
Hereditary Spastic Paraplegia: An Update


Abstract
:1. Introduction
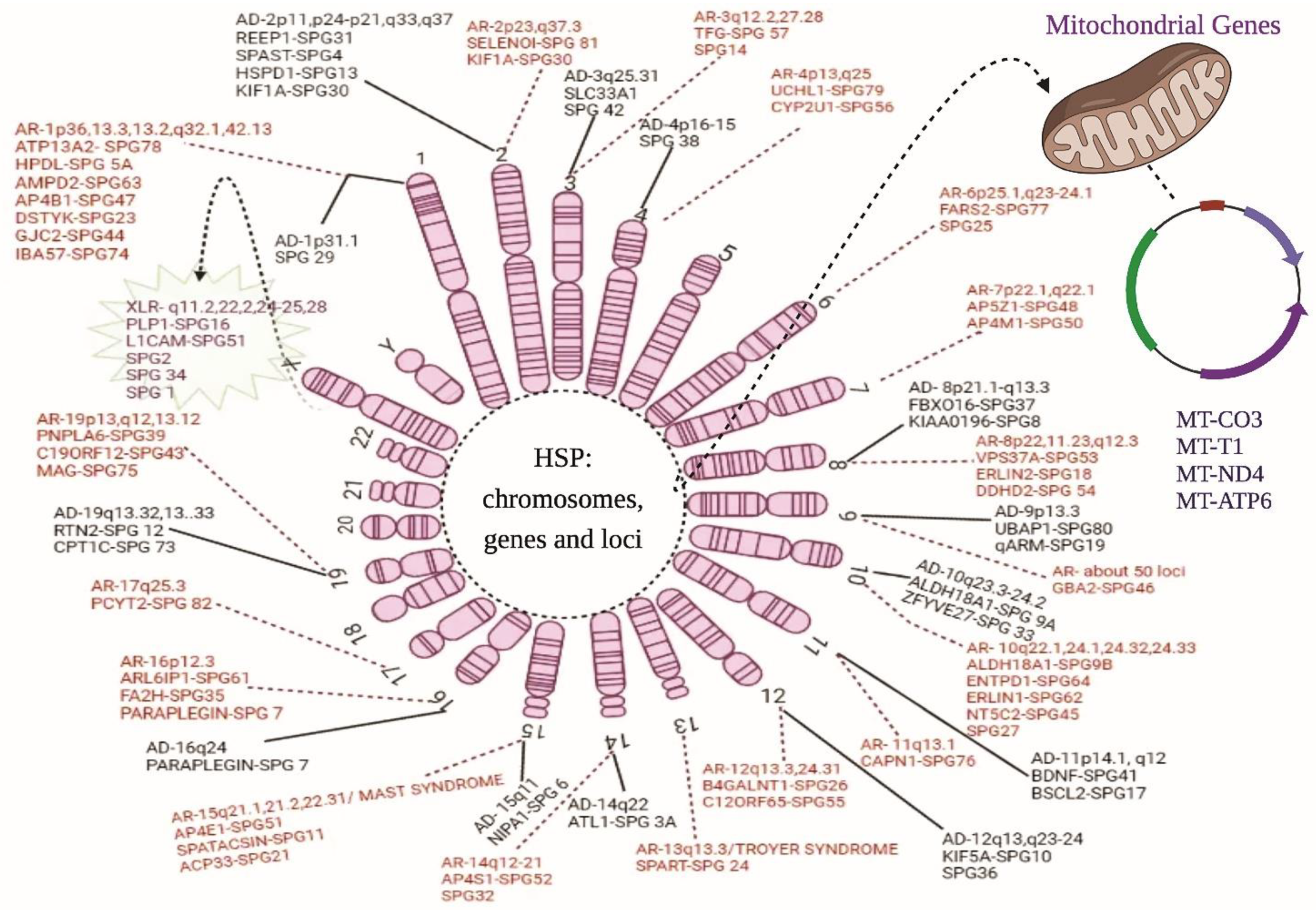
HSPs Stratification
2. Autosomal Dominant (AD) HSP
2.1. Chromosome 1
2.2. Chromosome 2
2.3. Chromosome 3
2.4. Chromosome 4
2.5. Chromosome 8
2.6. Chromosome 9
2.7. Chromosome 10
2.8. Chromosome 11
2.9. Chromosome 12
2.10. Chromosome 14
2.11. Chromosome 15
2.12. Chromosome 16
2.13. Chromosome 19
3. Autosomal Recessive (AR) HSPs
3.1. Chromosome 1
3.2. Chromosome 2
3.3. Chromosome 3
3.4. Chromosome 4
3.5. Chromosome 6
3.6. Chromosome 7
3.7. Chromosome 8
3.8. Chromosome 9
3.9. Chromosome 10
3.10. Chromosome 11
3.11. Chromosome 12
3.12. Chromosome 13
3.13. Chromosome 14
3.14. Chromosome 15
3.15. Chromosome 16
3.16. Chromosome 17
3.17. Chromosome 19
4. X-Linked Recessive (XLR) HSPs
Chromosome X
5. Maternal Inheritance (Mitochondrial HSPs)
5.1. Predictive Genes for the Various HSP Forms:
5.1.1. Chromosome 1
5.1.2. Chromosome 4
5.1.3. Chromosome 11
5.1.4. Chromosome 12
5.1.5. Chromosome X
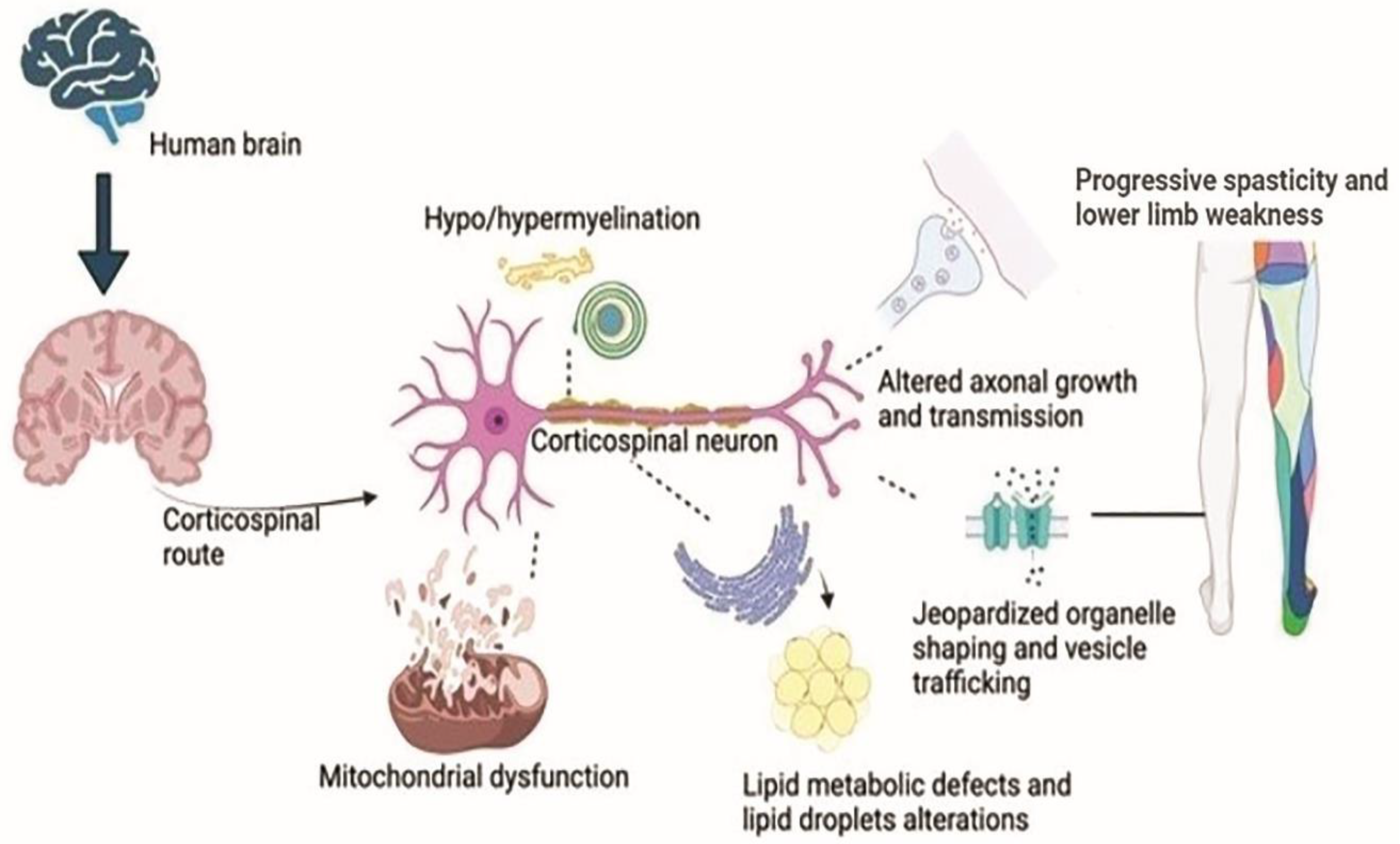
6. Neuron’s Pathology in HSP
7. Epidemiology of the HSP Genes
8. The Typical Cellular Models in HSP Pathogenesis
9. Organelles and Membrane Morphogenesis
9.1. An ATPase Integrated with Microtubule and Membrane Structure—Spastin
9.2. A Big GTPase Amalgamating ER Tubules—Atlastin
9.3. Maintaining ER Morphology
9.4. Defects in Metabolism of Lipid and Its Droplet
9.5. Lysosomal Regeneration by Spatacsin and Spastizin
9.6. HSP Adaptor Proteins
9.7. Endosome Tubule Formation and Strumpellin
9.8. Organelles Shape Defects Inducing Axon Pathology
10. Signalling of Bone Morphogenetic Protein (BMP)
11. Transport through Motor Proteins
12. Mitochondrial Dysfunction
13. Path of Axon
14. Defects in the Myelination Process (Hypo or Dysmyelination)
15. Metabolism of the Nucleotides
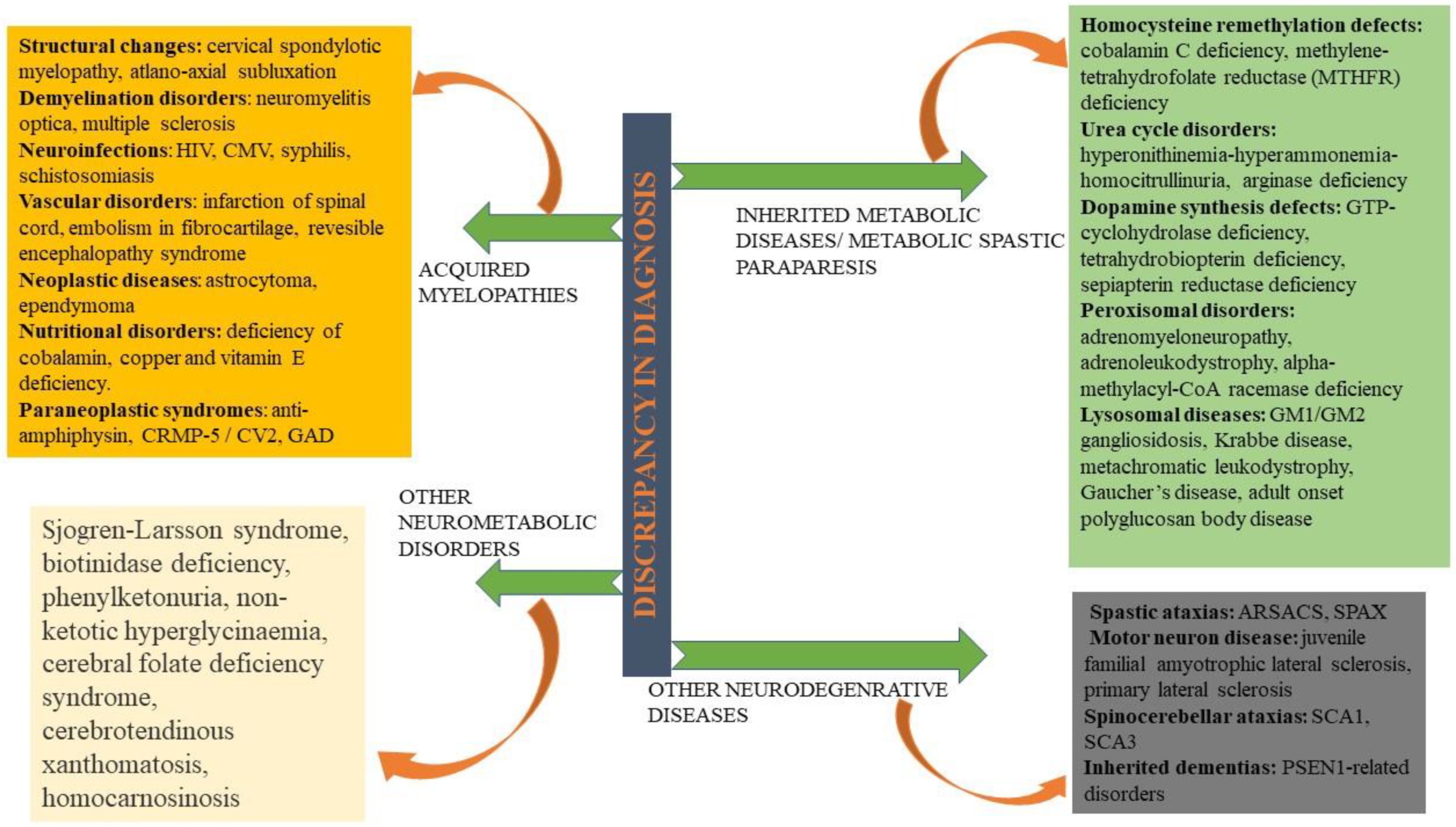
16. Diagnosis and Its Discrepancy in HSPs
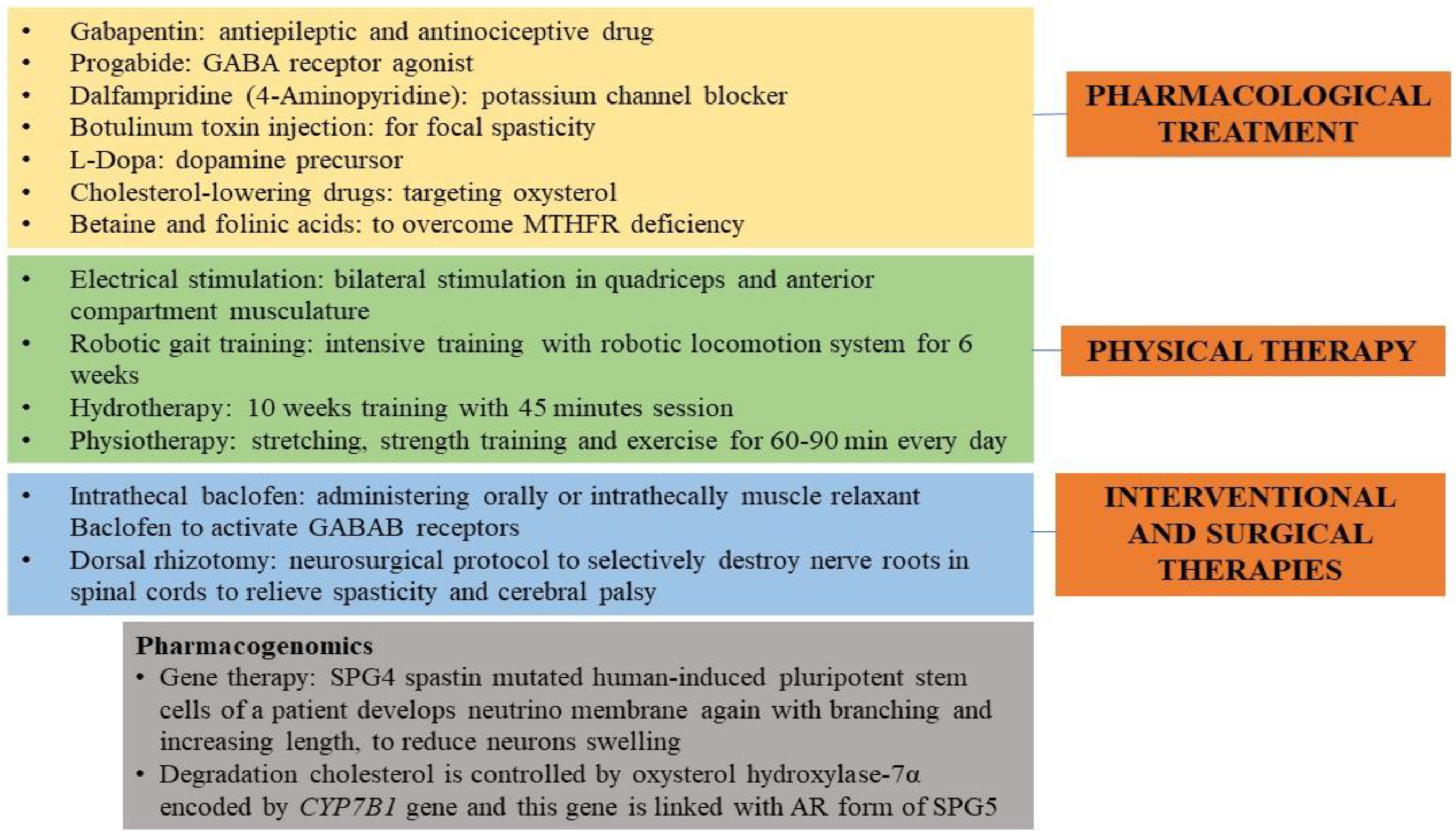
17. Therapeutic for HSP
18. Present-Day Treatment Mode
19. Futuristic Approaches
20. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shribman, S.; Reid, E.; Crosby, A.H.; Houlden, H.; Warner, T.T. Hereditary spastic paraplegia: From diagnosis to emerging therapeutic approaches. Lancet Neurol. 2019, 18, 1136–1146. [Google Scholar] [CrossRef]
- De Beukelaer, N.; Bar-On, L.; Hanssen, B.; Peeters, N.; Prinsen, S.; Ortibus, E.; Desloovere, K.; Van Campenhout, A. Muscle characteristics in pediatric hereditary spastic paraplegia vs. bilateral spastic cerebral palsy: An exploratory study. Front. Neurol. 2021, 12, 635032. [Google Scholar] [CrossRef] [PubMed]
- Koh, K.; Ishiura, H.; Tsuji, S.; Takiyama, Y. JASPAC: Japan spastic paraplegia research consortium. Brain Sci. 2018, 8, 153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schüle, R.; Wiethoff, S.; Martus, P.; Karle, K.N.; Otto, S.; Klebe, S.; Klimpe, S.; Gallenmüller, C.; Kurzwelly, D.; Henkel, D.; et al. Hereditary spastic paraplegia: Clinicogenetic lessons from 608 patients. Ann. Neurol. 2016, 79, 646–658. [Google Scholar] [CrossRef]
- Carosi, L.; Giudice, T.L.; Lullo, M.; Di Lombardi, F.; Babalini, C.; Gaudiello, F.; Marfia, G.A.; Massa, R.; Kawarai, T.; Orlacchio, A. Hereditary spastic paraplegia:A novel mutation and expansion of the phenotype variability in SPG10. J. Neurol. Neurosurg. Psychiatry 2015, 86, 702–704. [Google Scholar] [CrossRef] [Green Version]
- Lo Giudice, T.; Lombardi, F.; Santorelli, F.M.; Kawarai, T.; Orlacchio, A. Hereditary spastic paraplegia: Clinical-genetic characteristics and evolving molecular mechanisms. Exp. Neurol. 2014, 261, 518–539. [Google Scholar] [CrossRef]
- Montecchiani, C.; Pedace, L.; Lo Giudice, T.; Casella, A.; Mearini, M.; Gaudiello, F.; Pedroso, J.L.; Terracciano, C.; Caltagirone, C.; Massa, R.; et al. ALS5/SPG11/KIAA1840 mutations cause autosomal recessive axonal Charcot-Marie-Tooth disease. Brain 2016, 139, 73–85. [Google Scholar] [CrossRef] [Green Version]
- Orlacchio, A.; Kawarai, T.; Gaudiello, F.; St. George-Hyslop, P.H.; Floris, R.; Bernardi, G. New locus for hereditary spastic paraplegia maps to chromosome 1p31.1-1p21.1. Ann. Neurol. 2005, 58, 423–429. [Google Scholar] [CrossRef]
- Orlacchio, A.; Kawarai, T.; Gaudiello, F.; Torato, A.; Schillaci, O.; Stefani, A.; Floris, R.; St. George-Hyslop, P.H.; Sorbi, S.; Bernardi, G. Clinical and genetic study of a large SPG4 Italian family. Mov. Disord. 2005, 20, 1055–1059. [Google Scholar] [CrossRef]
- Orlacchio, A.; Patrono, C.; Borreca, A.; Babalini, C.; Bernardi, G.; Kawarai, T. Spastic paraplegia in Romania: High prevalence of SPG4 mutations. J. Neurol. Neurosurg. Psychiatry 2008, 79, 606–607. [Google Scholar] [CrossRef]
- Orlacchio, A.; Babalini, C.; Borreca, A.; Patrono, C.; Massa, R.; Basaran, S.; Munhoz, R.P.; Rogaeva, E.A.; St George-Hyslop, P.H.; Bernardi, G.; et al. SPATACSIN mutations cause autosomal recessive juvenile amyotrophic lateral sclerosis. Brain 2010, 133, 591–598. [Google Scholar] [CrossRef] [Green Version]
- Chrestian, N.; Dupré, N.; Gan-Or, Z.; Szuto, A.; Chen, S.; Venkitachalam, A.; Brisson, J.D.; Warman-Chardon, J.; Ahmed, S.; Ashtiani, S.; et al. Clinical and genetic study of hereditary spastic paraplegia in Canada. Neurol. Genet. 2016, 3, e122. [Google Scholar] [CrossRef] [Green Version]
- Balicza, P.; Grosz, Z.; Gonzalez, M.A.; Bencsik, R.; Pentelenyi, K.; Gal, A.; Varga, E.; Klivenyi, P.; Koller, J.; Züchner, S.; et al. Genetic background of the hereditary spastic paraplegia phenotypes in Hungary—An analysis of 58 probands. J. Neurol. Sci. 2016, 364, 116–121. [Google Scholar] [CrossRef]
- Namekawa, M.; Ribai, P.; Nelson, I.; Forlani, S.; Fellmann, F.; Goizet, C.; Depienne, C.; Stevanin, G.; Ruberg, M.; Dürr, A.; et al. SPG3A is the most frequent cause of hereditary spastic paraplegia with onset before age 10 years. Neurology 2006, 66, 112–114. [Google Scholar] [CrossRef]
- Reed, J.A.; Wilkinson, P.A.; Patel, H.; Simpson, M.A.; Chatonnet, A.; Robay, D.; Patton, M.A.; Crosby, A.H.; Warner, T.T. A novel NIPA1 mutation associated with a pure form of autosomal dominant hereditary spastic paraplegia. Neurogenetics 2005, 6, 79–84. [Google Scholar] [CrossRef]
- Valdmanis, P.N.; Meijer, I.A.; Reynolds, A.; Lei, A.; MacLeod, P.; Schlesinger, D.; Zatz, M.; Reid, E.; Dion, P.A.; Drapeau, P.; et al. Mutations in the KIAA0196 gene at the SPG8 locus cause hereditary spastic paraplegia. Am. J. Hum. Genet. 2007, 80, 152–161. [Google Scholar] [CrossRef] [Green Version]
- Salinas, S.; Proukakis, C.; Crosby, A.; Warner, T.T. Hereditary spastic paraplegia: Clinical features and pathogenetic mechanisms. Lancet Neurol. 2008, 7, 1127–1138. [Google Scholar] [CrossRef]
- Beetz, C.; Schüle, R.; Deconinck, T.; Tran-Viet, K.N.; Zhu, H.; Kremer, B.P.H.; Frints, S.G.M.; Van Zelst-Stams, W.A.G.; Byrne, P.; Otto, S.; et al. REEP1 mutation spectrum and genotype/phenotype correlation in hereditary spastic paraplegia type 31. Brain 2008, 131, 1078–1086. [Google Scholar] [CrossRef] [Green Version]
- Martignoni, M.; Riano, E.; Rugarli, E.I. The Role of ZFYVE27/Protrudin in Hereditary Spastic Paraplegia. Am. J. Hum. Genet. 2008, 83, 127–128. [Google Scholar] [CrossRef] [Green Version]
- Hansen, J.; Svenstrup, K.; Ang, D.; Nielsen, M.N.; Christensen, J.H.; Gregersen, N.; Nielsen, J.E.; Georgopoulos, C.; Bross, P. A novel mutation in the HSPD1 gene in a patient with hereditary spastic paraplegia. J. Neurol. 2007, 254, 897–900. [Google Scholar] [CrossRef]
- Shoukier, M.; Neesen, J.; Sauter, S.M.; Argyriou, L.; Doerwald, N.; Pantakani, K.D.V.; Mannan, A.U. Expansion of mutation spectrum, determination of mutation cluster regions and predictive structural classification of SPAST mutations in hereditary spastic paraplegia. Eur. J. Hum. Genet. 2009, 17, 187–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemani, T.; Steel, D.; Kaliakatsos, M.; DeVile, C.; Ververi, A.; Scott, R.; Getov, S.; Sudhakar, S.; Male, A.; Mankad, K.; et al. KIF1A-related disorders in children: A wide spectrum of central and peripheral nervous system involvement. J. Peripher. Nerv. Syst. 2020, 25, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.; Li, J.; Liu, Q.; Mao, F.; Li, J.; Qiu, R.; Hu, H.; Song, Y.; Yang, Y.; Gao, G.; et al. A Missense Mutation in SLC33A1, which encodes the Acetyl-CoA Transporter, causes autosomal-dominant spastic paraplegia (SPG42). Am. J. Hum. Genet. 2008, 83, 752–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orlacchio, A.; Patrono, C.; Gaudiello, F.; Rocchi, C.; Moschella, V.; Floris, R.; Bernardi, G.; Kawarai, T. Silver syndrome variant of hereditary spastic paraplegia: A locus to 4p and allelism with SPG4. Neurology 2008, 70, 1959–1966. [Google Scholar] [CrossRef]
- Hanein, S.; Dürr, A.; Ribai, P.; Forlani, S.; Leutenegger, A.L.; Nelson, I.; Babron, M.C.; Elleuch, N.; Depienne, C.; Charon, C.; et al. A novel locus for autosomal dominant “uncomplicated” hereditary spastic paraplegia maps to chromosome 8p21.1-q13.3. Hum. Genet. 2007, 122, 261–273. [Google Scholar] [CrossRef]
- Zendedel, R.; Schouten, B.C.; van Weert, J.C.M.; van den Putte, B. Informal interpreting in general practice: The migrant patient’s voice. Ethn. Health 2018, 23, 158–173. [Google Scholar] [CrossRef] [Green Version]
- Farazi Fard, M.A.; Rebelo, A.P.; Buglo, E.; Nemati, H.; Dastsooz, H.; Gehweiler, I.; Reich, S.; Reichbauer, J.; Quintáns, B.; Ordóñez-Ugalde, A.; et al. Truncating mutations in UBAP1 cause hereditary spastic paraplegia. Am. J. Hum. Genet. 2019, 104, 767–773. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Su, H.Z.; Dong, E.L.; Lin, X.H.; Zhao, M.; Yang, C.; Wang, C.; Wang, J.; Chen, Y.J.; Yu, H.; et al. Stop-gain mutations in UBAP1 cause pure autosomal-dominant spastic paraplegia. Brain 2019, 142, 2238–2252. [Google Scholar] [CrossRef] [Green Version]
- Valente, E.M.; Brancati, F.; Caputo, V.; Bertini, E.; Patrono, C.; Costanti, D.; Dallapiccola, B. Novel locus for autosomal dominant pure hereditary spastic paraplegia (SPG19) maps to chromosome 9q33-q34. Ann. Neurol. 2002, 51, 681–685. [Google Scholar] [CrossRef]
- Coutelier, M.; Goizet, C.; Durr, A.; Habarou, F.; Morais, S.; Dionne-Laporte, A.; Tao, F.; Konop, J.; Stoll, M.; Charles, P.; et al. Alteration of ornithine metabolism leads to dominant and recessive hereditary spastic paraplegia. Brain 2015, 138, 2191–2205. [Google Scholar] [CrossRef] [Green Version]
- Seri, M.; Cusano, R.; Forabosco, P.; Cinti, R.; Caroli, F.; Picco, P.; Bini, R.; Morra, V.B.; De Michele, G.; Lerone, M.; et al. Genetic mapping to 10q23.3-q24.2, in a large Italian pedigree, of a new syndrome showing bilateral cataracts, gastroesophageal reflux, and spastic paraparesis with amyotrophy. Am. J. Hum. Genet. 1999, 64, 586–593. [Google Scholar] [CrossRef] [Green Version]
- Mannan, A.U.; Krawen, P.; Sauter, S.M.; Boehm, J.; Chronowska, A.; Paulus, W.; Neesen, J.; Engel, W. ZFYVE27 (SPG33), a novel spastin-binding protein, is mutated in hereditary spastic paraplegia. Am. J. Hum. Genet. 2006, 79, 351–357. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Li, D.; Ma, Y.; Yan, J.; Yang, B.; Li, P.; Yu, A.; Lu, C.; Ma, X. Role of spastin and protrudin in neurite outgrowth. J. Cell. Biochem. 2012, 113, 2296–2307. [Google Scholar] [CrossRef]
- Zhao, G.H.; Hu, Z.M.; Shen, L.; Jiang, H.; Ren, Z.J.; Liu, X.M.; Xia, K.; Guo, P.; Pan, Q.; Tang, B.S. A novel candidate locus on chromosome 11p14.1-p11.2 for autosomal dominant hereditary spastic paraplegia. Chin. Med. J. 2008, 121, 430–434. [Google Scholar] [CrossRef]
- Chaudhry, R.; Kidambi, A.; Brewer, M.H.; Antonellis, A.; Mathews, K.; Nicholson, G.; Kennerson, M. Re-analysis of an original CMTX3 family using exome sequencing identifies a known BSCL2 mutation. Muscle Nerve 2013, 47, 922–924. [Google Scholar] [CrossRef] [Green Version]
- Crimella, C.; Baschirotto, C.; Arnoldi, A.; Tonelli, A.; Tenderini, E.; Airoldi, G.; Martinuzzi, A.; Trabacca, A.; Losito, L.; Scarlato, M.; et al. Mutations in the motor and stalk domains of KIF5A in spastic paraplegia type 10 and in axonal Charcot-Marie-Tooth type 2. Clin. Genet. 2012, 82, 157–164. [Google Scholar] [CrossRef]
- Fink, J.K. Hereditary spastic paraplegia: Clinico-pathologic features and emerging molecular mechanisms. Acta Neuropathol. 2013, 126, 307–328. [Google Scholar] [CrossRef] [Green Version]
- Schüle, R.; Bonin, M.; Dürr, A.; Forlani, S.; Sperfeld, A.D.; Klimpe, S.; Mueller, J.C.; Seibel, A.; Van De Warrenburg, B.P.; Bauer, P.; et al. Autosomal dominant spastic paraplegia with peripheral neuropathy maps to chr12q23-24. Neurology 2009, 72, 1893–1898. [Google Scholar] [CrossRef]
- Zhao, G.H.; Liu, X.M. Clinical features and genotype-phenotype correlation analysis in patients with ATL1 mutations: A literature reanalysis. Transl. Neurodegener. 2017, 6, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Orso, G.; Pendin, D.; Liu, S.; Tosetto, J.; Moss, T.J.; Faust, J.E.; Micaroni, M.; Egorova, A.; Martinuzzi, A.; McNew, J.A.; et al. Homotypic fusion of ER membranes requires the dynamin-like GTPase Atlastin. Nature 2010, 460, 978–983. [Google Scholar] [CrossRef]
- Tsang, H.T.H.; Edwards, T.L.; Wang, X.; Connell, J.W.; Davies, R.J.; Durrington, H.J.; O’kane, C.J.; Luzio, J.P.; Reid, E. The hereditary spastic paraplegia proteins NIPA1, spastin and spartin are inhibitors of mammalian BMP signalling. Hum. Mol. Genet. 2009, 18, 3805–3821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warnecke, T.; Duning, T.; Schwan, A.; Lohmann, H.; Deppe, M.; Epplen, J.T.; Young, P. Erratum: A novel form of autosomal recessive hereditary spastic paraplegia caused by a new SPG7 mutation. Neurology 2007, 69, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Lin, Z.; Fan, G.; Lu, J.; Hou, Y.; Habai, G.; Sun, L.; Yu, P.; Shen, Y.; Wen, M.; et al. The AAA protein spastin possesses two levels of basal ATPase activity. FEBS Lett. 2018, 592, 1625–1633. [Google Scholar] [CrossRef] [PubMed]
- Montenegro, G.; Rebelo, A.P.; Connell, J.; Allison, R.; Babalini, C.; D’Aloia, M.; Montieri, P.; Schüle, R.; Ishiura, H.; Price, J.; et al. Mutations in the ER-shaping protein reticulon 2 cause the axon-degenerative disorder hereditary spastic paraplegia type 12. J. Clin. Investig. 2012, 122, 538–544. [Google Scholar] [CrossRef]
- Van De Warrenburg, B.P.; Schouten, M.I.; De Bot, S.T.; Vermeer, S.; Meijer, R.; Pennings, M.; Gilissen, C.; Willemsen, M.A.; Scheffer, H.; Kamsteeg, E.J. Clinical exome sequencing for cerebellar ataxia and spastic paraplegia uncovers novel gene-disease associations and unanticipated rare disorders. Eur. J. Hum. Genet. 2016, 24, 1460–1466. [Google Scholar] [CrossRef] [Green Version]
- Rinaldi, C.; Schmidt, T.; Situ, A.J.; Johnson, J.O.; Lee, P.R.; Chen, K.-L.; Bott, L.C.; Fadó, R.; Harmison, G.H.; Parodi, S.; et al. Mutation inCPT1CAssociated with Pure Autosomal Dominant Spastic Paraplegia. JAMA Neurol. 2015, 72, 561–570. [Google Scholar] [CrossRef] [Green Version]
- Kara, E.; Tucci, A.; Manzoni, C.; Lynch, D.S.; Elpidorou, M.; Bettencourt, C.; Chelban, V.; Manole, A.; Hamed, S.A.; Haridy, N.A.; et al. Genetic and phenotypic characterization of complex hereditary spastic paraplegia. Brain 2016, 139, 1904–1918. [Google Scholar] [CrossRef]
- Estiar, M.A.; Leveille, E.; Spiegelman, D.; Dupre, N.; Trempe, J.F.; Rouleau, G.A.; Gan-Or, Z. Clinical and genetic analysis of ATP13A2 in hereditary spastic paraplegia expands the phenotype. Mol. Genet. Genom. Med. 2020, 8, e1052. [Google Scholar] [CrossRef] [Green Version]
- Estrada-Cuzcano, A.; Martin, S.; Chamova, T.; Synofzik, M.; Timmann, D.; Holemans, T.; Andreeva, A.; Reichbauer, J.; De Rycke, R.; Chang, D.I.; et al. Loss-of-function mutations in the ATP13A2/PARK9 gene cause complicated hereditary spastic paraplegia (SPG78). Brain 2017, 140, 287–305. [Google Scholar] [CrossRef] [Green Version]
- Husain, R.A.; Grimmel, M.; Wagner, M.; Hennings, J.C.; Marx, C.; Feichtinger, R.G.; Saadi, A.; Rostásy, K.; Radelfahr, F.; Bevot, A.; et al. Bi-allelic HPDL variants cause a neurodegenerative disease ranging from neonatal encephalopathy to adolescent-onset spastic paraplegia. Am. J. Hum. Genet. 2020, 107, 364–373. [Google Scholar] [CrossRef]
- Novarino, G.; Fenstermaker, A.G.; Zaki, M.S.; Hofree, M.; Silhavy, J.L.; Heiberg, A.D.; Abdellateef, M.; Rosti, B.; Scott, E.; Mansour, L.; et al. Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders. Science 2014, 343, 506–511. [Google Scholar] [CrossRef] [Green Version]
- Abdollahpour, H.; Alawi, M.; Kortüm, F.; Beckstette, M.; Seemanova, E.; Komárek, V.; Rosenberger, G.; Kutsche, K. An AP4B1 frameshift mutation in siblings with intellectual disability and spastic tetraplegia further delineates the AP-4 deficiency syndrome. Eur. J. Hum. Genet. 2015, 23, 256–259. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.W.; Hsu, C.K.; Michael, M.; Nanda, A.; Liu, L.; McMillan, J.R.; Pourreyron, C.; Takeichi, T.; Tolar, J.; Reid, E.; et al. Large intragenic deletion in DSTYK underlies autosomal-recessive complicated spastic paraparesis, SPG23. Am. J. Hum. Genet. 2017, 100, 364–370. [Google Scholar] [CrossRef] [Green Version]
- Orthmann-Murphy, J.L.; Salsano, E.; Abrams, C.K.; Bizzi, A.; Uziel, G.; Freidin, M.M.; Lamantea, E.; Zeviani, M.; Scherer, S.S.; Pareyson, D. Hereditary spastic paraplegia is a novel phenotype for GJA12/GJC2 mutations. Brain 2009, 132, 426–438. [Google Scholar] [CrossRef] [Green Version]
- Lossos, A.; Stümpfig, C.; Stevanin, G.; Gaussen, M.; Zimmerman, B.; El Mundwiller, E.; Asulin, M.; Chamma, L.; Sheffer, R.; Misk, A.; et al. Fe/S protein assembly gene IBA57 mutation causes hereditary spastic paraplegia. Neurology 2015, 84, 659–667. [Google Scholar] [CrossRef]
- Horibata, Y.; Elpeleg, O.; Eran, A.; Hirabayashi, Y.; Savitzki, D.; Tal, G.; Mandel, H.; Sugimoto, H. EPT1 (selenoprotein I) is critical for the neural development and maintenance of plasmalogen in humans. J. Lipid Res. 2018, 59, 1015–1026. [Google Scholar] [CrossRef] [Green Version]
- Spagnoli, C.; Rizzi, S.; Salerno, G.G.; Frattini, D.; Fusco, C. Long-term follow-up until early adulthood in autosomal dominant, complex SPG30 with a novel KIF1A variant: A case report. Ital. J. Pediatr. 2019, 45, 1–4. [Google Scholar] [CrossRef]
- Slosarek, E.L.; Schuh, A.L.; Pustova, I.; Johnson, A.; Bird, J.; Johnson, M.; Frankel, E.B.; Bhattacharya, N.; Hanna, M.G.; Burke, J.E.; et al. Pathogenic TFG mutations underlying hereditary spastic paraplegia impair secretory protein trafficking and axon fasciculation. Cell Rep. 2018, 24, 2248–2260. [Google Scholar] [CrossRef] [Green Version]
- Hanna, M.G.; Block, S.; Frankel, E.B.; Hou, F.; Johnson, A.; Yuan, L.; Knight, G.; Moresco, J.J.; Yates, J.R.; Ashton, R.; et al. TFG facilitates outer coat disassembly on COPII transport carriers to promote tethering and fusion with ER–Golgi intermediate compartments. Proc. Natl. Acad. Sci. USA 2017, 114, E7707–E7716. [Google Scholar] [CrossRef] [Green Version]
- Vazza, G.; Zortea, M.; Boaretto, F.; Micaglio, G.F.; Sartori, V.; Mostacciuolo, M.L. A new locus for autosomal recessive spastic paraplegia associated with mental retardation and distal motor neuropathy, SPG14, maps to chromosome 3q27-q28. Am. J. Hum. Genet. 2000, 67, 504–509. [Google Scholar] [CrossRef] [Green Version]
- Ji, Z.S.; Liu, Q.L.; Zhang, J.F.; Yang, Y.H.; Li, J.; Zhang, G.W.; Tan, M.H.; Lin, H.S.; Guo, G.Q. SUMOylation of spastin promotes the internalization of GluA1 and regulates dendritic spine morphology by targeting microtubule dynamics. Neurobiol. Dis. 2020, 146, 105133. [Google Scholar] [CrossRef]
- Kumar, R.; Jangir, D.K.; Verma, G.; Shekhar, S.; Hanpude, P.; Kumar, S.; Kumari, R.; Singh, N.; Bhavesh, N.S.; Jana, N.R.; et al. S-nitrosylation of UCHL1 induces its structural instability and promotes α-synuclein aggregation. Sci. Rep. 2017, 7, 1–16. [Google Scholar] [CrossRef]
- Rydning, S.L.; Backe, P.H.; Sousa, M.M.L.; Iqbal, Z.; Øye, A.M.; Sheng, Y.; Yang, M.; Lin, X.; Slupphaug, G.; Nordenmark, T.H.; et al. Novel UCHL1 mutations reveal new insights into ubiquitin processing. Hum. Mol. Genet. 2017, 26, 1031–1040. [Google Scholar] [CrossRef] [Green Version]
- Bibi, F.; Efthymiou, S.; Bourinaris, T.; Tariq, A.; Zafar, F.; Rana, N.; Salpietro, V.; Houlden, H.; Raja, G.K.; Saeed, S.; et al. Rare novel CYP2U1 and ZFYVE26 variants identified in two Pakistani families with spastic paraplegia. J. Neurol. Sci. 2020, 411, 116669. [Google Scholar] [CrossRef]
- Vantroys, E.; Larson, A.; Friederich, M.; Knight, K.; Swanson, M.A.; Powell, C.A.; Smet, J.; Vergult, S.; De Paepe, B.; Seneca, S.; et al. New insights into the phenotype of FARS2 deficiency. Mol. Genet. Metab. 2017, 122, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Hotait, M.; Nasreddine, W.; El-Khoury, R.; Dirani, M.; Nawfal, O.; Beydoun, A. FARS2 Mutations: More Than Two Phenotypes? A Case Report. Front. Genet. 2020, 11, 787. [Google Scholar] [CrossRef] [PubMed]
- Zortea, M.; Vettori, A.; Trevisan, C.P.; Bellini, S.; Vazza, G.; Armani, M.; Simonati, A.; Mostacciuolo, M.L. Genetic mapping of a susceptibility locus for disc herniation and spastic paraplegia on 6q23.3-q24.1. J. Med. Genet. 2002, 39, 387–390. [Google Scholar] [CrossRef] [PubMed]
- Rebolledo, D.L.; Lipson, K.E.; Brandan, E. Driving fibrosis in neuromuscular diseases: Role and regulation of connective tissue growth factor (CCN2/CTGF). Matrix Biol. Plus 2021, 11, 100059. [Google Scholar] [CrossRef] [PubMed]
- Hirst, J.; Edgar, J.R.; Esteves, T.; Darios, F.; Madeo, M.; Chang, J.; Roda, R.H.; Dürr, A.; Anheim, M.; Gellera, C.; et al. Loss of AP-5 results in accumulation of aberrant endolysosomes: Defining a new type of lysosomal storage disease. Hum. Mol. Genet. 2015, 24, 4984–4996. [Google Scholar] [CrossRef] [Green Version]
- Tüysüz, B.; Bilguvar, K.; Koçer, N.; Yalçinkaya, C.; Çaǧlayan, O.; Gül, E.; Şahin, S.; Çomu, S.; Günel, M. Autosomal recessive spastic tetraplegia caused by AP4M1 and AP4B1 gene mutation: Expansion of the facial and neuroimaging features. Am. J. Med. Genet. Part A 2014, 164, 1677–1685. [Google Scholar] [CrossRef]
- Hirst, J.; Madeo, M.; Smets, K.; Edgar, J.R.; Schols, L.; Li, J.; Yarrow, A.; Deconinck, T.; Baets, J.; Van Aken, E.; et al. Complicated spastic paraplegia in patients with AP5Z1 mutations (SPG48). Neurol. Genet. 2016, 2, 98. [Google Scholar] [CrossRef] [Green Version]
- Duerinckx, S.; Verhelst, H.; Perazzolo, C.; David, P.; Desmyter, L.; Pirson, I.; Abramowicz, M. Severe congenital microcephaly with AP4M1 mutation, a case report. BMC Med. Genet. 2017, 18, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Zivony-Elboum, Y.; Westbroek, W.; Kfir, N.; Savitzki, D.; Shoval, Y.; Bloom, A.; Rod, R.; Khayat, M.; Gross, B.; Samri, W.; et al. A founder mutation in Vps37A causes autosomal recessive complex hereditary spastic paraparesis. J. Med. Genet. 2012, 49, 462–472. [Google Scholar] [CrossRef]
- Park, J.M.; Lee, B.; Kim, J.H.; Park, S.Y.; Yu, J.; Kim, U.K.; Park, J.S. An autosomal dominant ERLIN2 mutation leads to a pure HSP phenotype distinct from the autosomal recessive ERLIN2 mutations (SPG18). Sci. Rep. 2020, 10, 1–6. [Google Scholar] [CrossRef]
- Darios, F.; Mochel, F.; Stevanin, G. Lipids in the physiopathology of hereditary spastic paraplegias. Front. Neurosci. 2020, 14, 74. [Google Scholar] [CrossRef] [Green Version]
- Hauser, S.; Poenisch, M.; Schelling, Y.; Höflinger, P.; Schuster, S.; Teegler, A.; Betten, R.; Gustafsson, J.Å.; Hübener-Schmid, J.; Schlake, T.; et al. mRNA as a novel treatment strategy for hereditary spastic paraplegia type 5. Mol. Ther. Methods Clin. Dev. 2019, 15, 359–370. [Google Scholar] [CrossRef] [Green Version]
- Nakamura-Shindo, K.; Ono, K.; Koh, K.; Ishiura, H.; Tsuji, S.; Takiyama, Y.; Yamada, M. A novel mutation in the GBA2 gene in a Japanese patient with SPG46: A case report. eNeurologicalSci 2020, 19, 100238. [Google Scholar] [CrossRef]
- Liu, Y.J.; Chen, J.; Li, X.; Zhou, X.; Hu, Y.M.; Chu, S.F.; Peng, Y.; Chen, N.H. Research progress on adenosine in central nervous system diseases. CNS Neurosci. Ther. 2019, 25, 899–910. [Google Scholar] [CrossRef]
- Su, W.; Zhou, Q.; Wang, Y.; Chishti, A.; Li, Q.Q.; Dayal, S.; Shiehzadegan, S.; Cheng, A.; Moore, C.; Bi, X.; et al. Deletion of the Capn1 gene results in alterations in signaling pathways related to Alzheimer’s disease, protein quality control and synaptic plasticity in mouse brain. Front. Genet. 2020, 11, 334. [Google Scholar] [CrossRef]
- Gan-Or, Z.; Bouslam, N.; Birouk, N.; Lissouba, A.; Chambers, D.B.; Vérièpe, J.; Androschuck, A.; Laurent, S.B.; Rochefort, D.; Spiegelman, D.; et al. Mutations in CAPN1 cause autosomal-recessive hereditary spastic paraplegia. Am. J. Hum. Genet. 2016, 98, 1038–1046. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Zhang, Y.J.; Xu, C.H.; De, L.; Liu, Z.J.; Wu, Y. The investigation of genetic and clinical features in patients with hereditary spastic paraplegia in central-Southern China. Mol. Genet. Genom. Med. 2021, 9, e1627. [Google Scholar] [CrossRef] [PubMed]
- Nishihara, H.; Omoto, M.; Takao, M.; Higuchi, Y.; Koga, M.; Kawai, M.; Kawano, H.; Ikeda, E.; Takashima, H.; Kanda, T. Autopsy case of the C12orf65 mutation in a patient with signs of mitochondrial dysfunction. Neurol. Genet. 2017, 3, 171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Hedera, P. Strumpellin and spartin, hereditary spastic paraplegia proteins, are binding partners. J. Exp. Neurosci. 2015, 9, 15–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, S.; Helbig, K.L.; Alcaraz, W.; Seaver, L.H.; Hsieh, D.T.; Rohena, L. Three cases of Troyer syndrome in two families of Filipino descent. Am. J. Med. Genet. Part A 2016, 170, 1780–1785. [Google Scholar] [CrossRef]
- Hodgkinson, C.A.; Bohlega, S.; Abu-Amero, S.N.; Cupler, E.; Kambouris, M.; Meyer, B.F.; Bharucha, V.A. A novel form of autosomal recessive pure hereditary spastic paraplegia maps to chromosome 13q14. Neurology 2002, 59, 1905–1909. [Google Scholar] [CrossRef]
- Stevanin, G.; Paternotte, C.; Coutinho, P.; Klebe, S.; Elleuch, N.; Loureiro, J.L.; Denis, E.; Cruz, V.T.; Dürr, A.; Prud’homme, J.F.; et al. A new locus for autosomal recessive spastic paraplegia (SPG32) on chromosome 14q12-q21. Neurology 2007, 68, 1837–1840. [Google Scholar] [CrossRef]
- Abou Jamra, R.; Philippe, O.; Raas-Rothschild, A.; Eck, S.H.; Graf, E.; Buchert, R.; Borck, G.; Ekici, A.; Brockschmidt, F.F.; Nöthen, M.M.; et al. Adaptor protein complex 4 deficiency causes severe autosomal-recessive intellectual disability, progressive spastic paraplegia, shy character, and short stature. Am. J. Hum. Genet. 2011, 88, 788–795. [Google Scholar] [CrossRef] [Green Version]
- Hardies, K.; May, P.; Djémié, T.; Tarta-Arsene, O.; Deconinck, T.; Craiu, D.; Helbig, I.; Suls, A.; Balling, R.; Weckhuysen, S.; et al. Recessive loss-of-function mutations in AP4S1 cause mild fever-sensitive seizures, developmental delay and spastic paraplegia through loss of AP-4 complex assembly. Hum. Mol. Genet. 2015, 24, 2218–2227. [Google Scholar] [CrossRef] [Green Version]
- Maemoto, Y.; Maruyama, T.; Nemoto, K.; Baba, T.; Motohashi, M.; Ito, A.; Tagaya, M.; Tani, K. DDHD1, but not DDHD2, suppresses neurite outgrowth in SH-SY5Y and PC12 cells by regulating protein transport from recycling endosomes. Front. Cell Dev. Biol. 2020, 8, 670. [Google Scholar] [CrossRef]
- Özdemir, T.R.; Gençpınar, P.; Arıcan, P.; Öztekin, Ö.; Dündar, N.O.; Özyılmaz, B. A case of spastic paraplegia-15 with a novel pathogenic variant in ZFYVE26 gene. Int. J. Neurosci. 2019, 129, 1198–1202. [Google Scholar] [CrossRef]
- Oz-Levi, D.; Ben-Zeev, B.; Ruzzo, E.K.; Hitomi, Y.; Gelman, A.; Pelak, K.; Anikster, Y.; Reznik-Wolf, H.; Bar-Joseph, I.; Olender, T.; et al. Mutation in TECPR2 reveals a role for autophagy in hereditary spastic paraparesis. Am. J. Hum. Genet. 2012, 91, 1065–1072. [Google Scholar] [CrossRef] [Green Version]
- Pozner, T.; Regensburger, M.; Engelhorn, T.; Winkler, J.; Winner, B. Janus-faced spatacsin (SPG11): Involvement in neurodevelopment and multisystem neurodegeneration. Brain 2020, 143, 2369–2379. [Google Scholar] [CrossRef]
- De Pace, R.; Skirzewski, M.; Damme, M.; Mattera, R.; Mercurio, J.; Foster, A.M.; Cuitino, L.; Jarnik, M.; Hoffmann, V.; Morris, H.D.; et al. Altered distribution of ATG9A and accumulation of axonal aggregates in neurons from a mouse model of AP-4 deficiency syndrome. PLoS Genet. 2018, 14, e1007363. [Google Scholar] [CrossRef] [Green Version]
- Parodi, L.; Fenu, S.; Stevanin, G.; Durr, A. Hereditary spastic paraplegia: More than an upper motor neuron disease. Rev. Neurol. 2017, 173, 352–360. [Google Scholar] [CrossRef]
- Fowler, P.C.; Byrne, D.J.; O’Sullivan, N.C. Rare disease models provide insight into inherited forms of neurodegeneration. J. Rare Dis. Res. Treat. 2016, 1, 17–21. [Google Scholar]
- Incecik, F.; Besen, S.; Bozdogan, S.T. Hereditary spastic paraplegia type 35 with a novel mutation in fatty acid 2-hydroxylase gene and literature review of the clinical features. Ann. Indian Acad. Neurol. 2018, 21, 335–339. [Google Scholar] [CrossRef]
- Osmanovic, A.; Widjaja, M.; Förster, A.; Weder, J.; Wattjes, M.P.; Lange, I.; Sarikidi, A.; Auber, B.; Raab, P.; Christians, A.; et al. SPG7 mutations in amyotrophic lateral sclerosis: A genetic link to hereditary spastic paraplegia. J. Neurol. 2020, 267, 2732–2743. [Google Scholar] [CrossRef]
- Vaz, F.M.; McDermott, J.H.; Alders, M.; Wortmann, S.B.; Kölker, S.; Pras-Raves, M.L.; Vervaart, M.A.T.; Van Lenthe, H.; Luyf, A.C.M.; Elfrink, H.L.; et al. Mutations in PCYT2 disrupt etherlipid biosynthesis and cause a complex hereditary spastic paraplegia. Brain 2019, 142, 3382–3397. [Google Scholar] [CrossRef] [Green Version]
- De Winter, J.; Beijer, D.; De Ridder, W.; Synofzik, M.; Zuchner, S.L.; Van Damme, P.; Spileers, W.; Baets, J. PCYT2 mutations disrupting etherlipid biosynthesis: Phenotypes converging on the CDP-ethanolamine pathway. Brain 2021, 144, e17. [Google Scholar] [CrossRef]
- Sunderhaus, E.R.; Law, A.D.; Kretzschmar, D. Disease-associated PNPLA6 mutations maintain partial functions when analyzed in Drosophila. Front. Neurosci. 2019, 13, 1207. [Google Scholar] [CrossRef] [PubMed]
- Dong, E.L.; Wang, C.; Wu, S.; Lu, Y.Q.; Lin, X.H.; Su, H.Z.; Zhao, M.; He, J.; Ma, L.X.; Wang, N.; et al. Clinical spectrum and genetic landscape for hereditary spastic paraplegias in China. Mol. Neurodegener. 2018, 13, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Santos, M.; Damásio, J.; Kun-Rodrigues, C.; Barbot, C.; Sequeiros, J.; Brás, J.; Alonso, I.; Guerreiro, R. Novel MAG variant causes cerebellar ataxia with oculomotor apraxia: Molecular basis and expanded clinical phenotype. J. Clin. Med. 2020, 9, 1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cloake, N.; Yan, J.; Aminian, A.; Pender, M.; Greer, J. PLP1 mutations in patients with multiple sclerosis: Identification of a new mutation and potential pathogenicity of the mutations. J. Clin. Med. 2018, 7, 342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linneberg, C.; Toft, C.L.F.; Kjaer-Sorensen, K.; Laursen, L.S. L1cam-mediated developmental processes of the nervous system are differentially regulated by proteolytic processing. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Finsterer, J.; Löscher, W.; Quasthoff, S.; Wanschitz, J.; Auer-Grumbach, M.; Stevanin, G. Hereditary spastic paraplegias with autosomal dominant, recessive, X-linked, or maternal trait of inheritance. J. Neurol. Sci. 2012, 318, 1–18. [Google Scholar] [CrossRef]
- Murala, S.; Nagarajan, E.; Bollu, P.C. Hereditary spastic paraplegia. Neurol. Sci. 2021, 42, 883–894. [Google Scholar] [CrossRef]
- Schellino, R.; Boido, M.; Vercelli, A. JNK signaling pathway involvement in spinal cord neuron development and death. Cells 2019, 8, 1576. [Google Scholar] [CrossRef] [Green Version]
- Mellesmoen, A.; Sheeler, C.; Ferro, A.; Rainwater, O.; Cvetanovic, M. Brain derived neurotrophic factor (BDNF) delays onset of pathogenesis in transgenic mouse model of spinocerebellar ataxia type 1 (SCA1). Front. Cell. Neurosci. 2018, 12, 509. [Google Scholar] [CrossRef] [Green Version]
- Lasser, M.; Tiber, J.; Lowery, L.A. The role of the microtubule cytoskeleton in neurodevelopmental disorders. Front. Cell. Neurosci. 2018, 12, 165. [Google Scholar] [CrossRef] [Green Version]
- Mei, J.; Li, Z.; Gui, J.F. Cooperation of Mtmr8 with PI3K regulates actin filament modeling and muscle development in zebrafish. PLoS ONE 2009, 4, e4979. [Google Scholar] [CrossRef] [Green Version]
- Vangeel, L.; Janssens, A.; Lemmens, I.; Lievens, S.; Tavernier, J.; Voets, T. The zinc-finger domain containing protein ZC4H2 interacts with TRPV1466, 4, enhancing channel activity and turnover at the plasma membrane. Int. J. Mol. Sci. 2020, 21, 3556. [Google Scholar] [CrossRef]
- Zhang, W.; Gong, J.; Ding, L.; Zhang, Z.; Pan, X.; Chen, X.; Guo, W.; Zhang, X.; Yang, X.; Peng, G.; et al. Functional validation of a human GLUD2 variant in a murine model of Parkinson’s disease. Cell Death Dis. 2020, 11, 1–17. [Google Scholar] [CrossRef] [Green Version]
- White, K.D.; Ince, P.G.; Lusher, M.; Lindsey, J.; Cookson, M.; Bashir, R.; Shaw, P.J.; Bushby, K.M.D. Clinical and pathologic findings in hereditary spastic paraparesis with spastin mutation. Neurology 2000, 55, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Blackstone, C. Hereditary Spastic Paraplegia. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2018; pp. 633–652. [Google Scholar]
- Tesson, C.; Koht, J.; Stevanin, G. Delving into the complexity of hereditary spastic paraplegias: How unexpected phenotypes and inheritance modes are revolutionizing their nosology. Hum. Genet. 2015, 134, 511–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beetz, C.; Pieber, T.R.; Hertel, N.; Schabhüttl, M.; Fischer, C.; Trajanoski, S.; Graf, E.; Keiner, S.; Kurth, I.; Wieland, T.; et al. Exome sequencing identifies a REEP1 mutation involved in distal hereditary motor neuropathy type v. Am. J. Hum. Genet. 2012, 91, 139–145. [Google Scholar] [CrossRef] [Green Version]
- Ruano, L.; Melo, C.; Silva, M.C.; Coutinho, P. The global epidemiology of hereditary ataxia and spastic paraplegia: A systematic review of prevalence studies. Neuroepidemiology 2014, 42, 174–183. [Google Scholar] [CrossRef]
- Klebe, S.; Stevanin, G.; Depienne, C. Clinical and genetic heterogeneity in hereditary spastic paraplegias: From SPG1 to SPG72 and still counting. Rev. Neurol. 2015, 171, 505–530. [Google Scholar] [CrossRef] [Green Version]
- Schüle, R.; Siddique, S.; Deng, X.; Yang, Y.; Donkervoort, S.; Hansson, M.; Madrid, R.E.; Siddique, N.; Schöls, L.; Björkhem, I. Marked accumulation of 27-hydroxycholesterol in SPG5 patients with hereditary spastic paresis. J. Lipid Res. 2010, 51, 819–823. [Google Scholar] [CrossRef] [Green Version]
- Casari, G.; Marconi, R. Spastic Paraplegia. Br. Med. J. 2018, 1, 642. [Google Scholar] [CrossRef]
- Verny, C.; Guegen, N.; Desquiret, V.; Chevrollier, A.; Prundean, A.; Dubas, F.; Cassereau, J.; Ferre, M.; Amati-Bonneau, P.; Bonneau, D.; et al. Hereditary spastic paraplegia-like disorder due to a mitochondrial ATP6 gene point mutation. Mitochondrion 2011, 11, 70–75. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Ferrero, E.; Coto, E.; Corao, A.I.; Díaz, M.; Gámez, J.; Esteban, J.; Gonzalo, J.F.; Pascual-Pascual, S.I.; De Munaín, A.L.; Morís, G.; et al. Mitochondrial DNA polymorphisms/haplogroups in hereditary spastic paraplegia. J. Neurol. 2012, 259, 246–250. [Google Scholar] [CrossRef]
- Zhu, P.P.; Denton, K.R.; Pierson, T.M.; Li, X.J.; Blackstone, C. Pharmacologic rescue of axon growth defects in a human iPSC model of hereditary spastic paraplegia SPG3A. Hum. Mol. Genet. 2014, 23, 5638–5648. [Google Scholar] [CrossRef] [Green Version]
- Hooper, C.; Puttamadappa, S.S.; Loring, Z.; Shekhtman, A.; Bakowska, J.C. Spartin activates atrophin-1-interacting protein 4 (AIP4) E3 ubiquitin ligase and promotes ubiquitination of adipophilin on lipid droplets. BMC. Biol. 2010, 8, 72. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.; Lee, S.; Blackstone, C. Spastic paraplegia proteins spastizin and spatacsin mediate autophagic lysosome reformation. J. Clin. Investig. 2014, 124, 5249–5262. [Google Scholar] [CrossRef] [Green Version]
- Ropers, F.; Derivery, E.; Hu, H.; Garshasbi, M.; Karbasiyan, M.; Herold, M.; Nürnberg, G.; Ullmann, R.; Gautreau, A.; Sperling, K.; et al. Identification of a novel candidate gene for non-syndromic autosomal recessive intellectual disability: The WASH complex member swip. Hum. Mol. Genet. 2011, 20, 2585–2590. [Google Scholar] [CrossRef] [Green Version]
- Bechara, A.; Nawabi, H.; Moret, F.; Yaron, A.; Weaver, E.; Bozon, M.; Abouzid, K.; Guan, J.L.; Tessier-Lavigne, M.; Lemmon, V.; et al. FAK-MAPK-dependent adhesion disassembly downstream of L1 contributes to semaphorin3A-induced collapse. EMBO J. 2008, 27, 1549–1562. [Google Scholar] [CrossRef] [Green Version]
- Da Graça, F.F.; De Rezende, T.J.R.; Vasconcellos, L.F.R.; Pedroso, J.L.; Barsottini, O.G.P.; França, M.C. Neuroimaging in Hereditary Spastic Paraplegias: Current Use and Future Perspectives. Front. Neurol. 2019, 9, 1117. [Google Scholar] [CrossRef]
- Hensiek, A.; Kirker, S.; Reid, E. Diagnosis, investigation and management of hereditary spastic paraplegias in the era of next-generation sequencing. J. Neurol. 2015, 272, 1601–1612. [Google Scholar] [CrossRef] [Green Version]
- Boone, P.M.; Liu, P.; Zhang, F.; Carvalho, C.M.; Towne, C.F.; Batish, S.D.; Lupski, J.R. Alu-specific microhomology-mediated deletion of the final exon of SPAST in three unrelated subjects with hereditary spastic paraplegia. Genet. Med. 2011, 13, 582–592. [Google Scholar] [CrossRef] [Green Version]
- Orlacchio, A.; Kawarai, T.; Totaro, A.; Errico, A.; St George-Hyslop, P.H.; Rugarli, E.I.; Bernardi, G. Hereditary spastic paraplegia: Clinical genetic study of 15 families. Arch. Neurol. 2004, 61, 849–855. [Google Scholar] [CrossRef] [Green Version]
- Mackay-Sim, A. Hereditary spastic paraplegia: From genes, cells and networks to novel pathways for drug discovery. Brain Sci. 2021, 11, 403. [Google Scholar] [CrossRef] [PubMed]
- Margetis, K.; Korfias, S.; Boutos, N.; Gatzonis, S.; Themistocleous, M.; Siatouni, A.; Dalivigka, Z.; Flaskas, T.; Stranjalis, G.; Boviatsis, E.; et al. Intrathecal baclofen therapy for the symptomatic treatment of hereditary spastic paraplegia. Clin. Neurol. Neurosurg. 2014, 123, 142–145. [Google Scholar] [CrossRef] [PubMed]
- Béreau, M.; Anheim, M.; Chanson, J.B.; Tio, G.; Echaniz-Laguna, A.; Depienne, C.; Collongues, N.; de Sèze, J. Dalfampridine in hereditary spastic paraplegia: A prospective, open study. J. Neurol. 2015, 262, 1285–1288. [Google Scholar] [CrossRef]
- De Niet, M.; De Bot, S.T.; Van De Warrenburg, B.P.C.; Weerdesteyn, V.; Geurts, A.C. Functional efects of botulinum toxin type-A treatment and subsequent stretching of spastic calf muscles: A study in patients with hereditary spastic paraplegia. J. Rehabil. Med. 2015, 47, 147–153. [Google Scholar] [CrossRef] [Green Version]
- Servelhere, K.R.; Faber, I.; Martinez, A.; Nickel, R.; Moro, A.; Germiniani, F.M.B.; Moscovich, M.; Blume, T.R.; Munhoz, R.P.; Teive, H.A.G.; et al. Botulinum toxin for hereditary spastic paraplegia: Effects on motor and non-motor manifestations. Arq. Neuropsiquiatr. 2018, 76, 183–188. [Google Scholar] [CrossRef] [Green Version]
- Xue, H.; Yu, A.; Chen, X.; Lin, N.; Lin, M.; Huang, H.; Xu, L. Prenatal diagnosis of PLP1 duplication by single nucleotide polymorphism array in a family with Pelizaeus-Merzbacher disease. Aging 2021, 13, 1488–1497. [Google Scholar] [CrossRef]
- Mohan, N.; Qiang, L.; Morfini, G.; Baas, P.W. Therapeutic Strategies for Mutant SPAST-Based Hereditary Spastic Paraplegia. Brain Sci. 2021, 11, 1081. [Google Scholar] [CrossRef]
- Havlicek, S.; Kohl, Z.; Mishra, H.K.; Prots, I.; Eberhardt, E.; Denguir, N.; Wend, H.; Plötz, S.; Boyer, L.; Marchetto, M.C.N.; et al. Gene dosage-dependent rescue of HSP neurite defects in SPG4 patients’ neurons. Hum. Mol. Genet. 2014, 23, 2527–2541. [Google Scholar] [CrossRef]
- Pirozzi, M.; Quattrini, A.; Andolfi, G.; Dina, G.; Malaguti, M.C.; Auricchio, A.; Rugarli, E.I. Intramuscular viral delivery of paraplegin rescues peripheral axonopathy in a model of hereditary spastic paraplegia. J. Clin. Investig. 2006, 116, 202–208. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pure HSP | Complex HSP |
|---|---|
| Impairments present in uncomplicated HSP plus other neurologic findings such as: Ataxia Seizures Intellectual disability Dementia Muscle atrophy Extrapyramidal disturbance Peripheral neuropathy |
| Criteria for Classification | Types |
|---|---|
| Symptoms and signs (Harding’s classification) | Pure HSP Complex HSP |
| Age and onset of spasticity (Harding’s classification) | Type I HSP (Early onset < 35 years) Type II HSP (Classical/late onset > 35 years) |
| Inheritance pattern | Autosomal dominant HSP Autosomal recessive HSP X-linked HSP Mitochondrial HSP De Novo |
| Intracellular involvement | Membrane/organelle trafficking Axonal transport Dysfunction of mitochondria Defective lipid metabolism Abnormalities in the myelination process |
| Inheritance Mode | Chromosome Number | Locus | Genes | HSP Subtype |
|---|---|---|---|---|
| Autosomal dominance (AD) | 1 | 1p31.1 | NERG1 * | SPG29 |
| 2 | 2p11.2 2p22.3 2q33.1 2q37.3 | REEP1 SPAST HSPD1 KIF1A | SPG31 SPG4 SPG13 SPG30 | |
| 3 | 3q25.31 | SLC33A1 | SPG42 | |
| 4 | 4p16-15 | JAKM1P1 * | SPG38 | |
| 8 | 8p21.1-q13.3 8q24.13 | FBXO16 KIAA0196 | SPG37 SPG8 | |
| 9 | 9p13.3 9q | UBAP1 - | SPG80 SPG19 | |
| 10 | 10q24.1 10q24.2 | ALDH18A1 ZFYVE27 | SPG9A SPG33 | |
| 11 | 11p14.1-p11.2 11q12.3 | BDNF * BSCL2 | SPG41 SPG17 | |
| 12 | 12q13.3 12q23-24 | KIF5A CKAP4 * | SPG10 SPG36 | |
| 14 | 14q22.1 | ATL1 | SPG3A | |
| 15 | 15q11.2 | NIPA1 | SPG6 | |
| 16 | 16q24.3 | PARAPLEGIN | SPG7 | |
| 19 | 19q13.32 19q13.33 | RTN2 CPT1C | SPG12 SPG73 | |
| Autosomal recessive (AR) | 1 | 1p36.13 1p34.1 1p13.3 1p13.2 1q32.1 1q42.13 1q42.13 | ATP13A2 HPDL AMPD2 AP4B1 DSTYK GJC2 IBA57 | SPG78 SPG83 SPG63 SPG47 SPG21 SPG44 SPG74 |
| 2 | 2p23.3 2q37.3 | SELENOI KIF1A | SPG81 SPG30 | |
| 3 | 3q12.2 3q27-q28 | TFG - | SPG57 SPG14 | |
| 4 | 4p13 4q25 | UCHL1 CYP2U1 | SPG79 SPG56 | |
| 6 | 6p25.1 6q23-24.1 | FARS2 - | SPG77 SPG25 | |
| 7 | 7p22.1 7q22.1 | AP5Z1 AP4M1 | SPG48 SPG50 | |
| 8 | 8p11.23 8p11.23 8p22 8q12.3 | ERLIN2 DDHD2 VPS37A CYP7B1 | SPG18 SPG54 SPG53 SPG5A | |
| 9 | 9p13.3 | GBA2 | SPG46 | |
| 10 | 10q22.1-q24.1 10q24.1 10q24.1 10q24.31 10q24.31-10q24.33 |
- ALDH18A1 ENTPD1 ERLIN1 NT5C2 | SPG27 SPG9B SPG64 SPG62 SPG45 | |
| 11 | 11q13.1 | CAPN1 | SPG76 | |
| 12 | 12q13.3 12q24.31 | B4GALNT1 C12ORF65 | SPG26 SPG55 | |
| 13 | 13q13.3 (TROYER SYNDROME) 13q14 | SPART - | SPG20 SPG24 | |
| 14 | 14q22.1 14q24.1 14q32.31 | DDHD1 ZFYVE26 TECPR2 | SPG28 SPG15 SPG49 | |
| 15 | 15q21.1 15q21.2 15q22.31 (MAST SYNDROME) | KIAA1840 AP4E1 ACP33 | SPG11 SPG51 SPG21 | |
| 16 | 16p12.3 16q23.1 16q24.3 | ARL6IP1 FA2H PARAPLEGIN | SPG61 SPG35 SPG7 | |
| 17 | 17q25.3 | PCYT2 | SPG 82 | |
| 19 | 19p13.2 19q12 19q13.12 | PNPLA6 C19ORF12 MAG | SPG39 SPG43 SPG75 | |
| X-Linked Inheritance (XLR) | X | Xq11.2 Xq11.2 Xq22.2 Xq24-25 Xq28 | MTMR8 * ZC4H2 * PLP1 GLUD2 L1CAM | SPG16 SPG16 SPG2 SPG34 SPG1 |
| Mitochondrial/Maternal Inheritance | Mitochondria | - - - - | MT-CO3 MT-T1 MT-ND4 MT-ATP6 | - - - - |
| Cellular Models Involved in HSP Pathogenesis | Proteins | SPG Forms |
|---|---|---|
1. Organelle’s morphogenesis/membrane structure
| SPAST Atlastin SPAST Atlastin REEP1 Reticulon 2 ARL6IP1 RAB3GAP2 Protrudin REEP2 AIP4/Spartin SLC33A1 Spatacsin Spastizin AP-4 for trafficking precursors of amyloids Strumpellin REEP1 Atlastin Spastizin Spatacsin Strumpellin | SPG4 SPG3A SPG4 SPG3A SPG31 SPG12 SPG61 SPG69 SPG33 SPG72 SPG20 SPG43 SPG11 SPG15 SPG47 SPG50 SPG52 SPG8 SPG31 SPG3A SPG15 SPG11 SPG8 |
| 2. Bone morphogenic proteins | NIPA1 Atlastin-1 Spastin Acetyl-CoA transporter | SPG6 SPG42 SPG3A SPG4 |
| 3. Motor proteins transportation | KIF5A KIF1C KIF1A | SPG10 SPG58 SPG30 |
| 4. Mitochondrial failure | Paraplegin HSP60 IBA57 Spartin | SPG7 SPG13 SPG74/SPG77 SPG20 |
| 5. Axon elongation path | L1CAM | SPG1 |
| 6. Myelination errors | PLP1 GJC2 MAG | SPG2 SPG44 SPG75 |
| 7. Nucleotide’s metabolism | AMPD2 NT5C2 ENTPD1 | SPG63 SPG65 SPG64 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meyyazhagan, A.; Orlacchio, A. Hereditary Spastic Paraplegia: An Update. Int. J. Mol. Sci. 2022, 23, 1697. https://doi.org/10.3390/ijms23031697
Meyyazhagan A, Orlacchio A. Hereditary Spastic Paraplegia: An Update. International Journal of Molecular Sciences. 2022; 23(3):1697. https://doi.org/10.3390/ijms23031697
Chicago/Turabian StyleMeyyazhagan, Arun, and Antonio Orlacchio. 2022. "Hereditary Spastic Paraplegia: An Update" International Journal of Molecular Sciences 23, no. 3: 1697. https://doi.org/10.3390/ijms23031697
APA StyleMeyyazhagan, A., & Orlacchio, A. (2022). Hereditary Spastic Paraplegia: An Update. International Journal of Molecular Sciences, 23(3), 1697. https://doi.org/10.3390/ijms23031697