
The Platelet Collagen Receptor GPVI Is Cleaved by Tspan15/ADAM10 and Tspan33/ADAM10 Molecular Scissors

 , , and
, , and 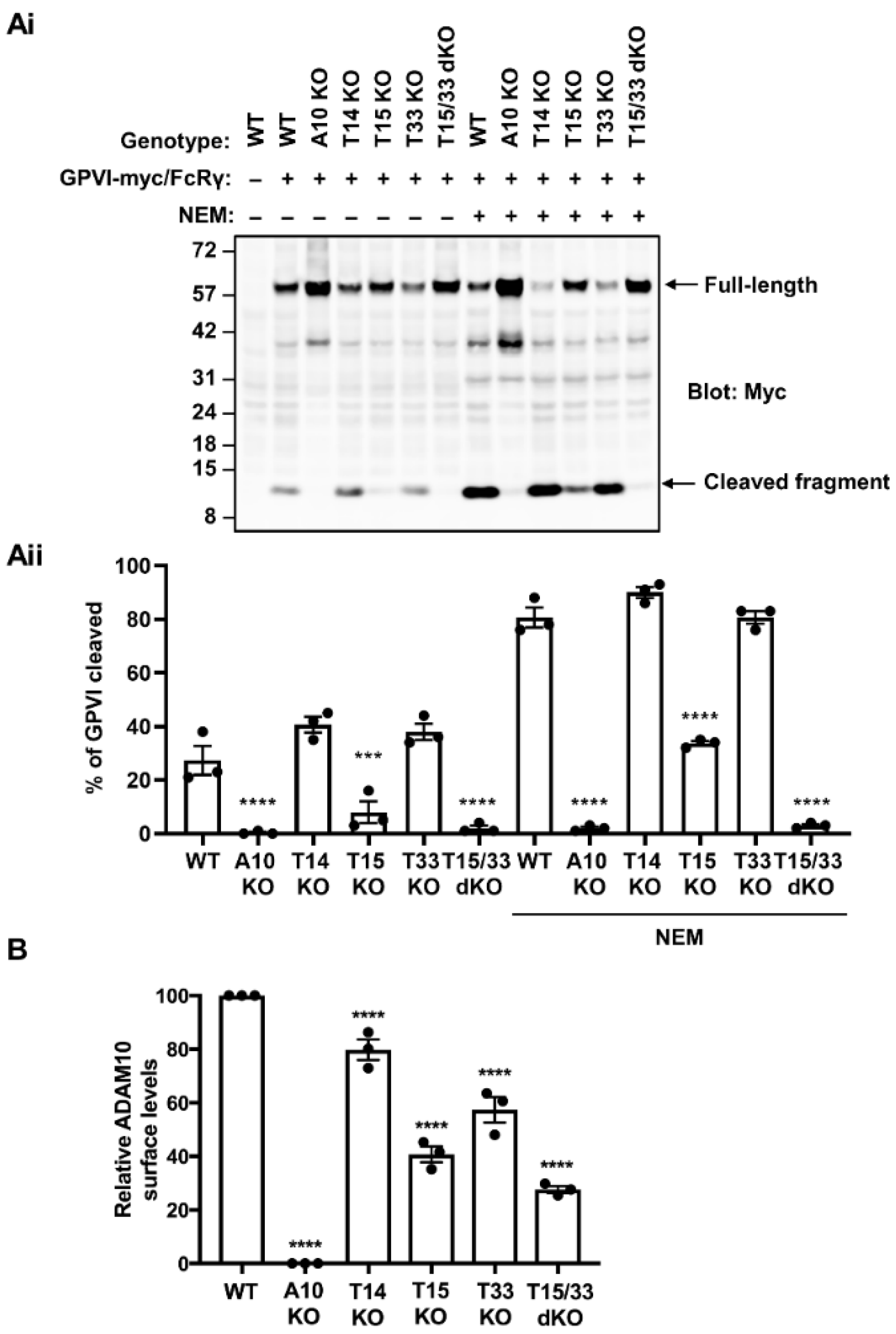{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. GPVI Cleavage Is Dependent on Tspan15 and Tspan33 in Transfected HEK-293T Cells
2.2. Tspan15 and Tspan33 Are Required for Cleavage of Endogenous GPVI in HEL Cells
2.3. Tspan15/ADAM10 Is the Most Efficient Scissor for GPVI in HEK-293T Cells
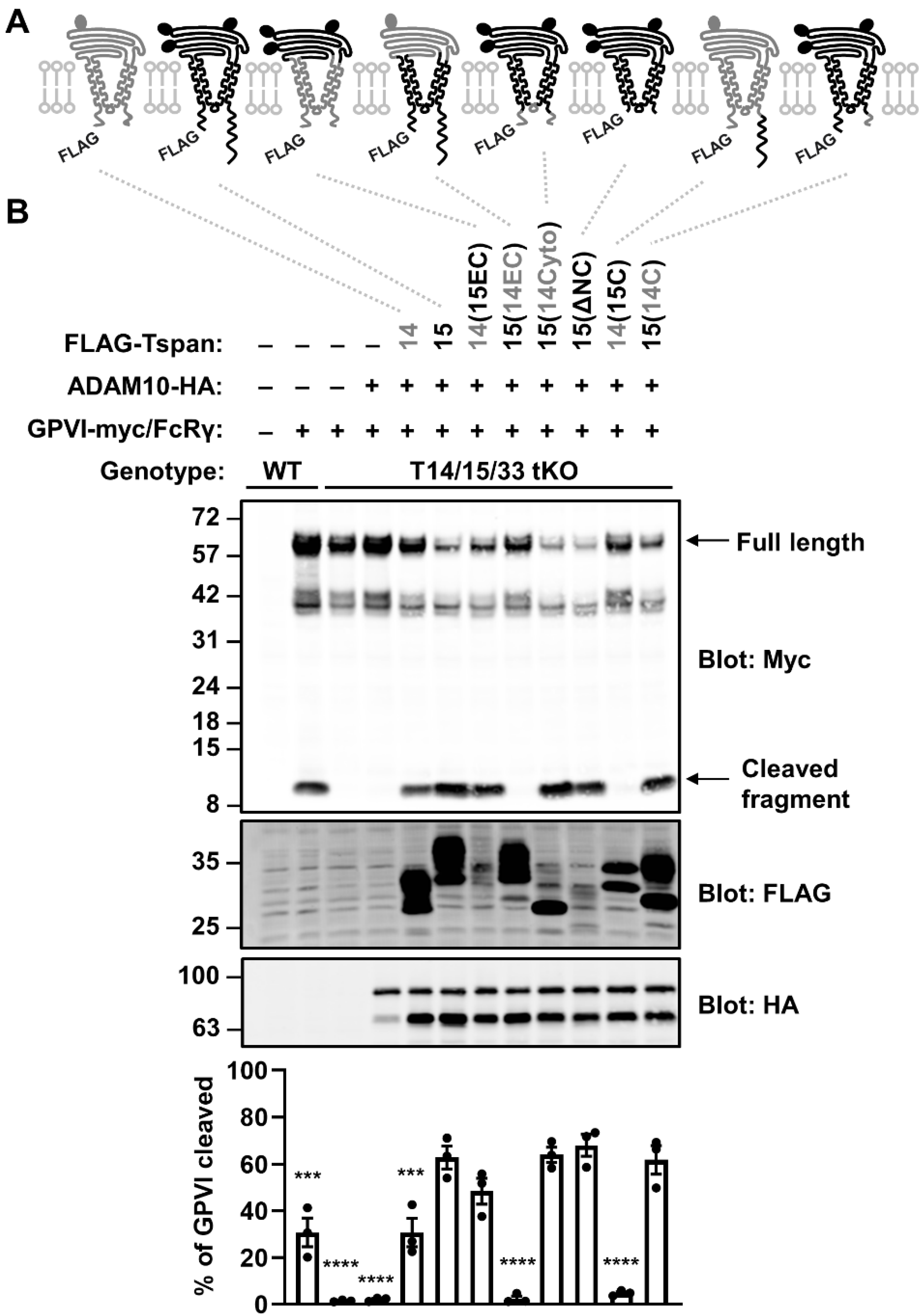
2.4. The Extracellular Region of Tspan15 Is Required for Efficient GPVI Cleavage and the C-terminus Is Inhibitory
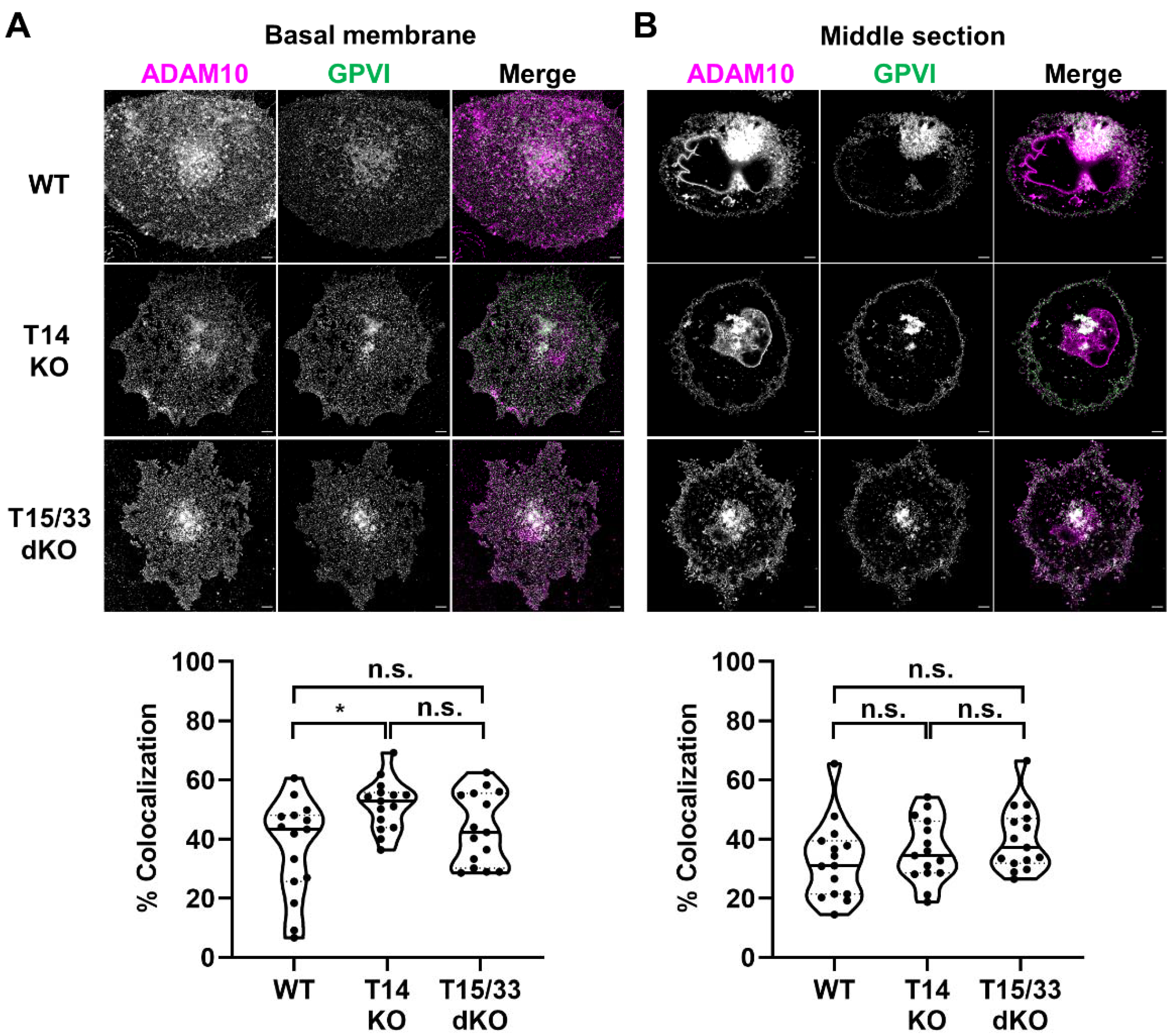
2.5. The Capacity of Tspan15 Wild-Type and Mutant Forms to Promote GPVI Cleavage Is Unrelated to the Extent to Which They Co-Localize with GPVI
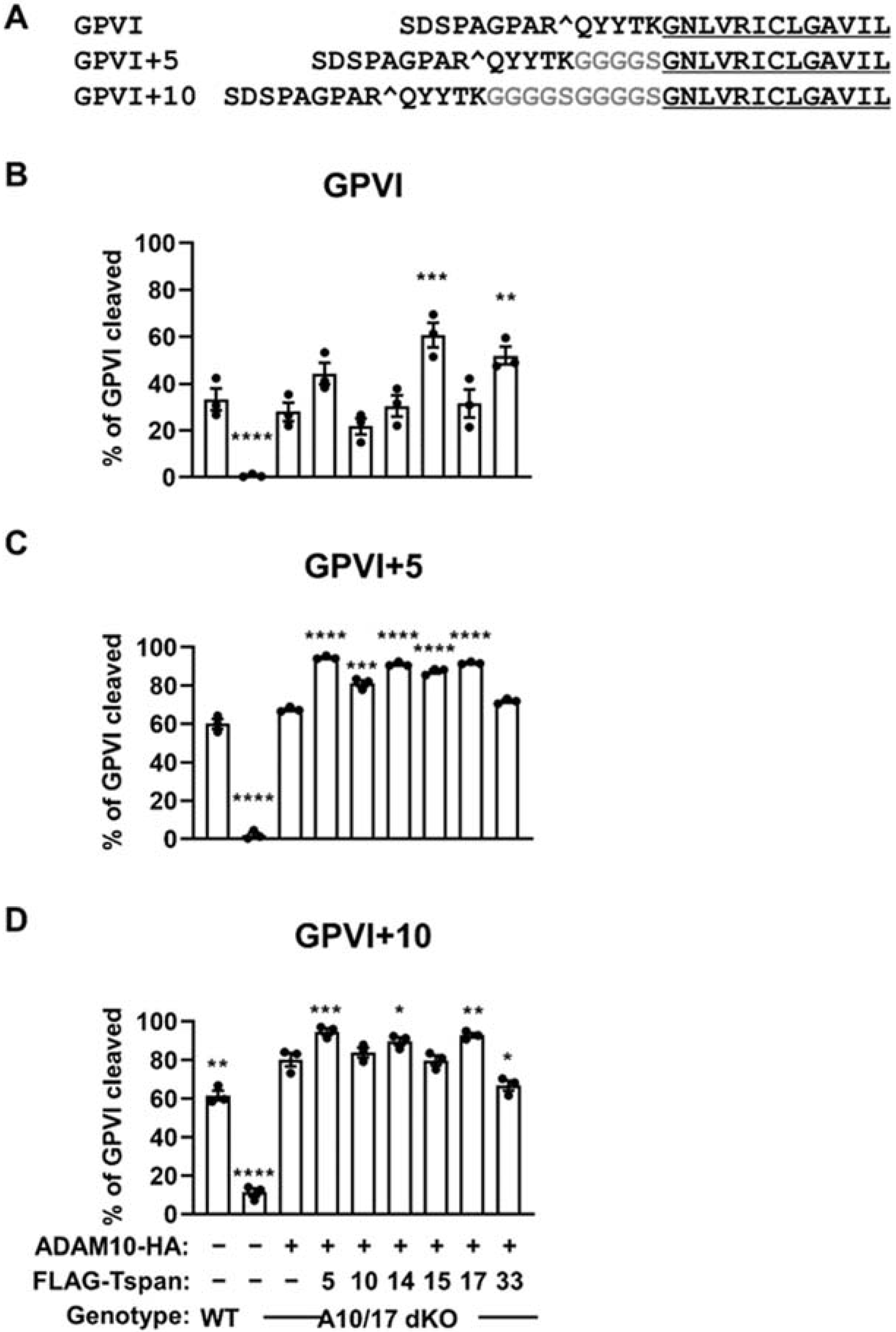
2.6. Evidence That Cut Site Position on GPVI Contributes to Specific Cleavage by Tspan15/ADAM10 and Tspan33/ADAM10
3. Discussion
4. Materials and Methods
4.1. Antibodies
4.2. Expression Constructs
4.3. CRISPR Guide RNA Design
- ADAM17 guide 1: 5′ CACCGCCGAAGCCCGGGTCATCCGG 3′ and 5′ AAACCCGGATGACCCGGGCTTCGGC 3′
- ADAM17 guide 2: 5′ CACCGCGAAAGGAACCACGCTGGTC 3′ and 5′ AAACGACCAGCGTGGTTCCTTTCGC 3′
- Tspan14 guide 1: 5′ CACCGTTATAGATACTCTAACGCCA 3′ and 5′ AAACTGGCGTTAGAGTATCTATAAC 3′
- Tspan14 guide 2: 5′ CACCGCAGATCGATATCGTCCCGGT 3′ and 5′ AAACACCGGGACGATATCGATCTGC 3′
- Tspan33 guide 1: 5′ CACCGGCTCGGCTAATGAAGCATGC 3′ and 5′ AAACGCATGCTTCATTAGCCGAGCC 3′
- Tspan33 guide 2: 5′ CACCGGAAGGAGAACTCCTCCCCGG 3′ and 5′ AAACCCGGGGAGGAGTTCTCCTTCC 3′
- GPVI guide: 5′ CACCGCCGACCGCCCTCTTCTGTCT 3′ and 5′ AAACAGACAGAAGAGGGCGGTCGGC 3′
4.4. Cell Culture and Transfections
4.5. GPVI Cleavage Assay
4.6. Western Blotting
4.7. Flow Cytometry
4.8. Confocal Microscopy
4.9. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jourdi, G.; Lordkipanidze, M.; Philippe, A.; Bachelot-Loza, C.; Gaussem, P. Current and Novel Antiplatelet Therapies for the Treatment of Cardiovascular Diseases. Int. J. Mol. Sci. 2021, 22, 13079. [Google Scholar] [CrossRef] [PubMed]
- Palacios-Acedo, A.L.; Mege, D.; Crescence, L.; Dignat-George, F.; Dubois, C.; Panicot-Dubois, L. Platelets, Thrombo-Inflammation, and Cancer: Collaborating With the Enemy. Front. Immunol. 2019, 10, 1805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stark, K.; Massberg, S. Interplay between inflammation and thrombosis in cardiovascular pathology. Nat. Rev. Cardiol. 2021, 18, 666–682. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.C.; Damaskinaki, F.N.; Cheung, Y.F.H.; Slater, A.; Watson, S.P. Structure-function relationship of the platelet glycoprotein VI (GPVI) receptor: Does it matter if it is a dimer or monomer? Platelets 2021, 32, 724–732. [Google Scholar] [CrossRef] [PubMed]
- Perrella, G.; Nagy, M.; Watson, S.P.; Heemskerk, J.W.M. Platelet GPVI (Glycoprotein VI) and Thrombotic Complications in the Venous System. Arter. Thromb. Vasc. Biol. 2021, 41, 2681–2692. [Google Scholar] [CrossRef]
- Harbi, M.H.; Smith, C.W.; Nicolson, P.L.R.; Watson, S.P.; Thomas, M.R. Novel antiplatelet strategies targeting GPVI, CLEC-2 and tyrosine kinases. Platelets 2021, 32, 29–41. [Google Scholar] [CrossRef]
- Harrison, N.; Koo, C.Z.; Tomlinson, M.G. Regulation of ADAM10 by the TspanC8 Family of Tetraspanins and Their Therapeutic Potential. Int. J. Mol. Sci. 2021, 22, 6707. [Google Scholar] [CrossRef]
- Bender, M.; Hofmann, S.; Stegner, D.; Chalaris, A.; Bosl, M.; Braun, A.; Scheller, J.; Rose-John, S.; Nieswandt, B. Differentially regulated GPVI ectodomain shedding by multiple platelet-expressed proteinases. Blood 2010, 116, 3347–3355. [Google Scholar] [CrossRef]
- Gardiner, E.E.; Karunakaran, D.; Shen, Y.; Arthur, J.F.; Andrews, R.K.; Berndt, M.C. Controlled shedding of platelet glycoprotein (GP)VI and GPIb-IX-V by ADAM family metalloproteinases. J. Thromb. Haemost. 2007, 5, 1530–1537. [Google Scholar] [CrossRef]
- Gardiner, E.E.; Arthur, J.F.; Kahn, M.L.; Berndt, M.C.; Andrews, R.K. Regulation of platelet membrane levels of glycoprotein VI by a platelet-derived metalloproteinase. Blood 2004, 104, 3611–3617. [Google Scholar] [CrossRef] [Green Version]
- Montague, S.J.; Delierneux, C.; Lecut, C.; Layios, N.; Dinsdale, R.J.; Lee, C.S.; Poulter, N.S.; Andrews, R.K.; Hampson, P.; Wearn, C.M.; et al. Soluble GPVI is elevated in injured patients: Shedding is mediated by fibrin activation of GPVI. Blood Adv. 2018, 2, 240–251. [Google Scholar] [CrossRef] [PubMed]
- Al-Tamimi, M.; Grigoriadis, G.; Tran, H.; Paul, E.; Servadei, P.; Berndt, M.C.; Gardiner, E.E.; Andrews, R.K. Coagulation-induced shedding of platelet glycoprotein VI mediated by factor Xa. Blood 2011, 117, 3912–3920. [Google Scholar] [CrossRef] [Green Version]
- Al-Tamimi, M.; Tan, C.W.; Qiao, J.; Pennings, G.J.; Javadzadegan, A.; Yong, A.S.; Arthur, J.F.; Davis, A.K.; Jing, J.; Mu, F.T.; et al. Pathologic shear triggers shedding of vascular receptors: A novel mechanism for down-regulation of platelet glycoprotein VI in stenosed coronary vessels. Blood 2012, 119, 4311–4320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pallini, C.; Pike, J.A.; O’Shea, C.; Andrews, R.K.; Gardiner, E.E.; Watson, S.P.; Poulter, N.S. Immobilized collagen prevents shedding and induces sustained GPVI clustering and signaling in platelets. Platelets 2021, 32, 59–73. [Google Scholar] [CrossRef] [PubMed]
- Al-Tamimi, M.; Gardiner, E.E.; Thom, J.Y.; Shen, Y.; Cooper, M.N.; Hankey, G.J.; Berndt, M.C.; Baker, R.I.; Andrews, R.K. Soluble glycoprotein VI is raised in the plasma of patients with acute ischemic stroke. Stroke 2011, 42, 498–500. [Google Scholar] [CrossRef] [Green Version]
- Stack, J.R.; Madigan, A.; Helbert, L.; Dunne, E.; Gardiner, E.E.; Andrews, R.K.; Finan, R.; Smyth, E.; Kenny, D.; McCarthy, G.M. Soluble glycoprotein VI, a specific marker of platelet activation is increased in the plasma of subjects with seropositive rheumatoid arthritis. PLoS ONE 2017, 12, e0188027. [Google Scholar] [CrossRef] [Green Version]
- Vulliamy, P.; Montague, S.J.; Gillespie, S.; Chan, M.V.; Coupland, L.A.; Andrews, R.K.; Warner, T.D.; Gardiner, E.E.; Brohi, K.; Armstrong, P.C. Loss of GPVI and GPIbalpha contributes to trauma-induced platelet dysfunction in severely injured patients. Blood Adv. 2020, 4, 2623–2630. [Google Scholar] [CrossRef]
- Pishko, A.M.; Andrews, R.K.; Gardiner, E.E.; Lefler, D.S.; Cuker, A. Soluble glycoprotein VI is a predictor of major bleeding in patients with suspected heparin-induced thrombocytopenia. Blood Adv. 2020, 4, 4327–4332. [Google Scholar] [CrossRef]
- Matthews, A.L.; Koo, C.Z.; Szyroka, J.; Harrison, N.; Kanhere, A.; Tomlinson, M.G. Regulation of Leukocytes by TspanC8 Tetraspanins and the "Molecular Scissor" ADAM10. Front. Immunol. 2018, 9, 1451. [Google Scholar] [CrossRef]
- Matthews, A.L.; Noy, P.J.; Reyat, J.S.; Tomlinson, M.G. Regulation of A disintegrin and metalloproteinase (ADAM) family sheddases ADAM10 and ADAM17: The emerging role of tetraspanins and rhomboids. Platelets 2017, 28, 333–341. [Google Scholar] [CrossRef] [Green Version]
- Saint-Pol, J.; Eschenbrenner, E.; Dornier, E.; Boucheix, C.; Charrin, S.; Rubinstein, E. Regulation of the trafficking and the function of the metalloprotease ADAM10 by tetraspanins. Biochem. Soc. Trans. 2017, 45, 937–944. [Google Scholar] [CrossRef] [PubMed]
- Dornier, E.; Coumailleau, F.; Ottavi, J.F.; Moretti, J.; Boucheix, C.; Mauduit, P.; Schweisguth, F.; Rubinstein, E. TspanC8 tetraspanins regulate ADAM10/Kuzbanian trafficking and promote Notch activation in flies and mammals. J. Cell Biol. 2012, 199, 481–496. [Google Scholar] [CrossRef] [Green Version]
- Haining, E.J.; Yang, J.; Bailey, R.L.; Khan, K.; Collier, R.; Tsai, S.; Watson, S.P.; Frampton, J.; Garcia, P.; Tomlinson, M.G. The TspanC8 subgroup of tetraspanins interacts with A disintegrin and metalloprotease 10 (ADAM10) and regulates its maturation and cell surface expression. J. Biol. Chem. 2012, 287, 39753–39765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eschenbrenner, E.; Jouannet, S.; Clay, D.; Chaker, J.; Boucheix, C.; Brou, C.; Tomlinson, M.G.; Charrin, S.; Rubinstein, E. TspanC8 tetraspanins differentially regulate ADAM10 endocytosis and half-life. Life Sci. Alliance 2020, 3, e201900444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koo, C.Z.; Harrison, N.; Noy, P.J.; Szyroka, J.; Matthews, A.L.; Hsia, H.E.; Muller, S.A.; Tushaus, J.; Goulding, J.; Willis, K.; et al. The tetraspanin Tspan15 is an essential subunit of an ADAM10 scissor complex. J. Biol. Chem. 2020, 295, 12822–12839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipper, C.H.; Gabriel, K.H.; Seegar, T.C.M.; Durr, K.L.; Tomlinson, M.G.; Blacklow, S.C. Crystal structure of the Tspan15 LEL domain reveals a conserved ADAM10 binding site. Structure 2021, 30, 206–214.e4. [Google Scholar] [CrossRef]
- Jouannet, S.; Saint-Pol, J.; Fernandez, L.; Nguyen, V.; Charrin, S.; Boucheix, C.; Brou, C.; Milhiet, P.E.; Rubinstein, E. TspanC8 tetraspanins differentially regulate the cleavage of ADAM10 substrates, Notch activation and ADAM10 membrane compartmentalization. Cell. Mol. Life Sci. 2016, 73, 1895–1915. [Google Scholar] [CrossRef] [Green Version]
- Noy, P.J.; Yang, J.; Reyat, J.S.; Matthews, A.L.; Charlton, A.E.; Furmston, J.; Rogers, D.A.; Rainger, G.E.; Tomlinson, M.G. TspanC8 Tetraspanins and A Disintegrin and Metalloprotease 10 (ADAM10) Interact via Their Extracellular Regions: EVIDENCE FOR DISTINCT BINDING MECHANISMS FOR DIFFERENT TspanC8 PROTEINS. J. Biol. Chem. 2016, 291, 3145–3157. [Google Scholar] [CrossRef] [Green Version]
- Prox, J.; Willenbrock, M.; Weber, S.; Lehmann, T.; Schmidt-Arras, D.; Schwanbeck, R.; Saftig, P.; Schwake, M. Tetraspanin15 regulates cellular trafficking and activity of the ectodomain sheddase ADAM10. Cell. Mol. Life Sci. 2012, 69, 2919–2932. [Google Scholar] [CrossRef]
- Seipold, L.; Altmeppen, H.; Koudelka, T.; Tholey, A.; Kasparek, P.; Sedlacek, R.; Schweizer, M.; Bar, J.; Mikhaylova, M.; Glatzel, M.; et al. In vivo regulation of the A disintegrin and metalloproteinase 10 (ADAM10) by the tetraspanin 15. Cell. Mol. Life Sci. 2018, 75, 3251–3267. [Google Scholar] [CrossRef]
- Reyat, J.S.; Chimen, M.; Noy, P.J.; Szyroka, J.; Rainger, G.E.; Tomlinson, M.G. ADAM10-Interacting Tetraspanins Tspan5 and Tspan17 Regulate VE-Cadherin Expression and Promote T Lymphocyte Transmigration. J. Immunol. 2017, 199, 666–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saint-Pol, J.; Billard, M.; Dornier, E.; Eschenbrenner, E.; Danglot, L.; Boucheix, C.; Charrin, S.; Rubinstein, E. New insights into the tetraspanin Tspan5 using novel monoclonal antibodies. J. Biol. Chem. 2017, 292, 9551–9566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Q.; Guo, H.; He, P.; Deng, H.; Gao, Y.; Dong, N.; Niu, W.; Liu, T.; Li, M.; Wang, S.; et al. Tspan5 promotes epithelial-mesenchymal transition and tumour metastasis of hepatocellular carcinoma by activating Notch signalling. Mol. Oncol. 2021, 15, 3184–3202. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Fujiwara, T.; Ye, S.; Li, X.; Zhao, H. Downregulation of Notch modulators, tetraspanin 5 and 10, inhibits osteoclastogenesis in vitro. Calcif. Tissue Int. 2014, 95, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Jadoui, S.; Le Chapelain, O.; Ollivier, V.; Mostefa-Kara, A.; Di Meglio, L.; Dupont, S.; Gros, A.; Nomenjanahary, M.S.; Desilles, J.P.; Mazighi, M.; et al. Glenzocimab does not impact glycoprotein VI-dependent inflammatory haemostasis. Haematologica 2021, 106, 2000–2003. [Google Scholar] [CrossRef] [PubMed]
- Voors-Pette, C.; Lebozec, K.; Dogterom, P.; Jullien, L.; Billiald, P.; Ferlan, P.; Renaud, L.; Favre-Bulle, O.; Avenard, G.; Machacek, M.; et al. Safety and Tolerability, Pharmacokinetics, and Pharmacodynamics of ACT017, an Antiplatelet GPVI (Glycoprotein VI) Fab. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 956–964. [Google Scholar] [CrossRef]
- Al-Tamimi, M.; Arthur, J.F.; Gardiner, E.; Andrews, R.K. Focusing on plasma glycoprotein VI. Thromb. Haemost. 2012, 107, 648–655. [Google Scholar]
- Al-Tamimi, M.; Qiao, J.; Gardiner, E.E. The utility of platelet activation biomarkers in thrombotic microangiopathies. Platelets 2022, in press. [Google Scholar]
- Matthews, A.L.; Szyroka, J.; Collier, R.; Noy, P.J.; Tomlinson, M.G. Scissor sisters: Regulation of ADAM10 by the TspanC8 tetraspanins. Biochem. Soc. Trans. 2017, 45, 719–730. [Google Scholar] [CrossRef] [Green Version]
- Huff, J.; Bergter, A.; Birkenbeil, J.; Kleppe, I.; Engelmann, R.; Krzic, U. The new 2D Superresolution mode for ZEISS Airyscan. Nat. Methods 2017, 14, 1223. [Google Scholar] [CrossRef]
- Uemura, K.; Kihara, T.; Kuzuya, A.; Okawa, K.; Nishimoto, T.; Ninomiya, H.; Sugimoto, H.; Kinoshita, A.; Shimohama, S. Characterization of sequential N-cadherin cleavage by ADAM10 and PS1. Neurosci. Lett. 2006, 402, 278–283. [Google Scholar] [CrossRef] [PubMed]
- Sunnarborg, S.W.; Hinkle, C.L.; Stevenson, M.; Russell, W.E.; Raska, C.S.; Peschon, J.J.; Castner, B.J.; Gerhart, M.J.; Paxton, R.J.; Black, R.A.; et al. Tumor necrosis factor-alpha converting enzyme (TACE) regulates epidermal growth factor receptor ligand availability. J. Biol. Chem. 2002, 277, 12838–12845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mumm, J.S.; Schroeter, E.H.; Saxena, M.T.; Griesemer, A.; Tian, X.; Pan, D.J.; Ray, W.J.; Kopan, R. A ligand-induced extracellular cleavage regulates gamma-secretase-like proteolytic activation of Notch1. Mol. Cell 2000, 5, 197–206. [Google Scholar] [CrossRef]
- Shah, J.; Rouaud, F.; Guerrera, D.; Vasileva, E.; Popov, L.M.; Kelley, W.L.; Rubinstein, E.; Carette, J.E.; Amieva, M.R.; Citi, S. A Dock-and-Lock Mechanism Clusters ADAM10 at Cell-Cell Junctions to Promote alpha-Toxin Cytotoxicity. Cell Rep. 2018, 25, 2132–2147.e7. [Google Scholar] [CrossRef] [Green Version]
- Sidahmed-Adrar, N.; Ottavi, J.F.; Benzoubir, N.; Ait Saadi, T.; Bou Saleh, M.; Mauduit, P.; Guettier, C.; Desterke, C.; Le Naour, F. Tspan15 Is a New Stemness-Related Marker in Hepatocellular Carcinoma. Proteomics 2019, 19, e1900025. [Google Scholar] [CrossRef] [PubMed]
- Boylan, B.; Chen, H.; Rathore, V.; Paddock, C.; Salacz, M.; Friedman, K.D.; Curtis, B.R.; Stapleton, M.; Newman, D.K.; Kahn, M.L.; et al. Anti-GPVI-associated ITP: An acquired platelet disorder caused by autoantibody-mediated clearance of the GPVI/FcRgamma-chain complex from the human platelet surface. Blood 2004, 104, 1350–1355. [Google Scholar] [CrossRef]
- Arduise, C.; Abache, T.; Li, L.; Billard, M.; Chabanon, A.; Ludwig, A.; Mauduit, P.; Boucheix, C.; Rubinstein, E.; Le Naour, F. Tetraspanins regulate ADAM10-mediated cleavage of TNF-alpha and epidermal growth factor. J. Immunol. 2008, 181, 7002–7013. [Google Scholar] [CrossRef] [Green Version]
- Tomlinson, M.G.; Calaminus, S.D.; Berlanga, O.; Auger, J.M.; Bori-Sanz, T.; Meyaard, L.; Watson, S.P. Collagen promotes sustained glycoprotein VI signaling in platelets and cell lines. J. Thromb. Haemost. 2007, 5, 2274–2283. [Google Scholar] [CrossRef]
- Horiuchi, K.; Le Gall, S.; Schulte, M.; Yamaguchi, T.; Reiss, K.; Murphy, G.; Toyama, Y.; Hartmann, D.; Saftig, P.; Blobel, C.P. Substrate selectivity of epidermal growth factor-receptor ligand sheddases and their regulation by phorbol esters and calcium influx. Mol. Biol. Cell 2007, 18, 176–188. [Google Scholar] [CrossRef] [Green Version]
- Hodgkins, A.; Farne, A.; Perera, S.; Grego, T.; Parry-Smith, D.J.; Skarnes, W.C.; Iyer, V. WGE: A CRISPR database for genome engineering. Bioinformatics 2015, 31, 3078–3080. [Google Scholar] [CrossRef] [Green Version]
- Ehrhardt, C.; Schmolke, M.; Matzke, A.; Knoblauch, A.; Will, C.; Wixler, V.; Ludwig, S. Polyethylenimine, a cost-effective transfection reagent. Signal Transduct. 2006, 6, 179–184. [Google Scholar] [CrossRef]
- Noy, P.J.; Lodhia, P.; Khan, K.; Zhuang, X.; Ward, D.G.; Verissimo, A.R.; Bacon, A.; Bicknell, R. Blocking CLEC14A-MMRN2 binding inhibits sprouting angiogenesis and tumour growth. Oncogene 2015, 34, 5821–5831. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koo, C.Z.; Matthews, A.L.; Harrison, N.; Szyroka, J.; Nieswandt, B.; Gardiner, E.E.; Poulter, N.S.; Tomlinson, M.G. The Platelet Collagen Receptor GPVI Is Cleaved by Tspan15/ADAM10 and Tspan33/ADAM10 Molecular Scissors. Int. J. Mol. Sci. 2022, 23, 2440. https://doi.org/10.3390/ijms23052440
Koo CZ, Matthews AL, Harrison N, Szyroka J, Nieswandt B, Gardiner EE, Poulter NS, Tomlinson MG. The Platelet Collagen Receptor GPVI Is Cleaved by Tspan15/ADAM10 and Tspan33/ADAM10 Molecular Scissors. International Journal of Molecular Sciences. 2022; 23(5):2440. https://doi.org/10.3390/ijms23052440
Chicago/Turabian StyleKoo, Chek Ziu, Alexandra L. Matthews, Neale Harrison, Justyna Szyroka, Bernhard Nieswandt, Elizabeth E. Gardiner, Natalie S. Poulter, and Michael G. Tomlinson. 2022. "The Platelet Collagen Receptor GPVI Is Cleaved by Tspan15/ADAM10 and Tspan33/ADAM10 Molecular Scissors" International Journal of Molecular Sciences 23, no. 5: 2440. https://doi.org/10.3390/ijms23052440
APA StyleKoo, C. Z., Matthews, A. L., Harrison, N., Szyroka, J., Nieswandt, B., Gardiner, E. E., Poulter, N. S., & Tomlinson, M. G. (2022). The Platelet Collagen Receptor GPVI Is Cleaved by Tspan15/ADAM10 and Tspan33/ADAM10 Molecular Scissors. International Journal of Molecular Sciences, 23(5), 2440. https://doi.org/10.3390/ijms23052440