S.; et al. Nonsteroidal anti-inflammatory drug use chronic liver disease and hepatocellular carcinoma. J. Natl. Cancer Inst. 2012, 104, 1808–1814. [Google Scholar] [CrossRef] [Green Version]
- Clouser, M.C.; Roe, D.J.; Foote, J.A.; Harrism, R.B. Effect of non-steroidal anti-inflammatory drugs on non-melanoma skin cancer incidence in the SKICAP-AK trial. Pharm. Drug Saf. 2009, 18, 276–283. [Google Scholar] [CrossRef]
- Elmets, C.A.; Viner, J.L.; Pentland, A.P.; Cantrell, W.; Lin, H.-Y.; Bailey, H.; Kang, S.; Linden, K.G.; Heffernan, M.; Duvic, M.; et al. Chemoprevention of nonmelanoma skin cancer with celecoxib: A randomized double-blind placebo-controlled trial. J. Natl. Cancer Inst. 2010, 102, 1835–1844. [Google Scholar] [CrossRef]
- Sun, L.; Yu, S. Meta-analysis: Non-steroidal anti-inflammatory drug use and the risk of esophageal squamous Cell carcinoma. Dis. Esophagus 2011, 24, 544–549. [Google Scholar] [CrossRef]
- Cui, X.J.; He, Q.; Zhang, J.-M.; Fan, H.-J.; Wen, Z.-F.; Qin, Y.-R. High-dose aspirin consumption contributes to decreased risk for pancreatic cancer in a systematic review and meta-analysis. Pancreas 2014, 43, 135–140. [Google Scholar] [CrossRef]
- Yiannakopoulou, E. Aspirin and NSAIDs for breast cancer chemoprevention. Eur. J. Cancer Prev. 2015, 24, 416–421. [Google Scholar] [CrossRef]
- de Pedro, M.; Baeza, S.; Escudero, M.T.; Dierssen-Sotos, T.; Gómez-Acebo, I.; Pollán, M.; Llorca, J. Effect of COX-2 inhibitors and other non-steroidal inflammatory drugs on breast cancer risk: A meta-analysis. Breast Cancer Res. Treat. 2015, 149, 525–536. [Google Scholar] [CrossRef]
- Daugherty, S.E.; Pfeiffer, R.M.; Sigurdson, A.J.; Hayes, R.B.; Leitzmann, M.; Schatzkin, A.; Hollenbeck, A.R.; Silverman, D.T. Nonsteroidal antiinflammatory drugs and bladder cancer: A pooled analysis. Am. J. Epidemiol. 2011, 173, 721–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicastro, H.L.; Grubbs, C.J.; Juliana, M.M.; Bode, A.M.; Kim, M.-S.; Lu, Y.; You, M.; Milne, G.; Boring, D.; Steele, V.E.; et al. Preventive effects of NSAIDs NO-NSAIDs and NSAIDs plus difluoromethylornithine in a chemically induced urinary bladder cancer model. Cancer Prev. Res. 2014, 7, 246–254. [Google Scholar] [CrossRef] [Green Version]
- Becker, C.; Wilson, J.C.; Jick, S.S.; Meier, C.R. Non-steroidal anti-inflammatory drugs and the risk of head and neck cancer: A case-control analysis. Int. J. Cancer 2015, 137, 2424–2431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothwell, P.M.; Price, J.F.; Fowkes, F.G.R.; Zanchetti, A.; Roncaglioni, M.C.; Tognoni, G.; Lee, R.; Belch, J.J.; Wilson, M.; Mehta, Z.; et al. Short-term effects of daily aspirin on cancer incidence mortality and non-vascular death: Analysis of the time course of risks and benefits in 51 randomised controlled trials. Lancet 2012, 379, 1602–1612. [Google Scholar] [CrossRef]
- Algra, A.M.; Rothwell, P.M. Effects of regular aspirin on long-term cancer incidence and metastasis: A systematic comparison of evidence from observational studies versus randomised trials. Lancet Oncol. 2012, 13, 518–527. [Google Scholar] [CrossRef]
- Simmons, D.L.; Botting, R.M.; Hla, T. Cyclooxygenase Isozymes: The Biology of Prostaglandin Synthesis and Inhibition. Pharm. Rev. 2004, 56, 387–437. [Google Scholar] [CrossRef] [Green Version]
- Brunton, L.; Knollman, B.; Hilal-Dandan, R. Goodman and Gilman’s the Pharmacological Basis of Therapeutics; McGraw-Hill Education/Medical: London, UK, 2017. [Google Scholar]
- Booker, R. NSAIDs: Uses effects risks and benefits. Pract. Nurse 2013, 43, 34–37. [Google Scholar]
- Gryglewski, R.J.; Dembínska-Kieć, A.; Korbut, R. A possible role of thromboxane A2 (TXA2) and prostacyclin (PGI2) in circulation. Acta Biol. Med. Ger. 1978, 37, 715–723. [Google Scholar]
- Fiorucci, S.; Antonelli, E. Cyclo-oxygenase isoenzymes. Structural basis for selective inhibition of cyclo-oxygenases by anti-inflammatory agents. Dig. Liver Dis. 2001, 33 (Suppl. S2), S2–S7. [Google Scholar] [CrossRef]
- Kune, G.; Kune, S.; Watson, L. Colorectal cancer risk chronic illnesses operations and medications: Case-control results from the Melbourne Colorectal Cancer Study. Cancer Res. 1988, 48, 4399–4404. [Google Scholar] [CrossRef] [Green Version]
- Ashok, V.; Dash, C.; Rohan, T.E.; Sprafka, J.M.; Terry, P.D. Selective cyclooxygenase-2 (COX-2) inhibitors and breast cancer risk. Breast 2011, 20, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Rozic, J.G.; Chakraborty, C.; Lala, P.K. Cyclooxygenase inhibitors retard murine mammary tumor progression by reducing tumor Cell migration invasiveness and angiogenesis. Int. J. Cancer 2001, 93, 497–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Stevens, J.; Hilton, M.B.; Seaman, S.; Conrads, T.P.; Veenstra, T.D.; Logsdon, D.; Morris, H.; Swing, D.A.; Patel, N.L.; et al. COX-2 Inhibition Potentiates Antiangiogenic Cancer Therapy and Prevents Metastasis in Preclinical Models. Sci. Trans. Med. 2014, 6, 242ra84. [Google Scholar] [CrossRef] [PubMed]
- Barnard, M.E.; Poole, E.M.; Curhan, G.C.; Eliassen, A.H.; Rosner, B.A.; Terry, K.L.; Tworoger, S.S. Association of Analgesic Use With Risk of Ovarian Cancer in the Nurses’ Health Studies. JAMA Oncol. 2018, 4, 1675–1682. [Google Scholar] [CrossRef] [Green Version]
- Bjorkman, D.J. Current status of nonsteroidal anti-inflammatory drug (NSAID) use in the United States: Risk factors and frequency of complications. Am. J. Med. 1999, 107, 3S–8S; discussion 8S-10S. [Google Scholar] [CrossRef]
- Wallace, J.L. Prostaglandins NSAIDs and gastric mucosal protection: Why doesn’t the stomach digest itself? Physiol. Rev. 2008, 88, 1547–1565. [Google Scholar] [CrossRef]
- Kashfi, K.; Rigas, B. Non-COX-2 targets and cancer: Expanding the molecular target repertoire of chemoprevention. Biochem. Pharm. 2005, 70, 969–986. [Google Scholar] [CrossRef]
- Bennett, A.; Civier, A.; Hensby, C.N.; Melhuish, P.B.; Stamford, I.F. Measurement of arachidonate and its metabolites extracted from human normal and malignant gastrointestinal tissues. Gut 1987, 28, 315–318. [Google Scholar] [CrossRef] [Green Version]
- Rigas, B.; Goldman, I.S.; Levine, L. Altered eicosanoid levels in human colon cancer. J. Lab. Clin. Med. 1993, 122, 518–523. [Google Scholar]
- Eberhart, C.E.; Coffey, R.J.; Radhika, A.; Giardiello, F.M.; Ferrenbach, S.; Dubois, R.N. Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and carcinomas. Gastroenterology 1994, 107, 1183–1188. [Google Scholar] [CrossRef]
- Wu, Q.-B.; Sun, G.-P. Expression of COX-2 and HER-2 in colorectal cancer and their correlation. World J. Gastroenterol. Gastroenterol. 2015, 21, 6206–6214. [Google Scholar] [CrossRef]
- Sheng, J.; Sun, H.; Yu, F.-B.; Li, B.; Zhang, Y.; Zhu, Y.-T. The Role of Cyclooxygenase-2 in Colorectal Cancer. Int. J. Med. Sci. 2020, 17, 1095–1101. [Google Scholar] [CrossRef]
- Dannenberg, A.J.; Subbaramaiah, K. Targeting cyclooxygenase-2 in human neoplasia: Rationale and promise. Cancer Cancer Cell Cell 2003, 4, 431–436. [Google Scholar] [CrossRef] [Green Version]
- Oshima, M.; Dinchuk, J.E.; Kargman, S.L.; Oshima, H.; Hancock, B.; Kwong, E.; Trzaskos, J.M.; Evans, J.F.; Taketo, M.M. Suppression of intestinal polyposis in ApcÐ716 knockout mice by inhibition of cyclooxygenase 2 (COX-2). Cell 1996, 87, 803–809. [Google Scholar] [CrossRef] [Green Version]
- North, G.L. Celecoxib as adjunctive therapy for treatment of colorectal cancer. Ann. Pharm. 2001, 35, 1638–1643. [Google Scholar] [CrossRef]
- Hanif, R.; Pittas, A.; Feng, Y.; Koutsos, M.I.; Qiao, L.; Staiano-Coico, L.; Shiff, S.I.; Rigas, B. Effects of nonsteroidal anti-inflammatory drugs on proliferation and on induction of apoptosis in colon cancer Cells by a prostaglandin-independent pathway. Biochem. Pharm. 1996, 52, 237–245. [Google Scholar] [CrossRef]
- Elder, D.J.E.; Halton, D.E.; Hague, A.; Paraskeva, C. Induction of apoptotic cell death in human colorectal carcinoma Cell lines by a cyclooxygenase-2 (COX-2)-selective nonsteroidal anti-inflammatory drug—Independence from COX-2 protein expression. Clin. Cancer Res. 1997, 3, 1679–1683. [Google Scholar]
- Dang, C.T.; Dannenberg, A.J.; Subbaramaiah, K.; Dickler, M.N.; Moasser, M.M.; Seidman, A.D.; D’Andrea, G.M.; Theodoulou, M. Phase II study of celecoxib and trastuzumab in metastatic breast cancer patients who have progressed after prior trastuzumab-based treatments. Clin. Cancer Res. 2004, 10, 4062–4067. [Google Scholar] [CrossRef] [Green Version]
- Becerra, C.R.; Frenkel, E.P.; Ashfaq, R.; Gaynor, R.B. Increased toxicity and lack of efficacy of Rofecoxib in combination with chemotherapy for treatment of metastatic colorectal cancer: A phase II study. Int. J. Cancer 2003, 105, 868–872. [Google Scholar] [CrossRef]
- Park, A.; Lee, Y.; Kim, M.S.; Kang, Y.J.; Park, Y.J.; Jung, H.; Kim, T.D.; Lee, H.G.; Choi, I.; Yoon, S.R. Prostaglandin E2 Secreted by Thyroid Cancer Cells Contributes to Immune Escape Through the Suppression of Natural Killer (NK) Cell Cytotoxicity and NK Cell Differentiation. Front. Immunol. 2018, 9, 1859. [Google Scholar] [CrossRef]
- Bacchi, S.; Palumbo, P.; Sponta, A.; Coppolino, M.F. Clinical pharmacology of non-steroidal anti-inflammatory drugs: A review. Antiinflamm. Antiallergy Agents Med. Chem. 2012, 11, 52–64. [Google Scholar] [CrossRef]
- Fiorucci, S. Prevention of nonsteroidal anti-inflammatory drug-induced ulcer: Looking to the future. Gastroenterol. Clin. N. Am. 2009, 38, 315–332. [Google Scholar] [CrossRef]
- Wallace, J.L.; Viappiani, S.; Bolla, M. Cyclooxygenase-inhibiting nitric oxide donators for osteoarthritis. Trends Pharm. Sci. 2009, 30, 112–117. [Google Scholar] [CrossRef]
- Chinthapalli, K. High dose NSAIDs may double the risk of heart attacks and heart failure says new study. BMJ 2013, 346, f3533. [Google Scholar] [CrossRef]
- Fiorucci, S.; Distrutti, E. COXIBs CINODs and HS-releasing NSAIDs: Current perspectives in the development of safer non steroidal anti-inflammatory drugs. Curr. Med. Chem. 2011, 18, 3494–3505. [Google Scholar] [CrossRef]
- Pepine, C.J.; Gurbel, P.A. Cardiovascular safety of NSAIDs: Additional insights after PRECISION and point of view. Clin. Cardiol. 2017, 40, 1352–1356. [Google Scholar] [CrossRef] [Green Version]
- Walker, C.; Biasucci, L.M. Cardiovascular safety of non-steroidal anti-inflammatory drugs revisited. Postgrad. Med. 2018, 130, 55–71. [Google Scholar] [CrossRef]
- Bombardier, C.; Laine, L.; Reicin, A.; Shapiro, D.; Burgos-Vargas, R.; Davis, B.; Day, R.; Ferraz, M.B.; Hawkey, C.J.; Hochberg, M.C.; et al. Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis. VIGOR Study Group. N. Engl. J. Med. 2000, 343, 1520–1528. [Google Scholar] [CrossRef]
- Silverstein, F.E.; Faich, G.; Goldstein, J.L.; Simon, L.S.; Pincus, T.; Whelton, A.; Makuch, R.; Eisen, G.; Agrawal, N.M.; Stenson, W.F.; et al. Gastrointestinal toxicity with celecoxib vs. nonsteroidal anti-inflammatory drugs for osteoarthritis and rheumatoid arthritis: The CLASS study: A randomized controlled trial. Celecoxib Long-term Arthritis Safety Study. JAMA 2000, 284, 1247–1255. [Google Scholar] [CrossRef] [Green Version]
- Inotai, A.; Mészáros, A. Economic evaluation of nonsteroidal anti-inflammatory drug strategies in rheumatoid arthritis. Int. J. Technol. Assess Health Care 2009, 25, 190–195. [Google Scholar] [CrossRef]
- Chen, L.C.; Ashcroft, D.M. Risk of myocardial infarction associated with selective COX-2 inhibitors: Meta-analysis of randomised controlled trials. Pharm. Drug Saf. 2007, 16, 762–772. [Google Scholar] [CrossRef]
- Bresalier, R.S.; Sandler, R.S.; Quan, H.; Bolognese, J.A.; Oxenius, B.; Horgan, K.; Lines, C.; Riddell, R.; Morton, D.; Lanas, A.; et al. Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. N. Engl. J. Med. 2005, 352, 1092–1102. [Google Scholar] [CrossRef] [Green Version]
- FitzGerald, G.A.; Patrono, C. The coxibs selective inhibitors of cyclooxygenase-2. N. Engl. J. Med. 2001, 345, 433–442. [Google Scholar] [CrossRef]
- Singh, G.; Miller, J.D.; Huse, D.M.; Pettitt, D.; D’Agostino, R.B.; Russell, M.W. Consequences of increased systolic blood pressure in patients with osteoarthritis and rheumatoid arthritis. J. Rheumatol. 2003, 30, 714–719. [Google Scholar]
- Kearney, P.M.; Baigent, C.; Godwin, J.; Halls, H.; Emberson, J.R.; Patrono, C. Do selective cyclo-oxygenase-2 inhibitors and traditional non-steroidal anti-inflammatory drugs increase the risk of atherothrombosis? Meta-analysis of randomised trials. BMJ 2006, 332, 1302–1308. [Google Scholar] [CrossRef] [Green Version]
- Park, M.H.; Hong, J.T. Roles of NF-κB in cancer and inflammatory diseases and their therapeutic approaches. Cells 2016, 5, 15. [Google Scholar] [CrossRef]
- Kopp, E.; Ghosh, S. Inhibition of NF-kappa B by sodium salicylate and aspirin. Science 1994, 265, 956–959. [Google Scholar] [CrossRef]
- Stark, L.A.; Din, F.V.N.; Zwacka, R.M.; Dunlop, M.G. Aspirin-induced activation of the NF-kappaB signaling pathway: A novel mechanism for aspirin-mediated apoptosis in colon cancer cells. FASEB J. 2001, 15, 1273–1275. [Google Scholar] [CrossRef]
- Cho, M.; Gwak, J.; Park, S.; Won, J.; Kim, D.E.; Yea, S.S.; Cha, I.J.; Kim, T.K.; Shin, J.G.; Oh, S. Diclofenac attenuates Wnt/β-catenin signaling in colon cancer cells by activation of NF-κB. FEBS Lett. 2005, 579, 4213–4218. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, Y.; Gaynor, R.B. Role of the NF-kappaB pathway in the pathogenesis of human disease states. Mol. Med. Curr. Mol. Med. 2001, 1, 287–296. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Yin, M.J.; Lin, K.M.; Gaynor, R. BSulindac inhibits activation of the NF-kappaB pathway. J. Biol. Chem. 1999, 274, 27307–27314. [Google Scholar] [CrossRef] [Green Version]
- Arico, S.; Pattingre, S.; Bauvy, C.; Gane, P.; Barbat, A.; Codogno, P.; Ogier-Denis, E. Celecoxib induces apoptosis by inhibiting 3-phosphoinositide-dependent protein kinase-1 activity in the human colon cancer HT-29 cell line. J. Biol. Chem. 2002, 277, 27613–27621. [Google Scholar] [CrossRef] [Green Version]
- Benelli, R.; Barboro, P.; Costa, D.; Astigiano, S.; Barbieri, O.; Capaia, M.; Poggi, A.; Ferrari, N. Multifocal signal modulation therapy by celecoxib: A strategy for managing castration-resistant prostate cancer. Int. J. Mol. Sci. 2019, 20, 6091. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.S.; Kim, J.E.; Huang, Z.; Chen, H.; Langfald, A.; Lubet, R.A.; Grubbs, C.J.; Dong, Z.; Bode, A.M. Naproxen Induces Cell-Cycle Arrest and Apoptosis in Human Urinary Bladder Cancer Cell Lines and Chemically Induced Cancers by Targeting PI3K. Cancer Prev. Res. 2014, 7, 236–245. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, J.M.; Lenhard, J.M.; Oliver, B.B.; Ringold, G.M.; Kliewer, S.A. Peroxisome proliferator-activated receptors α and γ are activated by indomethacin and other non-steroidal anti-inflammatory drugs. J. Biol. Chem. 1997, 272, 3406–3410. [Google Scholar] [CrossRef] [Green Version]
- He, T.-C.; Chan, T.A.; Vogelstein, B.; Kinzler, K.W. PPARδ Is an APC-regulated target of nonsteroidal anti-inflammatory drugs. Cell 1999, 99, 335–345. [Google Scholar] [CrossRef] [Green Version]
- Park, B.H.; Vogelstein, B.; Kinzler, K.W. Genetic disruption of Genetic disruption of PPARδ decreases the tumorigenicity of human colon cancer cells. Proc. Natl. Acad. Sci. USA 2001, 98, 2598. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.H.; Chen, G.S.; Ou, X.L.; Yang, Y.; Luo, C.; Zhang, Y.; Shao, Y.; Xu, H.C.; Xiao, B.; Xue, Y.P.; et al. Inhibition of COX-2 and activation of peroxisome proliferator-activated receptor gamma synergistically inhibits proliferation and induces apoptosis of human pancreatic carcinoma cells. Cancer Lett. 2009, 275, 247–255. [Google Scholar] [CrossRef]
- Narayanan, N.K.; Narayanan, B.A.; Reddy, B.S. A combination of docosahexaenoic acid and celecoxib prevents prostate cancer cell growth in vitro and is associated with modulation of nuclear factor-kappaB and steroid hormone receptors. Int. J. Oncol. 2005, 26, 785–792. [Google Scholar] [CrossRef]
- Husain, S.S.; Szabo, I.L.; Pai, R.; Soreghan, B.; Jones, M.K.; Tarnawski, A.S. MAPK (ERK2) kinase—A key target for NSAIDs-induced inhibition of gastric cancer cell proliferation and growth. Life Sci. 2001, 69, 3045–3054. [Google Scholar] [CrossRef]
- Ou, Y.C.; Yang, C.R.; Cheng, C.L.; Raung, S.L.; Hung, Y.Y.; Chen, C.J. Indomethacin induces apoptosis in 786-O renal cell carcinoma cells by activating mitogen-activated protein kinases and AKT. Eur. J. Pharmacol. 2007, 563, 49–60. [Google Scholar] [CrossRef]
- Kim, T.; Jin, S.; Kim, W.; Kang, E.; Choi, K.; Kim, H.; Shin, S.; Kang, J. Prolonged activation of mitogen-activated protein kinases during NSAID-induced apoptosis in HT-29 colon cancer cells. Int. J. Colorectal Dis. 2001, 16, 167–173. [Google Scholar] [CrossRef]
- Elder, D.J.E.; Halton, D.E.; Playle, L.C.; Paraskeva, C. The MEK/ERK pathway mediates COX-2-selective NSAID-induced apoptosis and induced COX-2 protein expression in colorectal carcinoma cells. Int. J. Cancer 2002, 99, 323–327. [Google Scholar] [CrossRef]
- Sun, Y.; Sinicrope, F.A. Selective inhibitors of MEK1/ERK44/42 and p38 mitogen-activated protein kinases potentiate apoptosis induction by sulindac sulfide in human colon carcinoma cells. Mol. Cancer Therap. 2005, 4, 51–59. [Google Scholar]
- Setia, S.; Nehru, B.; Sanyal, S.N. Upregulation of MAPK/Erk and PI3K/Akt pathways in ulcerative colitis-associated colon cancer. Biomed. Pharm. 2014, 68, 1023–1029. [Google Scholar] [CrossRef]
- Jia, Z.; Zhang, H.; Ma, C.; Li, N.; Wang, M. Celecoxib enhances apoptosis of the liver cancer cells via regulating ERK/JNK/P38 pathway. J. Buon 2021, 26, 875–881. [Google Scholar]
- Park, S.W.; Kim, H.S.; Hah, J.W.; Jeong, W.J.; Kim, K.H.; Sung, M.W. Celecoxib inhibits cell proliferation through the activation of ERK and p38 MAPK in head and neck squamous cell carcinoma cell lines. Anticancer Drugs 2010, 21, 823–830. [Google Scholar] [CrossRef]
- Heidel Florian, H.; Bullinger, L.; Feng, Z.; Wang, Z.; Neff, T.A.; Stein, L.; Kalaitzidis, D.; Lane, S.W.; Armstrong, S.A. Genetic and pharmacologic inhibition of β-catenin targets imatinib-resistant leukemia stem cells in CML. Cell Stem Cell 2012, 10, 412–424. [Google Scholar] [CrossRef] [Green Version]
- Han, A.; Song, Z.; Tong, C.; Hu, D.; Bi, X.; Augenlicht, L.H.; Yang, W. Sulindac suppresses β-catenin expression in human cancer cells. Eur. J. Pharm. 2008, 583, 26–31. [Google Scholar] [CrossRef] [Green Version]
- Rice, P.L.; Kelloff, J.; Sullivan, H.; Driggers, L.J.; Beard, K.S.; Kuwada, S.; Piazza, G.; Ahnen, D.J. Sulindac metabolites induce caspase- and proteasome-dependent degradation of β-catenin protein in human colon cancer cells. Mol. Cancer Therap. 2003, 2, 885–892. [Google Scholar]
- Boon, E.M.J.; Keller, J.J.; Wormhoudt, T.A.M.; Giardiello, F.M.; Offerhaus, G.J.A.; Van Der Neut, R.; Pals, S.T. Sulindac targets nuclear β-catenin accumulation and Wnt signalling in adenomas of patients with familial adenomatous polyposis and in human colorectal cancer cell lines. Br. J. Cancer 2004, 90, 224–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sareddy, G.R.; Kesanakurti, D.; Kirti, P.B.; Babu, P.P. Nonsteroidal anti-inflammatory drugs diclofenac and celecoxib attenuates Wnt/β-catenin/Tcf signaling pathway in human glioblastoma cells. Neurochem. Res. 2013, 38, 2313–2322. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Chen, Y.; Liu, H.; Yang, J.; Song, X.; Zhao, J.; He, N.; Zhou, C.J.; Wang, Y.; Huang, C.; et al. Celecoxib targets breast cancer stem cells by inhibiting the synthesis of prostaglandin E and down-regulating the Wnt pathway activity. Oncotarget 2017, 8, 115254–115269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tinsley, H.N.; Gary, B.D.; Keeton, A.B.; Zhang, W.; Abadi, A.H.; Reynolds, R.C.; Piazza, G.A. Sulindac sulfide selectively inhibits growth and induces apoptosis of human breast tumor cells by phosphodiesterase 5 inhibition elevation of cyclic GMP and activation of protein kinase G. Mol. Cancer Therap. 2009, 8, 3331–3340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tinsley, H.N.; Gary, B.D.; Thaiparambil, J.; Li, N.; Lu, W.; Li, Y.; Maxuitenko, Y.Y.; Keeton, A.B.; Piazza, G.A. Colon tumor cell growth-inhibitory activity of sulindac sulfide and other nonsteroidal anti-inflammatory drugs is associated with phosphodiesterase 5 inhibition. Cancer Prev. Res. 2010, 3, 1303–1313. [Google Scholar] [CrossRef] [Green Version]
- Tinsley, H.N.; Gary, B.D.; Keeton, A.B.; Lu, W.; Li, Y.; Piazza, G.A. Inhibition of PDE5 by sulindac sulfide selectively induces apoptosis and attenuates oncogenic Wnt/β-catenin-mediated transcription in human breast tumor cells. Cancer Prev. Res. 2011, 4, 1275–1284. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Xi, Y.; Tinsley, H.N.; Gurpinar, E.; Gary, B.D.; Zhu, B.; Li, Y.; Chen, X.; Keeton, A.B.; Abadi, A.H.; et al. Sulindac selectively inhibits colon tumor cell growth by activating the cGMP/PKG pathway to suppress Wnt/β-catenin signaling. Mol. Cancer Therap. 2013, 12, 1848–1859. [Google Scholar] [CrossRef] [Green Version]
- Din, F.V.; Valanciute, A.; Houde, V.P.; Zibrova, D.; Green, K.A.; Sakamoto, K.; Alessi, D.R.; Dunlop, M.G. Aspirin inhibits mTOR signaling activates amp-activated protein kinase and induces autophagy in colorectal cancer cells. Gastroenterology 2012, 142, 1504–1515. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; He, D.; Song, E.; Jiang, M.; Song, Y. Celecoxib enhances the sensitivity of non-small-cell lung cancer Cells to radiation-induced apoptosis through downregulation of the Akt/mTOR signaling pathway and COX-2 expression. PLoS ONE 2019, 14, e0223760. [Google Scholar] [CrossRef]
- Lu, Y.; Liu, X.F.; Liu, T.R.; Fan, R.F.; Xu, Y.C.; Zhang, X.Z.; Liu, L.L. Celecoxib exerts antitumor effects in HL-60 acute leukemia cells and inhibits autophagy by affecting lysosome function. Biomed. Pharm. 2016, 84, 1551–1557. [Google Scholar] [CrossRef]
- Huang, S.; Sinicrope, F.A. Celecoxib-induced apoptosis is enhanced by ABT-737 and by inhibition of autophagy in human colorectal cancer cells. Autophagy 2010, 6, 256–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, K.H.; Kuo, K.L.; Ho, I.L.; Chang, H.C.; Chuang, Y.T.; Lin, W.C.; Lee, P.Y.; Chang, S.C.; Chiang, C.K.; Pu, Y.S.; et al. Celecoxib-induced cytotoxic effect is potentiated by inhibition of autophagy in human urothelial carcinoma cells. PLoS ONE 2013, 8, e82034. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Dong, X.; Xiu, P.; Wang, F.; Liu, J.; Wei, H.; Xu, Z.; Liu, F.; Li, T.; Li, J. Blocking autophagy enhances meloxicam lethality to hepatocellular carcinoma by promotion of endoplasmic reticulum stress. Cell Prolif. 2015, 48, 691–704. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Li, R.; Xiu, P.; Dong, X.; Xu, Z.; Zhai, B.; Liu, F.; Jiang, H.; Sun, X.; Li, J.; et al. Meloxicam executes its antitumor effects against hepatocellular carcinoma in COX-2- dependent and -independent pathways. PLoS ONE 2014, 9, e92864. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Shi, J.; Mao, S.Y.; Xu, Y.S.; Zhang, D.; Feng, L.Y.; Zhang, B.; Yan, Y.Y.; Wang, S.C.; Pan, J.P.; et al. Role of p38 MAPK in enhanced human cancer cells killing by the combination of aspirin and ABT-737. J. Cell Mol. Med. 2015, 19, 408–417. [Google Scholar] [CrossRef]
- Grösch, S.; Tegeder, I.; Niederberger, E.; Bräutigam, L.; Geisslinger, G. COX-2 independent induction of cell cycle arrest and apoptosis in colon cancer cells by the selective COX-2 inhibitor celecoxib. FASEB J. 2001, 15, 1–22. [Google Scholar] [CrossRef]
- Kim, J.S.; Baek, S.J.; Sali, T.; Eling, T.E. The conventional nonsteroidal anti-inflammatory drug sulindac sulfide arrests ovarian cancer cell growth via the expression of NAG-1/MIC-1/GDF-15. Mol. Cancer Therap. 2005, 4, 487–493. [Google Scholar] [CrossRef]
- Carrasco-Pozo, C.; Pastene, E.; Vergara, C.; Zapata, M.; Sandoval, C.; Gotteland, M. Stimulation of cytosolic and mitochondrial calcium mobilization by indomethacin in Caco-2 cells: Modulation by the polyphenols quercetin resveratrol and rutin. Biochim. Biophys. Acta (Bba)—Gen. Subj. 2012, 1820, 2052–2061. [Google Scholar] [CrossRef]
- Aggarwal, S.; Taneja, N.; Lin, L.; Orringer, M.B.; Rehemtulla, A.; Beer, D.G. Indomethacin-Induced Apoptosis in Esophageal Adenocarcinoma Cells Involves Upregulation of Bax and Translocation of Mitochondrial Cytochrome C Independent of COX-2 Expression. Neoplasia 2000, 2, 346–356. [Google Scholar] [CrossRef] [Green Version]
- Maier, T.J.; Schilling, K.; Schmidt, R.; Geisslinger, G.; Grösch, S. Cyclooxygenase-2 (COX-2)-dependent and -independent anticarcinogenic effects of celecoxib in human colon carcinoma cells. Biochem. Pharm. 2004, 67, 1469–1478. [Google Scholar] [CrossRef]
- Wang, Y.J.; Niu, X.P.; Yang, L.; Han, Z.; Ma, Y.J. Effects of celecoxib on cycle kinetics of gastric cancer cells and protein expression of cytochrome C and caspase-9. Asian Pac. J. Cancer Prev. 2013, 14, 2343–2347. [Google Scholar]
- Li, M.; Wu, X.; Xu, X.-C. Induction of apoptosis in colon cancer cells by cyclooxygenase-2 inhibitor NS398 through a cytochrome c-dependent pathway. Clinical Cancer Res. 2001, 7, 1010–1016. [Google Scholar]
- Zimmermann, K.C.; Waterhouse, N.J.; Goldstein, J.C.; Schuler, M.; Green, D.R. Aspirin induces apoptosis through release of cytochrome c from mitochondria. Neoplasia 2000, 2, 505–513. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.S.; Baek, S.J.; Flake, G.P.; Loftin, C.D.; Calvo, B.F.; Eling, T.E. Expression and regulation of nonsteroidal anti-inflammatory drug–activated gene (NAG-1) in human and mouse tissue. Gastroenterology 2002, 122, 1388–1398. [Google Scholar] [CrossRef] [PubMed]
- Baek, S.J.; Kim, K.S.; Nixon, J.B.; Wilson, L.C.; Eling, T.E. Cyclooxygenase inhibitors regulate the expression of a TGF-β superfamily member that has proapoptotic and antitumorigenic activities. Mol. Pharm. 2001, 59, 901–908. [Google Scholar] [CrossRef] [Green Version]
- Kadowaki, M.; Yoshioka, H.; Kamitani, H.; Watanabe, T.; Wade, P.A.; Eling, T.E. DNA methylation-mediated silencing of nonsteroidal anti-inflammatory drug-activated gene (NAG-1/GDF15) in glioma cell lines. Int. J. Cancer 2012, 130, 267–277. [Google Scholar] [CrossRef]
- Jang, T.J.; Kang, H.J.; Kim, J.R.; Yang, C.H. Non-steroidal anti-inflammatory drug activated gene (NAG-1) expression is closely related to death receptor-4 and -5 induction which may explain sulindac sulfide induced gastric Cancer Cell apoptosis. Carcinogenesis 2004, 25, 1853–1858. [Google Scholar] [CrossRef] [Green Version]
- Baek, S.J.; Wilson, L.C.; Lee, C.H.; Eling, T.E. Dual function of nonsteroidal anti-inflammatory drugs (nsaids): Inhibition of cyclooxygenase and induction of NSAID-activated gene. J. Pharm. Exp. Therap. 2002, 301, 1126–1131. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.T.; Chen, Z.X.; Wei, B.; Zhang, B.; Wang, C.H.; Huang, M.H.; Liu, R.; Tang, C.W. Preoperative growth inhibition of human gastric adenocarcinoma treated with a combination of celecoxib and octreotide. Acta Pharm. Sin. 2007, 28, 1842–1850. [Google Scholar] [CrossRef] [Green Version]
- Iguchi, G.; Chrysovergis, K.; Lee, S.H.; Baek, S.J.; Langenbach, R.; Eling, T.E. A reciprocal relationship exists between non-steroidal anti-inflammatory drug-activated gene-1 (NAG-1) and cyclooxygenase-2. Cancer Lett. 2009, 282, 152–158. [Google Scholar] [CrossRef] [Green Version]
- Youns, M.; Efferth, T.; Hoheisel, J.D. Transcript profiling identifies novel key players mediating the growth inhibitory effect of NS-398 on human pancreatic cancer cells. Eur. J. Pharm. 2011, 650, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Wynne, S.; Djakiew, D. NSAID Inhibition of Prostate Cancer Cell Migration Is Mediated by Nag-1 Induction via the p38 MAPK-p75NTR Pathway. Molecular Cancer Res. 2010, 8, 1656–1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.C.; Chang, C.M.; Hsu, W.L.; Chiu, S.J.; Tsai, Y.T.; Chou, Y.H.; Hou, M.F.; Wang, J.Y.; Lee, M.H.; Tsai, K.L.; et al. Indomethacin inhibits cancer cell migration via attenuation of cellular calcium mobilization. Molecules 2013, 18, 6584–6596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, A.J.; Hsu, A.L.; Lin, H.P.; Song, X.; Chen, C.S. The cyclo-oxygenase-2 inhibitor celecoxib perturbs intracellular calcium by inhibiting endoplasmic reticulum Ca2+-ATPases: A plausible link with its anti-tumour effect and cardiovascular risks. Biochem. J. 2002, 366 Pt 3, 831–837. [Google Scholar] [CrossRef] [Green Version]
- Pyrko, P.; Kardosh, A.; Liu, Y.T.; Soriano, N.; Xiong, W.; Chow, R.H.; Uddin, J.; Petasis, N.A.; Mircheff, A.K.; Farley, R.A.; et al. Calcium-activated endoplasmic reticulum stress as a major component of tumor cell death induced by 2,5-dimethyl-celecoxib a non-coxib analogue of celecoxib. Mol. Cancer Therap. 2007, 6, 1262–1275. [Google Scholar] [CrossRef] [Green Version]
- Coca, R.; Soler, F.; Cortes-Castell, E.; Gil-Guillen, V.; Fernandez-Belda, F. Inhibition mechanism of the intracellular transporter Ca2+-Pump from sarco-endoplasmic reticulum by the antitumor agent dimethyl-celecoxib. PLoS ONE 2014, 9, e102083. [Google Scholar] [CrossRef] [Green Version]
- Dai, X.; Yan, J.; Fu, X.; Pan, Q.; Sun, D.; Xu, Y.; Wang, J.; Nie, L.; Tong, L.; Shen, A.; et al. Aspirin inhibits cancer metastasis and angiogenesis via targeting heparanase. Clin. Cancer Res. 2017, 23, 6267–6278. [Google Scholar] [CrossRef] [Green Version]
- Maity, G.; Chakraborty, J.; Ghosh, A.; Haque, I.; Banerjee, S.; Banerjee, S.K. Aspirin suppresses tumor cell-induced angiogenesis and their incongruity. J. Cell Commun. Signal. 2019, 13, 491–502. [Google Scholar] [CrossRef]
- Akrami, H.; Aminzadeh, S.; Fallahi, H. Inhibitory effect of ibuprofen on tumor survival and angiogenesis in gastric cancer cell. Tumor Biol. 2015, 36, 3237–3243. [Google Scholar] [CrossRef]
- Yao, M.; Zhou, W.; Sangha, S.; Albert, A.; Chang, A.J.; Liu, T.C.; Wolfe, M.M. Effects of nonselective cyclooxygenase inhibition with low-dose ibuprofen on tumor growth angiogenesis metastasis and survival in a mouse model of colorectal cancer. Clin. Cancer Res. 2005, 11, 1618–1628. [Google Scholar] [CrossRef] [Green Version]
- Vlad, A.M.; Colţău, M.I.; Pușcaș, I. Relationship between NSAIDs gastric antisecretory and carbonic anhydrase isoenzyme. Acta Med. Trans. 2015, 20, 73. [Google Scholar]
- Weber, A.; Casini, A.; Heine, A.; Kuhn, D.; Supuran, C.T.; Scozzafava, A.; Klebe, G. Unexpected nanomolar inhibition of carbonic anhydrase by COX-2-selective celecoxib: New pharmacological opportunities due to related binding site recognition. J. Med. Chem. 2004, 47, 550–557. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, J.F.; Carlsson, U.; Hammarström, P.; Sokol, G.H.; Cantilena, L.R. the cyclooxygenase-2 inhibitor celecoxib is a potent inhibitor of human carbonic anhydrase II. Inflammation 2004, 28, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Clària, J.; Lee, M.H.; Serhan, C.N. Aspirin-triggered lipoxins (15-epi-LX) are generated by the human lung adenocarcinoma cell line (A549)-neutrophil interactions and are potent inhibitors of cell proliferation. Mol. Med. 1996, 2, 583–596. [Google Scholar] [CrossRef] [Green Version]
- Gilligan, M.M.; Gartung, A.; Sulciner, M.L.; Norris, P.C.; Sukhatme, V.P.; Bielenberg, D.R.; Huang, S.; Kieran, M.W.; Serhan, C.N.; Panigrahy, D. Aspirin-triggered proresolving mediators stimulate resolution in cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 6292–6297. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Yuan, X.; Li, W.; Cao, Q.; Shu, Y. Aspirin-triggered resolvin D1 inhibits TGF-β1-induced EMT through the inhibition of the mTOR pathway by reducing the expression of PKM2 and is closely linked to oxidative stress. Int. J. Mol. Med. 2016, 38, 1235–1242. [Google Scholar] [CrossRef] [Green Version]
- Vannitamby, A.; Saad, M.I.; Aloe, C.; Wang, H.; Kumar, B.; Vlahos, R.; Selemidis, S.; Irving, L.; Steinfort, D.; Jenkins, B.J.; et al. Aspirin-triggered resolvin D1 reduces proliferation and the neutrophil to lymphocyte ratio in a mutant KRAS-driven lung adenocarcinoma model. Cancers 2021, 13, 3224. [Google Scholar] [CrossRef]
- Lawrence, T.; Gilroy, D.W.; Colville-Nash, P.R.; Willoughby, D.A. Possible new role for NF-κB in the resolution of inflammation. Nat. Med. 2001, 7, 1291–1297. [Google Scholar] [CrossRef]
- Huang, H.Y.; Zhang, Z.J.; Cao, C.B.; Wang, N.; Liu, F.F.; Peng, J.Q.; Ren, X.J.; Qian, J. The TLR4/NF-κB signaling pathway mediates the growth of colon cancer. Eur. Rev. Med. Pharm. Sci. 2014, 18, 3834–3843. [Google Scholar]
- Nomura, A.; Majumder, K.; Giri, B.; Dauer, P.; Dudeja, V.; Roy, S.; Banerjee, S.; Saluja, A.K. Inhibition of NF-kappa B pathway leads to deregulation of epithelial–mesenchymal transition and neural invasion in pancreatic cancer. Lab. Investig. 2016, 96, 1268–1278. [Google Scholar] [CrossRef] [Green Version]
- Pires, B.R.B.; Mencalha, A.L.; Ferreira, G.M.; de Souza, W.F.; Morgado-Díaz, J.A.; Maia, A.M.; Corrêa, S.; Abdelhay, E.S. NF-kappaB is involved in the regulation of EMT genes in breast cancer cells. PLoS ONE 2017, 12, e0169622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Annunziata, C.M.; Stavnes, H.T.; Kleinberg, L.; Berner, A.; Hernandez, L.F.; Birrer, M.J.; Steinberg, S.M.; Davidson, B.; Kohn, E.C. Nuclear factor κB transcription factors are coexpressed and convey a poor outcome in ovarian cancer. Cancer 2010, 116, 3276–3284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen Charlie, D.; Sawyers, L. Charles NF-κB activates prostate-specific antigen expression and is upregulated in androgen-independent prostate cancer. Mol. Cell. Biol. 2002, 22, 2862–2870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ismail A, H.; Lessard, L.; Mes-Masson, A.M.; Saad, F. Expression of NF-κB in prostate cancer lymph node metastases. Prostate 2004, 58, 308–313. [Google Scholar] [CrossRef]
- Nishio, H.; Yaguchi, T.; Sugiyama, J.; Sumimoto, H.; Umezawa, K.; Iwata, T.; Susumu, N.; Fujii, T.; Kawamura, N.; Kobayashi, A.; et al. Immunosuppression through constitutively activated NF-κB signalling in human ovarian cancer and its reversal by an NF-κB inhibitor. Br. J. Cancer 2014, 110, 2965–2974. [Google Scholar] [CrossRef] [Green Version]
- Longnecker, D.S.; Wiebkin, P.; Schaeffer, B.K.; Roebuck, B.D. Experimental carcinogenesis in the pancreas. Int. Rev. Exp. Pathol. 1984, 26, 177–229. [Google Scholar]
- Kaltschmidt, B.; Greiner, J.F.; Kadhim, H.M.; Kaltschmidt, C. Subunit-Specific Role of NF-κB in Cancer. Biomedicines 2018, 6, 44. [Google Scholar] [CrossRef] [Green Version]
- Naugler, W.E.; Karin, M. NF-κB and cancer—Identifying targets and mechanisms. Curr. Opin. Genet. Dev. 2008, 18, 19–26. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2018, 9, 7204–7218. [Google Scholar] [CrossRef] [Green Version]
- Hoesel, B.; Schmid, J.A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef] [Green Version]
- Sethi, G.; Sung, B.; Aggarwal, B.B. TNF: A master switch for inflammation to cancer. Front. Biosci. 2008, 13, 5094–5107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Withoff, S.; Verma, I.M. Inflammation-associated cancer: NF-κB is the lynchpin. Trends Immunol. 2005, 26, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhou, B.P. TNF-α/NF-κB/Snail pathway in cancer cell migration and invasion. Br. J. Cancer 2010, 102, 639–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Li, J.; Zhang, L.; Huang, S.; Zhao, X.; Zhao, X. Celecoxib induced apoptosis against different breast cancer cell lines by down-regulated NF-κB pathway. Biochem. Biophys. Res. Commun. 2017, 490, 969–976. [Google Scholar] [CrossRef] [PubMed]
- Hemmings, B.A.; Restuccia, D.F. Pi3k-PKB/AKT pathway. Cold Spring Harbor Perspect. Biol. 2012, 4, a011189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nitulescu, G.M.; Van De Venter, M.; Nitulescu, G.; Ungurianu, A.; Juzenas, P.; Peng, Q.; Olaru, O.T.; Grădinaru, D.; Tsatsakis, A.; Tsoukalas, D.; et al. The Akt pathway in oncology therapy and beyond. Int. J. Oncol. 2018, 53, 2319–2331. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Chen, X.; Hay, N. Akt as a target for cancer therapy: More is not always better (lessons from studies in mice). Br. J. Cancer 2017, 117, 159–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalhoub, N.; Baker, S.J. PTEN and the PI3-kinase pathway in cancer. Annu. Rev. Pathol. 2009, 4, 127–150. [Google Scholar] [CrossRef] [Green Version]
- Nakatani, K.; Thompson, D.A.; Barthel, A.; Sakaue, H.; Liu, W.; Weigel, R.J.; Roth, R.A. Up-regulation of Akt3 in estrogen receptor-deficient breast cancers and androgen-independent Prostate cancer lines. J. Biol. Chem. 1999, 274, 21528–21532. [Google Scholar] [CrossRef] [Green Version]
- Cristiano, B.E.; Chan, J.C.; Hannan, K.M.; Lundie, N.A.; Marmy-Conus, N.J.; Campbell, I.G.; Phillips, W.A.; Robbie, M.; Hannan, R.D.; Pearson, R.B. A specific role for AKT3 in the genesis of ovarian cancer through modulation of G-M phase transition. Cancer Res. 2006, 66, 11718–11725. [Google Scholar] [CrossRef] [Green Version]
- Osaki, M.; Oshimura, M.; Ito, H. PI3K-Akt pathway: Its functions and alterations in human cancer. Apoptosis 2004, 9, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Hennessy, B.T.; Smith, D.L.; Ram, P.T.; Lu, Y.; Mills, G.B. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat. Rev. Drug Discov. 2005, 4, 988–1004. [Google Scholar] [CrossRef] [PubMed]
- Hou, C.C.; Hung, S.L.; Kao, S.H.; Chen, T.H.; Lee, H.M. Celecoxib induces heme-oxygenase expression in glomerular mesangial cells. Ann. New York Acad. Sci. 2005, 1042, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wang, C.; Dong, H.; Wang, X.; Gao, F.; Zhang, S.; Zhang, X. Aspirin has a better effect on PIK3CA mutant colorectal cancer cells by PI3K/Akt/raptor pathway. Mol. Med. 2020, 26, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, J.; Moller, D.E. The mechanisms of action of PPARs. Ann. Rev. Med. 2002, 53, 409–435. [Google Scholar] [CrossRef] [Green Version]
- Michalik, L.; Auwerx, J.; Berger, J.P.; Chatterjee, V.K.; Glass, C.K.; Gonzalez, F.J.; Grimaldi, P.A.; Kadowaki, T.; Lazar, M.A.; O’Rahilly, S.; et al. International Union of Pharmacology. LXI. Peroxisome Proliferator-Activated Receptors. Pharm. Rev. 2006, 58, 726. [Google Scholar] [CrossRef]
- Sarraf, P.; Mueller, E.; Jones, D.; King, F.J.; DeAngelo, D.J.; Partridge, J.B.; Holden, S.A.; Chen, L.B.; Singer, S.; Fletcher, C.; et al. Differentiation and reversal of malignant changes in colon cancer through PPARgamma. Nat. Med. 1998, 4, 1046–1052. [Google Scholar] [CrossRef]
- Hase, T.; Yoshimura, R.; Mitsuhashi, M.; Segawa, Y.; Kawahito, Y.; Wada, S.; Nakatani, T.; Sano, H. Expression of peroxisome proliferator-activated receptors in human testicular cancer and growth inhibition by its agonists. Urology 2002, 60, 542–547. [Google Scholar] [CrossRef]
- Wang, X.; Wang, G.; Shi, Y.; Sun, L.; Gorczynski, R.; Li, Y.J.; Xu, Z.; Spaner, D.E. PPAR-delta promotes survival of breast cancer cells in harsh metabolic conditions. Oncogenesis 2016, 5, e232. [Google Scholar] [CrossRef] [Green Version]
- Brockman, J.A.; Gupta, R.A.; DuBois, R.N. Activation of PPARγ leads to inhibition of anchorage-independent growth of human colorectal cancer cells. Gastroenterology 1998, 115, 1049–1055. [Google Scholar] [CrossRef]
- Michalik, L.; Desvergne, B.; Wahli, W. Peroxisome-proliferator-activated receptors and cancers: Complex stories. Nat. Nat. Rev. Cancer 2004, 4, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Kohno, H.; Yoshitani, S.I.; Takashima, S.; Okumura, A.; Murakami, A.; Hosokawa, M. Ligands for peroxisome proliferator-activated receptors alpha and gamma inhibit chemically induced colitis and formation of aberrant crypt foci in rats. Cancer Res. 2001, 61, 2424–2428. [Google Scholar] [PubMed]
- Osawa, E.; Nakajima, A.; Wada, K.; Ishimine, S.; Fujisawa, N.; Kawamori, T.; Matsuhashi, N.; Kadowaki, T.; Ochiai, M.; Sekihara, H.; et al. Peroxisome proliferator-activated receptor gamma ligands suppress colon carcinogenesis induced by azoxymethane in mice. Gastroenterology 2003, 124, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Saez, E.; Tontonoz, P.; Nelson, M.C.; Alvarez, J.G.; Baird, S.M.; Thomazy, V.A.; Evans, R.M. Activators of the nuclear receptor PPARgamma enhance colon polyp formation. Nat. Med. 1998, 4, 1058–1061. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, A.M.; Chen, I.; Desreumaux, P.; Najib, J.; Fruchart, J.C.; Geboes, K.; Briggs, M.; Heyman, R.; Auwerx, J. Activation of the peroxisome proliferator-activated receptor gamma promotes the development of colon tumors in C57BL/6J-APCMin/+ mice. Nat. Med. 1998, 4, 1053–1057. [Google Scholar] [CrossRef]
- Wasan, H.S.; Novelli, M.; Bee, J.; Bodmer, W.F. Dietary fat influences on polyp phenotype in multiple intestinal neoplasia mice. Proc. Natl. Acad. Sci. USA 1997, 94, 3308–3313. [Google Scholar] [CrossRef] [Green Version]
- Peters, J.M.; Hollingshead, H.E.; Gonzalez, F.J. Role of peroxisome-proliferator-activated receptor beta/delta (PPARbeta/delta) in gastrointestinal tract function and disease. Clin. Sci. 2008, 115, 107–127. [Google Scholar] [CrossRef] [Green Version]
- Peters, J.M.; Aoyama, T.; Cattley, R.C.; Nobumitsu, U.; Hashimoto, T.; Gonzalez, F.J. Role of peroxisome proliferator-activated receptor alpha in altered cell cycle regulation in mouse liver. Carcinogenesis 1998, 19, 1989–1994. [Google Scholar] [CrossRef] [Green Version]
- Reed, K.R.; Sansom, O.J.; Hayes, A.J.; Gescher, A.J.; Winton, D.J.; Peters, J.M.; Clarke, A.R. PPARδ status and Apc-mediated tumourigenesis in the mouse intestine. Oncogene 2004, 23, 8992–8996. [Google Scholar] [CrossRef] [Green Version]
- Shureiqi, I.; Jiang, W.; Zuo, X.; Wu, Y.; Stimmel, J.B.; Leesnitzer, L.M.; Morris, J.S.; Fan, H.Z.; Fischer, S.M.; Lippman, S.M. The 15-lipoxygenase-1 product 13-S-hydroxyoctadecadienoic acid down-regulates PPAR-δ to induce apoptosis in colorectal cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 9968. [Google Scholar] [CrossRef] [Green Version]
- Brash, A.R.; Boeglin, W.E.; Chang, M.S. Discovery of a second 15S-lipoxygenase in humans. Proc. Natl. Acad. Sci. USA 1997, 94, 6148–6152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baer, A.N.; Costello, P.B.; Green, F.A. In vivo activation of an omega-6 oxygenase in human skin. Biochem. Biophys. Res. Commun. 1991, 180, 98–104. [Google Scholar] [CrossRef]
- Ikawa, H.; Kamitani, H.; Calvo, B.F.; Foley, J.F.; Eling, T.E. Expression of 15-lipoxygenase-1 in human colorectal cancer. Cancer Res. 1999, 59, 360–366. [Google Scholar] [PubMed]
- Shureiqi, I.; Chen, D.; Lotan, R.; Yang, P.; Newman, R.A.; Fischer, S.M.; Lippman, S.M. 15-Lipoxygenase-1 mediates nonsteroidal anti-inflammatory drug-induced apoptosis independently of cyclooxygenase-2 in colon cancer cells. Cancer Res. 2000, 60, 6846–6850. [Google Scholar] [PubMed]
- Tachibana, K.; Yamasaki, D.; Ishimoto, K. The Role of PPARs in Cancer. PPAR Res. 2008, 2008, 102737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.K.; Choi, E.-J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta (Bba)–Mol. Basis Dis. 2010, 1802, 396–405. [Google Scholar] [CrossRef] [Green Version]
- Raman, M.; Chen, W.; Cobb, M.H. Differential regulation and properties of MAPKs. Oncogene 2007, 26, 3100–3112. [Google Scholar] [CrossRef] [Green Version]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef] [Green Version]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef] [Green Version]
- Thalhamer, T.; McGrath, M.A.; Harnett, M.M. MAPKs and their relevance to arthritis and inflammation. Rheumatology 2008, 47, 409–414. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Kim, S.C.; Yu, T.; Yi, Y.S.; Rhee, M.H.; Sung, G.H.; Yoo, B.C.; Cho, J.Y. Functional roles of p38 mitogen-activated protein kinase in macrophage-mediated inflammatory responses. Mediat. Inflamm. 2014, 2014, 352371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, P.; Han, J.; Hui, L. MAPK signaling in inflammation-associated cancer development. Protein Cell Cell 2010, 1, 218–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Zhang, Y.; Chen, S.; Liu, W.; Lin, Y.; Zhang, H.; Yu, F. Non-Steroidal Anti-Inflammatory Drugs (NSAIDs) sensitize melanoma cells to MEK inhibition and inhibit metastasis and relapse by inducing degradation of AXL. Pigment Cell Melanoma Res. 2021. [Google Scholar] [CrossRef] [PubMed]
- Torii, S.; Yamamoto, T.; Tsuchiya, Y.; Nishida, E. ERK MAP kinase in G1 cell cycle progression and cancer. Cancer Sci. 2006, 97, 697–702. [Google Scholar] [CrossRef] [PubMed]
- Papachristou, D.J.; Batistatou, A.; Sykiotis, G.P.; Varakis, I.; Papavassiliou, A.G. Activation of the JNK–AP-1 signal transduction pathway is associated with pathogenesis and progression of human osteosarcomas. Bone 2003, 32, 364–371. [Google Scholar] [CrossRef]
- Avisetti, D.R.; Babu, K.S.; Kalivendi, S.V. Activation of p38/JNK pathway is responsible for embelin induced apoptosis in lung cancer cells: Transitional role of reactive oxygen species. PLoS ONE 2014, 9, e87050. [Google Scholar] [CrossRef]
- Hardwick, J.C.H.; Van Den Brink, G.R.; Offerhaus, G.J.; Van Deventer, S.J.; Peppelenbosch, M.P. NF-kappaB p38 MAPK and JNK are highly expressed and active in the stroma of human colonic adenomatous polyps. Oncogene 2001, 20, 819–827. [Google Scholar] [CrossRef] [Green Version]
- Malumbres, M.; Barbacid, M. RAS oncogenes: The first 30 years. Nat. Nat. Rev. Cancer 2003, 3, 459–465. [Google Scholar] [CrossRef]
- Haigis, K.M.; Kendall, K.R.; Wang, Y.; Cheung, A.; Haigis, M.C.; Glickman, J.N.; Niwa-Kawakita, M.; Sweet-Cordero, A.; Sebolt-Leopold, J.; Shannon, K.M.; et al. Differential effects of oncogenic K-Ras and N-Ras on proliferation differentiation and tumor progression in the colon. Nat. Genet. 2008, 40, 600–608. [Google Scholar] [CrossRef] [Green Version]
- Bos, J.L.; Fearon, E.R.; Hamilton, S.R.; Verlaan–de Vries, M.; van Boom, J.H.; van der Eb, A.J.; Vogelstein, B. Prevalence of ras gene mutations in human colorectal cancers. Nature 1987, 327, 293–297. [Google Scholar] [CrossRef]
- Forrester, K.; Almoguera, C.; Han, K.; Grizzle, W.E.; Perucho, M. Detection of high incidence of K-ras oncogenes during human colon tumorigenesis. Nature 1987, 327, 298–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodenhuis, S.; van de Wetering, M.L.; Mooi, W.J.; Evers, S.G.; van Zandwijk, N.; Bos, J.L. Mutational Activation of the K-ras Oncogene. N. Engl. J. Med. 1987, 317, 929–935. [Google Scholar] [CrossRef] [PubMed]
- Almoguera, C.; Shibata, D.; Forrester, K.; Martin, J.; Arnheim, N.; Perucho, M. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell 1988, 53, 549–554. [Google Scholar] [CrossRef] [Green Version]
- Campbell, S.L.; Khosravi-Far, R.; Rossman, K.L.; Clark, G.J.; Der, C.J. Increasing complexity of Ras signaling. Oncogene 1998, 17, 1395–1413. [Google Scholar] [CrossRef] [Green Version]
- Vogelstein, B.; Fearon, E.R.; Hamilton, S.R.; Kern, S.E.; Preisinger, A.C.; Leppert, M.; Smits, A.M.; Bos, J.L. Genetic alterations during colorectal-tumor development. N. Engl. J. Med. 1988, 319, 525–532. [Google Scholar] [CrossRef] [Green Version]
- Calcagno, S.R.; Li, S.; Colon, M.; Kreinest, P.A.; Thompson, E.A.; Fields, A.P.; Murray, N.R. Oncogenic K-ras promotes early carcinogenesis in the mouse proximal colon. Int. J. Cancer 2008, 122, 2462–2470. [Google Scholar] [CrossRef] [Green Version]
- Schwenger, P.; Bellosta, P.; Vietor, I.; Basilico, C.; Skolnik, E.Y.; Vilček, J. Sodium salicylate induces apoptosis via p38 mitogen-activated protein kinase but inhibits tumor necrosis factor-induced c-Jun N-terminal kinase/stress-activated protein kinase activation. Proc. Natl. Acad. Sci. USA 1997, 94, 2869–2873. [Google Scholar] [CrossRef] [Green Version]
- Schwenger, P.; Alpert, D.; Skolnik, E.Y.; Vilček, J. Cell-type-specific activation of c-Jun N-terminal kinase by salicylates. J. Cell. Physiol. 1999, 179, 109–114. [Google Scholar] [CrossRef]
- Yuan, Z.; Zhao, J.; Wang, Z.; Ren, G.; Zhang, Z.; Ma, G. Effects of aspirin on hepatocellular carcinoma and its potential molecular mechanism. J. Buon 2020, 25, 981–986. [Google Scholar]
- Sun, J.; Guo, C.; Zheng, W.; Zhang, X. Aspirin inhibits proliferation and promotes apoptosis of hepatocellular carcinoma Cells via wnt/β-catenin signaling pathway. Panminerva Med. 2019. [Google Scholar] [CrossRef]
- Li, X.; Xiang, Y.; Li, F.; Yin, C.; Li, B.; Ke, X. WNT/β-catenin signaling pathway regulating t cell-inflammation in the tumor microenvironment. Front. Immunol. 2019, 10, 2293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva-García, O.; Valdez-Alarcón, J.J.; Baizabal-Aguirre, V.M. The Wntβ-Catenin signaling pathway controls the inflammatory response in infections caused by pathogenic bacteria. Mediat. Inflamm. 2014, 2014, 310183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doucas, H.; Garcea, G.; Neal, C.P.; Manson, M.M.; Berry, D.P. Changes in the Wnt signalling pathway in gastrointestinal cancers and their prognostic significance. Eur. J. Cancer 2005, 41, 365–379. [Google Scholar] [CrossRef] [PubMed]
- Pai, S.G.; Carneiro, B.A.; Mota, J.M.; Costa, R.; Leite, C.A.; Barroso-Sousa, R.; Kaplan, J.B.; Chae, Y.K.; Giles, F.J. Wnt/beta-catenin pathway: Modulating anticancer immune response. J. Hematol. Oncol. 2017, 10, 101. [Google Scholar] [CrossRef] [Green Version]
- Suryawanshi, A.; Tadagavadi, R.K.; Swafford, D.; Manicassamy, S. Modulation of inflammatory responses by wnt/β-catenin signaling in dendritic cells: A novel immunotherapy target for autoimmunity and cancer. Front. Immunol. 2016, 7, 460. [Google Scholar] [CrossRef] [Green Version]
- Ma, B.; Hottiger, M.O. Crosstalk between Wnt/β-Catenin and NF-κB Signaling Pathway during Inflammation. Front. Immunol. 2016, 7, 378. [Google Scholar] [CrossRef]
- Manoharan, I.; Hong, Y.; Suryawanshi, A.; Angus-Hill, M.L.; Sun, Z.; Mellor, A.L.; Munn, D.H.; Manicassamy, S. TLR2-dependent activation of β-catenin pathway in dendritic cells induces regulatory responses and attenuates autoimmune inflammation. J. Immunol. 2014, 193, 4203–4213. [Google Scholar] [CrossRef] [Green Version]
- Hadjihannas, M.V.; Brückner, M.; Jerchow, B.; Birchmeier, W.; Dietmaier, W.; Behrens, J. Aberrant Wnt/β-catenin signaling can induce chromosomal instability in colon cancer. Proc. Natl. Acad. Sci. USA 2006, 103, 10747–10752. [Google Scholar] [CrossRef] [Green Version]
- Jung, Y.-S.; Jun, S.; Lee, S.H.; Sharma, A.; Park, J.I. Wnt2 complements Wnt/β-catenin signaling in colorectal cancer. Oncotarget 2015, 6, 37257–37268. [Google Scholar] [CrossRef] [Green Version]
- Ying, J.; Li, H.; Yu, J.; Ng, K.M.; Poon, F.F.; Wong, S.C.C.; Chan, A.T.; Sung, J.J.; Tao, Q. wnt5a exhibits tumor-suppressive activity through antagonizing the Wnt/β-catenin signaling and is frequently methylated in colorectal cancer. Clin. Cancer Res. 2008, 14, 55–61. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.; Guan, H.; Fang, L.; Yang, Y.; Zhu, X.; Yuan, J.; Wu, J.; Li, M. MicroRNA-374a activates Wnt/β-catenin signaling to promote breast cancer metastasis. J. Clin. Investig. 2013, 123, 566–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, T.D.; Suto, M.J.; Li, Y. The wnt/β-catenin signaling pathway: A potential therapeutic target in the treatment of triple negative breast cancer. J. Cell. Biochem. 2012, 113, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Jang, G.-B.; Kim, J.Y.; Cho, S.D.; Park, K.S.; Jung, J.Y.; Lee, H.Y.; Hong, I.S.; Nam, J.S. Blockade of Wnt/β-catenin signaling suppresses breast cancer metastasis by inhibiting CSC-like phenotype. Sci. Rep. 2015, 5, 12465. [Google Scholar] [CrossRef] [Green Version]
- Lucijanic, M.; Livun, A.; Tomasovic-Loncaric, C.; Stoos-Veic, T.; Pejsa, V.; Jaksic, O.; Prka, Z.; Kusec, R. Canonical Wnt/β-catenin signaling pathway is dysregulated in patients with primary and secondary myelofibrosis. Clin. Lymphoma Myeloma Leuk. 2016, 16, 523–526. [Google Scholar] [CrossRef] [Green Version]
- Mathur, R.; Sehgal, L.; Braun, F.K.; Berkova, Z.; Romaguerra, J.; Wang, M.; Rodriguez, M.A.; Fayad, L.; Neelapu, S.S.; Samaniego, F. Targeting Wnt pathway in mantle cell lymphoma-initiating cells. J. Hematol. Oncol. 2015, 8, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geduk, A.; Atesoglu, E.B.; Tarkun, P.; Mehtap, O.; Hacihanefioglu, A.; Demirsoy, E.T.; Baydemir, C. The Role of β-catenin in Bcr/Abl negative myeloproliferative neoplasms: An immunohistochemical study. Clin. Lymphoma Myeloma Leuk. 2015, 15, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Manoharan, I.; Suryawanshi, A.; Majumdar, T.; Angus-Hill, M.L.; Koni, P.A.; Manicassamy, B.; Mellor, A.L.; Munn, D.H.; Manicassamy, S. β-catenin promotes regulatory T-cell responses in tumors by inducing vitamin a metabolism in dendritic cells. Cancer Res. 2015, 75, 656–665. [Google Scholar] [CrossRef] [Green Version]
- Damsky William, E.; Damsky, W.E.; Curley, D.P.; Santhanakrishnan, M.; Rosenbaum, L.E.; Platt, J.T.; Rothberg, B.E.G.; Taketo, M.M.; Dankort, D.; Rimm, D.L.; et al. β-Catenin signaling controls metastasis in Braf-activated Pten-deficient melanomas. Cancer Cell 2011, 20, 741–754. [Google Scholar] [CrossRef] [Green Version]
- Gallagher, S.J.; Rambow, F.; Kumasaka, M.; Champeval, D.; Bellacosa, A.; Delmas, V.; Larue, L. Beta-catenin inhibits melanocyte migration but induces melanoma metastasis. Oncogene 2013, 32, 2230–2238. [Google Scholar] [CrossRef] [Green Version]
- Hoseong Yang, S.; Andl, T.; Grachtchouk, V.; Wang, A.; Liu, J.; Syu, L.J.; Ferris, J.; Wang, T.S.; Glick, A.B.; Millar, S.E.; et al. Pathological responses to oncogenic Hedgehog signaling in skin are dependent on canonical Wnt/β-catenin signaling. Nat. Genet. 2008, 40, 1130–1135. [Google Scholar] [CrossRef]
- Vallée, A.; Lecarpentier, Y.; Vallée, J.N. Targeting the canonical WNT/β-catenin pathway in cancer treatment using non-steroidal anti-inflammatory drugs. Cells 2019, 8, 726. [Google Scholar] [CrossRef] [Green Version]
- Akrami, H.; Moradi, B.; Borzabadi Farahani, D.; Mehdizadeh, K. Ibuprofen reduces Cell Prolif.eration through inhibiting Wnt/β catenin signaling pathway in gastric cancer stem cells. Cell Biol. Int. 2018, 42, 949–958. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, M.F.; Faucz, F.R.; Bimpaki, E.; Horvath, A.; Levy, I.; de Alexandre, R.B.; Ahmad, F.; Manganiello, V.; Stratakis, C.A. Clinical and molecular genetics of the phosphodiesterases (PDEs). Endocr. Rev. 2014, 35, 195–233. [Google Scholar] [CrossRef]
- Yamanaka, Y.; Mammoto, T.; Kirita, T.; Mukai, M.; Mashimo, T.; Sugimura, M.; Kishi, Y.; Nakamura, H. Epinephrine inhibits invasion of oral squamous carcinoma cells by modulating intracellular cAMP. Cancer Lett. 2002, 176, 143–148. [Google Scholar] [CrossRef]
- Hirsh, L.; Dantes, A.; Suh, B.S.; Yoshida, Y.; Hosokawa, K.; Tajima, K.; Kotsuji, F.; Merimsky, O.; Amsterdam, A. Phosphodiesterase inhibitors as anti-cancer drugs. Biochem. Pharm. 2004, 68, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Sarfati, M.; Mateo, V.; Baudet, S.; Rubio, M.; Fernandez, C.; Davi, F.; Binet, J.L.; Delic, J.; Merle-Béral, H. Sildenafil and vardenafil types 5 and 6 phosphodiesterase inhibitors induce caspase-dependent apoptosis of B-chronic lymphocytic leukemia cells. Blood 2003, 101, 265–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Lee, K.; Xi, Y.; Zhu, B.; Gary, B.D.; Ramírez-Alcántara, V.; Gurpinar, E.; Canzoneri, J.C.; Fajardo, A.; Sigler, S.; et al. Phosphodiesterase 10A: A novel target for selective inhibition of colon tumor cell growth and β-catenin-dependent TCF transcriptional activity. Oncogene 2015, 34, 1499–1509. [Google Scholar] [CrossRef] [Green Version]
- Piazza, G.A.; Ward, A.; Chen, X.; Maxuitenko, Y.; Coley, A.; Aboelella, N.S.; Buchsbaum, D.J.; Boyd, M.R.; Keeton, A.B.; Zhou, G. PDE5 and PDE10 inhibition activates cGMP/PKG signaling to block Wnt/β-catenin transcription cancer cell growth and tumor immunity. Drug Discov. Today 2020, 25, 1521–1527. [Google Scholar] [CrossRef]
- Zhu, B.; Lindsey, A.; Li, N.; Lee, K.; Ramirez-Alcantara, V.; Canzoneri, J.C.; Fajardo, A.; da Silva, L.M.; Thomas, M.; Piazza, J.T.; et al. Phosphodiesterase 10A is overexpressed in lung tumor cells and inhibitors selectively suppress growth by blocking β-catenin and MAPK signaling. Oncotarget 2017, 8, 69264–69280. [Google Scholar] [CrossRef] [Green Version]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR Signaling in growth and metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef] [Green Version]
- Zou, Z.; Tao, T.; Li, H.; Zhu, X. mTOR signaling pathway and mTOR inhibitors in cancer: Progress and challenges. Cell Biosci. 2020, 10, 31. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.J.; Crowe, P.; Yang, J.L. Current clinical regulation of PI3K/PTEN/Akt/mTOR signalling in treatment of human cancer. J. Cancer Res. Clin. Oncol. 2015, 141, 671–689. [Google Scholar] [CrossRef] [PubMed]
- Ricoult, S.J.; Yecies, J.L.; Ben-Sahra, I.; Manning, B.D. Oncogenic PI3K and K-Ras stimulate de novo lipid synthesis through mTORC1 and SREBP. Oncogene 2016, 35, 1250–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Qian, J.; He, Q.; Zhao, H.; Toral-Barza, L.; Shi, C.; Zhang, X.; Wu, J.; Yu, K. mTOR complex-2 stimulates acetyl-CoA and de novo lipogenesis through ATP citrate lyase in HER2/PIK3CA-hyperactive breast cancer. Oncotarget 2016, 7, 25224–25240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, A.C.; Liu, Y.; Edlind, M.P.; Ingolia, N.T.; Janes, M.R.; Sher, A.; Shi, E.Y.; Stumpf, C.R.; Christensen, C.; Bonham, M.J.; et al. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature 2012, 485, 55–61. [Google Scholar] [CrossRef] [Green Version]
- Guri, Y.; Colombi, M.; Dazert, E.; Hindupur, S.K.; Roszik, J.; Moes, S.; Jenoe, P.; Heim, M.H.; Riezman, I.; Riezman, H.; et al. mTORC2 Promotes Tumorigenesis via Lipid Synthesis. Cancer Cell 2017, 32, 807–823.e12. [Google Scholar] [CrossRef] [Green Version]
- Di Malta, C.; Siciliano, D.; Calcagni, A.; Monfregola, J.; Punzi, S.; Pastore, N.; Eastes, A.N.; Davis, O.; De Cegli, R.; Zampelli, A.; et al. Transcriptional activation of RagD GTPase controls mTORC1 and promotes cancer growth. Science 2017, 356, 1188–1192. [Google Scholar] [CrossRef] [Green Version]
- Ogier-Denis, E.; Codogno, P. Autophagy: A barrier or an adaptive response to cancer. Biochim. Biophys. Acta (Bba)—Rev. Cancer 2003, 1603, 113–128. [Google Scholar] [CrossRef]
- Kim, J.; Klionsky, D.J. Autophagy cytoplasm-to-vacuole targeting pathway and pexophagy in yeast and mammalian cells. Annu. Rev. Biochem. 2000, 69, 303–342. [Google Scholar] [CrossRef]
- Kanzawa, T.; Germano, I.M.; Komata, T.; Ito, H.; Kondo, Y.; Kondo, S. Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ 2004, 11, 448–457. [Google Scholar] [CrossRef] [Green Version]
- Dickstein, R.J.; Nitti, G.; Dinney, C.P.; Davies, B.R.; Kamat, A.M.; McConkey, D.J. Autophagy limits the cytotoxic effects of the AKT inhibitor AZ7328 in human bladder cancer cells. Cancer Biol. Ther. 2012, 13, 1325–1338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paglin, S.; Hollister, T.; Delohery, T.; Hackett, N.; McMahill, M.; Sphicas, E.; Domingo, D.; Yahalom, J. A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Cancer Res. 2001, 61, 439–444. [Google Scholar] [PubMed]
- Yu, C.; Li, W.B.; Liu, J.B.; Lu, J.W.; Feng, J.F. Autophagy: Novel applications of nonsteroidal anti-inflammatory drugs for primary cancer. Cancer Med. 2018, 7, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Luo, R.Z.; Lu, Y.; Zhang, X.; Yu, Q.; Khare, S.; Kondo, S.; Kondo, Y.; Yu, Y.; Mills, G.B. The tumor suppressor gene ARHI regulates autophagy and tumor dormancy in human ovarian cancer cells. J. Clin. Investig. 2008, 118, 3917–3929. [Google Scholar] [CrossRef] [Green Version]
- Butler, D.E.; Marlein, C.; Walker, H.F.; Frame, F.M.; Mann, V.M.; Simms, M.S.; Davies, B.R.; Collins, A.T.; Maitland, N.J. Inhibition of the PI3K/AKT/mTOR pathway activates autophagy and compensatory Ras/Raf/MEK/ERK signalling in prostate cancer. Oncotarget 2017, 8, 56698–56713. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.W.; Mutter, R.W.; Cao, C.; Albert, J.M.; Freeman, M.; Hallahan, D.E.; Lu, B. Autophagy for cancer therapy through inhibition of pro-apoptotic proteins and mammalian target of rapamycin signaling. J. Biol. Chem. 2006, 281, 36883–36890. [Google Scholar] [CrossRef] [Green Version]
- Cory, S.; Adams, J.M. The Bcl2 family: Regulators of the cellular life-or-death switch. Nat. Rev. Cancer 2002, 2, 647–656. [Google Scholar] [CrossRef]
- Green, D.R.; Reed, J.C. Mitochondria and apoptosis. Science 1998, 281, 1309–1312. [Google Scholar] [CrossRef]
- Jürgensmeier, J.M.; Xie, Z.; Deveraux, Q.; Ellerby, L.; Bredesen, D.; Reed, J.C. Bax directly induces release of cytochrome c from isolated mitochondria. Proc. Natl. Acad. Sci. USA 1998, 95, 4997–5002. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.; Yang, J.; Jones, D. Mitochondrial control of apoptosis: The role of cytochrome c. Biochim. Biophys. Acta (Bba)—Bioenerg. 1998, 1366, 139–149. [Google Scholar] [CrossRef] [Green Version]
- Zhivotovsky, B.; Orrenius, S.; Brustugun, O.T.; Døskeland, S.O. Injected cytochrome c induces apoptosis. Nature 1998, 391, 449–450. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Baek, S.J.; Eling, T.E. The diverse roles of nonsteroidal anti-inflammatory drug activated gene (NAG-1/GDF15) in cancer. Biochem. Pharm. 2013, 85, 597–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, J.M.; Sali, T.; Okazaki, R.; Anna, C.; Hollingshead, M.; Hose, C.; Monks, A.; Walker, N.J.; Baek, S.J.; Eling, T.E. Drug-induced expression of nonsteroidal anti-inflammatory drug-activated gene/macrophage inhibitory cytokine-1/prostate-derived factor a putative tumor suppressor inhibits tumor growth. J. Pharm. Exp. Therap. 2006, 318, 899–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawahara, T.; Ishiguro, H.; Hoshino, K.; Teranishi, J.I.; Miyoshi, Y.; Kubota, Y.; Uemura, H. Analysis of NSAID-activated gene 1 expression in prostate cancer. Urol. Int. 2010, 84, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Monteith, G.R.; McAndrew, D.; Faddy, H.M.; Roberts-Thomson, S.J. Calcium and cancer: Targeting Ca2+ transport. Nat. Rev. Cancer 2007, 7, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Panda, S.; Chatterjee, O.; Roy, L.; Chatterjee, S. Targeting Ca2+ signaling: A new arsenal against cancer. Drug Discov. Today 2021. [Google Scholar] [CrossRef]
- Ibrahim, S.; Dakik, H.; Vandier, C.; Chautard, R.; Paintaud, G.; Mazurier, F.; Lecomte, T.; Guéguinou, M.; Raoul, W. Expression profiling of calcium channels and calcium-activated potassium channels in colorectal cancer. Cancers 2019, 11, 561. [Google Scholar] [CrossRef] [Green Version]
- Phan, N.N.; Wang, C.Y.; Chen, C.F.; Sun, Z.; Lai, M.D.; Lin, Y.C. Voltage-gated calcium channels: Novel targets for cancer therapy. Oncol. Lett. 2017, 14, 2059–2074. [Google Scholar] [CrossRef] [Green Version]
- Sritangos, P.; Pena Alarcon, E.; James, A.D.; Sultan, A.; Richardson, D.A.; Bruce, J.I. Plasma Membrane Ca(2+) ATPase isoform 4 (PMCA4) has an important role in numerous hallmarks of pancreatic cancer. Cancers 2020, 12, 218. [Google Scholar] [CrossRef] [Green Version]
- Silvestri, R.; Pucci, P.; Venalainen, E.; Matheou, C.; Mather, R.; Chandler, S.; Aceto, R.; Rigas, S.H.; Wang, Y.; Rietdorf, K.; et al. T-type calcium channels drive the proliferation of androgen-receptor negative prostate cancer cells. Prostate 2019, 79, 1580–1586. [Google Scholar] [CrossRef]
- Das, A.; Pushparaj, C.; Bahí, N.; Sorolla, A.; Herreros, J.; Pamplona, R.; Vilella, R.; Matias-Guiu, X.; Marti, R.M.; Cantí, C. Functional expression of voltage-gated calcium channels in human melanoma. Pigment Cell Melanoma Res. 2012, 25, 200–212. [Google Scholar] [CrossRef]
- Cabanas, H.; Harnois, T.; Magaud, C.; Cousin, L.; Constantin, B.; Bourmeyster, N.; Déliot, N. Deregulation of calcium homeostasis in Bcr-Abl-dependent chronic myeloid leukemia. Oncotarget 2018, 9, 26309–26327. [Google Scholar] [CrossRef] [Green Version]
- Nishida, N.; Yano, H.; Nishida, T.; Kamura, T.; Kojiro, M. Angiogenesis in cancer. Vasc. Health Risk Manag. 2006, 2, 213–219. [Google Scholar] [CrossRef]
- Kerbel, R.S. Tumor Angiogenesis. N. Engl. J. Med. 2008, 358, 2039–2049. [Google Scholar] [CrossRef] [Green Version]
- Holmgren, L.; O’Reilly, M.S.; Folkman, J. Dormancy of micrometastases: Balanced proliferation and apoptosis in the presence of angiogenesis suppression. Nat. Med. 1995, 1, 149–153. [Google Scholar] [CrossRef]
- Streit, M.; Riccardi, L.; Velasco, P.; Brown, L.F.; Hawighorst, T.; Bornstein, P.; Detmar, M. Thrombospondin-2: A potent endogenous inhibitor of tumor growth and angiogenesis. Proc. Natl. Acad. Sci. USA 1999, 96, 14888–14893. [Google Scholar] [CrossRef] [Green Version]
- Dews, M.; Homayouni, A.; Yu, D.; Murphy, D.; Sevignani, C.; Wentzel, E.; Furth, E.E.; Lee, W.M.; Enders, G.H.; Mendell, J.T.; et al. Augmentation of tumor angiogenesis by a Myc-activated microRNA cluster. Nat. Genet. 2006, 38, 1060–1065. [Google Scholar] [CrossRef] [Green Version]
- Tiwari, M. Apoptosis angiogenesis and cancer therapies. J. Cancer Ther. Res. 2012, 1, 3. [Google Scholar] [CrossRef] [Green Version]
- Tarnawski, A.S.; Jones, M.K. Inhibition of angiogenesis by NSAIDs: Molecular mechanisms and clinical implications. J. Mol. Med. 2003, 81, 627–636. [Google Scholar] [CrossRef]
- Pastorekova, S.; Parkkila, S.; Pastorek, J.; Supuran, C.T. Carbonic anhydrases: Current state of the art, therapeutic applications and future prospects. J. Enzym. Inhib. Med. Chem. 2004, 19, 199–229. [Google Scholar] [CrossRef]
- Chiche, J.; Ilc, K.; Laferriere, J.; Trottier, E.; Dayan, F.; Mazure, N.M.; Brahimi-Horn, M.C.; Pouysségur, J. Hypoxia-inducible carbonic anhydrase IX and XII promote tumor cell growth by counteracting acidosis through the regulation of the intracellular pH. Cancer Res. 2009, 69, 358–368. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Lomelino, C.L.; Mboge, M.Y.; Frost, S.C.; McKenna, R. Cancer drug development of carbonic anhydrase inhibitors beyond the active site. Molecules 2018, 23, 1045. [Google Scholar] [CrossRef] [Green Version]
- Pastorek, J.; Pastorekova, S. Hypoxia-induced carbonic anhydrase IX as a target for cancer therapy: From biology to clinical use. Semin. Cancer Biol. 2015, 31, 52–64. [Google Scholar] [CrossRef]
- Hynninen, P.; Vaskivuo, L.; Saarnio, J.; Haapasalo, H.; Kivelä, J.; Pastorekova, S.; Pastorek, J.; Waheed, A.; Sly, W.S.; Puistola, U.; et al. Expression of transmembrane carbonic anhydrases IX and XII in ovarian tumours. Histopathology 2006, 49, 594–602. [Google Scholar] [CrossRef]
- Kim, J.Y.; Shin, H.J.; Kim, T.H.; Cho, K.H.; Shin, K.H.; Kim, B.K.; Roh, J.W.; Lee, S.; Park, S.Y.; Hwang, Y.J.; et al. Tumor-associated carbonic anhydrases are linked to metastases in primary cervical cancer. J. Cancer Res. Clin. Oncol. 2006, 132, 302–308. [Google Scholar] [CrossRef]
- Tafreshi, N.K.; Bui, M.M.; Bishop, K.; Lloyd, M.C.; Enkemann, S.A.; Lopez, A.S.; Abrahams, D.; Carter, B.W.; Vagner, J.; Grobmyer, S.R.; et al. Noninvasive Detection of Breast Cancer Lymph Node Metastasis Using Carbonic Anhydrases IX and XII Targeted Imaging Probes. Clin. Cancer Res. 2012, 18, 207. [Google Scholar] [CrossRef] [Green Version]
- Hussain, S.A.; Ganesan, R.; Reynolds, G.; Gross, L.; Stevens, A.; Pastorek, J.; Murray, P.G.; Perunovic, B.; Anwar, M.S.; Billingham, L.; et al. Hypoxia-regulated carbonic anhydrase IX expression is associated with poor survival in patients with invasive breast cancer. Br. J. Cancer 2007, 96, 104–109. [Google Scholar] [CrossRef]
- Kivelä, A.J.; Parkkila, S.; Saarnio, J.; Karttunen, T.J.; Kivelä, J.; Parkkila, A.K.; Pastoreková, S.; Pastorek, J.; Waheed, A.; Sly, W.S.; et al. Expression of transmembrane carbonic anhydrase isoenzymes IX and XII in normal human pancreas and pancreatic tumours. Histochem. Cell Biol. 2000, 114, 197–204. [Google Scholar] [CrossRef]
- Järvelä, S.; Parkkila, S.; Bragge, H.; Kähkönen, M.; Parkkila, A.K.; Soini, Y.; Pastorekova, S.; Pastorek, J.; Haapasalo, H. Carbonic anhydrase IX in oligodendroglial brain tumors. BMC Cancer 2008, 8, 1. [Google Scholar] [CrossRef] [Green Version]
- Buckley, C.D.; Gilroy, D.W.; Serhan, C.N. Serhan Proresolving Lipid Mediators and Mechanisms in the Resolution of Acute Inflammation. Immunity 2014, 40, 315–327. [Google Scholar] [CrossRef] [Green Version]
- Janakiram, N.B.; Mohammed, A.; Rao, C.V. Role of lipoxins resolvins and other bioactive lipids in colon and pancreatic cancer. Cancer Metastasis Rev. 2011, 30, 507–523. [Google Scholar] [CrossRef] [PubMed]
- Weylandt, K.H.; Chiu, C.Y.; Gomolka, B.; Waechter, S.F.; Wiedenmann, B. Omega-3 fatty acids and their lipid mediators: Towards an understanding of resolvin and protectin formation. Prostaglandins Other Lipid Mediat. 2012, 97, 73–82. [Google Scholar] [CrossRef]
- Panigrahy, D.; Gilligan, M.M.; Serhan, C.N.; Kashfi, K. Resolution of inflammation: An organizing principle in biology and medicine. Pharm. Ther. 2021, 227, 107879. [Google Scholar] [CrossRef]
- Serhan, C.N.; Petasis, N.A. Resolvins and protectins in inflammation resolution. Chem. Rev. 2011, 111, 5922–5943. [Google Scholar] [CrossRef] [Green Version]
- Serhan, C.N.; Hamberg, M.; Samuelsson, B. Lipoxins: Novel series of biologically active compounds formed from arachidonic acid in human leukocytes. Proc. Natl. Acad. Sci. USA 1984, 81, 5335–5339. [Google Scholar] [CrossRef] [Green Version]
- Serhan, C.N.; Dalli, J.; Colas, R.A.; Winkler, J.W.; Chiang, N. Protectins and maresins: New pro-resolving families of mediators in acute inflammation and resolution bioactive metabolome. Biochim. Biophys. Acta (Bba)—Mol. Cell Biol. Lipids 2015, 1851, 397–413. [Google Scholar] [CrossRef] [Green Version]
- Serhan, C.N. Pro-resolving lipid mediators are leads for resolution physiology. Nature 2014, 510, 92–101. [Google Scholar] [CrossRef] [Green Version]
- Berquin, I.M.; Edwards, I.J.; Chen, Y.Q. Multi-targeted therapy of cancer by omega-3 fatty acids. Cancer Lett. 2008, 269, 363–377. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Mullapudi, B.; Torres, C.; Mascariñas, E.; Mancinelli, G.; Diaz, A.M.; McKinney, R.; Barron, M.; Schultz, M.; Heiferman, M.; et al. Omega-3 fatty acids prevent early pancreatic carcinogenesis via repression of the AKT pathway. Nutrients 2018, 10, 1289. [Google Scholar] [CrossRef] [Green Version]
- Nowak, J.; Weylandt, K.H.; Habbel, P.; Wang, J.; Dignass, A.; Glickman, J.N.; Kang, J.X. Colitis-associated colon tumorigenesis is suppressed in transgenic mice rich in endogenous n-3 fatty acids. Carcinogenesis 2007, 28, 1991–1995. [Google Scholar] [CrossRef] [Green Version]
- Weylandt, K.H.; Krause, L.F.; Gomolka, B.; Chiu, C.Y.; Bilal, S.; Nadolny, A.; Waechter, S.F.; Fischer, A.; Rothe, M.; Kang, J.X. Suppressed liver tumorigenesis in fat-1 mice with elevated omega-3 fatty acids is associated with increased omega-3 derived lipid mediators and reduced TNF-α. Carcinogenesis 2011, 32, 897–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fishbein, A.; Hammock, B.D.; Serhan, C.N.; Panigrahy, D. Carcinogenesis: Failure of resolution of inflammation? Pharm. Ther. 2021, 218, 107670. [Google Scholar] [CrossRef] [PubMed]
- Filippou, P.S.; Karagiannis, G.S. Cytokine storm during chemotherapy: A new companion diagnostic emerges? Oncotarget 2020, 11, 213–215. [Google Scholar] [CrossRef] [PubMed]
- Hammock, B.D. Eicosanoids: The overlooked storm in coronavirus disease 2019 (COVID-19)? Am. J. Pathol. 2020, 190, 1782–1788. [Google Scholar] [CrossRef]
- Panigrahy, D.; Gilligan, M.M.; Huang, S.; Gartung, A.; Cortés-Puch, I.; Sime, P.J.; Phipps, R.P.; Serhan, C.N.; Hammock, B.D. Inflammation resolution: A dual-pronged approach to averting cytokine storms in COVID-19? Cancer Metastasis Rev. 2020, 39, 337–340. [Google Scholar] [CrossRef]
- Recchiuti, A.; Serhan, C.N. Pro-resolving lipid mediators (SPMs) and their actions in regulating mirna in novel resolution circuits in inflammation. Front. Immunol. 2012, 3, 298. [Google Scholar] [CrossRef] [Green Version]
- Levy, B.D.; Clish, C.B.; Schmidt, B.; Gronert, K.; Serhan, C.N. Lipid mediator class switching during acute inflammation: Signals in resolution. Nat. Immunol. 2001, 2, 612–619. [Google Scholar] [CrossRef]
- Petasis, N.A.; Akritopoulou-Zanze, I.; Fokin, V.V.; Bernasconi, G.; Keledjian, R.; Yang, R.; Uddin, J.; Nagulapalli, K.C.; Serhan, C.N. Design synthesis and bioactions of novel stable mimetics of lipoxins and aspirin-triggered lipoxins. Prostaglandins Leukot. Essent. Fat. Acids 2005, 73, 301–321. [Google Scholar] [CrossRef]
- Romano, M. Lipoxin and aspirin-triggered lipoxins. ScientificWorldJournal 2010, 10, 1048–1064. [Google Scholar] [CrossRef] [Green Version]
- Clària, J.; Serhan, C.N. Aspirin triggers previously undescribed bioactive eicosanoids by human endothelial cell-leukocyte interactions. Proc. Natl. Acad. Sci. USA 1995, 92, 9475–9479. [Google Scholar] [CrossRef] [Green Version]
- Colgan, S.P.; Colgan, S.P.; Serhan, C.N.; Parkos, C.A.; Delp-Archer, C.; Madara, J.L. Lipoxin A4 modulates transmigration of human neutrophils across intestinal epithelial monolayers. J. Clin. Investig. 1993, 92, 75–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, S.F.; Pillai, P.S.; Recchiuti, A.; Yang, R.; Serhan, C.N. Pro-resolving actions and stereoselective biosynthesis of 18S E-series resolvins in human leukocytes and murine inflammation. J. Clin. Investig. 2011, 121, 569–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capdevila, J.H.; Wei, S.; Helvig, C.; Falck, J.R.; Belosludtsev, Y.; Truan, G.; Graham-Lorence, S.E.; Peterson, J.A. The highly stereoselective oxidation of polyunsaturated fatty acids by cytochrome P450BM-3. J. Biol. Chem. 1996, 271, 22663–22671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tjonahen, E.; Oh, S.F.; Siegelman, J.; Elangovan, S.; Percarpio, K.B.; Hong, S.; Arita, M.; Serhan, C.N. Resolvin E2: Identification and anti-inflammatory actions: Pivotal role of human 5-lipoxygenase in resolvin E series biosynthesis. Chem. Biol. 2006, 13, 1193–1202. [Google Scholar] [CrossRef] [Green Version]
- Serhan, C.N.; Clish, C.B.; Brannon, J.; Colgan, S.P.; Chiang, N.; Gronert, K. Novel functional sets of lipid-derived mediators with antiinflammatory actions generated from omega-3 fatty acids via cyclooxygenase 2-nonsteroidal antiinflammatory drugs and transcellular processing. J. Exp. Med. 2000, 192, 1197–1204. [Google Scholar] [CrossRef] [Green Version]
- Serhan, C.N.; Hong, S.; Gronert, K.; Colgan, S.P.; Devchand, P.R.; Mirick, G.; Moussignac, R.L. Resolvins: A Family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J. Exp. Med. 2002, 196, 1025–1037. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.P.; Oh, S.F.; Uddin, J.; Yang, R.; Gotlinger, K.; Campbell, E.; Colgan, S.P.; Petasis, N.A.; Serhan, C.N. Resolvin D1 and its aspirin-triggered 17R epimer. Stereochemical assignments anti-inflammatory properties and enzymatic inactivation. J. Biol. Chem. 2007, 282, 9323–9334. [Google Scholar] [CrossRef] [Green Version]
- Dalli, J.; Chiang, N.; Serhan, C.N. Elucidation of novel 13-series resolvins that increase with atorvastatin and clear infections. Nat. Med. 2015, 21, 1071–1075. [Google Scholar] [CrossRef]
- Fullerton, J.N.; O’Brien, A.J.; Gilroy, D.W. Lipid mediators in immune dysfunction after severe inflammation. Trends Immunol. 2014, 35, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Serhan, C.N.; Gotlinger, K.; Hong, S.; Lu, Y.; Siegelman, J.; Baer, T.; Yang, R.; Colgan, S.P.; Petasis, N.A. Anti-inflammatory actions of neuroprotectin D1/Protectin D1 and its natural stereoisomers: Assignments of dihydroxy-containing docosatrienes. J. Immunol. 2006, 176, 1848–1859. [Google Scholar] [CrossRef] [Green Version]
- Deng, B.; Wang, C.W.; Arnardottir, H.H.; Li, Y.; Cheng, C.Y.C.; Dalli, J.; Serhan, C.N. Maresin Biosynthesis and identification of Maresin 2 a new anti-inflammatory and pro-resolving mediator from human macrophages. PLoS ONE 2014, 9, e102362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serhan, C.N.; Yang, R.; Martinod, K.; Kasuga, K.; Pillai, P.S.; Porter, T.F.; Oh, S.F.; Spite, M. Maresins: Novel macrophage mediators with potent antiinflammatory and proresolving actions. J. Exp. Med. 2009, 206, 15–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serhan Charles, N.; Fredman, G.; Yang, R.; Karamnov, S.; Belayev, L.S.; Bazan, N.G.; Zhu, M.; Winkler, J.W.; Petasis, N.A. Novel proresolving aspirin-triggered DHA pathway. Chem. Biol. 2011, 18, 976–987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalli, J.; Zhu, M.; Vlasenko, N.A.; Deng, B.; Haeggström, J.Z.; Petasis, N.A.; Serhan, C.N. The novel 13S,14S-epoxy-maresin is converted by human macrophages to maresin 1 (MaR1) inhibits leukotriene A4 hydrolase (LTA4H) and shifts macrophage phenotype. FASEB J. 2013, 27, 2573–2583. [Google Scholar] [CrossRef] [Green Version]
- Serhan, C.N.; Dalli, J.; Karamnov, S.; Choi, A.; Park, C.K.; Xu, Z.Z.; Ji, R.R.; Zhu, M.; Petasis, N.A. Macrophage proresolving mediator maresin 1 stimulates tissue regeneration and controls pain. FASEB J. 2012, 26, 1755–1765. [Google Scholar] [CrossRef] [Green Version]
- Mudge, D.; Kieran, M.W.; Bielenberg, D.; Benny, O.; Dalli, J.; Huang, S.; Serhan, C.N.; Panigrahy, D. Maresin 1: A potent endogenous anti-inflammatory and pro-resolving inhibitor of primary tumor growth and metastasis. In Proceedings of the AACR Annual Meeting, San Diego, CA, USA, 5–9 April 2014. [Google Scholar]
- Vatnick, D.R.; Lehner, K.; Gilligan, M.; Panigrahy, D.; Gus-Brautbar, Y.; Ramon, S.; Huang, S.; Serhan, C. Control of breast cancer through the resolution of inflammation. FASEB J. 2016, 30, 698.3. [Google Scholar]



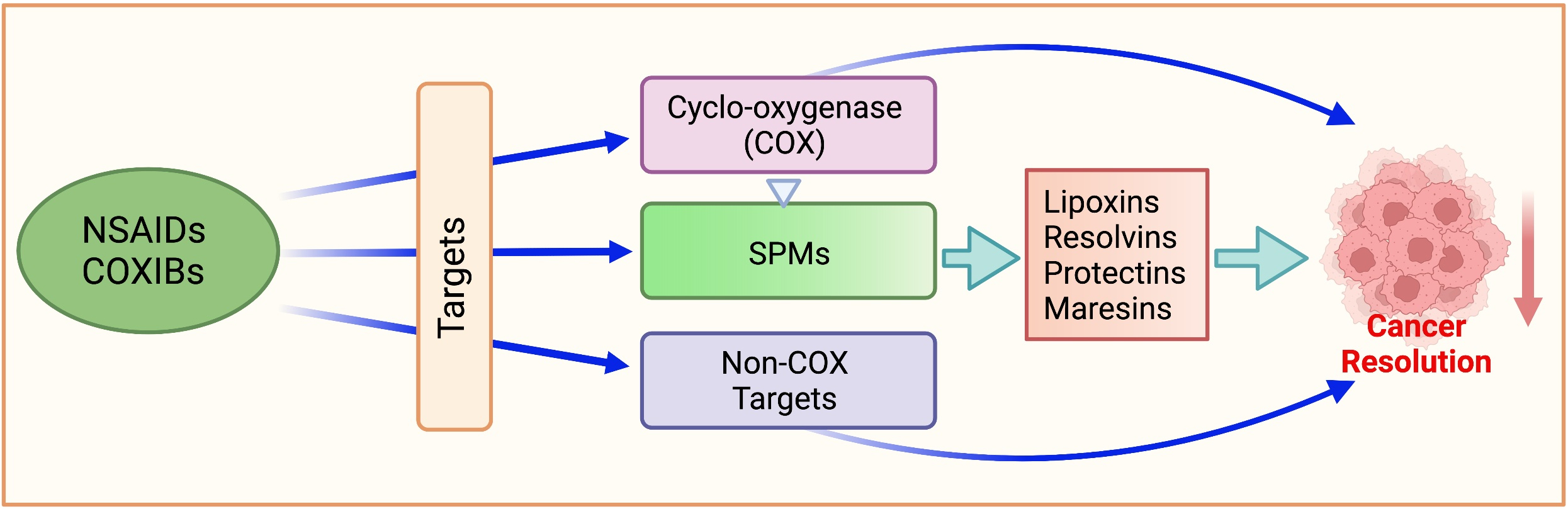{kind=link}
{kind=link}
{kind=link}
| Target | NSAID | Reference |
|---|---|---|
| NF-κB | Aspirin | [67,68,69] |
| Diclofenac | [70] | |
| Sulindac | [71,72] | |
| PDK-1/Akt | Celecoxib | [73,74] |
| Naproxen | [75] | |
| PPAR | Aspirin | [76] |
| Ibuprofen | [76] | |
| Indomethacin | [76,77] | |
| Sulindac | [78] | |
| NS398 | [79] | |
| Celecoxib | [80] | |
| MAPKs | Indomethacin | [81,82,83] |
| NS398 | [81,84,85] | |
| Celecoxib | [86,87,88] | |
| Sulindac sulfide | [85] | |
| Wnt/β-catenin | Indomethacin | [89] |
| Sulindac | [90,91,92] | |
| Diclofenac | [93] | |
| Celecoxib | [93,94] | |
| PDEs | Sulindac sulfide | [95,96,97,98] |
| mTOR | Aspirin | [99] |
| Celecoxib | [100] | |
| Autophagy | Celecoxib | [101,102,103] |
| Meloxicam | [104,105] | |
| Aspirin | [99,106] | |
| Cell kinetics | Sulindac | [47] |
| Piroxicam | [47] | |
| Celecoxib | [107] | |
| SC560 | [107] | |
| Naproxen | [75] | |
| Sulindac sulfide | [108] | |
| Cytochrome c | Indomethacin | [109,110] |
| Celecoxib | [111,112] | |
| NS398 | [113] | |
| Aspirin | [114] | |
| NAG-1 | Sulindac | [115] |
| Sulindac sulfide | [108,116,117,118] | |
| Indomethacin | [116,118,119] | |
| Piroxicam | [119] | |
| Diclofenac | [119] | |
| Aspirin | [118] | |
| Celecoxib | [120,121] | |
| NS398 | [122] | |
| Ibuprofen, Flurbiprofen | [123] | |
| Ca2+ mobilization | Indomethacin | [109,124] |
| Celecoxib | [125] | |
| 2,3-Dimethylcelecoxib (DMC, celecoxib analog) | [126,127] | |
| Angiogenesis | Aspirin | [128,129] |
| Ibuprofen | [130,131] | |
| Carbonic Anhydrase | Indomethacin | [132] |
| Celecoxib | [132,133,134] | |
| Valdecoxib | [133,134] | |
| SPMs Lipoxins Resolvins Protectins Maresins | ||
| Aspirin | [135] | |
| Aspirin | [136,137,138] | |
| Not studied | ||
| Not studied |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kolawole, O.R.; Kashfi, K. NSAIDs and Cancer Resolution: New Paradigms beyond Cyclooxygenase. Int. J. Mol. Sci. 2022, 23, 1432. https://doi.org/10.3390/ijms23031432
Kolawole OR, Kashfi K. NSAIDs and Cancer Resolution: New Paradigms beyond Cyclooxygenase. International Journal of Molecular Sciences. 2022; 23(3):1432. https://doi.org/10.3390/ijms23031432
Chicago/Turabian StyleKolawole, Oluwafunke R., and Khosrow Kashfi. 2022. "NSAIDs and Cancer Resolution: New Paradigms beyond Cyclooxygenase" International Journal of Molecular Sciences 23, no. 3: 1432. https://doi.org/10.3390/ijms23031432
APA StyleKolawole, O. R., & Kashfi, K. (2022). NSAIDs and Cancer Resolution: New Paradigms beyond Cyclooxygenase. International Journal of Molecular Sciences, 23(3), 1432. https://doi.org/10.3390/ijms23031432