
Up-Regulation of Cyclooxygenase-2 (COX-2) Expression by Temozolomide (TMZ) in Human Glioblastoma (GBM) Cell Lines


 ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. COX-2 Expression and TMZ Sensitivity of GBM Cell Lines
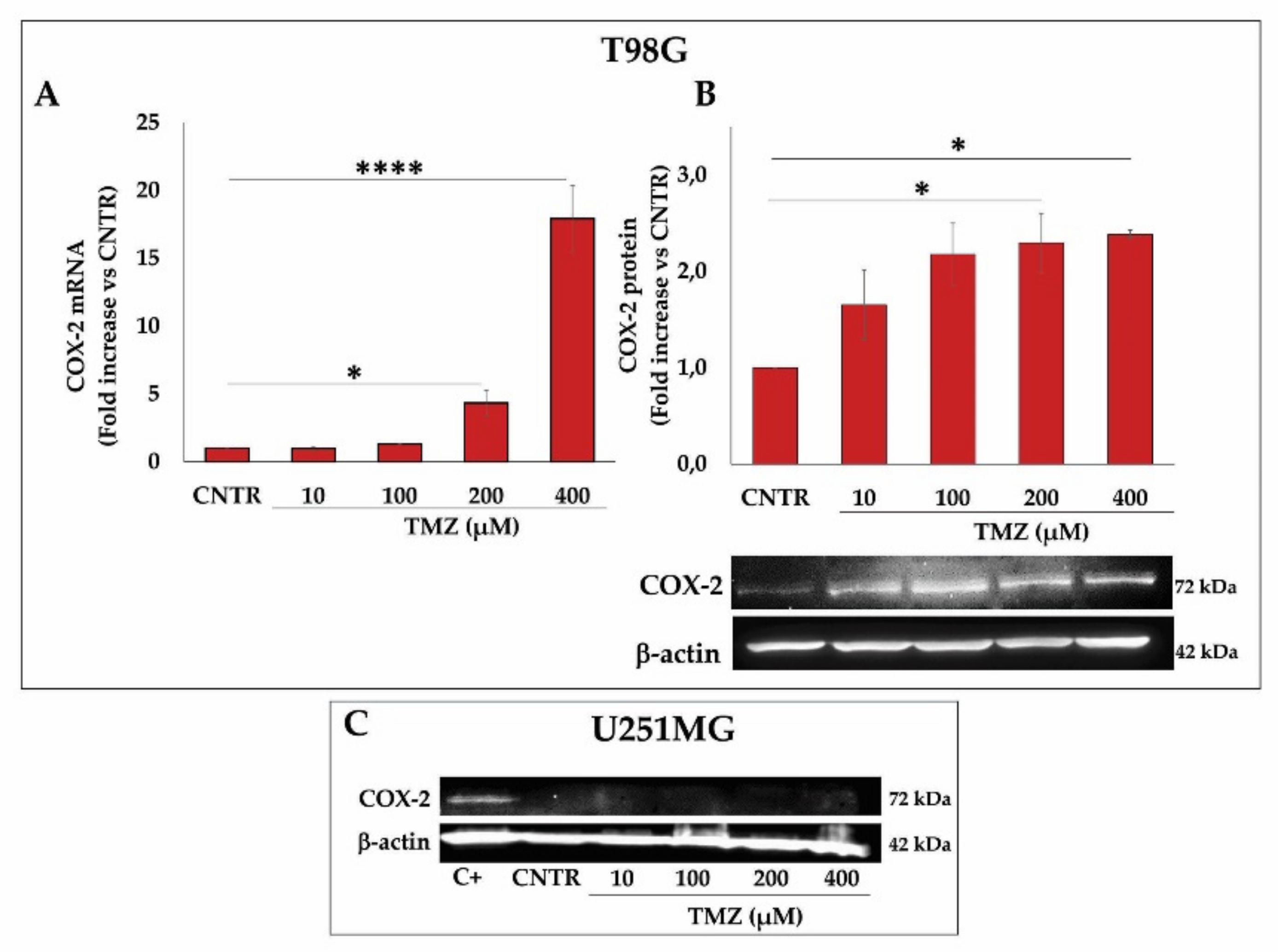
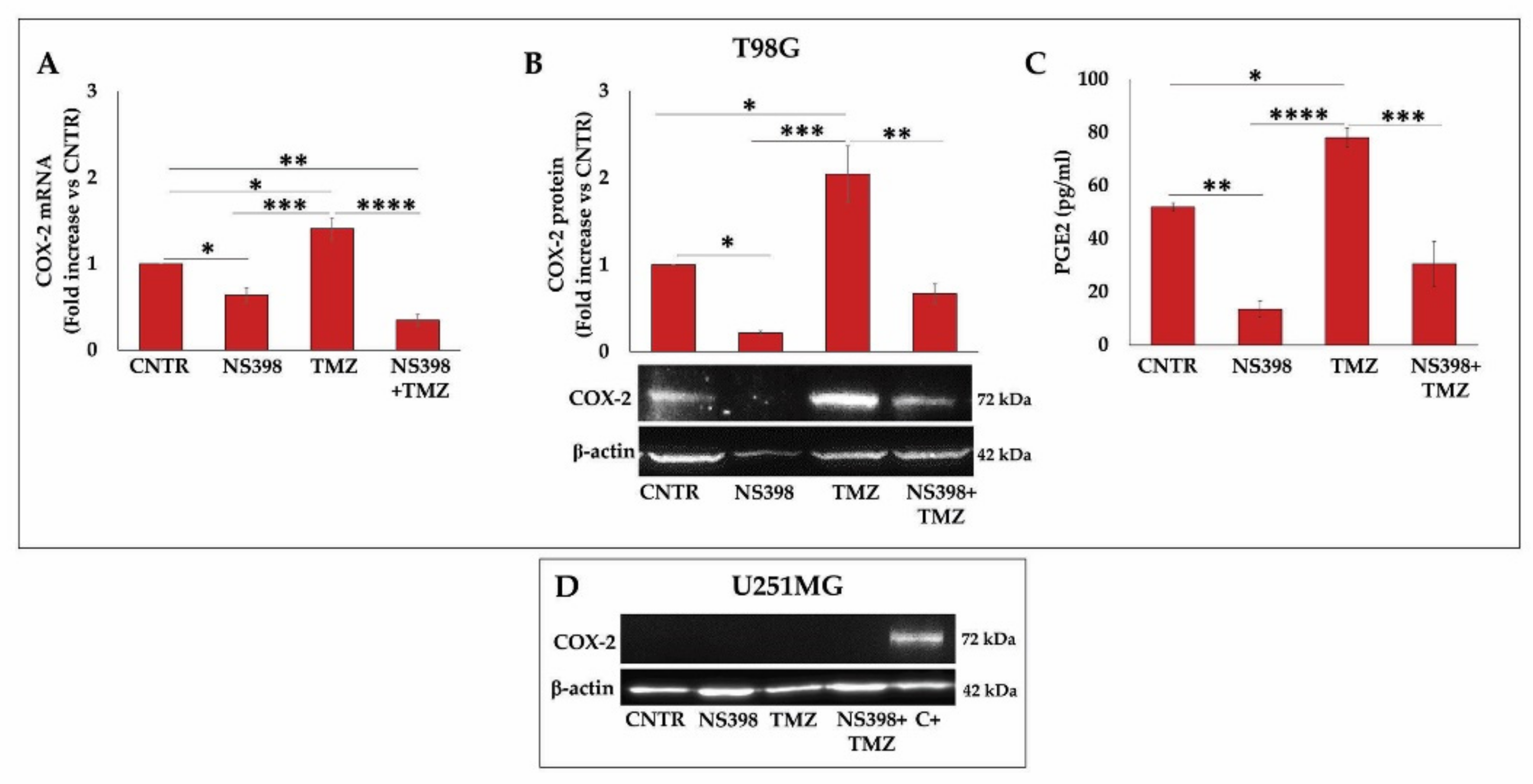
2.2. Effect of TMZ on COX-2 Expression in GBM Cell Lines
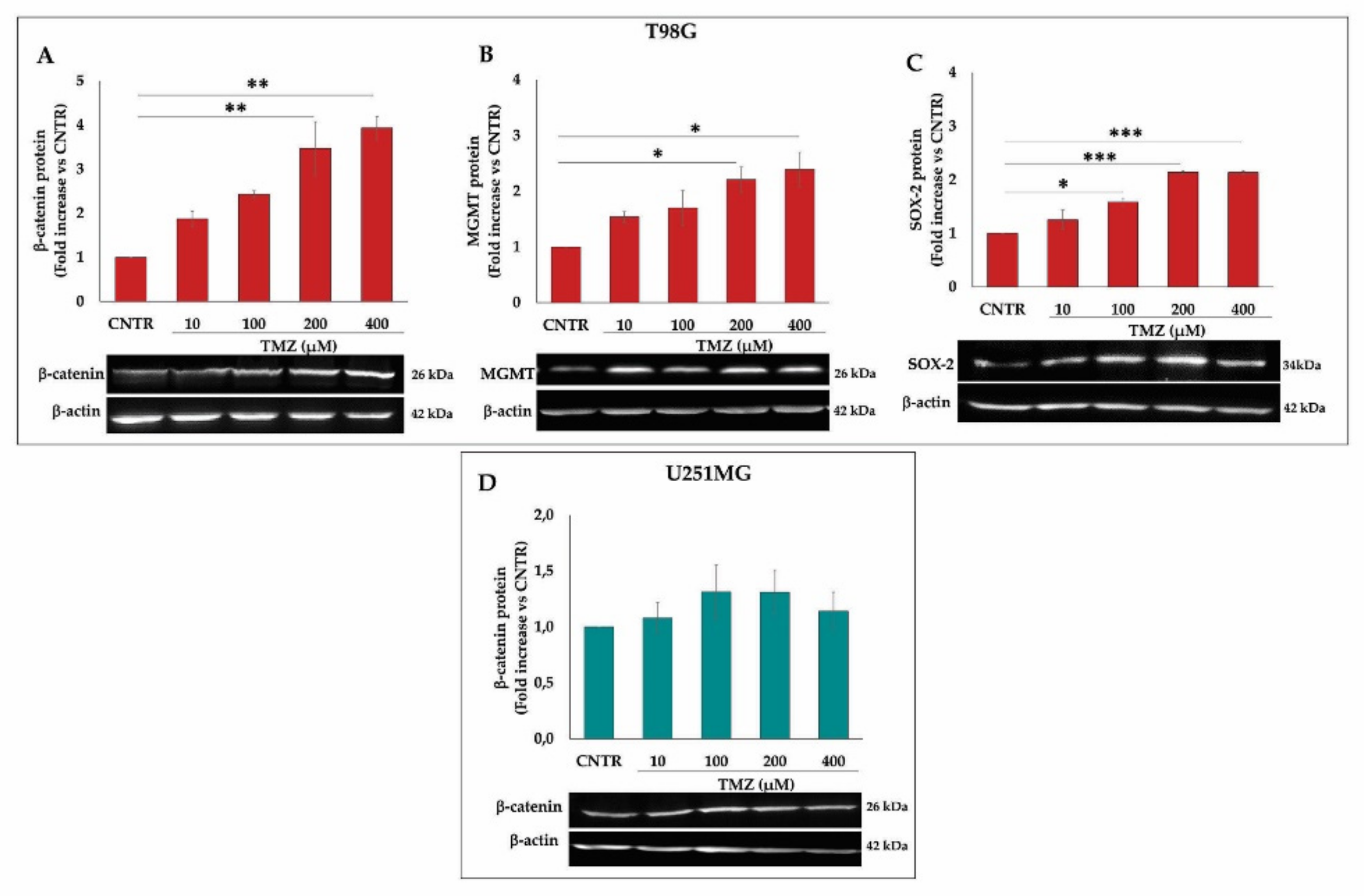
2.3. Influence of TMZ Treatment on β-Catenin, MGMT, and SOX-2 Levels in GBM Cells
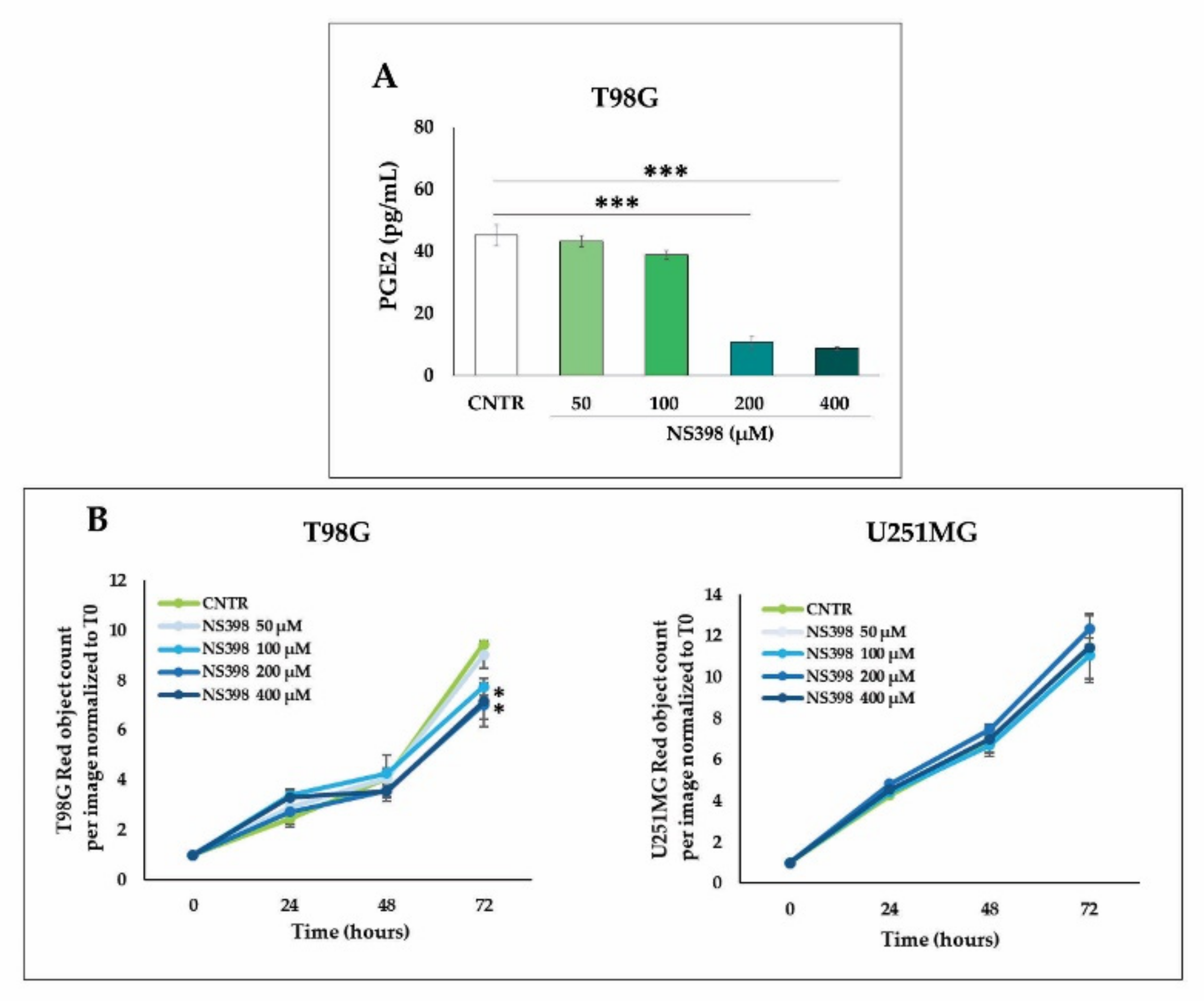
2.4. Effects of NS398 on PGE2 Release and Cell Proliferation Rate in GBM Cell Lines
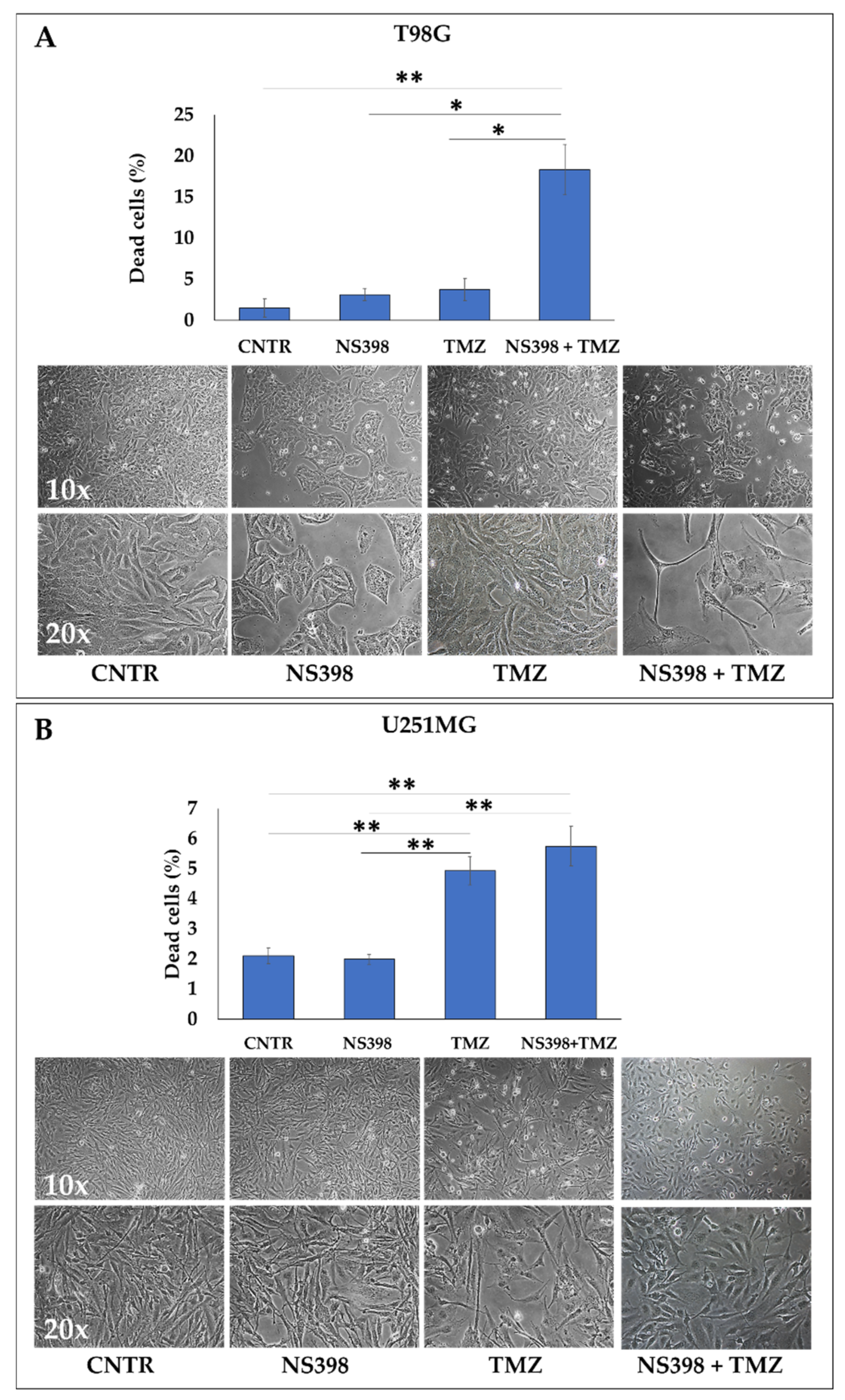
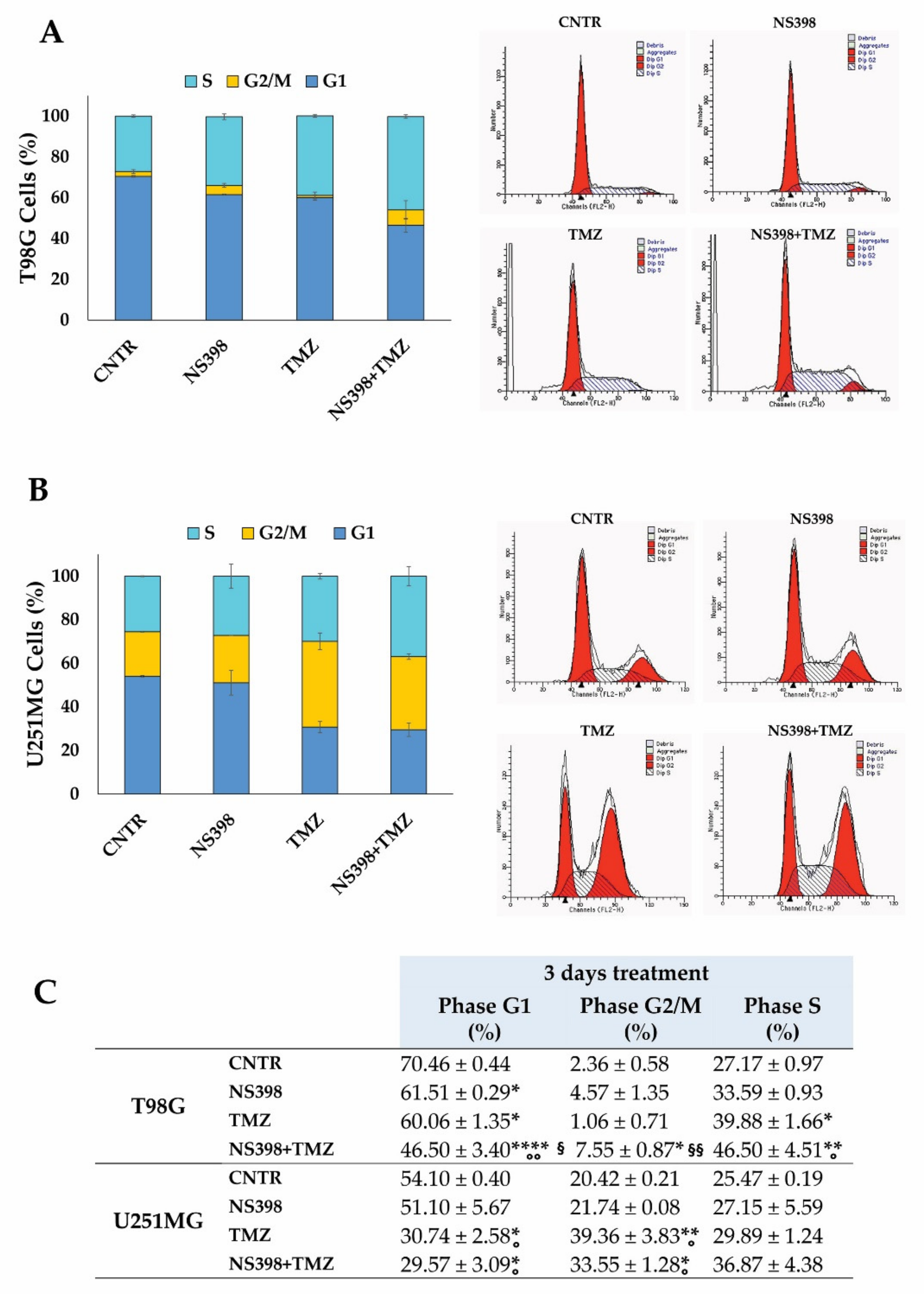
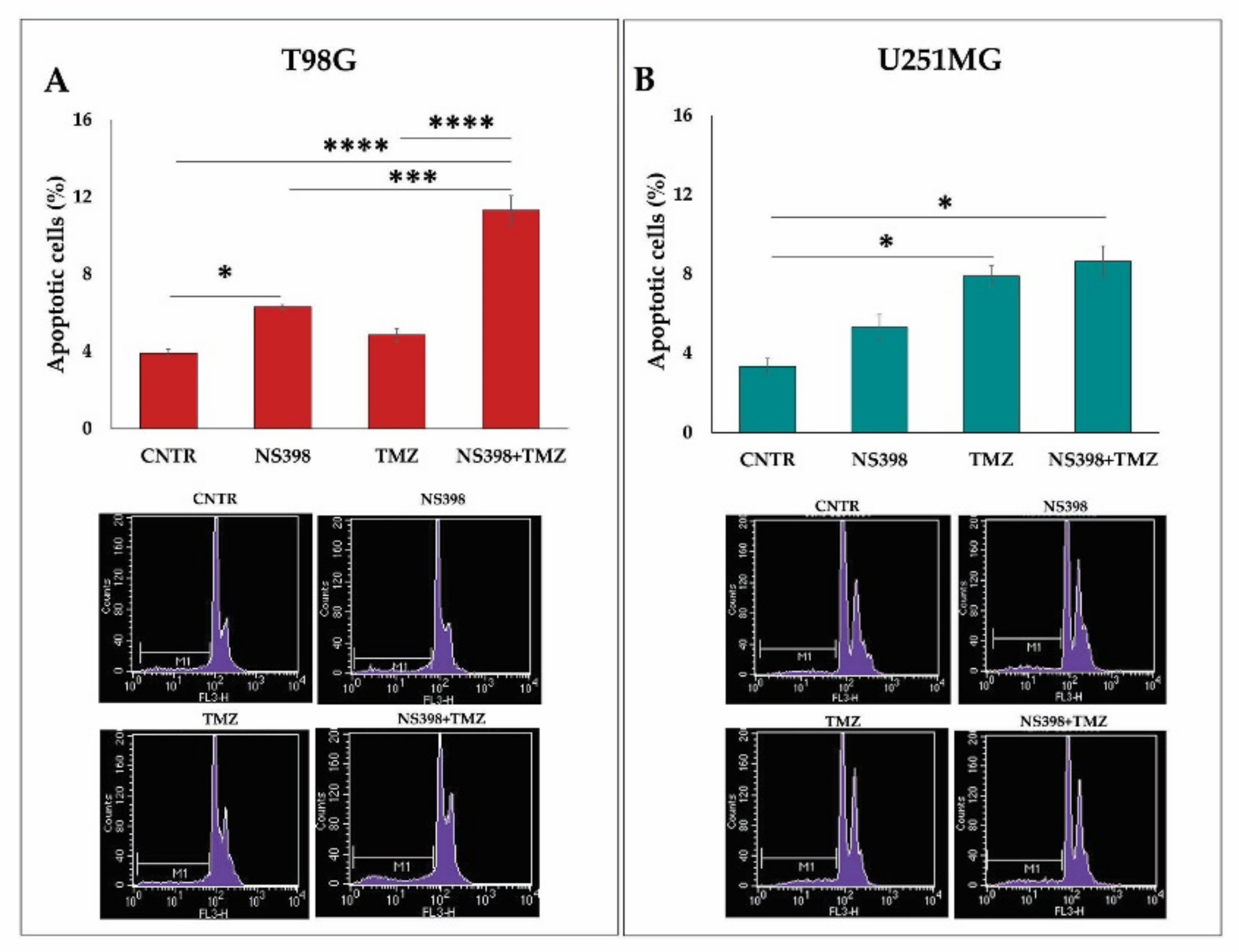
2.5. Effects of NS398 Alone or Combined with TMZ on GBM Cell Viability, Cell Cycle Distribution, and Apoptosis
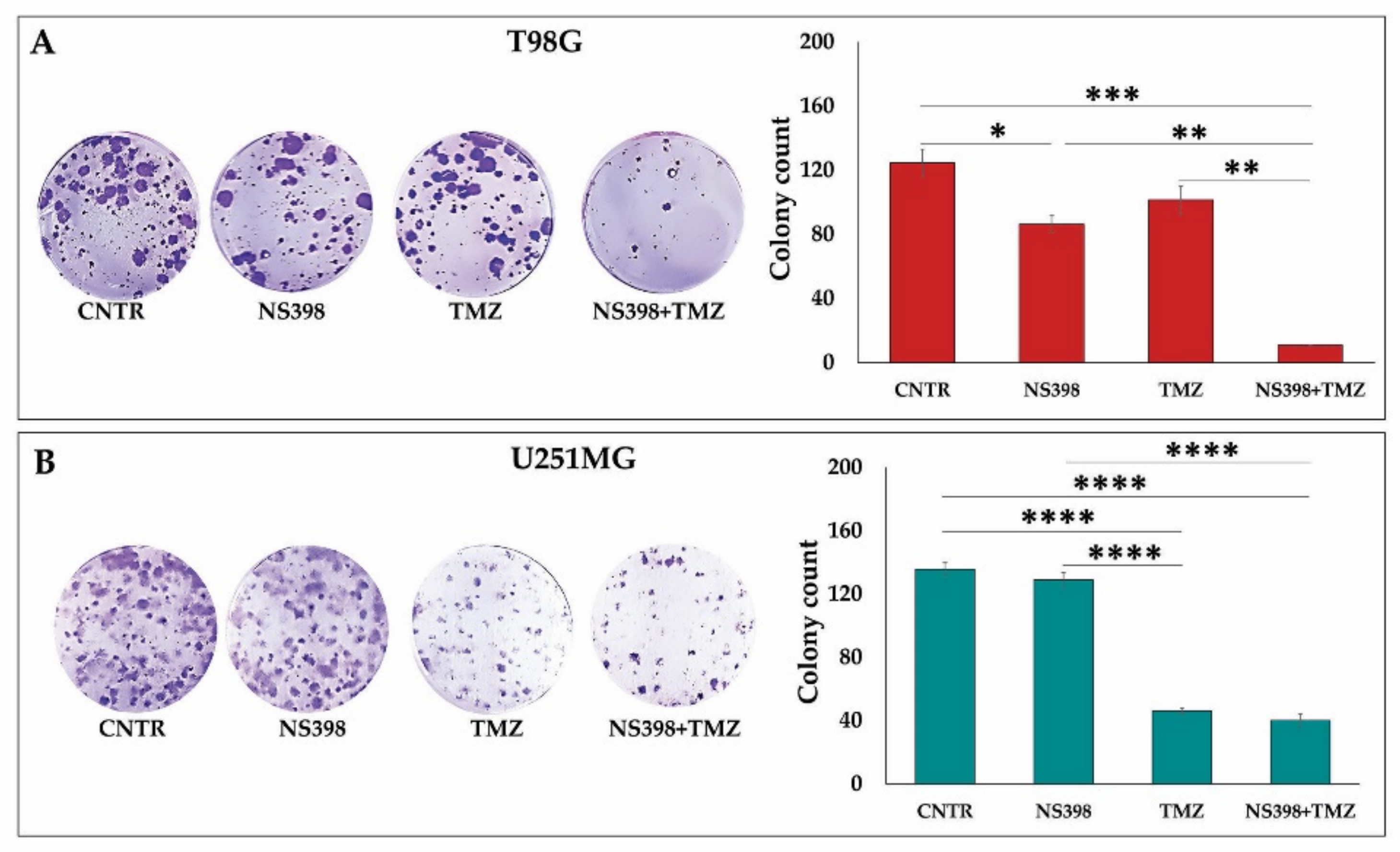
2.6. Effects of NS398 Alone or Combined with TMZ on Colony Formation Ability of GBM Cell Lines
2.7. NS398 Counteracted TMZ-Induced COX-2 Overexpression in T98G Cells
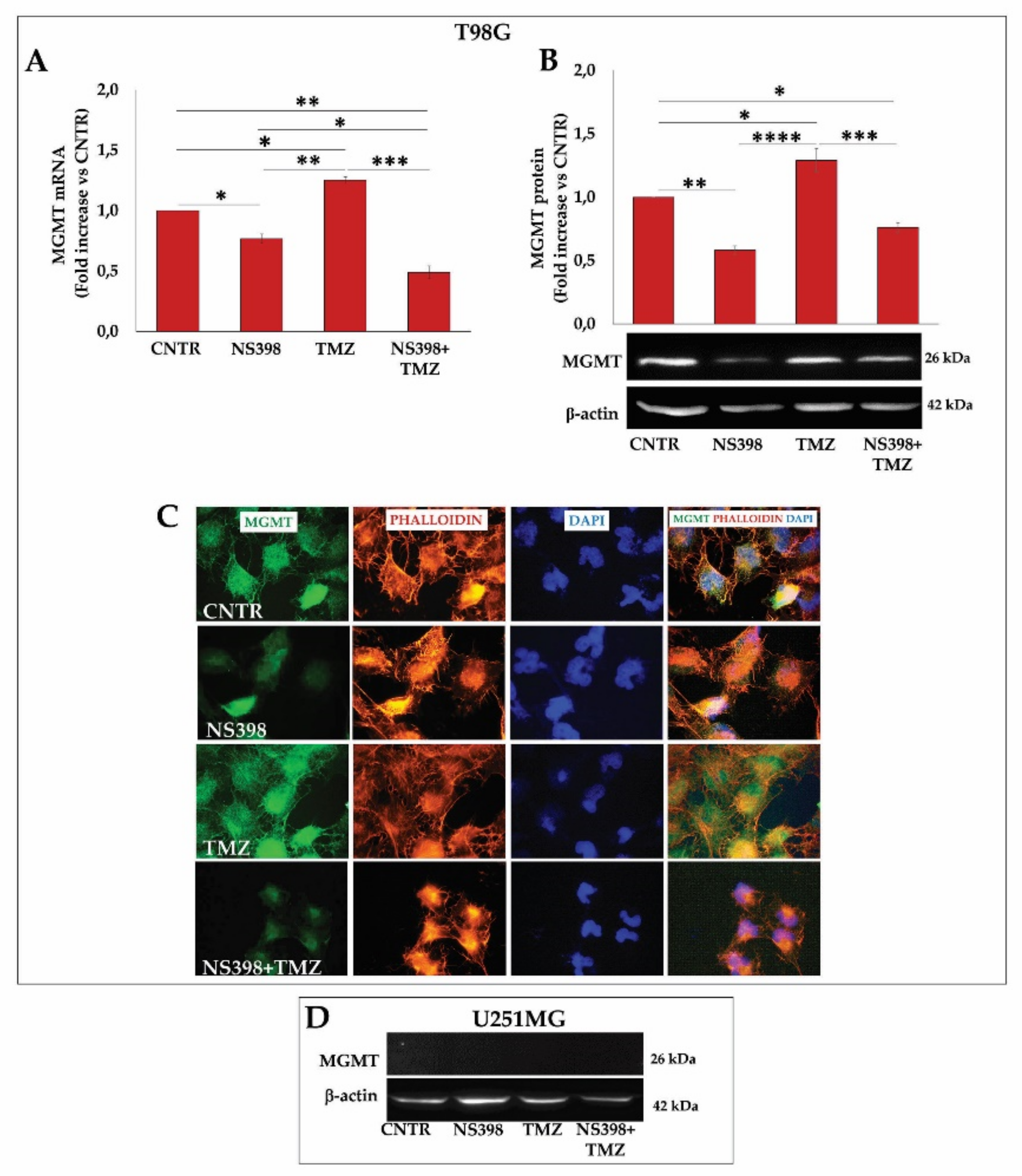
2.8. NS398 Counteracted TMZ-Induced MGMT Overexpression in T98G Cells
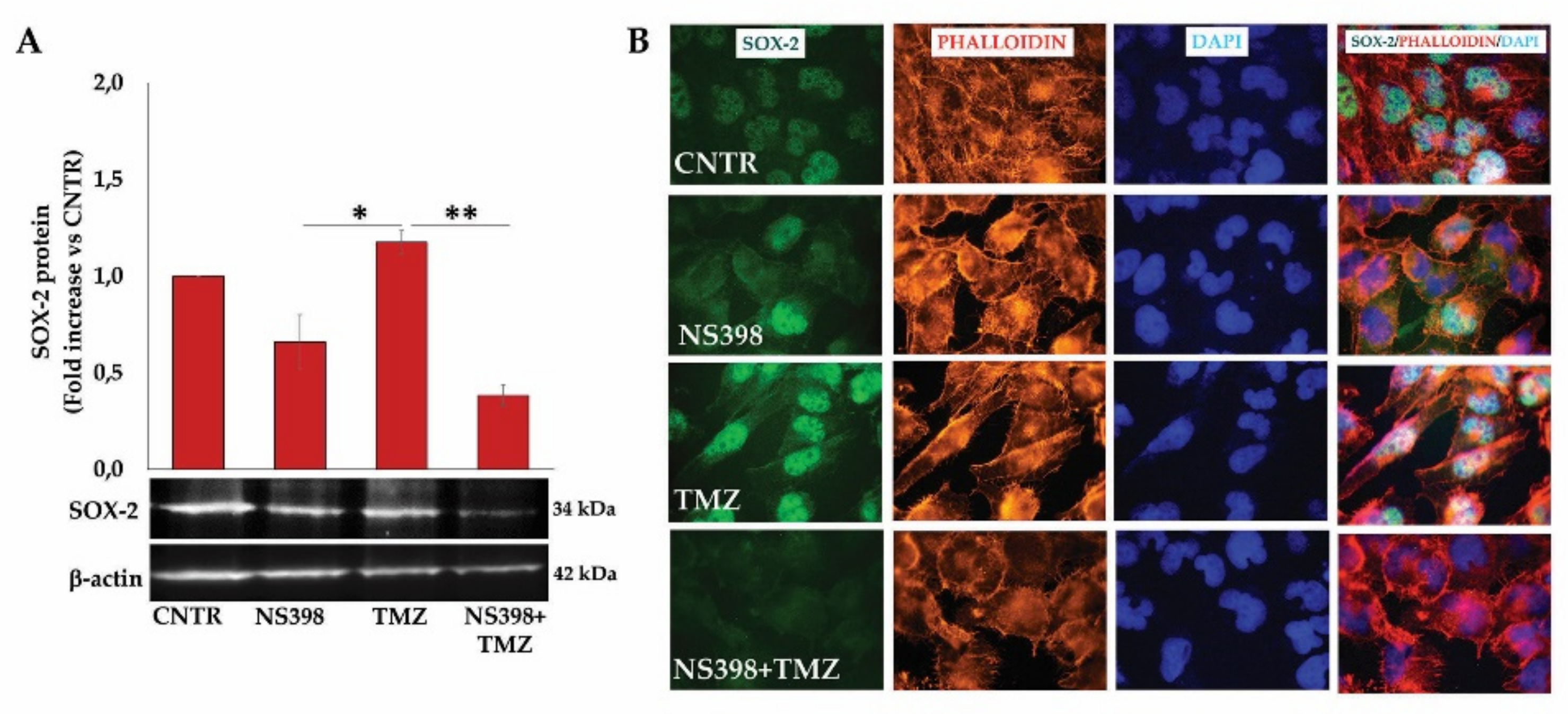
2.9. NS398 Counteracted TMZ-Induced β-Atenin and SOX-2 Overexpression in T98G Cells
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Culture Conditions
4.2. TMZ, and NS398 Preparation and Analysis of Cell Viability and Growth
4.3. Total RNA Extraction and Gene Expression
4.4. Western Blot (WB) Analysis
4.5. Prostaglandin 2 (PGE2) Level Assay
4.6. Cell Cycle Distribution and Apoptotic Cells Detection
4.7. Colony Formation Assay
4.8. Immunofluorescence Staining
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shergalis, A.; Bankhead, A.; Luesakul, U.; Muangsin, N.; Neamati, N. Current Challenges and Opportunities in Treating Glioblastoma. Pharmacol. Rev. 2018, 70, 412–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birzu, C.; French, P.; Caccese, M.; Cerretti, G.; Idbaih, A.; Zagonel, V.; Lombardi, G. Recurrent Glioblastoma: From Molecular Landscape to New Treatment Perspectives. Cancers 2021, 13, 47. [Google Scholar] [CrossRef] [PubMed]
- Taylor, O.G.; Brzozowski, J.S.; Skelding, K.A. Glioblastoma Multiforme: An Overview of Emerging Therapeutic Targets. Front. Oncol. 2019, 9, 963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oldrini, B.; Vaquero-Siguero, N.; Mu, Q.H.; Kroon, P.; Zhang, Y.; Galan-Ganga, M.; Bao, Z.S.; Wang, Z.; Liu, H.J.; Sa, J.K.; et al. MGMT genomic rearrangements contribute to chemotherapy resistance in gliomas. Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef]
- Serrano-Heras, G.; Castro-Robles, B.; Romero-Sanchez, C.M.; Carrion, B.; Barbella-Aponte, R.; Sandoval, H.; Segura, T. Involvement of N-methylpurine DNA glycosylase in resistance to temozolomide in patient-derived glioma cells. Sci. Rep. 2020, 10, 1–10. [Google Scholar] [CrossRef]
- Butler, M.; Pongor, L.; Su, Y.T.; Xi, L.Q.; Raffeld, M.; Quezado, M.; Trepel, J.; Aldape, K.; Pommier, Y.; Wu, J. MGMT Status as a Clinical Biomarker in Glioblastoma. Trends Cancer 2020, 6, 380–391. [Google Scholar] [CrossRef]
- Yu, W.; Zhang, L.L.; Wei, Q.C.; Shao, A.W. O-6-Methylguanine-DNA Methyltransferase (MGMT): Challenges and New Opportunities in Glioma Chemotherapy. Front. Oncol. 2020, 9, 1547. [Google Scholar] [CrossRef] [Green Version]
- Joki, T.; Heese, O.; Nikas, D.C.; Bello, L.; Zhang, J.P.; Kraeft, S.K.; Seyfried, N.T.; Abe, T.; Chen, L.B.; Carroll, R.S.; et al. Expression of cyclooxygenase 2 (COX-2) in human glioma and in Vitro inhibition by a specific COX-2 inhibitor, NS-398. Cancer Res. 2000, 60, 4926–4931. [Google Scholar] [PubMed]
- Shono, T.; Tofilon, P.J.; Bruner, J.M.; Owolabi, O.; Lang, F.F. Cyclooxygenase-2 expression in human gliomas: Prognostic significance and molecular correlations. Cancer Res. 2001, 61, 4375–4381. [Google Scholar]
- Karim, A.; Mccarthy, K.; Jawahar, A.; Smith, D.; Willis, B.; Nanda, A. Differential cyclooxygenase-2 enzyme expression in radiosensitive versus radioresistant glioblastoma multiforme cell lines. Anticancer Res. 2005, 25, 675–679. [Google Scholar] [PubMed]
- Kuipers, G.K.; Slotman, B.J.; Wedekind, L.E.; Stoter, T.R.; Van Den Berg, J.; Sminia, P.; Vincent, M.; Lafleur, M. Radiosensitization of human glioma cells by cyclooxygenase-2 (COX-2) inhibition: Independent on COX-2 expression and dependent on the COX-2 inhibitor and sequence of administration. Int. J. Radiat. Biol. 2007, 83, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, P.; Lombardi, F.; Augello, F.R.; Giusti, I.; Dolo, V.; Leocata, P.; Cifone, M.G.; Cinque, B. Biological effects of selective COX-2 inhibitor NS398 on human glioblastoma cell lines. Cancer Cell Int. 2020, 20, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.X.; Qiu, J.G.; Li, Q.Q.; Shi, Z. Prostaglandin E2 Signaling: Alternative Target for Glioblastoma? Trends Cancer 2017, 3, 75–78. [Google Scholar] [CrossRef] [Green Version]
- Cook, P.J.; Thomas, R.; Kingsley, P.J.; Shimizu, F.; Montrose, D.C.; Marnett, L.J.; Tabar, V.S.; Dannenberg, A.J.; Benezra, R. Cox-2-derived PGE(2) induces Id1-dependent radiation resistance and self-renewal in experimental glioblastoma. Neuro-Oncology 2016, 18, 1379–1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Valenzuela, F.; Escobar, E.; Perez-Tomas, R.; Montecinos, V.P. The Inflammatory Profile of the Tumor Microenvironment, Orchestrated by Cyclooxygenase-2, Promotes Epithelial-Mesenchymal Transition. Front. Oncol. 2021, 11, 686792. [Google Scholar] [CrossRef]
- Tong, D.L.; Liu, Q.L.; Wang, L.A.; Xie, Q.B.; Pang, J.; Huang, Y.Q.; Wang, L.F.; Liu, G.L.; Zhang, D.Z.; Lan, W.H.; et al. The roles of the COX2/PGE2/EP axis in therapeutic resistance. Cancer Metastasis Rev. 2018, 37, 355–368. [Google Scholar] [CrossRef]
- Sachdeva, R.; Wu, M.; Smiljanic, S.; Kaskun, O.; Ghannad-Zadeh, K.; Celebre, A.; Isaev, K.; Morrissy, A.S.; Guan, J.; Tong, J.F.; et al. ID1 Is Critical for Tumorigenesis and Regulates Chemoresistance in Glioblastoma. Cancer Res. 2019, 79, 4057–4071. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.X.; Bo, Z.Y.; Gong, W.Y.; Guo, Y. Inhibitor of Differentiation 1 (Id1) in Cancer and Cancer Therapy. Int. J. Med. Sci. 2020, 17, 995–1005. [Google Scholar] [CrossRef] [Green Version]
- Jalota, A.; Kumar, M.; Das, B.C.; Yadav, A.K.; Chosdol, K.; Sinha, S. A drug combination targeting hypoxia induced chemoresistance and stemness in glioma cells. Oncotarget 2018, 9, 18351–18366. [Google Scholar] [CrossRef] [Green Version]
- Qiu, J.G.; Shi, Z.; Jiang, J.X. Cyclooxygenase-2 in glioblastoma multiforme. Drug Discov. Today 2017, 22, 148–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, K.; Gerelchuluun, A.; Hong, Z.S.; Sun, L.; Zenkoh, J.; Moritake, T.; Tsuboi, K. Celecoxib enhances radiosensitivity of hypoxic glioblastoma cells through endoplasmic reticulum stress. Neuro-Oncology 2013, 15, 1186–1199. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.I.; Chiou, S.H.; Hueng, D.Y.; Tai, L.K.; Huang, P.I.; Kao, C.L.; Chen, Y.W.; Sytwu, H.K. Celecoxib and radioresistant glioblastoma-derived CD133(+) cells: Improvement in radiotherapeutic effects Laboratory investigation. J. Neurosurg. 2011, 114, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.G.; Guan, J.; Li, C.; Gunter, S.; Nusrat, L.; Ng, S.; Dhand, K.; Morshead, C.; Kim, A.; Das, S. Aberrantly activated Cox-2 and Wnt signaling interact to maintain cancer stem cells in glioblastoma. Oncotarget 2017, 8, 82217–82230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stockhammer, F.; Misch, M.; Koch, A.; Czabanka, M.; Plotkin, M.; Blechschmidt, C.; Tuettenberg, J.; Vajkoczy, P. Continuous low-dose temozolomide and celecoxib in recurrent glioblastoma. J. Neuro-Oncol. 2010, 100, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Kast, R.E.; Karpel-Massler, G.; Halatsch, M.E. CUSP9*treatment protocol for recurrent glioblastoma: Aprepitant, artesunate, auranofin, captopril, celecoxib, disulfiram, itraconazole, ritonavir, sertraline augmenting continuous low dose temozolomide. Oncotarget 2014, 5, 8052–8082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickstrom, M.; Dyberg, C.; Milosevic, J.; Einvik, C.; Calero, R.; Sveinbjornsson, B.; Sanden, E.; Darabi, A.; Siesjo, P.; Kool, M.; et al. Wnt/beta-catenin pathway regulates MGMT gene expression in cancer and inhibition of Wnt signalling prevents chemoresistance. Nat. Commun. 2015, 6, 1–10. [Google Scholar] [CrossRef]
- Zuccarini, M.; Giuliani, P.; Ziberi, S.; Carluccio, M.; Di Iorio, P.; Caciagli, F.; Ciccarelli, R. The Role of Wnt Signal in Glioblastoma Development and Progression: A Possible New Pharmacological Target for the Therapy of This Tumor. Genes 2018, 9, 105. [Google Scholar] [CrossRef] [Green Version]
- Johnsen, J.I.; Wickstrom, M.; Baryawno, N. Wingless/beta-catenin signaling as a modulator of chemoresistance in cancer. Mol. Cell. Oncol. 2016, 3, e1131356. [Google Scholar] [CrossRef] [Green Version]
- Roos, J.; Grosch, S.; Werz, O.; Schroder, P.; Ziegler, S.; Fulda, S.; Paulus, P.; Urbschat, A.; Kuhn, B.; Maucher, I.; et al. Regulation of tumorigenic Wnt signaling by cyclooxygenase-2, 5-lipoxygenase and their pharmacological inhibitors: A basis for novel drugs targeting cancer cells? Pharmacol. Ther. 2016, 157, 43–64. [Google Scholar] [CrossRef]
- Buchanan, F.G.; DuBois, R.N. Connecting COX-2 and Wnt in cancer. Cancer Cell 2006, 9, 6–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, X.Y.; Zang, J.; Zheng, M.H.; Zhang, Y.F.; Yue, K.Y.; Cao, X.L.; Cao, Y.; Li, X.X.; Han, H.; Jiang, X.F.; et al. Temozolomide Treatment Induces HMGB1 to Promote the Formation of Glioma Stem Cells via the TLR2/NEAT1/Wnt Pathway in Glioblastoma. Front. Cell Dev. Biol. 2021, 9, 883. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.Y.; Song, L.B.; Wu, W.J.; Zhou, Y.Q.; Zhu, J.R.; Wu, G.Y.; Cao, L.X.; Song, J.W.; Li, J.; Zhang, W. TRIM14 promotes chemoresistance in gliomas by activating Wnt/beta-catenin signaling via stabilizing Dvl2. Oncogene 2018, 37, 5476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomar, V.S.; Patil, V.; Somasundaram, K. Temozolomide induces activation of Wnt/beta-catenin signaling in glioma cells via PI3K/Akt pathway: Implications in glioma therapy. Cell Biol. Toxicol. 2020, 36, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Ulasov, I.V.; Nandi, S.; Dey, M.; Sonabend, A.M.; Lesniak, M.S. Inhibition of Sonic Hedgehog and Notch Pathways Enhances Sensitivity of CD133(+) Glioma Stem Cells to Temozolomide Therapy. Mol. Med. 2011, 17, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, M.T.; Miyake, J.A.; Gomes, R.N.; Feitoza, F.; Stevannato, P.B.; da Cunha, A.S.; Serachi, F.D.; Panagopoulos, A.T.; Colquhoun, A. Cyclooxygenase Inhibition Alters Proliferative, Migratory, and Invasive Properties of Human Glioblastoma Cells In Vitro. Int. J. Mol. Sci. 2021, 22, 4297. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Jo, S.; Lee, H.; Kim, T.U.; Kim, I.C.; Yim, J.H.; Chung, H. Lobarstin Enhances Chemosensitivity in Human Glioblastoma T98G Cells. Anticancer. Res. 2013, 33, 5445–5451. [Google Scholar] [PubMed]
- Lee, S.Y. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 2016, 3, 198–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montaldi, A.P.; Godoy, P.R.D.V.; Sakamoto-Hojo, E.T. APE1/REF-1 down-regulation enhances the cytotoxic effects of temozolomide in a resistant glioblastoma cell line. Mutat. Res. Genet. Toxicol. Environ. Mutagenesis 2015, 793, 19–29. [Google Scholar] [CrossRef]
- Gurpinar, E.; Grizzle, W.E.; Piazza, G.A. COX-Independent Mechanisms of Cancer Chemoprevention by Anti-Inflammatory Drugs. Front. Oncol. 2013, 3, 181. [Google Scholar] [CrossRef] [Green Version]
- Ponten, F.; Jirstrom, K.; Uhlen, M. The Human Protein Atlas-A tool for pathology. J. Pathol. 2008, 216, 387–393. [Google Scholar] [CrossRef]
- Chahal, M.; Abdulkarim, B.; Xu, Y.X.; Guiot, M.C.; Easaw, J.C.; Stifani, N.; Sabri, S. O(6)-Methylguanine-DNA Methyltransferase Is a Novel Negative Effector of Invasion in Glioblastoma Multiforme. Mol. Cancer Ther. 2012, 11, 2440–2450. [Google Scholar] [CrossRef] [Green Version]
- Latour, M.; Her, N.G.; Kesari, S.; Nurmemmedov, E. WNT Signaling as a Therapeutic Target for Glioblastoma. Int. J. Mol. Sci. 2021, 22, 8428. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, P.; Miconi, G.; Cinque, B.; Lombardi, F.; La Torre, C.; Dehcordi, S.R.; Galzio, R.; Cimini, A.; Giordano, A.; Cifone, M.G. NOS2 expression in glioma cell lines and glioma primary cell cultures: Correlation with neurosphere generation and SOX-2 expression. Oncotarget 2017, 8, 25582–25598. [Google Scholar] [CrossRef] [Green Version]
- Valtorta, S.; Lo Dico, A.; Raccagni, I.; Gaglio, D.; Belloli, S.; Politi, L.S.; Martelli, C.; Diceglie, C.; Bonanomi, M.; Ercoli, G.; et al. Metformin and temozolomide, a synergic option to overcome resistance in glioblastoma multiforme models. Oncotarget 2017, 8, 113090–113104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herbener, V.J.; Burster, T.; Goreth, A.; Pruss, M.; von Bandemer, H.; Baisch, T.; Fitzel, R.; Siegelin, M.D.; Karpel-Massler, G.; Debatin, K.M.; et al. Considering the Experimental use of Temozolomide in Glioblastoma Research. Biomedicines 2020, 8, 151. [Google Scholar] [CrossRef] [PubMed]
- Ostermann, S.; Csajka, C.; Buclin, T.; Leyvraz, S.; Lejeune, F.; Decosterd, L.A.; Stupp, R. Plasma and cerebrospinal fluid population pharmacokinetics of temozolomide in malignant glioma patients. Clin. Cancer Res. 2004, 10, 3728–3736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosso, L.; Brock, C.S.; Gallo, J.M.; Saleem, A.; Price, P.M.; Turkheimer, F.E.; Aboagye, E.O. A new model for prediction of drug distribution in tumor and normal tissues: Pharmacokinetics of temozolomide in glioma patients. Cancer Res. 2009, 69, 120–127. [Google Scholar] [CrossRef] [Green Version]
- Stepanenko, A.A.; Chekhonin, V.P. On the Critical Issues in Temozolomide Research in Glioblastoma: Clinically Relevant Concentrations and MGMT-independent Resistance. Biomedicines 2019, 7, 92. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.C.; Kunnumakkara, A.B.; Aggarwal, S.; Aggarwal, B.B. Inflammation, a Double-Edge Sword for Cancer and Other Age-Related Diseases. Front. Immunol. 2018, 9, 2160. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.; Li, D.; Fu, J.L.; Sun, Y.; Li, Y.R.; Qu, R.F.; Jin, X.; Li, D.F. Upregulation of cyclooxygenase-2 is associated with activation of the alternative nuclear factor kappa B signaling pathway in colonic adenocarcinoma. Am. J. Transl. Res. 2015, 7, 1612–1620. [Google Scholar] [PubMed]
- Soubannier, V.; Stifani, S. NF-kappaB Signalling in Glioblastoma. Biomedicines 2017, 5, 29. [Google Scholar] [CrossRef] [Green Version]
- Munoz, J.L.; Rodriguez-Cruz, V.; Greco, S.J.; Nagula, V.; Scotto, K.W.; Rameshwar, P. Temozolomide induces the production of epidermal growth factor to regulate MDR1 expression in glioblastoma cells. Mol. Cancer Ther. 2014, 13, 2399–2411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouzer, C.A.; Marnett, L.J. Structural and Chemical Biology of the Interaction of Cyclooxygenase with Substrates and Non-Steroidal Anti-Inflammatory Drugs. Chem. Rev. 2020, 120, 7592–7641. [Google Scholar] [CrossRef] [PubMed]
- Bergqvist, F.; Ossipova, E.; Idborg, H.; Raouf, J.; Checa, A.; Englund, K.; Englund, P.; Khoonsari, P.E.; Kultima, K.; Wheelock, C.E.; et al. Inhibition of mPGES-1 or COX-2 Results in Different Proteomic and Lipidomic Profiles in A549 Lung Cancer Cells. Front. Pharmacol. 2019, 10, 636. [Google Scholar] [CrossRef] [Green Version]
- Majchrzak-Celinska, A.; Misiorek, J.O.; Kruhlenia, N.; Przybyl, L.; Kleszcz, R.; Rolle, K.; Krajka-Kuzniak, V. COXIBs and 2,5-dimethylcelecoxib counteract the hyperactivated Wnt/beta-catenin pathway and COX-2/PGE2/EP4 signaling in glioblastoma cells. BMC Cancer 2021, 21, 493. [Google Scholar] [CrossRef]
- Authier, A.; Farrand, K.J.; Broadley, K.W.; Ancelet, L.R.; Hunn, M.K.; Stone, S.; McConnell, M.J.; Hermans, I.F. Enhanced immunosuppression by therapy-exposed glioblastoma multiforme tumor cells. Int. J. Cancer 2015, 136, 2566–2578. [Google Scholar] [CrossRef] [PubMed]
- Pu, D.; Yin, L.; Huang, L.; Qin, C.; Zhou, Y.; Wu, Q.; Li, Y.; Zhou, Q.; Li, L. Cyclooxygenase-2 Inhibitor: A Potential Combination Strategy with Immunotherapy in Cancer. Front. Oncol. 2021, 11, 637504. [Google Scholar] [CrossRef]
- Buque, A.; Galluzzi, L. Modeling Tumor Immunology and Immunotherapy in Mice. Trends Cancer 2018, 4, 599–601. [Google Scholar] [CrossRef]
- Haddad, A.F.; Young, J.S.; Amara, D.; Berger, M.S.; Raleigh, D.R.; Aghi, M.K.; Butowski, N.A. Mouse models of glioblastoma for the evaluation of novel therapeutic strategies. Neurooncol. Adv. 2021, 3, vdab100. [Google Scholar] [CrossRef] [PubMed]
- Ishii, N.; Maier, D.; Merlo, A.; Tada, M.; Sawamura, Y.; Diserens, A.C.; Van Meir, E.G. Frequent co-alterations of TP53, p16/CDKN2A, p14(ARF), PTEN tumor suppressor genes in human glioma cell lines. Brain Pathol. 1999, 9, 469–479. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Lin, H.; Zhang, X.; Li, J. Resveratrol reverses temozolomide resistance by downregulation of MGMT in T98G glioblastoma cells by the NF-kappaB-dependent pathway. Oncol. Rep. 2012, 27, 2050–2056. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.Y.; Jiang, R.; Lian, Z.W.; Zhang, W.J.; Hu, Z.Q.; Hu, D.L. miR-222/GAS5 is involved in DNA damage and cytotoxic effects induced by temozolomide in T98G cell line. J. Appl. Toxicol. 2019, 39, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Milani, R.; Brognara, E.; Fabbri, E.; Finotti, A.; Borgatti, M.; Lampronti, I.; Marzaro, G.; Chilin, A.; Lee, K.K.; Kok, S.H.; et al. Corilagin Induces High Levels of Apoptosis in the Temozolomide-Resistant T98G Glioma Cell Line. Oncol. Res. 2018, 26, 1307–1315. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(T)(-Delta Delta C) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Balla, M.M.S.; Yadav, H.D.; Pandey, B.N. Tumorsphere assay provides a better in vitro method for cancer stem-like cells enrichment in A549 lung adenocarcinoma cells. Tissue Cell 2019, 60, 21–24. [Google Scholar] [CrossRef] [PubMed]
- Maurya, D.K. ColonyCountJ: A User-Friendly Image J Add-on Program for Quantification of Different Colony Parameters in Clonogenic Assay. J. Clin. Toxicol. 2017, 7, 2161. [Google Scholar] [CrossRef] [Green Version]












Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lombardi, F.; Augello, F.R.; Artone, S.; Gugu, M.K.; Cifone, M.G.; Cinque, B.; Palumbo, P. Up-Regulation of Cyclooxygenase-2 (COX-2) Expression by Temozolomide (TMZ) in Human Glioblastoma (GBM) Cell Lines. Int. J. Mol. Sci. 2022, 23, 1545. https://doi.org/10.3390/ijms23031545
Lombardi F, Augello FR, Artone S, Gugu MK, Cifone MG, Cinque B, Palumbo P. Up-Regulation of Cyclooxygenase-2 (COX-2) Expression by Temozolomide (TMZ) in Human Glioblastoma (GBM) Cell Lines. International Journal of Molecular Sciences. 2022; 23(3):1545. https://doi.org/10.3390/ijms23031545
Chicago/Turabian StyleLombardi, Francesca, Francesca Rosaria Augello, Serena Artone, Mitilda Karoli Gugu, Maria Grazia Cifone, Benedetta Cinque, and Paola Palumbo. 2022. "Up-Regulation of Cyclooxygenase-2 (COX-2) Expression by Temozolomide (TMZ) in Human Glioblastoma (GBM) Cell Lines" International Journal of Molecular Sciences 23, no. 3: 1545. https://doi.org/10.3390/ijms23031545
APA StyleLombardi, F., Augello, F. R., Artone, S., Gugu, M. K., Cifone, M. G., Cinque, B., & Palumbo, P. (2022). Up-Regulation of Cyclooxygenase-2 (COX-2) Expression by Temozolomide (TMZ) in Human Glioblastoma (GBM) Cell Lines. International Journal of Molecular Sciences, 23(3), 1545. https://doi.org/10.3390/ijms23031545