
Structure-Based Cyclic Glycoprotein Ibα-Derived Peptides Interfering with von Willebrand Factor-Binding, Affecting Platelet Aggregation under Shear

 , , ,
, , ,  , , , ,
, , , ,  and
and
Abstract
:1. Introduction
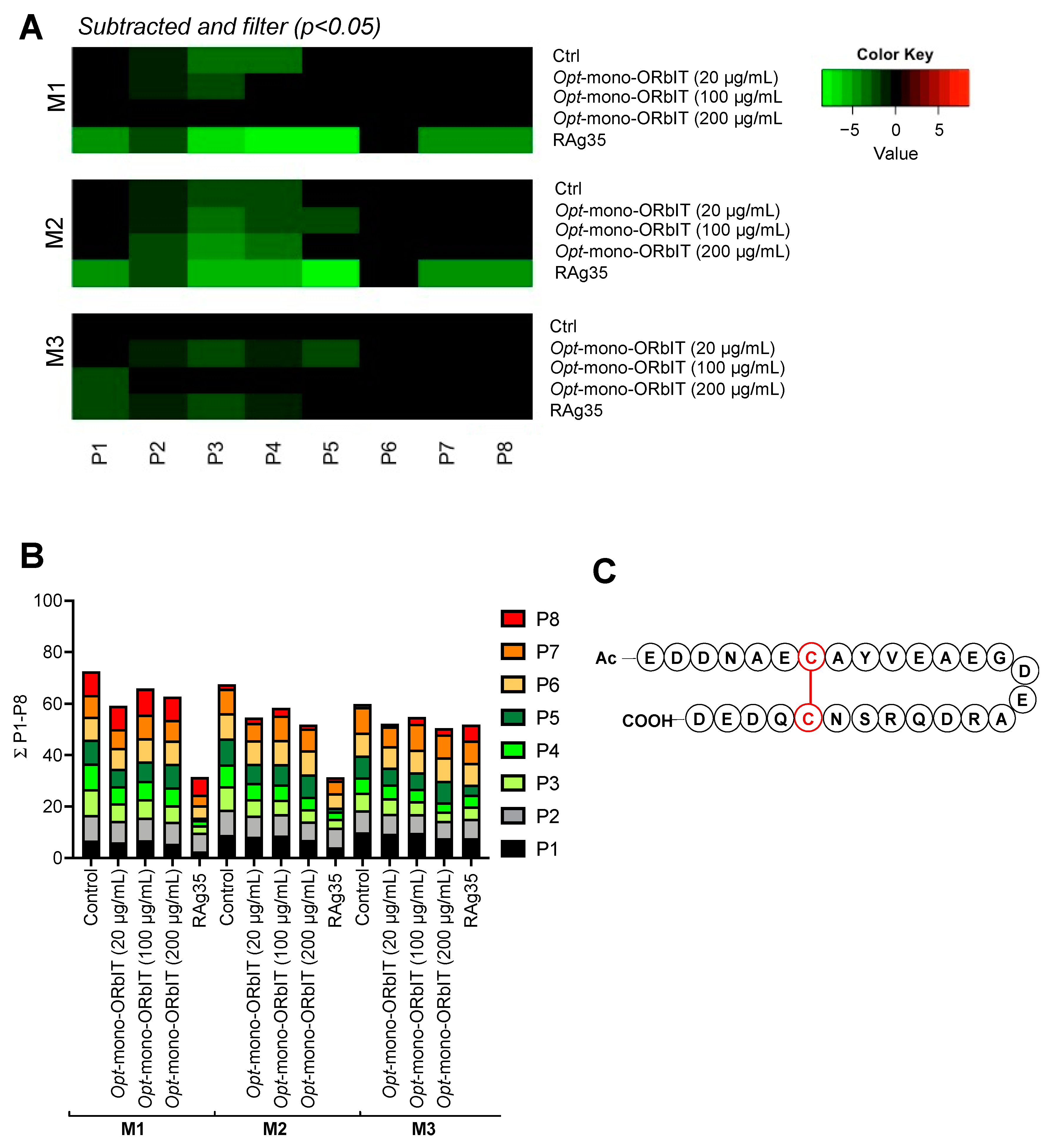
2. Results
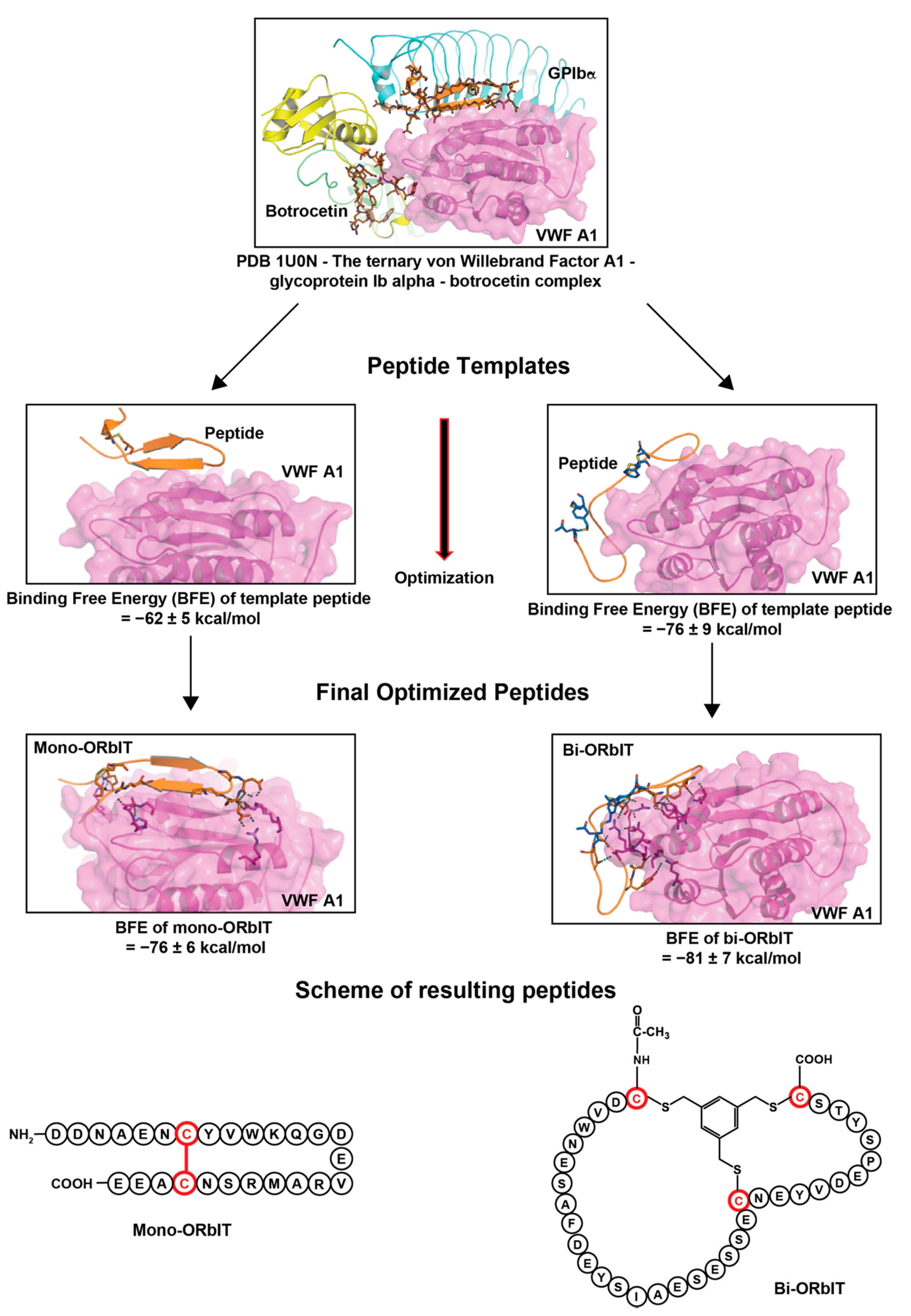
2.1. Design and Synthesis of Cyclic GPIbα-Mimicking Peptides Binding to the VWF A1 Domain
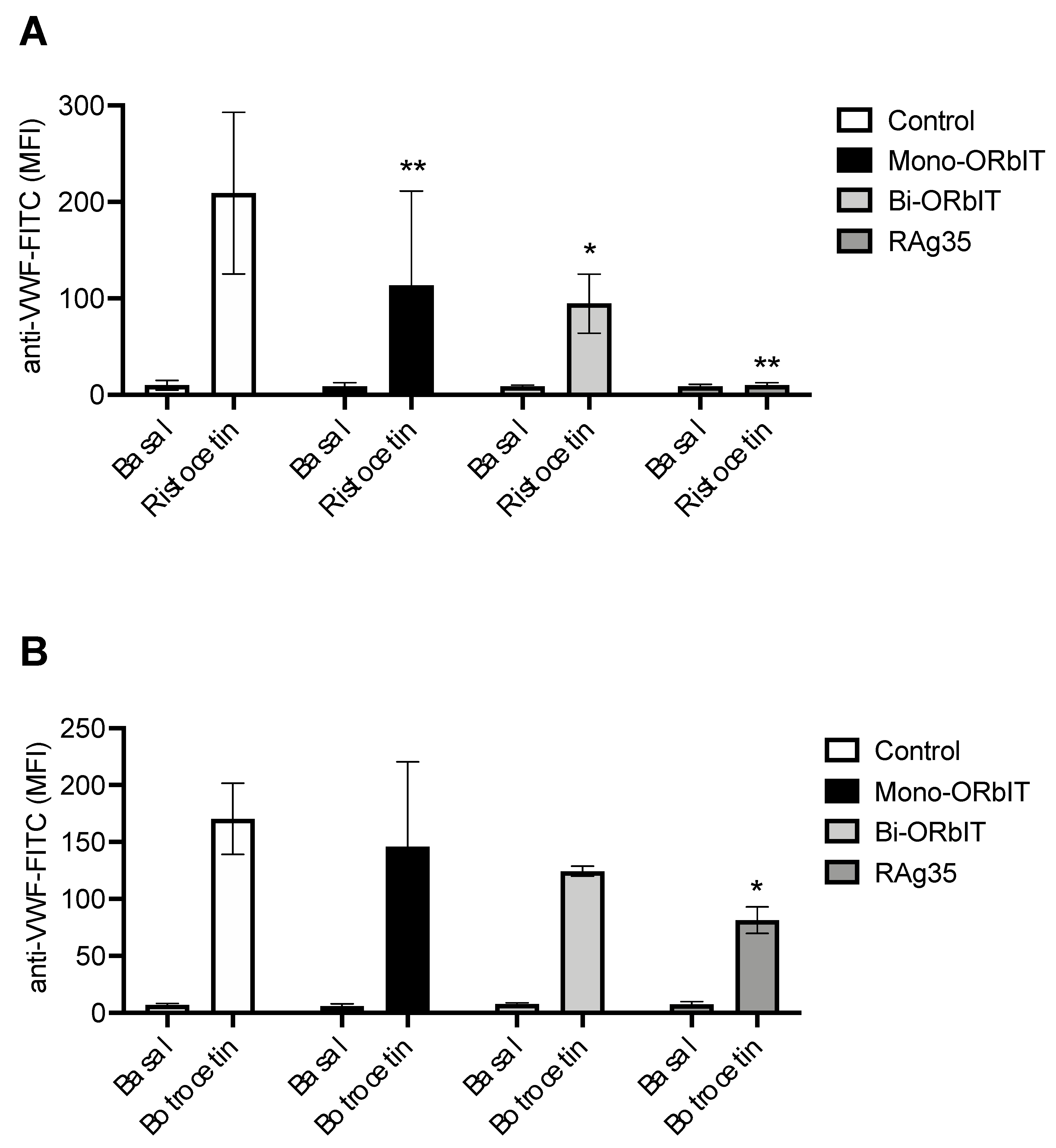
2.2. GPIbα-Derived Inhibitory Peptides Interfere with Ristocetin and Botrocetin-Induced VWF-Binding
2.3. Interference in the VWF A1 Domain Interaction with GPIbα, Affecting Thrombus Formation at High-Shear Flow
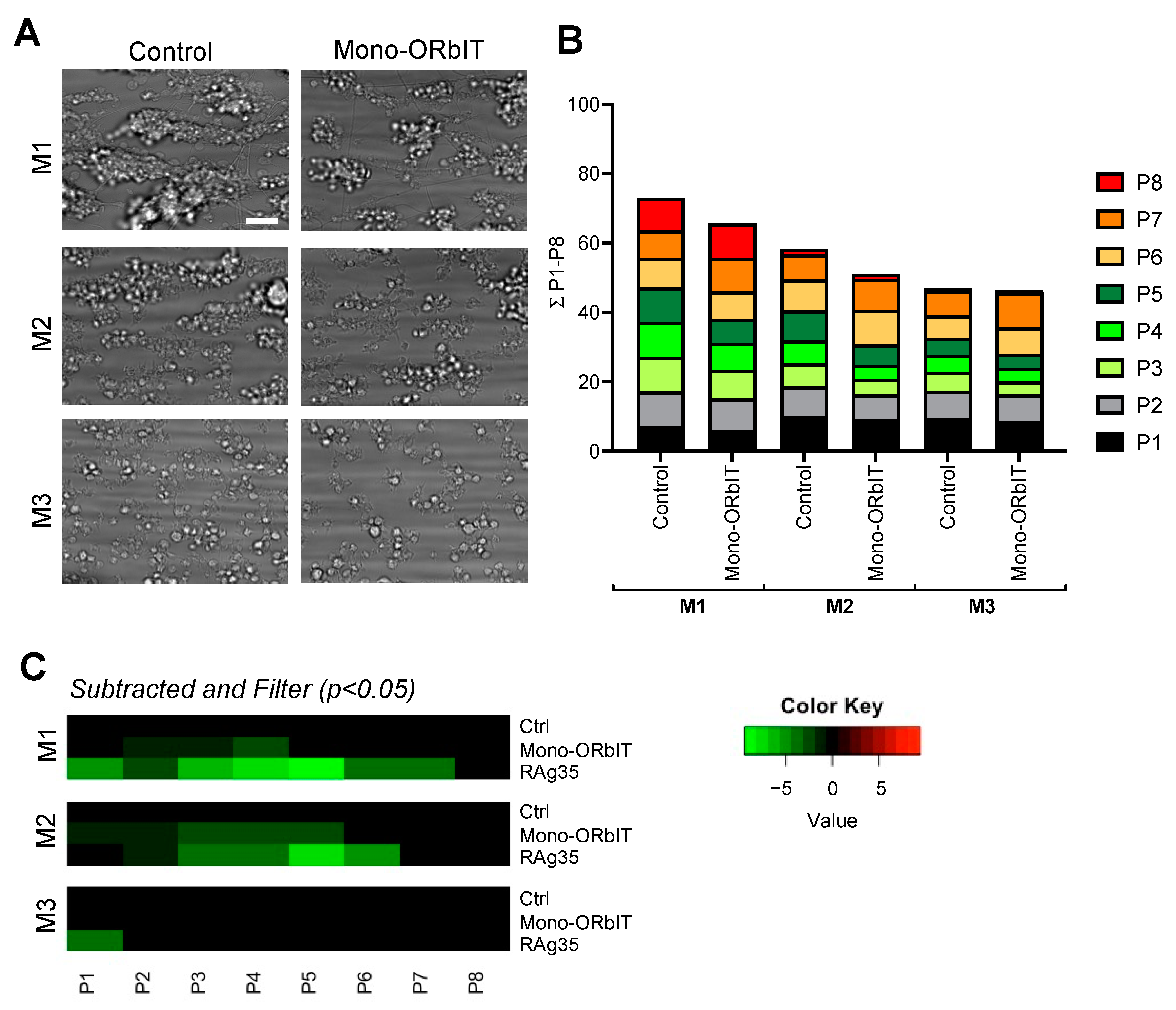
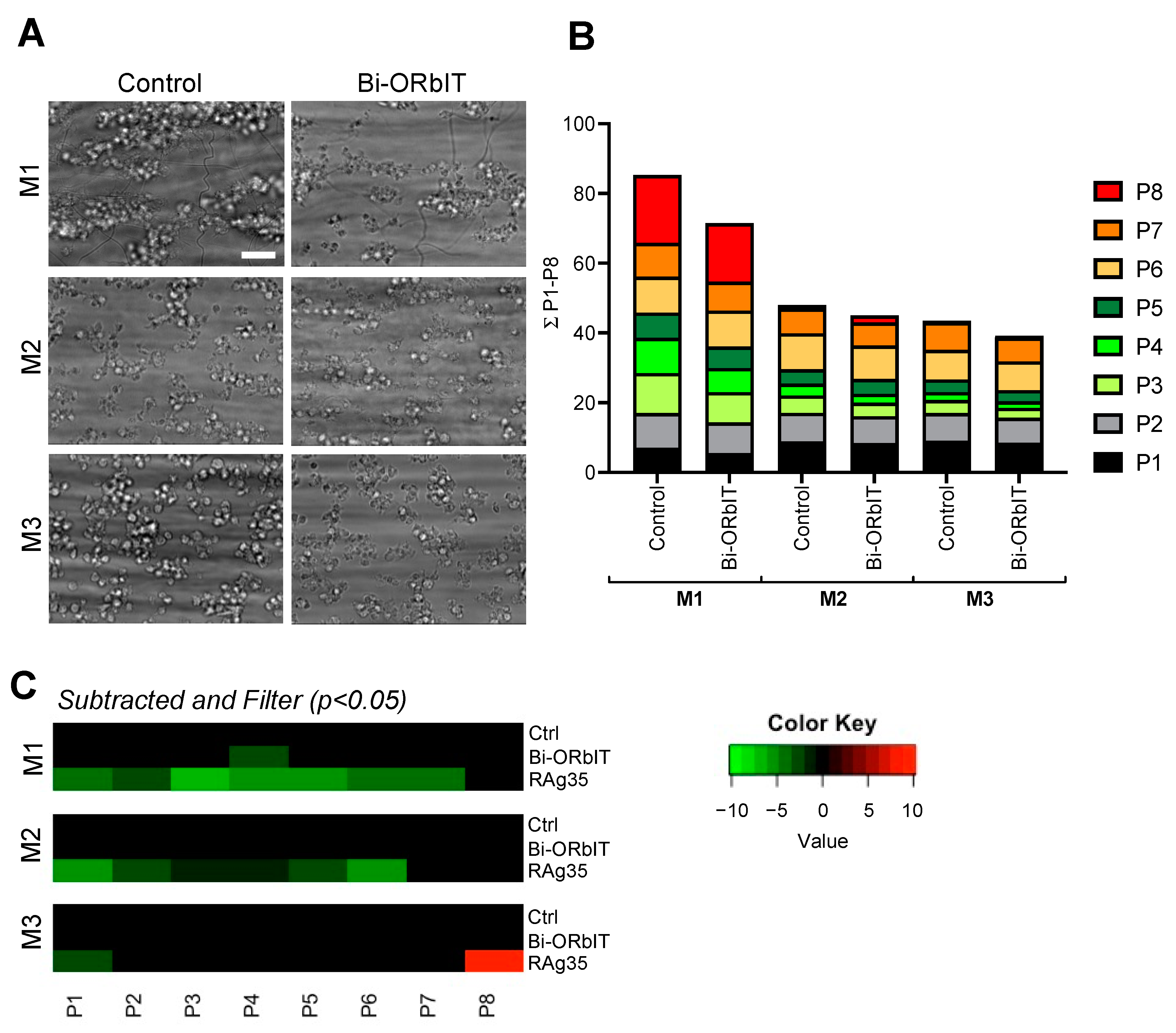
2.4. Designed GPIbα-Derived Peptides Interfering with Platelet Aggregation and Thrombus Formation under High-Shear Flow
2.5. Additional Optimization of Mono-ORbIT Peptide to Opt-Mono-ORbIT
3. Discussion
3.1. Functional Efficacy of In Silico Designed GPIba Peptides Interfering in the Binding to the VWF A1 Domain
3.2. Role of VWF A1 Domain in Shear-Dependent Platelet Aggregation and Thrombus Formation on Collagen Surfaces
3.3. Conclusions
4. Materials and Methods
4.1. Materials
4.2. Molecular Dynamics and Binding Free-Energy Calculations
4.3. Blood and Platelet Isolation
4.4. Flow Cytometric Assessment of Platelet GPIbα-VWF A1 Interaction
4.5. Assessment of Shear-Dependent Whole-Blood Thrombus Formation
4.6. Real-Time Brightfield and Fluorescence Microscopy
4.7. Statistics and Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sadler, J.E. Biochemistry and genetics of von Willebrand factor. Annu. Rev. Biochem. 1998, 67, 395–424. [Google Scholar] [CrossRef] [PubMed]
- Wagner, D.D. Cell biology of von Willebrand factor. Annu. Rev. Cell Biol. 1990, 6, 217–246. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Moake, J.L.; Nolasco, L.; Bernardo, A.; Arceneaux, W.; Shrimpton, C.N.; Schade, A.J.; McIntire, L.V.; Fujikawa, K.; Lopez, J.A. ADAMTS-13 rapidly cleaves newly secreted ultralarge von Willebrand factor multimers on the endothelial surface under flowing conditions. Blood 2002, 100, 4033–4039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crawley, J.T.; de Groot, R.; Xiang, Y.; Luken, B.M.; Lane, D.; de Groot, R. Unravelling the scissile bond: How ADAMTS13 recognises and cleaves von Willebrand factor. Blood 2011, 118, 3212–3221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slayter, H.; Loscalzo, J.; Bockenstedt, P.; Handin, R.I. Native conformation of human von Willebrand protein. Analysis by electron microscopy and quasi-elastic light scattering. J. Biol. Chem. 1985, 260, 8559–8563. [Google Scholar] [CrossRef]
- Siedlecki, C.A.; Lestini, B.J.; Kottke-Marchant, K.K.; Eppell, S.J.; Wilson, D.L.; Marchant, R.E. Shear-dependent changes in the three-dimensional structure of human von Willebrand factor. Blood 1996, 88, 2939–2950. [Google Scholar] [CrossRef] [Green Version]
- Savage, B.; Saldivar, E.; Ruggeri, Z.M. Initiation of platelet adhesion by arrest onto fibrinogen or translocation on von Willebrand factor. Cell 1996, 84, 289–297. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, B.; de Witt, S.; Solecka, B.A.; Kröning, M.; Obser, T.; Cosemans, J.M.; Schneppenheim, R.; Heemskerk, J.W.; Kannicht, C. Distinct role of von Willebrand factor triplet bands in glycoprotein Ib-dependent platelet adhesion and thrombus formation under flow. Semin. Thromb. Hemost. 2013, 39, 306–314. [Google Scholar] [CrossRef]
- Read, M.S.; Smith, S.V.; Lamb, M.A.; Brinkhous, K.M. Role of botrocetin in platelet agglutination: Formation of an activated complex of botrocetin and von Willebrand factor. Blood 1989, 74, 1031–1035. [Google Scholar] [CrossRef]
- Trabold, K.; Makhoul, S.; Gambaryan, S.; van Ryn, J.; Walter, U.; Jurk, K. The direct thrombin inhibitors dabigatran and lepirudin inhibit GPIbα-mediated platelet aggregation. Thromb. Haemost. 2019, 119, 916–929. [Google Scholar] [CrossRef]
- Fukuda, K.; Doggett, T.; Laurenzi, I.J.; Liddington, R.C.; Diacovo, T.G. The snake venom protein botrocetin acts as a biological brace to promote dysfunctional platelet aggregation. Nat. Struct. Mol. Biol. 2005, 12, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Ruggeri, Z.M.; Orje, J.N.; Habermann, R.; Federici, A.B.; Reininger, A.J. Activation-independent platelet adhesion and aggregation under elevated shear stress. Blood 2006, 108, 1903–1910. [Google Scholar] [CrossRef] [PubMed]
- Van der Meijden, P.E.; Heemskerk, J.W. Platelet biology and functions: New concepts and future clinical perspectives Nat. Rev. Cardiol. 2019, 16, 166–179. [Google Scholar]
- Versteeg, H.H.; Heemskerk, J.W.; Levi, M.; Reitsma, P.S. New fundamentals in hemostasis. Physiol. Rev. 2013, 93, 327–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savage, B.; Sixma, J.J.; Ruggeri, Z.M. Functional self-association of von Willebrand factor during platelet adhesion under flow. Proc. Natl. Acad. Sci. USA 2002, 99, 425–430. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.P.; Vink, T.; Schiphorst, M.; van Zanten, G.H.; IJsseldijk, M.J.W.; de Groot, P.G.; Sixma, J.J. Platelet thrombus formation on collagen at high shear rates is mediated by von Willebrand factor-glycoprotein Ib interaction and inhibited by von Willebrand factor-glycoprotein IIb/IIIa interaction. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1661–1667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brouns, S.; van Geffen, J.P.; Heemskerk, J.W. High throughput measurement of platelet aggregation under flow. Platelets 2018, 29, 662–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, W.; Xu, Y.; Chen, W.; Paul, D.S.; Syed, A.K.; Dragovich, M.A.; Liang, X.; Zakas, P.; Berndt, M.C.; Di Paola, J.; et al. Platelet clearance via shear-induced unfolding of a membrane mechanoreceptor. Nat. Commun. 2016, 7, e12863. [Google Scholar] [CrossRef]
- Broos, K.; Feys, H.B.; De Meyer, S.F.; Vanhoorelbeke, K.; Deckmyn, H. Platelets at work in primary hemostasis. Blood Rev. 2011, 25, 155–167. [Google Scholar] [CrossRef]
- Crawley, J.T.; Scully, M.A. Thrombotic thrombocytopenic purpura: Basic pathophysiology and therapeutic strategies. Hematology 2013, 2013, 292–299. [Google Scholar] [CrossRef]
- Kremer Hovinga, J.A.; Coppo, P.; Lammle, B.; Moake, J.L.; Miyata, T.; Vanhoorelbeke, K. Thrombotic thrombocytopenic purpura. Nat. Rev. Dis. Primers 2017, 3, 17020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayme, G.; Adam, F.; Legendre, P.; Bazaa, A.; Proulle, V.; Denis, C.V.; Christophe, O.D.; Lenting, P.J. A novel single-domain antibody against von Willebrand factor A1 domain resolves leukocyte recruitment and vascular leakage during inflammation. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1736–1740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scully, M.; Cataland, S.R.; Peyvandi, F.; Coppo, P.; Knobl, P.; Kremer Hovinga, J.A.; Metjian, A.; de la Rubia, J.; Pavenski, K.; Callewaert, F.; et al. Caplacizumab treatment for acquired thrombotic thrombocytopenic purpura. N. Engl. J. Med. 2019, 380, 335–346. [Google Scholar] [CrossRef]
- Jilma-Stohlawetz, P.; Knöbl, P.; Gilbert, J.C.; Jilma, B. The anti-von Willebrand factor aptamer ARC1779 increases von Willebrand factor levels and platelet counts in patients with type 2B von Willebrand disease. Thromb. Haemost. 2012, 108, 284–290. [Google Scholar] [CrossRef] [Green Version]
- Siller-Matula, J.M.; Merhi, Y.; Tanguay, J.F.; Duerschmied, D.; Wagner, D.D.; McGinness, K.E.; Pendergrast, P.S.; Chung, J.K.; Tian, X.; Schaub, R.G.; et al. ARC15105 is a potent antagonist of von Willebrand factor mediated platelet activation and adhesion. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 902–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovacevic, K.D.; Buchtele, N.; Schoergenhofer, C.; Derhaschnig, U.; Gelbenegger, G.; Brostjan, C.; Zhu, S.; Gilbert, J.C.; Jilma, B. The aptamer BT200 effectively inhibits von Willebrand factor (VWF) dependent platelet function after stimulated VWF release by desmopressin or endotoxin. Sci. Rep. 2020, 10, 11180. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Reheman, A.; Hou, Y.; Zhou, H.; Wang, Y.; Marshall, A.H.; Liang, C.; Dai, X.; Li, B.X.; Vanhoorelbeke, K.; et al. Anfibatide, a novel GPIb complex antagonist, inhibits platelet adhesion and thrombus formation in vitro and in vivo in murine models of thrombosis. Thromb. Haemost. 2014, 111, 279–289. [Google Scholar]
- Muttenthaler, M.; King, G.F.; Adams, D.J.; Alewood, P.F. Trends in peptide drug discovery. Nat. Rev. Drug Discov. 2021, 20, 309–325. [Google Scholar] [CrossRef]
- Henninot, A.; Collins, J.C.; Nuss, J.M. The current state of peptide drug discovery: Back to the future? J. Med. Chem. 2018, 61, 1382–1414. [Google Scholar] [CrossRef]
- Nicolaes, G.A.; Kulharia, M.; Voorberg, J.; Kaijen, P.H.; Wroblewska, A.; Wielders, S.; Schrijver, R.; Sperandio, O.; Villoutreix, B.O. Rational design of small molecules targeting the C2 domain of coagulation factor VIII. Blood 2014, 123, 113–120. [Google Scholar] [CrossRef] [Green Version]
- Van den Berg, S.M.; Seijkens, T.T.; Kusters, P.J.; Zarzycka, B.; Beckers, L.; den Toom, M.; Gijbels, M.J.; Chatzigeorgiou, A.; Weber, C.; de Winther, M.P.; et al. Blocking CD40-TRAF6 interactions by small-molecule inhibitor 6860766 ameliorates the complications of diet-induced obesity in mice. Int. J. Obes. (Lond) 2015, 39, 782–790. [Google Scholar] [CrossRef] [PubMed]
- Seijkens, T.T.; van Tiel, C.M.; Kusters, P.J.; Atzler, D.; Soehnlein, O.; Zarzycka, B.; Aarts, S.A.; Lameijer, M.; Gijbels, M.J.; Beckers, L.; et al. Targeting CD40-Induced TRAF6 signaling in macrophages reduces atherosclerosis. J. Am. Coll. Cardiol. 2018, 71, 527–542. [Google Scholar] [CrossRef] [PubMed]
- Alard, J.E.; Ortega-Gomez, A.; Wichapong, K.; Bongiovanni, M.; Horckmans, M.; Megens, R.T.; Leoni, G.; Ferraro, B.; Rossaint, J.; Paulin, N.; et al. Recruitment of classical monocytes can be inhibited by disturbing heteromers of neutrophil HNP1 and platelet CCL5. Sci. Transl. Med. 2015, 7, 317ra196. [Google Scholar] [CrossRef]
- Wichapong, K.; Poelman, H.; Ercig, B.; Hrdinova, J.; Liu, X.; Lutgens, E.; Nicolaes, G.A. Rational modulator design by exploitation of protein-protein complex structures. Future Med. Chem. 2019, 11, 1015–1033. [Google Scholar] [CrossRef] [PubMed]
- Schumski, A.; Ortega-Gomez, A.; Wichapong, K.; Winter, C.; Lemnitzer, P.; Viola, J.R.; Pinilla-Vera, M.; Folco, E.; Solis-Mezarino, V.; Volker-Albert, M.; et al. Endotoxinemia accelerates atherosclerosis through electrostatic charge-mediated monocyte adhesion. Circulation 2021, 143, 254–266. [Google Scholar] [CrossRef]
- Huizinga, E.G.; Tsuji, S.; Romijn, R.A.P.; Schiphorst, M.E.; de Groot, P.G.; Sixma, J.J.; Gros, P. Structures of glycoprotein Iba and its complex with von Willebrand factor A1 domain. Science 2002, 297, 1176–1179. [Google Scholar] [CrossRef] [PubMed]
- Wichapong, K.; Silvestre-Roig, C.; Braster, Q.; Schumski, A.; Soehnlein, O.; Nicolaes, G.A.F. Structure-based peptide design targeting intrinsically disordered proteins: Novel histone H4 and H2A peptidic inhibitors. Comput. Struct. Biotechnol. J. 2021, 19, 934–948. [Google Scholar] [CrossRef]
- Timmerman, P.; Puijk, W.C.; Boshuizen, R.S.; van Dijken, P.; Slootstra, J.W.; Beruskens, F.J.; Parren, P.W.; Huber, A.; Bachmann, M.F.; Meloen, R.H. Functional reconstruction of structurally complex epitopes using CLIPS technology. Open Vaccine J. 2009, 2, 56–67. [Google Scholar] [CrossRef] [Green Version]
- Sixma, J.J.; Sakariassen, K.S.; Stel, H.V.; Houdijk, W.P.; In der Maur, D.W.; Hamer, R.J.; de Groot, P.G.; van Mourik, J.A. Functional domains on von Willebrand factor. Recognition of discrete tryptic fragments by monoclonal antibodies that inhibit interaction of von Willebrand factor with platelets and with collagen. J. Clin. Investig. 1984, 74, 736–744. [Google Scholar] [CrossRef] [Green Version]
- De Witt, S.M.; Swieringa, F.; Cavill, R.; Lamers, M.M.; van Kruchten, R.; Mastenbroek, T.; Baaten, C.; Coort, S.; Pugh, N.; Schulz, A.; et al. Identification of platelet function defects by multi-parameter assessment of thrombus formation. Nat. Commun. 2014, 5, e4257. [Google Scholar] [CrossRef] [Green Version]
- Nagy, M.; Mastenbroek, T.G.; Mattheij, N.J.; De Witt, S.; Clemetson, K.J.; Kirschner, J.; Schulz, A.; Braun, A.; Cosemans, J.M.; Zieger, B.; et al. Variable impairment of platelet functions in patients with severe, genetically linked immune deficiencies. Haematologica 2018, 103, 540–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stel, H.V.; Sakariassen, K.S.; Scholte, B.J.; Veerman, E.C.; van der Kwast, T.H.; de Groot, P.G.; Sixma, J.J.; van Mourik, J.A. Characterization of 25 monoclonal antibodies to factor VIII-von Willebrand factor: Relationship between ristocetin-induced platelet aggregation and platelet adherence to subendothelium. Blood 1984, 63, 1408–1415. [Google Scholar] [CrossRef] [PubMed]
- Bladbjerg, E.M.; de Maat, M.P.; Christensen, K.; Bathum, L.; Jespersen, J.; Hjelmborg, J. Genetic influence on thrombotic risk markers in the elderly: A Danish twin study. J. Thromb. Haemost. 2006, 4, 599–607. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Yu, F.; Buchanan, A.; Fu, Y.; Campos, M.; Wu, K.K.; Chambless, L.E.; Folsom, A.R.; Boerwinkle, E.; Dong, J.F. Possible race and gender divergence in association of genetic variations with plasma von Willebrand factor: A study of ARIC and 1000 genome cohorts. PLoS ONE 2014, 9, e84810. [Google Scholar]
- Jastrzebska, M.; Marcinowska, Z.; Oledzki, S.; Chelstowski, K.; Siennicka, A.; Klysz, M.; Clark, J.S. Variable gender-dependent platelet responses to combined antiplatelet therapy in patients with stable coronary-artery disease. J. Physiol. Pharmacol. 2018, 69, 26402. [Google Scholar]
- Goto, S.; Ikeda, Y.; Saldívar, E.; Ruggeri, Z.M. Distinct mechanisms of platelet aggregation as a consequence of different shearing flow conditions. J. Clin. Investig. 1998, 101, 479–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westein, E.; van der Meer, A.D.; Kuijpers, M.J.; Frimat, J.P.; van den Berg, A.; Heemskerk, J.W. Atherosclerotic geometries spatially confine and exacerbate pathological thrombus formation poststenosis in a von Willebrand factor-dependent manner. Proc. Natl. Acad. Sci. USA 2013, 110, 1357–1362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoefer, T.; Rana, A.; Niego, B.; Jagdale, S.; Albers, H.J.; Gardiner, E.E.; Andrews, R.K.; Van der Meer, A.D.; Hagemeyer, C.E.; Westein, E. Targeting shear gradient activated von Willebrand factor by the novel single-chain antibody A1 reduces occlusive thrombus formation in vitro. Haematologica 2021, 106, 2874–2884. [Google Scholar] [CrossRef]
- Gonzalez-Muniz, R.; Bonache, M.A.; Perez de Vega, M.J. Modulating protein-protein interactions by cyclic and macrocyclic peptides. Prominent strategies and examples. Molecules 2021, 26, 445. [Google Scholar] [CrossRef]
- Elverdi, T.; Eskazan, A.E. Caplacizumab as an emerging treatment option for acquired thrombotic thrombocytopenic purpura. Drug Des. Devel. Ther. 2019, 13, 1251–1258. [Google Scholar] [CrossRef] [Green Version]
- Marqus, S.; Pirogova, E.; Piva, T.J. Evaluation of the use of therapeutic peptides for cancer treatment. J. Biomed. Sci. 2017, 24, 21. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Wichapong, K.; Lamers, S.; Reutelingsperger, C.P.M.; Nicolaes, G.A.F. Autocitrullination of PAD4 does not alter its enzymatic activity: In vitro and in silico studies. Int. J. Biochem. Cell Biol. 2021, 134, 105938. [Google Scholar] [CrossRef] [PubMed]
- Wichapong, K.; Alard, J.E.; Ortega-Gomez, A.; Weber, C.; Hackeng, T.M.; Soehnlein, O.; Nicolaes, G.A. Structure-based design of peptidic inhibitors of the interaction between CC chemokine ligand 5 (CCL5) and human neutrophil peptides 1 (HNP1). J. Med. Chem. 2016, 59, 4289–4301. [Google Scholar] [CrossRef]
- van Geffen, J.P.; Brouns, S.L.; Batista, J.; McKinney, H.; Kempster, C.; Nagy, M.; Sivapalaratnam, S.; Baaten, C.C.; Bourry, N.; Frontini, M.; et al. High-throughput elucidation of thrombus formation reveals sources of platelet function variability. Haematologica 2019, 104, 1256–1267. [Google Scholar] [CrossRef] [PubMed]
- Van Kruchten, R.; Cosemans, J.M.; Heemskerk, J.W. Measurement of whole blood thrombus formation using parallel-plate flow chambers: A practical guide. Platelets 2012, 23, 229–242. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequence | Peptide | BFE (kcal/mol) |
|---|---|---|
| Monocyclic | ||
| GPIbα | 221QDNAENVYVWKQGVDVKAMTSNVASV246 | −35 ± 6 |
| Template peptide | H-QDNAENCYVWKQGVDVKAMTSNCAEE-OH Disulfide bond between Cys7 and Cys23 | −62 ± 5 |
| Mono-ORbIT | H-DDNAENCYVWKQGDEVRAMRSNCAEE-OH Disulfide bond between Cys7 and Cys23 | −76 ± 6 |
| Bicyclic | ||
| Botrocetin | 2070DVWNKCRF2077 2089DYYLIAEYEC2098C2113-T2114 | - |
| GPIbα | 225ENVYVWK231 237KAMTS241 | |
| Template peptide | Ac-CDVWNESAFDYYSIAEYECSTENCYVWEPSDTSC-OH Disulfide bond between Cys1 and Cys19 as well as Cys24 and Cys34 | −76 ± 9 |
| Bi-ORbIT | Ac-CDVWNESAFDEYSIAESESSECNEYVDEPSYTSC-OH CLIPS(T3 *) connection between Cys1, Cys22, and Cys34 | −81 ± 7 |
| Parameter | Range | Scaling |
|---|---|---|
| Brightfield images (platelet adhesion) | ||
| P1 platelet deposition (% SAC) | 0–66 | 0–10 |
| P2 morphology score (0–5) | 0–3.8 | 0–10 |
| Brightfield images (platelet aggregation) | ||
| P3 thrombus contraction score (0–3) | 0–2.2 | 0–10 |
| P4 thrombus multilayer score (0–3) | 0–1.9 | 0–10 |
| P5 thrombus aggregate coverage (% SAC) | 0–12 | 0–10 |
| Fluorescence images (platelet activation) | ||
| P6 CD62P expression (% SAC) | 0–50 | 0–10 |
| P7 fibrinogen-binding (% SAC) | 0–21 | 0–10 |
| P8 PS exposure (% SAC) | 0–17 | 0–10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hrdinova, J.; Fernández, D.I.; Ercig, B.; Tullemans, B.M.E.; Suylen, D.P.L.; Agten, S.M.; Jurk, K.; Hackeng, T.M.; Vanhoorelbeke, K.; Voorberg, J.; et al. Structure-Based Cyclic Glycoprotein Ibα-Derived Peptides Interfering with von Willebrand Factor-Binding, Affecting Platelet Aggregation under Shear. Int. J. Mol. Sci. 2022, 23, 2046. https://doi.org/10.3390/ijms23042046
Hrdinova J, Fernández DI, Ercig B, Tullemans BME, Suylen DPL, Agten SM, Jurk K, Hackeng TM, Vanhoorelbeke K, Voorberg J, et al. Structure-Based Cyclic Glycoprotein Ibα-Derived Peptides Interfering with von Willebrand Factor-Binding, Affecting Platelet Aggregation under Shear. International Journal of Molecular Sciences. 2022; 23(4):2046. https://doi.org/10.3390/ijms23042046
Chicago/Turabian StyleHrdinova, Johana, Delia I. Fernández, Bogac Ercig, Bibian M. E. Tullemans, Dennis P. L. Suylen, Stijn M. Agten, Kerstin Jurk, Tilman M. Hackeng, Karen Vanhoorelbeke, Jan Voorberg, and et al. 2022. "Structure-Based Cyclic Glycoprotein Ibα-Derived Peptides Interfering with von Willebrand Factor-Binding, Affecting Platelet Aggregation under Shear" International Journal of Molecular Sciences 23, no. 4: 2046. https://doi.org/10.3390/ijms23042046
APA StyleHrdinova, J., Fernández, D. I., Ercig, B., Tullemans, B. M. E., Suylen, D. P. L., Agten, S. M., Jurk, K., Hackeng, T. M., Vanhoorelbeke, K., Voorberg, J., Reutelingsperger, C. P. M., Wichapong, K., Heemskerk, J. W. M., & Nicolaes, G. A. F. (2022). Structure-Based Cyclic Glycoprotein Ibα-Derived Peptides Interfering with von Willebrand Factor-Binding, Affecting Platelet Aggregation under Shear. International Journal of Molecular Sciences, 23(4), 2046. https://doi.org/10.3390/ijms23042046