
Mechanisms Underlying Dichotomous Procoagulant COAT Platelet Generation—A Conceptual Review Summarizing Current Knowledge

 ,
,  ,
,  ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Procoagulant Platelet Characterization
2.1. In Search for a Consensus for the Characterization of Procoagulant Platelets
2.2. Delayed Onset of Dichotomous Procoagulant Response
2.3. Externalization of Negatively Charged Phospholipids
2.4. Coating with α-Granule Proadhesive and Procoagulant Proteins
2.5. Surface Exposure of Cytosolic Factor XIII
2.6. Decreased Aggregatory Properties
2.7. Cell Membrane Remodeling
2.8. Mitochondrial Depolarization
2.9. Sustained “Supramaximal” Cytosolic Ca2+ Concentration
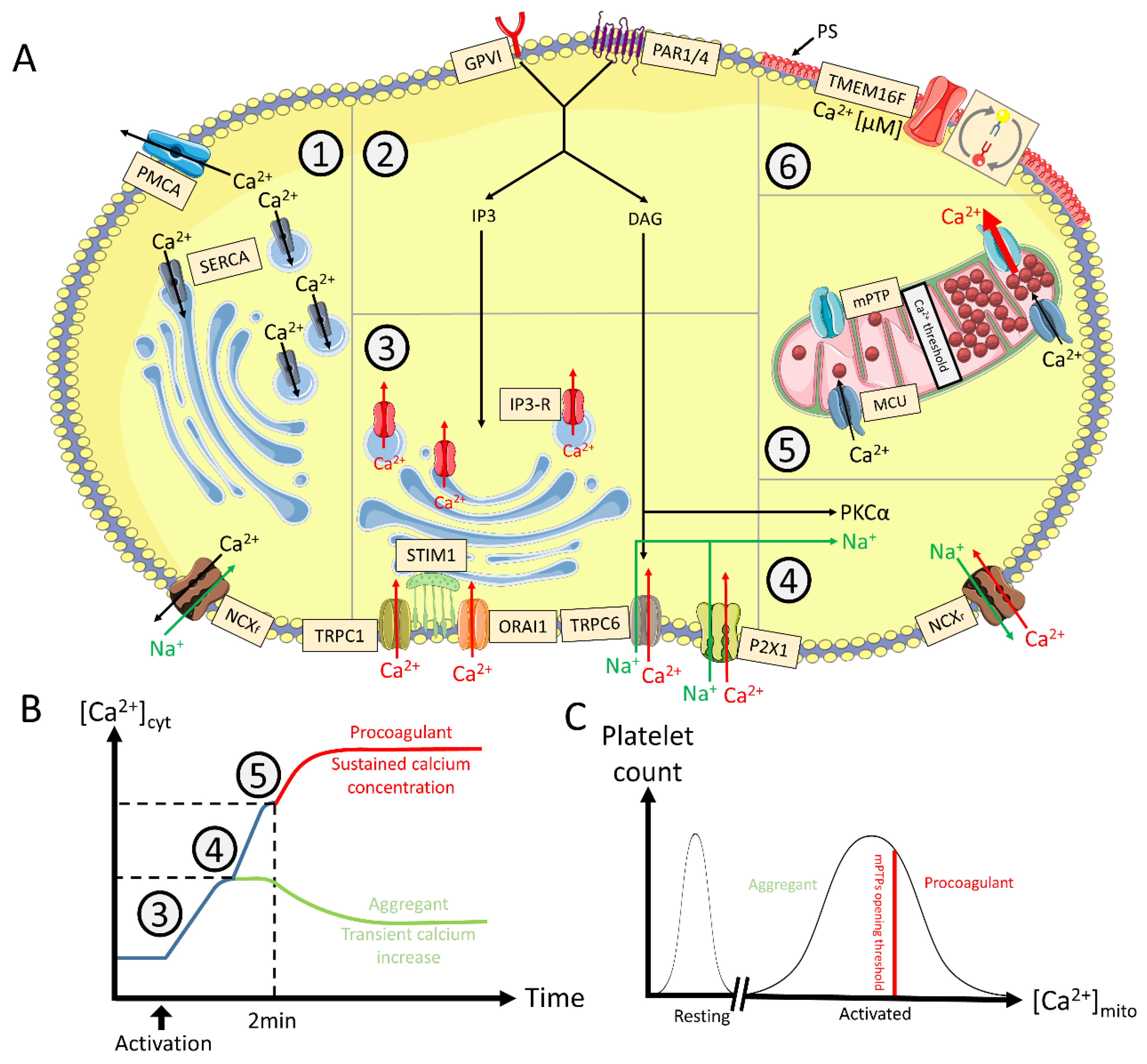
3. Cytosolic Ion Fluxes in Procoagulant COAT Platelets
3.1. Ca2+ Fluxes
3.1.1. Role of [Ca2+]cyt in the Procoagulant Response
3.1.2. Downregulation
3.1.3. Upregulation
3.1.4. Oscillating versus Sustained Increase of [Ca2+]cyt
3.2. Sodium Fluxes
3.2.1. Role of Cytosolic Sodium in the Procoagulant Response
3.2.2. Downregulation
3.2.3. Upregulation
3.3. Chloride Fluxes
3.4. Potassium Fluxes
4. Mechanisms Involved in Procoagulant COAT Platelet Formation
4.1. Surface Receptors
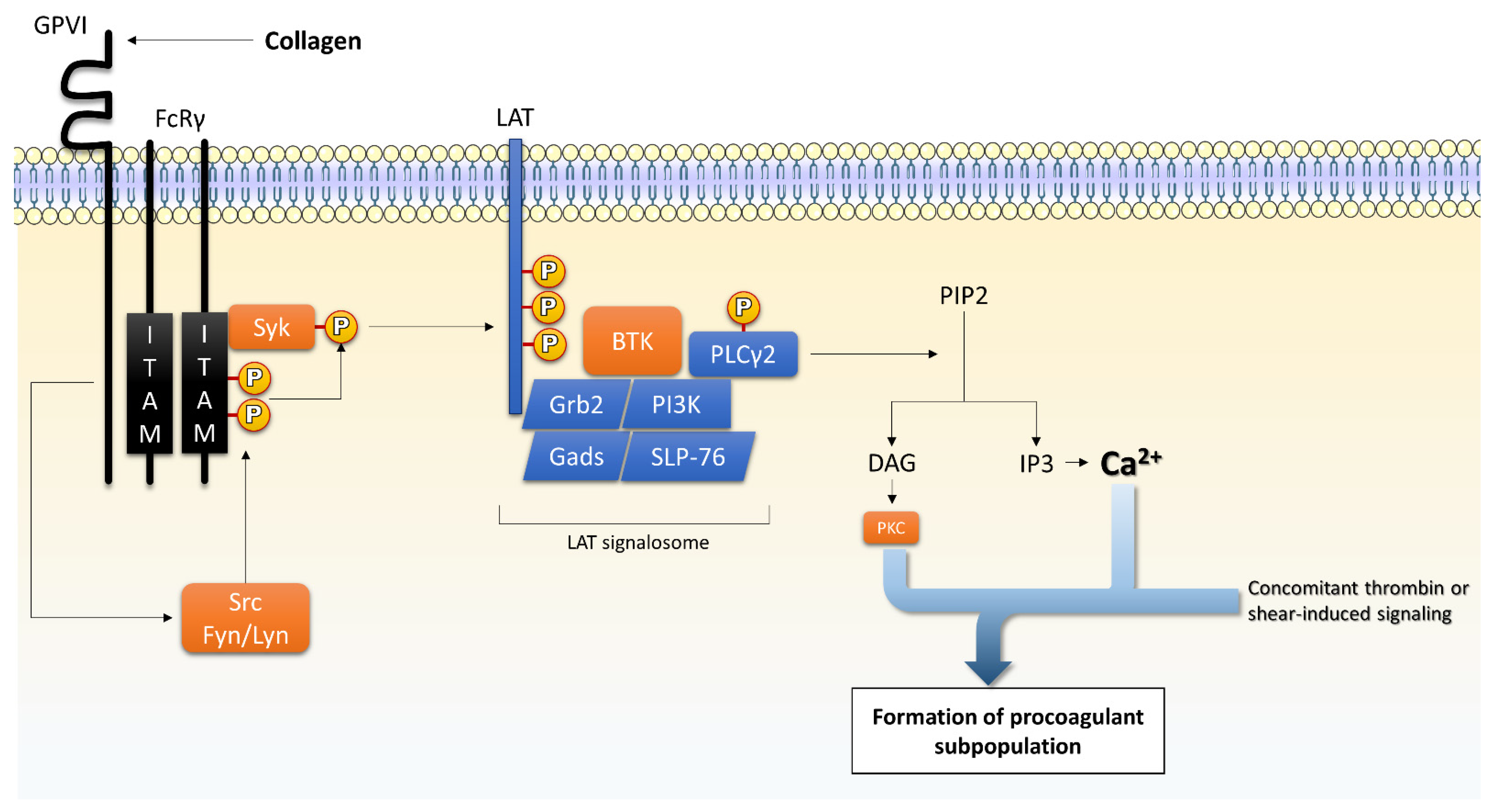
4.1.1. GPVI
4.1.2. Protease Activated Receptors (PAR) 1 and 4
4.1.3. GPIb-IX-V
4.1.4. P2Y12
4.1.5. Receptors That Are Regarded as Less Relevant for the Procoagulant Response
4.2. Key Regulators of Intracellular Signaling
4.2.1. PLC/PKC Isoforms
4.2.2. p38 Mitogen-Activated Protein Kinases (p38 MAPK)
4.2.3. NCX
4.2.4. Mitochondria
4.2.5. Scramblase
4.3. Extracellular Outside-In Signaling of GPIIb-IIIa in Aggregation
5. Potential Drivers of Platelet Phenotypic Diversification
5.1. Intrinsic Platelet Heterogeneity
5.1.1. Platelet Size
5.1.2. Number and Contents of Granules
5.1.3. Number of Mitochondria
5.1.4. Receptor Density and Reactivity
5.2. Age-Dependent Alterations
5.2.1. Apoptotic Platelets Exhibiting PS Exposure Are Not Functionally Procoagulant
5.2.2. ROS Accumulated during Aging and Their Role
5.2.3. Shedding of Surface Receptors with Aging and Strong Activation
5.3. Variability of Activation Pathways
5.3.1. NCX Reverse Mode
5.3.2. Protein Phosphorylation
5.3.3. GPIIb-IIIa Outside-In Signaling
5.4. Rheology and Cell–Cell Interactions
5.4.1. Local Agonist Concentration
5.4.2. Shear Stress
5.4.3. Architecture of the Clot
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alberio, L.; Safa, O.; Clemetson, K.J.; Esmon, C.T.; Dale, G.L. Surface expression and functional characterization of α-granule factor V in human platelets: Effects of ionophore A23187, thrombin, collagen, and convulxin. Blood 2000, 95, 1694–1702. [Google Scholar] [CrossRef] [PubMed]
- Aliotta, A.; Calderara, D.B.; Zermatten, M.G.; Marchetti, M.; Alberio, L. Thrombocytopathies: Not Just Aggregation Defects-The Clinical Relevance of Procoagulant Platelets. J. Clin. Med. 2021, 10, 894. [Google Scholar] [CrossRef] [PubMed]
- Agbani, E.O.; Poole, A.W. Procoagulant platelets: Generation, function, and therapeutic targeting in thrombosis. Blood 2017, 130, 2171–2179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, Y.; Guo, H.; Zhang, Y.; Qiao, R. Procoagulant platelets: Generation, characteristics, and therapeutic target. J. Clin. Lab. Anal. 2021, 35, e23750. [Google Scholar] [CrossRef]
- Podoplelova, N.A.; Nechipurenko, D.Y.; Ignatova, A.A.; Sveshnikova, A.N.; Panteleev, M.A. Procoagulant Platelets: Mechanisms of Generation and Action. Hämostaseologie 2021, 41, 146–153. [Google Scholar] [CrossRef]
- Saxena, K.; Pethe, K.; Dale, G.L. Coated-platelet levels may explain some variability in clinical phenotypes observed with severe hemophilia. J. Thromb. Haemost. 2010, 8, 1140–1142. [Google Scholar] [CrossRef]
- Daskalakis, M.; Colucci, G.; Keller, P.; Rochat, S.; Silzle, T.; Biasiutti, F.D.; Barizzi, G.; Alberio, L. Decreased generation of procoagulant platelets detected by flow cytometric analysis in patients with bleeding diathesis. Cytom. Part B Clin. Cytom. 2014, 86, 397–409. [Google Scholar] [CrossRef]
- Prodan, C.I.; Stoner, J.A.; Cowan, L.D.; Dale, G.L. Lower coated-platelet levels are associated with early hemorrhagic transformation in patients with non-lacunar brain infarction. J. Thromb. Haemost. 2010, 8, 1185–1190. [Google Scholar] [CrossRef]
- Prodan, C.I.; Stoner, J.A.; Dale, G.L. Lower Coated-Platelet Levels Are Associated With Increased Mortality After Spontaneous Intracerebral Hemorrhage. Stroke 2015, 46, 1819–1825. [Google Scholar] [CrossRef] [Green Version]
- Kirkpatrick, A.C.; Stoner, J.A.; Dale, G.L.; Prodan, C.I. Elevated coated-platelets in symptomatic large-artery stenosis patients are associated with early stroke recurrence. Platelets 2014, 25, 93–96. [Google Scholar] [CrossRef]
- Kirkpatrick, A.C.; Vincent, A.S.; Dale, G.L.; Prodan, C.I. Increased platelet procoagulant potential predicts recurrent stroke and TIA after lacunar infarction. J. Thromb. Haemost. 2020, 18, 660–668. [Google Scholar] [CrossRef]
- Khattab, M.H.; Prodan, C.I.; Vincent, A.S.; Xu, C.; Jones, K.R.; Thind, S.; Rabadi, M.; Mithilesh, S.; Mathews, E.; Guthery, L.; et al. Increased procoagulant platelet levels are predictive of death in COVID-19. GeroScience 2021, 43, 2055–2065. [Google Scholar] [CrossRef]
- Pasalic, L.; Wing-Lun, E.; Lau, J.K.; Campbell, H.; Pennings, G.J.; Lau, E.; Connor, D.; Liang, H.P.; Muller, D.; Kritharides, L.; et al. Novel assay demonstrates that coronary artery disease patients have heightened procoagulant platelet response. J. Thromb. Haemost. 2018, 16, 1198–1210. [Google Scholar] [CrossRef]
- Stalker, T.J.; Traxler, E.A.; Wu, J.; Wannemacher, K.M.; Cermignano, S.L.; Voronov, R.; Diamond, S.L.; Brass, L.F. Hierarchical organization in the hemostatic response and its relationship to the platelet-signaling network. Blood 2013, 121, 1875–1885. [Google Scholar] [CrossRef]
- Versteeg, H.H.; Heemskerk, J.W.M.; Levi, M.; Reitsma, P.H. New fundamentals in hemostasis. Physiol. Rev. 2013, 93, 327–358. [Google Scholar] [CrossRef] [Green Version]
- Welsh, J.D.; Stalker, T.J.; Voronov, R.; Muthard, R.W.; Tomaiuolo, M.; Diamond, S.L.; Brass, L.F. A systems approach to hemostasis: 1. The interdependence of thrombus architecture and agonist movements in the gaps between platelets. Blood 2014, 124, 1808–1815. [Google Scholar] [CrossRef]
- Van Der Meijden, P.E.J.; Heemskerk, J.W.M. Platelet biology and functions: New concepts and clinical perspectives. Nat. Rev. Cardiol. 2019, 16, 166–179. [Google Scholar] [CrossRef]
- Nechipurenko, D.Y.; Receveur, N.; Yakimenko, A.O.; Shepelyuk, T.O.; Yakusheva, A.A.; Kerimov, R.R.; Obydennyy, S.I.; Eckly, A.; Leon, C.; Gachet, C.; et al. Clot Contraction Drives the Translocation of Procoagulant Platelets to Thrombus Surface. Arter. Thromb. Vasc. Biol. 2019, 39, 37–47. [Google Scholar] [CrossRef]
- McFadyen, J.D.; Schaff, M.; Peter, K. Current and future antiplatelet therapies: Emphasis on preserving haemostasis. Nat. Rev. Cardiol. 2018, 15, 181–191. [Google Scholar] [CrossRef]
- Shi, J.; Pipe, S.W.; Rasmussen, J.T.; Heegaard, C.W.; Gilbert, G.E. Lactadherin blocks thrombosis and hemostasis in vivo: Correlation with platelet phosphatidylserine exposure. J. Thromb. Haemost. 2008, 6, 1167–1174. [Google Scholar] [CrossRef] [Green Version]
- Agbani, E.O.; Williams, C.M.; Li, Y.; Van Den Bosch, M.T.J.; Moore, S.F.; Mauroux, A.; Hodgson, L.; Verkman, A.S.; Hers, I.; Poole, A.W. Aquaporin-1 regulates platelet procoagulant membrane dynamics and in vivo thrombosis. JCI Insight 2018, 3, e99062. [Google Scholar] [CrossRef] [PubMed]
- Millington-Burgess, S.L.; Harper, M.T. Cytosolic and mitochondrial Ca2+ signaling in procoagulant platelets. Platelets 2021, 32, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Delaney, M.K.; Liu, J.; Kim, K.; Shen, B.; Stojanovic-Terpo, A.; Zheng, Y.; Cho, J.; Du, X. Agonist-induced platelet procoagulant activity requires shear and a Rac1-dependent signaling mechanism. Blood 2014, 124, 1957–1967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estevez, B.; Du, X. New Concepts and Mechanisms of Platelet Activation Signaling. Physiology 2017, 32, 162–177. [Google Scholar] [CrossRef] [Green Version]
- Raffaello, A.; Mammucari, C.; Gherardi, G.; Rizzuto, R. Calcium at the Center of Cell Signaling: Interplay between Endoplasmic Reticulum, Mitochondria, and Lysosomes. Trends Biochem. Sci. 2016, 41, 1035–1049. [Google Scholar] [CrossRef] [Green Version]
- Abbasian, N.; Millington-Burgess, S.L.; Chabra, S.; Malcor, J.-D.; Harper, M.T. Supramaximal calcium signaling triggers procoagulant platelet formation. Blood Adv. 2020, 4, 154–164. [Google Scholar] [CrossRef] [Green Version]
- Heemskerk, J.W.; Mattheij, N.J.; Cosemans, J.M. Platelet-based coagulation: Different populations, different functions. J. Thromb. Haemost. 2013, 11, 2–16. [Google Scholar] [CrossRef]
- Kulkarni, S.; Jackson, S.P. Platelet Factor XIII and Calpain Negatively Regulate Integrin αIIbβ3 Adhesive Function and Thrombus Growth. J. Biol. Chem. 2004, 279, 30697–30706. [Google Scholar] [CrossRef] [Green Version]
- Hua, V.M.; Abeynaike, L.; Glaros, E.; Campbell, H.; Pasalic, L.; Hogg, P.J.; Chen, V.M.Y. Necrotic platelets provide a procoagulant surface during thrombosis. Blood 2015, 126, 2852–2862. [Google Scholar] [CrossRef]
- Mazepa, M.; Hoffman, M.; Monroe, D. Superactivated Platelets. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1747–1752. [Google Scholar] [CrossRef] [Green Version]
- Podoplelova, N.A.; Sveshnikova, A.N.; Kotova, Y.N.; Eckly, A.; Receveur, N.; Nechipurenko, D.Y.; Obydennyi, S.I.; Kireev, I.I.; Gachet, C.; Ataullakhanov, F.I.; et al. Coagulation factors bound to procoagulant platelets concentrate in cap structures to promote clotting. Blood 2016, 128, 1745–1755. [Google Scholar] [CrossRef] [Green Version]
- Agbani, E.O.; Williams, C.M.; Hers, I.; Poole, A.W. Membrane Ballooning in Aggregated Platelets is Synchronised and Mediates a Surge in Microvesiculation. Sci. Rep. 2017, 7, 2770. [Google Scholar] [CrossRef] [Green Version]
- Storrie, B. A tip of the cap to procoagulant platelets. Blood 2016, 128, 1668–1669. [Google Scholar] [CrossRef] [Green Version]
- Dale, G.L. Coated-platelets: An emerging component of the procoagulant response. J. Thromb. Haemost. 2005, 3, 2185–2192. [Google Scholar] [CrossRef]
- Szasz, R.; Dale, G.L. COAT platelets. Curr. Opin. Hematol. 2003, 10, 351–355. [Google Scholar] [CrossRef]
- Dale, G.L.; Friese, P.; Batar, P.; Hamilton, S.F.; Reed, G.L.; Jackson, K.W.; Clemetson, K.J.; Alberio, L. Stimulated platelets use serotonin to enhance their retention of procoagulant proteins on the cell surface. Nature 2002, 415, 175–179. [Google Scholar] [CrossRef]
- Aliotta, A.; Krüsi, M.; Calderara, D.B.; Zermatten, M.G.; Gomez, F.J.; Batista Mesquita Sauvage, A.P.; Alberio, L. Characterization of Procoagulant COAT Platelets in Patients with Glanzmann Thrombasthenia. Int. J. Mol. Sci. 2020, 21, 9515. [Google Scholar] [CrossRef]
- Morton, L.F.; Hargreaves, P.G.; Farndale, R.W.; Young, R.D.; Barnes, M.J. Integrin alpha 2 beta 1-independent activation of platelets by simple collagen-like peptides: Collagen tertiary (triple-helical) and quaternary (polymeric) structures are sufficient alone for alpha 2 beta 1-independent platelet reactivity. Biochem. J. 1995, 306, 337–344. [Google Scholar] [CrossRef]
- Södergren, A.L.; Ramström, S. Platelet subpopulations remain despite strong dual agonist stimulation and can be characterised using a novel six-colour flow cytometry protocol. Sci. Rep. 2018, 8, 1441. [Google Scholar] [CrossRef] [Green Version]
- Polgar, J.; Clemetson, J.M.; Kehrel, B.E.; Wiedemann, M.; Magnenat, E.M.; Wells, T.N.; Clemetson, K.J. Platelet activation and signal transduction by convulxin, a C-type lectin from Crotalus durissus terrificus (tropical rattlesnake) venom via the p62/GPVI collagen receptor. J. Biol. Chem. 1997, 272, 13576–13583. [Google Scholar] [CrossRef] [Green Version]
- Munnix, I.C.A.; Kuijpers, M.J.E.; Auger, J.; Thomassen, C.M.L.G.D.; Panizzi, P.; van Zandvoort, M.A.M.; Rosing, J.; Bock, P.E.; Watson, S.P.; Heemskerk, J.W.M. Segregation of Platelet Aggregatory and Procoagulant Microdomains in Thrombus Formation. Arter. Thromb. Vasc. Biol. 2007, 27, 2484–2490. [Google Scholar] [CrossRef] [Green Version]
- Aliotta, A.; Calderara, D.B.; Zermatten, M.G.; Alberio, L. Sodium–Calcium Exchanger Reverse Mode Sustains Dichotomous Ion Fluxes Required for Procoagulant COAT Platelet Formation. Thromb. Haemost. 2021, 121, 309–321. [Google Scholar] [CrossRef]
- Harper, M.T.; Londoño, J.E.C.; Quick, K.; Londoño, J.C.; Flockerzi, V.; Philipp, S.E.; Birnbaumer, L.; Freichel, M.; Poole, A.W. Transient Receptor Potential Channels Function as a Coincidence Signal Detector Mediating Phosphatidylserine Exposure. Sci. Signal. 2013, 6, ra50. [Google Scholar] [CrossRef]
- Aliotta, A.; Calderara, D.B.; Alberio, L. Flow Cytometric Monitoring of Dynamic Cytosolic Calcium, Sodium, and Potassium Fluxes Following Platelet Activation. Cytometry. Part A J. Int. Soc. Anal. Cytol. 2020, 97, 933–944. [Google Scholar] [CrossRef]
- Keuren, J.F.; Wielders, S.J.; Ulrichts, H.; Hackeng, T.; Heemskerk, J.W.; Deckmyn, H.; Bevers, E.M.; Lindhout, T. Synergistic effect of thrombin on collagen-induced platelet procoagulant activity is mediated through protease-activated receptor-1. Arter. Thromb. Vasc. Biol. 2005, 25, 1499–1505. [Google Scholar] [CrossRef] [Green Version]
- Alberio, L.; Ravanat, C.; Hechler, B.; Mangin, P.H.; Lanza, F.; Gachet, C. Delayed-onset of procoagulant signalling revealed by kinetic analysis of COAT platelet formation. Thromb. Haemost. 2017, 117, 1101–1114. [Google Scholar] [CrossRef] [Green Version]
- Rukoyatkina, N.; Butt, E.; Subramanian, H.; Nikolaev, V.O.; Mindukshev, I.; Walter, U.; Gambaryan, S.; Benz, P.M. Protein kinase A activation by the anti-cancer drugs ABT-737 and thymoquinone is caspase-3-dependent and correlates with platelet inhibition and apoptosis. Cell Death Dis. 2017, 8, e2898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, E.C.; Wang, H.; Christensen, H.; McMillan-Ward, E.; Israels, S.J.; Bang, K.W.A.; Rand, M.L. Analysis of procoagulant phosphatidylserine-exposing platelets by imaging flow cytometry. Res. Pract. Thromb. Haemost. 2018, 2, 736–750. [Google Scholar] [CrossRef] [PubMed]
- Calderara, D.B.; Crettaz, D.; Aliotta, A.; Barelli, S.; Tissot, J.-D.; Prudent, M.; Alberio, L. Generation of procoagulant collagen- and thrombin-activated platelets in platelet concentrates derived from buffy coat: The role of processing, pathogen inactivation, and storage. Transfusion 2018, 58, 2395–2406. [Google Scholar] [CrossRef] [PubMed]
- Kirkpatrick, A.C.; Vincent, A.S.; Dale, G.L.; Prodan, C.I. Clopidogrel use and smoking cessation result in lower coated-platelet levels after stroke. Platelets 2020, 31, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zhang, P.; Solari, F.A.; Sickmann, A.; Garcia, A.; Jurk, K.; Heemskerk, J.W.M. Molecular Proteomics and Signalling of Human Platelets in Health and Disease. Int. J. Mol. Sci. 2021, 22, 9860. [Google Scholar] [CrossRef]
- Roka-Moiia, Y.; Walk, R.; Palomares, D.E.; Ammann, K.R.; Dimasi, A.; Italiano, J.E.; Sheriff, J.; Bluestein, D.; Slepian, M.J. Platelet Activation via Shear Stress Exposure Induces a Differing Pattern of Biomarkers of Activation versus Biochemical Agonists. Thromb. Haemost. 2020, 120, 776–792. [Google Scholar] [CrossRef]
- Aliotta, A.; Calderara, D.B.; Zermatten, M.G.; Alberio, L. High-Dose Epinephrine Enhances Platelet Aggregation at the Expense of Procoagulant Activity. Thromb. Haemost. 2021, 121, 1337–1344. [Google Scholar] [CrossRef]
- Mattheij, N.J.; Gilio, K.; van Kruchten, R.; Jobe, S.M.; Wieschhaus, A.J.; Chishti, A.H.; Collins, P.; Heemskerk, J.W.; Cosemans, J.M. Dual mechanism of integrin alphaIIbbeta3 closure in procoagulant platelets. J. Biol. Chem 2013, 288, 13325–13336. [Google Scholar] [CrossRef] [Green Version]
- Seigneuret, M.; Devaux, P.F. ATP-dependent asymmetric distribution of spin-labeled phospholipids in the erythrocyte membrane: Relation to shape changes. Proc. Natl. Acad. Sci. USA 1984, 81, 3751–3755. [Google Scholar] [CrossRef] [Green Version]
- Zwaal, R.F.A.; Schroit, A.J. Pathophysiologic Implications of Membrane Phospholipid Asymmetry in Blood Cells. Blood 1997, 89, 1121–1132. [Google Scholar] [CrossRef]
- Stoilova-McPhie, S. Factor VIII and Factor V Membrane Bound Complexes. In Macromolecular Protein Complexes III: Structure and Function; Harris, J.R., Marles-Wright, J., Eds.; Springer: Cham, Switzerland, 2021; Volume 96, pp. 153–175. [Google Scholar]
- Nagata, S.; Sakuragi, T.; Segawa, K. Flippase and scramblase for phosphatidylserine exposure. Curr. Opin. Immunol. 2020, 62, 31–38. [Google Scholar] [CrossRef]
- Millington-Burgess, S.L.; Harper, M.T. Gene of the issue: ANO6 and Scott Syndrome. Platelets 2020, 31, 964–967. [Google Scholar] [CrossRef]
- Baig, A.A.; Haining, E.J.; Geuss, E.; Beck, S.; Swieringa, F.; Wanitchakool, P.; Schuhmann, M.K.; Stegner, D.; Kunzelmann, K.; Kleinschnitz, C.; et al. TMEM16F-Mediated Platelet Membrane Phospholipid Scrambling Is Critical for Hemostasis and Thrombosis but not Thromboinflammation in Mice—Brief Report. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2152–2157. [Google Scholar] [CrossRef] [Green Version]
- Szasz, R.; Dale, G.L. Thrombospondin and fibrinogen bind serotonin-derivatized proteins on COAT-platelets. Blood 2002, 100, 2827–2831. [Google Scholar] [CrossRef]
- Mattheij, N.J.A.; Swieringa, F.; Mastenbroek, T.G.; Berny-Lang, M.A.; May, F.; Baaten, C.C.F.M.J.; Van Der Meijden, P.E.J.; Henskens, Y.M.C.; Beckers, E.A.M.; Suylen, D.P.L.; et al. Coated platelets function in platelet-dependent fibrin formation via integrin α IIb β 3 and transglutaminase factor XIII. Haematologica 2016, 101, 427–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alshehri, F.S.M.; Whyte, C.S.; Mutch, N.J. Factor XIII-A: An Indispensable “Factor” in Haemostasis and Wound Healing. Int. J. Mol. Sci. 2021, 22, 3055. [Google Scholar] [CrossRef] [PubMed]
- Somodi, L.; Debreceni, I.B.; Kis, G.; Cozzolino, M.; Kappelmayer, J.; Antal, M.; Panyi, G.; Bárdos, H.; Mutch, N.J.; Muszbek, L. Activation mechanism dependent surface exposure of cellular factor XIII on activated platelets and platelet microparticles. J. Thromb. Haemost. 2022. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.L.; Lionikiene, A.S.; Fraser, S.R.; Whyte, C.S.; Booth, N.A.; Mutch, N.J. Functional factor XIII-A is exposed on the stimulated platelet surface. Blood 2014, 124, 3982–3990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Li, X.; Shi, X.; Zhu, M.; Wang, J.; Huang, S.; Huang, X.; Wang, H.; Li, L.; Deng, H.; et al. Platelet integrin αIIbβ3: Signal transduction, regulation, and its therapeutic targeting. J. Hematol. Oncol. 2019, 12, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shattil, S.J.; Kashiwagi, H.; Pampori, N. Integrin signaling: The platelet paradigm. Blood 1998, 91, 2645–2657. [Google Scholar] [CrossRef] [Green Version]
- Mischnik, M.; Gambaryan, S.; Subramanian, H.; Geiger, J.; Schütz, C.; Timmer, J.; Dandekar, T. A comparative analysis of the bistability switch for platelet aggregation by logic ODE based dynamical modeling. Mol. BioSyst. 2014, 10, 2082–2089. [Google Scholar] [CrossRef]
- Aslan, J.E. Platelet Shape Change. In Platelets in Thrombotic and Non-Thrombotic Disorders; Springer: Cham, Switzerland, 2017; pp. 321–336. [Google Scholar]
- Siljander, P.; Farndale, R.W.; Feijge, M.A.; Comfurius, P.; Kos, S.; Bevers, E.M.; Heemskerk, J.W. Platelet adhesion enhances the glycoprotein VI-dependent procoagulant response: Involvement of p38 MAP kinase and calpain. Arter. Thromb. Vasc. Biol. 2001, 21, 618–627. [Google Scholar] [CrossRef]
- Agbani, E.O.; van den Bosch, M.T.; Brown, E.; Williams, C.M.; Mattheij, N.J.; Cosemans, J.M.; Collins, P.W.; Heemskerk, J.W.; Hers, I.; Poole, A.W. Coordinated Membrane Ballooning and Procoagulant Spreading in Human Platelets. Circulation 2015, 132, 1414–1424. [Google Scholar] [CrossRef] [Green Version]
- Cong, J.; Goll, D.E.; Peterson, A.M.; Kapprell, H.P. The role of autolysis in activity of the Ca2+-dependent proteinases (μ-calpain and m-calpain). J. Biol. Chem. 1989, 264, 10096–10103. [Google Scholar] [CrossRef]
- Nomura, S.; Shimizu, M. Clinical significance of procoagulant microparticles. J. Intensive Care 2015, 3, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, H.; Davies, J.E.; Harper, M.T. 2-Aminoethoxydiphenylborate (2-APB) inhibits release of phosphatidylserine-exposing extracellular vesicles from platelets. Cell Death Discov. 2020, 6, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morel, O.; Jesel, L.; Freyssinet, J.-M.; Toti, F. Cellular Mechanisms Underlying the Formation of Circulating Microparticles. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 15–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nawaz, M.; Camussi, G.; Valadi, H.; Nazarenko, I.; Ekström, K.; Wang, X.; Principe, S.; Shah, N.; Ashraf, N.M.; Fatima, F.; et al. The emerging role of extracellular vesicles as biomarkers for urogenital cancers. Nat. Rev. Urol. 2014, 11, 688–701. [Google Scholar] [CrossRef]
- Jackson, S.P.; Schoenwaelder, S.M. Procoagulant platelets: Are they necrotic? Blood 2010, 116, 2011–2018. [Google Scholar] [CrossRef] [Green Version]
- Kholmukhamedov, A.; Janecke, R.; Choo, H.J.; Jobe, S.M. The mitochondrial calcium uniporter regulates procoagulant platelet formation. J. Thromb. Haemost. 2018, 16, 2315–2321. [Google Scholar] [CrossRef] [Green Version]
- Abramov, A.Y.; Duchen, M.R. Measurements of Threshold of Mitochondrial Permeability Transition Pore Opening in Intact and Permeabilized Cells by Flash Photolysis of Caged Calcium. In Neurodegeneration: Methods and Protocols; Manfredi, G., Kawamata, H., Eds.; Humana Press: Totowa, NJ, USA, 2011; Volume 793, pp. 299–309. [Google Scholar]
- Obydennyy, S.I.; Sveshnikova, A.N.; Ataullakhanov, F.I.; Panteleev, M.A. Dynamics of calcium spiking, mitochondrial collapse and phosphatidylserine exposure in platelet subpopulations during activation. J. Thromb. Haemost. 2016, 14, 1867–1881. [Google Scholar] [CrossRef]
- Varga-Szabo, D.; Braun, A.; Nieswandt, B. Calcium signaling in platelets. J. Thromb. Haemost. 2009, 7, 1057–1066. [Google Scholar] [CrossRef]
- Aliotta, A.; Alberio, L. Another piece of knowledge in the puzzle of procoagulant COAT platelets. J. Thromb. Haemost. 2022. [Google Scholar] [CrossRef]
- Stafford, N.; Wilson, C.; Oceandy, D.; Neyses, L.; Cartwright, E.J. The Plasma Membrane Calcium ATPases and Their Role as Major New Players in Human Disease. Physiol. Rev. 2017, 97, 1089–1125. [Google Scholar] [CrossRef] [Green Version]
- Redondo, P.C.; Salido, G.M.; Pariente, J.A.; Sage, S.O.; Rosado, J.A. SERCA2b and 3 play a regulatory role in store-operated calcium entry in human platelets. Cell. Signal. 2008, 20, 337–346. [Google Scholar] [CrossRef]
- Wang, Y.; Shi, J.; Tong, X. Cross-Talk between Mechanosensitive Ion Channels and Calcium Regulatory Proteins in Cardiovascular Health and Disease. Int. J. Mol. Sci. 2021, 22, 8782. [Google Scholar] [CrossRef]
- Lu, W.; Xu, D.; Tu, R.; Hu, Z. Morphology of platelet Golgi apparatus and their significance after acute cerebral infarction. Neural Regen Res. 2013, 8, 2134–2143. [Google Scholar]
- Yadav, S.; Williamson, J.K.; Aronova, M.A.; Prince, A.A.; Pokrovskaya, I.D.; Leapman, R.D.; Storrie, B. Golgi proteins in circulating human platelets are distributed across non-stacked, scattered structures. Platelets 2017, 28, 400–408. [Google Scholar] [CrossRef]
- Heijnen, H.; Korporaal, S. Platelet Morphology and Ultrastructure. In Platelets in Thrombotic and Non-Thrombotic Disorders: Pathophysiology, Pharmacology and Therapeutics: An Update; Springer: Cham, Switzerland, 2017; pp. 21–37. [Google Scholar]
- Jardín, I.; López, J.J.; Pariente, J.A.; Salido, G.M.; Rosado, J.A. Intracellular Calcium Release from Human Platelets: Different Messengers for Multiple Stores. Trends Cardiovasc. Med. 2008, 18, 57–61. [Google Scholar] [CrossRef]
- Tykocki, N.R.; Jackson, W.F.; Watts, S.W. Reverse-mode Na+/Ca2+ exchange is an important mediator of venous contraction. Pharmacol. Res. 2012, 66, 544–554. [Google Scholar] [CrossRef] [Green Version]
- Atkinson, B.T.; Ellmeier, W.; Watson, S.P. Tec regulates platelet activation by GPVI in the absence of Btk. Blood 2003, 102, 3592–3599. [Google Scholar] [CrossRef] [Green Version]
- Quek, L.S.; Bolen, J.; Watson, S.P. A role for Bruton’s tyrosine kinase (Btk) in platelet activation by collagen. Curr. Biol. 1998, 8, 1137–1140. [Google Scholar] [CrossRef] [Green Version]
- Bye, A.P.; Unsworth, A.J.; Gibbins, J.M. Platelet signaling: A complex interplay between inhibitory and activatory networks. J. Thromb. Haemost. 2016, 14, 918–930. [Google Scholar] [CrossRef] [Green Version]
- Bourguignon, L.Y.W.; Iida, N.; Jin, H. The involvement of the cytoskeleton in regulating IP3 receptor-mediated internal Ca2+ release in human blood platelets. Cell Biol. Int. 1993, 17, 751–758. [Google Scholar] [CrossRef]
- Quinton, T.M.; Dean, W.L. Cyclic AMP-dependent phosphorylation of the inositol-1,4,5-trisphosphate receptor inhibits Ca2+ release from platelet membranes. Biochem. Biophys. Res. Commun. 1992, 184, 893–899. [Google Scholar] [CrossRef]
- Silva-Rojas, R.; Laporte, J.; Böhm, J. STIM1/ORAI1 Loss-of-Function and Gain-of-Function Mutations Inversely Impact on SOCE and Calcium Homeostasis and Cause Multi-Systemic Mirror Diseases. Front. Physiol. 2020, 11, 604941. [Google Scholar] [CrossRef]
- Ramanathan, G.; Gupta, S.; Thielmann, I.; Pleines, I.; Varga-Szabo, D.; May, F.; Mannhalter, C.; Dietrich, A.; Nieswandt, B.; Braun, A. Defective diacylglycerol-induced Ca2+ entry but normal agonist-induced activation responses in TRPC6-deficient mouse platelets. J. Thromb. Haemost. 2012, 10, 419–429. [Google Scholar] [CrossRef]
- Espinosa, E.V.P.; Lin, O.A.; Karim, Z.A.; Alshbool, F.Z.; Khasawneh, F.T. Mouse transient receptor potential channel type 6 selectively regulates agonist-induced platelet function. Biochem. Biophys. Rep. 2019, 20, 100685. [Google Scholar] [CrossRef] [PubMed]
- Gachet, C. P2 receptors, platelet function and pharmacological implications. Thromb. Haemost. 2008, 99, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Zitt, C.; Zobel, A.; Obukhov, A.G.; Harteneck, C.; Kalkbrenner, F.; Lückhoff, A.; Schultz, G. Cloning and Functional Expression of a Human Ca2+-Permeable Cation Channel Activated by Calcium Store Depletion. Neuron 1996, 16, 1189–1196. [Google Scholar] [CrossRef] [Green Version]
- Schaefer, M.; Plant, T.D.; Obukhov, A.G.; Hofmann, T.; Gudermann, T.; Schultz, G. Receptor-mediated Regulation of the Nonselective Cation Channels TRPC4 and TRPC5. J. Biol. Chem. 2000, 275, 17517–17526. [Google Scholar] [CrossRef] [Green Version]
- Tolstykh, G.P.; Cantu, J.C.; Tarango, M.; Ibey, B.L. Receptor- and store-operated mechanisms of calcium entry during the nanosecond electric pulse-induced cellular response. Biochim. Biophys. Acta-Biomembr. 2019, 1861, 685–696. [Google Scholar] [CrossRef]
- Pulcinelli, F.M.; Trifirò, E.; Massimi, I.; Di Renzo, L. A functional interaction between TRPC/NCKX induced by DAG plays a role in determining calcium influx independently from PKC activation. Platelets 2013, 24, 554–559. [Google Scholar] [CrossRef]
- Hayashi, T.; Mogami, H.; Murakami, Y.; Nakamura, T.; Kanayama, N.; Konno, H.; Urano, T. Real-time analysis of platelet aggregation and procoagulant activity during thrombus formation in vivo. Pflügers Arch.-Eur. J. Physiol. 2008, 456, 1239–1251. [Google Scholar] [CrossRef] [Green Version]
- Sveshnikova, A.N.; Ataullakhanov, F.I.; Panteleev, A.M. Compartmentalized calcium signaling triggers subpopulation formation upon platelet activation through PAR1. Mol. BioSyst. 2015, 11, 1052–1060. [Google Scholar] [CrossRef]
- Borin, M.; Siffert, W. Stimulation by thrombin increases the cytosolic free Na+ concentration in human platelets. Studies with the novel fluorescent cytosolic Na+ indicator sodium-binding benzofuran isophthalate. J. Biol. Chem. 1990, 265, 19543–19550. [Google Scholar] [CrossRef]
- Sage, S.O.; Rink, T.J.; Mahaut-Smith, M.P. Resting and ADP-evoked changes in cytosolic free sodium concentration in human platelets loaded with the indicator SBFI. J. Physiol. 1991, 441, 559–573. [Google Scholar] [CrossRef] [Green Version]
- Borin, M.; Siffert, W. Further characterization of the mechanisms mediating the rise in cytosolic free Na+ in thrombin-stimulated platelets. Evidence for inhibition of the Na+, K(+)-ATPase and for Na+ entry via a Ca2+ influx pathway. J. Biol. Chem. 1991, 266, 13153–13160. [Google Scholar] [CrossRef]
- Cerecedo, D.; Martinez-Vieyra, I.; Alonso-Rangel, L.; Benitez-Cardoza, C.; Ortega, A. Epithelial sodium channel modulates platelet collagen activation. Eur. J. Cell Biol. 2014, 93, 127–136. [Google Scholar] [CrossRef]
- Yamaguchi, A.; Azuma, H.; Sekizaki, S.; Suzuki, H.; Tanoue, K.; Yamazaki, H. Thrombin-induced membrane depolarization of platelets and its inhibition by cetiedil. J. Biochem. 1988, 103, 787–791. [Google Scholar]
- Roberts, D.E.; McNicol, A.; Bose, R. Mechanism of collagen activation in human platelets. J. Biol. Chem. 2004, 279, 19421–19430. [Google Scholar] [CrossRef] [Green Version]
- Colucci, G.; Stutz, M.; Rochat, S.; Conte, T.; Pavicic, M.; Reusser, M.; Giabbani, E.; Huynh, A.; Thurlemann, C.; Keller, P.; et al. The effect of desmopressin on platelet function: A selective enhancement of procoagulant COAT platelets in patients with primary platelet function defects. Blood 2014, 123, 1905–1916. [Google Scholar] [CrossRef] [Green Version]
- Tomasiak, M.; Stelmach, H.; Rusak, T.; Ciborowski, M.; Radziwon, P. The involvement of Na+/K(+)-ATPase in the development of platelet procoagulant response. Acta. Biochim. Pol. 2007, 54, 625–639. [Google Scholar] [CrossRef] [Green Version]
- Roberts, D.E.; Matsuda, T.; Bose, R. Molecular and functional characterization of the human platelet Na+/Ca2+ exchangers. Br. J. Pharmacol. 2012, 165, 922–936. [Google Scholar] [CrossRef] [Green Version]
- Tomasiak, M.; Ciborowski, M.; Stelmach, H. The role of Na+/H+ exchanger in serotonin secretion from porcine blood platelets. Acta Biochim. Pol. 2005, 52, 811–822. [Google Scholar] [CrossRef]
- Tomasiak, M.M.; Stelmach, H.; Bodzenta-Lukaszyk, A.; Tomasiak, M. Involvement of Na+/H+ exchanger in desmopressin-induced platelet procoagulant response. Acta Biochim. Pol. 2004, 51, 773–788. [Google Scholar] [CrossRef] [Green Version]
- Evans, R.J.; Lewis, C.; Virginio, C.; Lundstrom, K.; Buell, G.; Surprenant, A.; North, R.A. Ionic permeability of, and divalent cation effects on, two ATP-gated cation channels (P2X receptors) expressed in mammalian cells. J. Physiol. 1996, 497, 413–422. [Google Scholar] [CrossRef] [Green Version]
- Morrell, C.N.; Sun, H.; Ikeda, M.; Beique, J.C.; Swaim, A.M.; Mason, E.; Martin, T.V.; Thompson, L.E.; Gozen, O.; Ampagoomian, D.; et al. Glutamate mediates platelet activation through the AMPA receptor. J. Exp. Med. 2008, 205, 575–584. [Google Scholar] [CrossRef] [Green Version]
- Verkhratsky, A.; Trebak, M.; Perocchi, F.; Khananshvili, D.; Sekler, I. Crosslink between calcium and sodium signalling. Exp. Physiol. 2018, 103, 157–169. [Google Scholar] [CrossRef]
- Samson, J.; Stelmach, H.; Tomasiak, M. The importance of Na+/H+ exchanger for the generation of procoagulant activity by porcine blood platelets. Platelets 2001, 12, 436–442. [Google Scholar] [CrossRef]
- Wright, J.R.; Amisten, S.; Goodall, A.H.; Mahaut-Smith, M.P. Transcriptomic analysis of the ion channelome of human platelets and megakaryocytic cell lines. Thromb. Haemost. 2016, 116, 272–284. [Google Scholar] [CrossRef] [Green Version]
- Qiu, M.R.; Jiang, L.; Matthaei, K.I.; Schoenwaelder, S.M.; Kuffner, T.; Mangin, P.; Joseph, J.E.; Low, J.; Connor, D.; Valenzuela, S.M.; et al. Generation and characterization of mice with null mutation of the chloride intracellular channel 1 gene. Genesis 2010, 48, 127–136. [Google Scholar] [CrossRef]
- Taylor, K.A.; Wilson, D.G.S.; Harper, M.T.; Pugh, N. Extracellular chloride is required for efficient platelet aggregation. Platelets 2018, 29, 79–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harper, M.T.; Poole, A.W. Chloride channels are necessary for full platelet phosphatidylserine exposure and procoagulant activity. Cell Death Dis. 2013, 4, e969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, T.; Iehara, T.; Sato, K.; Fujii, T.; Sakai, H.; Okada, Y. TMEM16F is a component of a Ca2+-activated Cl- channel but not a volume-sensitive outwardly rectifying Cl- channel. Am. J. Physiol. Cell Physiol. 2013, 304, C748–C759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zieve, P.D.; Gamble, J.L., Jr.; Jackson, D.P. Effects of Thrombin on the Potassium and Atp Content of Platelets. J. Clin. Investig. 1964, 43, 2063–2069. [Google Scholar] [CrossRef] [PubMed]
- Wiley, J.S.; Kuchibhotla, J.; Shaller, C.C.; Colman, R.W. Potassium uptake and release by human blood platelets. Blood 1976, 48, 185–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCloskey, C.; Jones, S.; Amisten, S.; Snowden, R.T.; Kaczmarek, L.K.; Erlinge, D.; Goodall, A.H.; Forsythe, I.D.; Mahaut-Smith, M.P. Kv1.3 is the exclusive voltage-gated K+ channel of platelets and megakaryocytes: Roles in membrane potential, Ca2+ signalling and platelet count. J. Physiol. 2010, 588, 1399–1406. [Google Scholar] [CrossRef] [Green Version]
- Kerbiriou-Nabias, D.; Arachiche, A.; Dachary-Prigent, J. Phosphatidylserine exposure and calcium-activated potassium efflux in platelets. Br. J. Haematol. 2011, 155, 268–270. [Google Scholar] [CrossRef]
- Wolfs, J.L.; Wielders, S.J.; Comfurius, P.; Lindhout, T.; Giddings, J.C.; Zwaal, R.F.; Bevers, E.M. Reversible inhibition of the platelet procoagulant response through manipulation of the Gardos channel. Blood 2006, 108, 2223–2228. [Google Scholar] [CrossRef]
- Vergara, C.; Latorre, R.; Marrion, N.V.; Adelman, J.P. Calcium-activated potassium channels. Curr. Opin. Neurobiol. 1998, 8, 321–329. [Google Scholar] [CrossRef]
- Rapetti-Mauss, R.; Lacoste, C.; Picard, V.; Guitton, C.; Lombard, E.; Loosveld, M.; Nivaggioni, V.; Dasilva, N.; Salgado, D.; Desvignes, J.P.; et al. A mutation in the Gardos channel is associated with hereditary xerocytosis. Blood 2015, 126, 1273–1280. [Google Scholar] [CrossRef] [Green Version]
- Gessner, G.; Schonherr, K.; Soom, M.; Hansel, A.; Asim, M.; Baniahmad, A.; Derst, C.; Hoshi, T.; Heinemann, S.H. BKCa channels activating at resting potential without calcium in LNCaP prostate cancer cells. J. Membr. Biol. 2005, 208, 229–240. [Google Scholar] [CrossRef]
- Mattheij, N.J.; Braun, A.; van Kruchten, R.; Castoldi, E.; Pircher, J.; Baaten, C.C.; Wulling, M.; Kuijpers, M.J.; Kohler, R.; Poole, A.W.; et al. Survival protein anoctamin-6 controls multiple platelet responses including phospholipid scrambling, swelling, and protein cleavage. FASEB J. 2016, 30, 727–737. [Google Scholar] [CrossRef]
- Jobe, S.M.; Leo, L.; Eastvold, J.S.; Dickneite, G.; Ratliff, T.L.; Lentz, S.R.; Di Paola, J. Role of FcRgamma and factor XIIIA in coated platelet formation. Blood 2005, 106, 4146–4151. [Google Scholar] [CrossRef] [Green Version]
- Dutting, S.; Bender, M.; Nieswandt, B. Platelet GPVI: A target for antithrombotic therapy?! Trends Pharmacol. Sci. 2012, 33, 583–590. [Google Scholar] [CrossRef]
- Boulaftali, Y.; Noé, B.H.T.; Jandrot-Perrus, M.; Mangin, P.H. Gpvi. In Platelets in Thrombotic and Non-Thrombotic Disorders; Springer: Cham, Switzerland, 2017; pp. 113–127. [Google Scholar]
- Rayes, J.; Watson, S.P.; Nieswandt, B. Functional significance of the platelet immune receptors GPVI and CLEC-2. J. Clin. Investig. 2019, 129, 12–23. [Google Scholar] [CrossRef] [Green Version]
- Watson, S.P.; Auger, J.M.; McCarty, O.J.; Pearce, A.C. GPVI and integrin alphaIIb beta3 signaling in platelets. J. Thromb. Haemost. 2005, 3, 1752–1762. [Google Scholar] [CrossRef]
- Li, Z.; Delaney, M.K.; O’Brien, K.A.; Du, X. Signaling during platelet adhesion and activation. Arter. Thromb. Vasc. Biol. 2010, 30, 2341–2349. [Google Scholar] [CrossRef] [Green Version]
- Heemskerk, J.W.; Vuist, W.M.; Feijge, M.A.; Reutelingsperger, C.P.; Lindhout, T. Collagen but not fibrinogen surfaces induce bleb formation, exposure of phosphatidylserine, and procoagulant activity of adherent platelets: Evidence for regulation by protein tyrosine kinase-dependent Ca2+ responses. Blood 1997, 90, 2615–2625. [Google Scholar] [CrossRef]
- Thiagarajan, P.; Tait, J.F. Binding of annexin V/placental anticoagulant protein I to platelets. Evidence for phosphatidylserine exposure in the procoagulant response of activated platelets. J. Biol. Chem. 1990, 265, 17420–17423. [Google Scholar] [CrossRef]
- Topalov, N.N.; Kotova, Y.N.; Vasil’ev, S.A.; Panteleev, M.A. Identification of signal transduction pathways involved in the formation of platelet subpopulations upon activation. Br. J. Haematol. 2012, 157, 105–115. [Google Scholar] [CrossRef]
- Yau, J.W.; Teoh, H.; Verma, S. Endothelial cell control of thrombosis. BMC Cardiovasc. Disord. 2015, 15, 130. [Google Scholar] [CrossRef] [Green Version]
- De Candia, E. Mechanisms of platelet activation by thrombin: A short history. Thromb. Res. 2012, 129, 250–256. [Google Scholar] [CrossRef]
- Stalker, T.J.; Newman, D.K.; Ma, P.; Wannemacher, K.M.; Brass, L.F. Platelet signaling. Handb. Exp. Pharmacol. 2012, 210, 59–85. [Google Scholar]
- Andersen, H.; Greenberg, D.L.; Fujikawa, K.; Xu, W.; Chung, D.W.; Davie, E.W. Protease-activated receptor 1 is the primary mediator of thrombin-stimulated platelet procoagulant activity. Proc. Natl. Acad. Sci. USA 1999, 96, 11189–11193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, X.; Nieman, M.T. PAR4 (Protease-Activated Receptor 4). Arterioscler. Thromb. Vasc. Biol. 2018, 38, 287–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dormann, D.; Clemetson, K.J.; Kehrel, B.E. The GPIb thrombin-binding site is essential for thrombin-induced platelet procoagulant activity. Blood 2000, 96, 2469–2478. [Google Scholar] [CrossRef]
- Yin, H.; Liu, J.; Li, Z.; Berndt, M.C.; Lowell, C.A.; Du, X. Src family tyrosine kinase Lyn mediates VWF/GPIb-IX-induced platelet activation via the cGMP signaling pathway. Blood 2008, 112, 1139–1146. [Google Scholar] [CrossRef] [Green Version]
- Bryckaert, M.; Rosa, J.-P.; Denis, C.V.; Lenting, P.J. Of von Willebrand factor and platelets. Cell. Mol. Life Sci. 2015, 72, 307–326. [Google Scholar] [CrossRef] [Green Version]
- Adam, F.; Guillin, M.-C.; Jandrot-Perrus, M. Glycoprotein Ib-mediated platelet activation. Eur. J. Biochem. 2003, 270, 2959–2970. [Google Scholar] [CrossRef]
- Shapiro, M.J.; Weiss, E.J.; Faruqi, T.R.; Coughlin, S.R. Protease-activated Receptors 1 and 4 Are Shut Off with Distinct Kinetics after Activation by Thrombin. J. Biol. Chem. 2000, 275, 25216–25221. [Google Scholar] [CrossRef] [Green Version]
- Ravanat, C.; Strassel, C.; Hechler, B.; Schuhler, S.; Chicanne, G.; Payrastre, B.; Gachet, C.; Lanza, F. A central role of GPIb-IX in the procoagulant function of platelets that is independent of the 45-kDa GPIbalpha N-terminal extracellular domain. Blood 2010, 116, 1157–1164. [Google Scholar] [CrossRef] [Green Version]
- Weiss, H.J. Impaired platelet procoagulant mechanisms in patients with bleeding disorders. Semin. Thromb. Hemost. 2009, 35, 233–241. [Google Scholar] [CrossRef]
- Rand, M.L.; Wang, H.; Bang, K.W.A.; Teitel, J.M.; Blanchette, V.S.; Freedman, J.; Nurden, A.T. Phosphatidylserine exposure and other apoptotic-like events in Bernard-Soulier syndrome platelets. Am. J. Hematol. 2010, 85, 584–592. [Google Scholar] [CrossRef]
- Liu, J.; Fitzgerald, M.E.; Berndt, M.C.; Jackson, C.W.; Gartner, T.K. Bruton tyrosine kinase is essential for botrocetin/VWF-induced signaling and GPIb-dependent thrombus formation in vivo. Blood 2006, 108, 2596–2603. [Google Scholar] [CrossRef]
- Andrews, R.K.; Berndt, M.C. Chapter 10-The GPIb-IX-V Complex. In Platelets, 3rd ed.; Michelson, A.D., Ed.; Academic Press: Cambridge, MA, USA, 2013; pp. 195–213. [Google Scholar]
- Van der Meijden, P.E.; Feijge, M.A.; Giesen, P.L.; Huijberts, M.; van Raak, L.P.; Heemskerk, J.W. Platelet P2Y12 receptors enhance signalling towards procoagulant activity and thrombin generation. A study with healthy subjects and patients at thrombotic risk. Thromb. Haemost. 2005, 93, 1128–1136. [Google Scholar] [CrossRef]
- Mahaut-Smith, M.P.; Tolhurst, G.; Evans, R.J. Emerging roles for P2X1receptors in platelet activation. Platelets 2004, 15, 131–144. [Google Scholar] [CrossRef]
- Norgard, N.B.; Hann, C.L.; Dale, G.L. Cangrelor attenuates coated-platelet formation. Clin. Appl. Thromb. Hemost. 2009, 15, 177–182. [Google Scholar] [CrossRef] [Green Version]
- Norgard, N.B.; Saya, S.; Hann, C.L.; Hennebry, T.A.; Schechter, E.; Dale, G.L. Clopidogrel attenuates coated-platelet production in patients undergoing elective coronary catheterization. J. Cardiovasc. Pharmacol. 2008, 52, 536–539. [Google Scholar] [CrossRef]
- Kotova, Y.N.; Ataullakhanov, F.I.; Panteleev, M.A. Formation of coated platelets is regulated by the dense granule secretion of adenosine 5′diphosphate acting via the P2Y12 receptor. J. Thromb. Haemost. 2008, 6, 1603–1605. [Google Scholar] [CrossRef]
- Dorsam, R.T.; Tuluc, M.; Kunapuli, S.P. Role of protease-activated and ADP receptor subtypes in thrombin generation on human platelets. J. Thromb. Haemost. 2004, 2, 804–812. [Google Scholar] [CrossRef]
- Keularts, I.M.L.W.; Van Gorp, R.M.A.; Feijge, M.A.H.; Vuist, W.M.J.; Heemskerk, J.W.M. α2A-Adrenergic Receptor Stimulation Potentiates Calcium Release in Platelets by Modulating cAMP Levels. J. Biol. Chem. 2000, 275, 1763–1772. [Google Scholar] [CrossRef] [Green Version]
- Gąsecka, A.; Rogula, S.; Eyileten, C.; Postuła, M.; Jaguszewski, M.J.; Kochman, J.; Mazurek, T.; Nieuwland, R.; Filipiak, K.J. Role of P2Y Receptors in Platelet Extracellular Vesicle Release. Int. J. Mol. Sci. 2020, 21, 6065. [Google Scholar] [CrossRef]
- Mahaut-Smith, M.P.; Jones, S.; Evans, R.J. The P2X1 receptor and platelet function. Purinergic Signal. 2011, 7, 341–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burke, J.E.; Dennis, E.A. Phospholipase A2 structure/function, mechanism, and signaling. J. Lipid Res. 2009, 50, S237–S242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knezevic, I.; Borg, C.; Le Breton, G.C. Identification of Gq as one of the G-proteins which copurify with human platelet thromboxane A2/prostaglandin H2 receptors. J. Biol. Chem. 1993, 268, 26011–26017. [Google Scholar] [CrossRef]
- Djellas, Y.; Manganello, J.M.; Antonakis, K.; Le Breton, G.C. Identification of Gα13 as One of the G-proteins That Couple to Human Platelet Thromboxane A2 Receptors. J. Biol. Chem. 1999, 274, 14325–14330. [Google Scholar] [CrossRef] [Green Version]
- Prodan, C.I.; Joseph, P.M.; Vincent, A.S.; Dale, G.L. Coated-platelet levels are influenced by smoking, aspirin, and selective serotonin reuptake inhibitors. J. Thromb. Haemost. 2007, 5, 2149–2151. [Google Scholar] [CrossRef]
- Heemskerk, J.W.; Harper, M.T.; Cosemans, J.M.; Poole, A.W. Unravelling the different functions of protein kinase C isoforms in platelets. FEBS Lett. 2011, 585, 1711–1716. [Google Scholar] [CrossRef] [Green Version]
- Gilio, K.; Harper, M.T.; Cosemans, J.M.; Konopatskaya, O.; Munnix, I.C.; Prinzen, L.; Leitges, M.; Liu, Q.; Molkentin, J.D.; Heemskerk, J.W.; et al. Functional divergence of platelet protein kinase C (PKC) isoforms in thrombus formation on collagen. J. Biol. Chem. 2010, 285, 23410–23419. [Google Scholar] [CrossRef] [Green Version]
- Harper, M.T.; Molkentin, J.D.; Poole, A.W. Protein kinase C alpha enhances sodium-calcium exchange during store-operated calcium entry in mouse platelets. Cell Calcium. 2010, 48, 333–340. [Google Scholar] [CrossRef]
- Okat, Z. The molecular functions of protein kinase C (PKC) isoforms. Int. Phys. Med. Rehabil. J. 2018, 3, 1. [Google Scholar] [CrossRef] [Green Version]
- Lever, R.A.; Hussain, A.; Sun, B.B.; Sage, S.O.; Harper, A.G. Conventional protein kinase C isoforms differentially regulate ADP- and thrombin-evoked Ca2+ signalling in human platelets. Cell Calcium 2015, 58, 577–588. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, A.; Roy, S.; Saha, B.; Mukherjee, D. Spatio-Temporal Regulation of PKC Isoforms Imparts Signaling Specificity. Front. Immunol. 2016, 7, 45. [Google Scholar] [CrossRef] [Green Version]
- Zang, Q.; Lu, Z.; Curto, M.; Barile, N.; Shalloway, D.; Foster, D.A. Association between v-Src and Protein Kinase C δ in v-Src-transformed Fibroblasts. J. Biol. Chem. 1997, 272, 13275–13280. [Google Scholar] [CrossRef] [Green Version]
- Pula, G.; Crosby, D.; Baker, J.; Poole, A.W. Functional interaction of protein kinase Calpha with the tyrosine kinases Syk and Src in human platelets. J. Biol. Chem. 2005, 280, 7194–7205. [Google Scholar] [CrossRef] [Green Version]
- Makhoul, S.; Dorschel, S.; Gambaryan, S.; Walter, U.; Jurk, K. Feedback Regulation of Syk by Protein Kinase C in Human Platelets. Int. J. Mol. Sci. 2019, 21, 176. [Google Scholar] [CrossRef] [Green Version]
- Hall, K.J.; Harper, M.T.; Gilio, K.; Cosemans, J.M.; Heemskerk, J.W.; Poole, A.W. Genetic analysis of the role of protein kinase Ctheta in platelet function and thrombus formation. PLoS ONE 2008, 3, e3277. [Google Scholar] [CrossRef] [Green Version]
- Harper, M.T.; Poole, A.W. Protein kinase Ctheta negatively regulates store-independent Ca2+ entry and phosphatidylserine exposure downstream of glycoprotein VI in platelets. J. Biol. Chem. 2010, 285, 19865–19873. [Google Scholar] [CrossRef] [Green Version]
- Soriani, A.; Moran, B.; De Virgilio, M.; Kawakami, T.; Altman, A.; Lowell, C.; Eto, K.; Shattil, S.J. A role for PKCtheta in outside-in alphaIIbbeta3 signaling. J. Thromb. Haemost. 2006, 4, 648–655. [Google Scholar] [CrossRef]
- Sun, L.; Mao, G.; Rao, A.K. Association of CBFA2 mutation with decreased platelet PKC-theta and impaired receptor-mediated activation of GPIIb-IIIa and pleckstrin phosphorylation: Proteins regulated by CBFA2 play a role in GPIIb-IIIa activation. Blood 2004, 103, 948–954. [Google Scholar] [CrossRef] [Green Version]
- Cohen, S.; Braiman, A.; Shubinsky, G.; Ohayon, A.; Altman, A.; Isakov, N. PKCtheta is required for hemostasis and positive regulation of thrombin-induced platelet aggregation and alpha-granule secretion. Biochem. Biophys Res. Commun. 2009, 385, 22–27. [Google Scholar] [CrossRef]
- Cohen, S.; Braiman, A.; Shubinsky, G.; Isakov, N. Protein kinase C-theta in platelet activation. FEBS Lett. 2011, 585, 3208–3215. [Google Scholar] [CrossRef] [Green Version]
- Patel, P.; Naik, U.P. Platelet MAPKs—A 20+ year history: What do we really know? J. Thromb. Haemost. 2020, 18, 2087–2102. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Nebreda, A.R. Mechanisms and functions of p38 MAPK signalling. Biochem. J. 2010, 429, 403–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rukoyatkina, N.; Mindukshev, I.; Walter, U.; Gambaryan, S. Dual role of the p38 MAPK/cPLA2 pathway in the regulation of platelet apoptosis induced by ABT-737 and strong platelet agonists. Cell Death Dis. 2013, 4, e931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giladi, M.; Tal, I.; Khananshvili, D. Structural Features of Ion Transport and Allosteric Regulation in Sodium-Calcium Exchanger (NCX) Proteins. Front. Physiol. 2016, 7, 30. [Google Scholar] [CrossRef] [Green Version]
- Mahaut-Smith, M.P. A role for platelet TRPC channels in the Ca2+ response that induces procoagulant activity. Sci. Signal. 2013, 6, pe23. [Google Scholar] [CrossRef]
- Harper, A.G.; Sage, S.O. A key role for reverse Na+/Ca2+ exchange influenced by the actin cytoskeleton in store-operated Ca2+ entry in human platelets: Evidence against the de novo conformational coupling hypothesis. Cell Calcium 2007, 42, 606–617. [Google Scholar] [CrossRef]
- De Marchi, E.; Bonora, M.; Giorgi, C.; Pinton, P. The mitochondrial permeability transition pore is a dispensable element for mitochondrial calcium efflux. Cell Calcium 2014, 56, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Remenyi, G.; Szasz, R.; Friese, P.; Dale, G.L. Role of mitochondrial permeability transition pore in coated-platelet formation. Arter. Thromb. Vasc. Biol. 2005, 25, 467–471. [Google Scholar] [CrossRef] [Green Version]
- Choo, H.J.; Saafir, T.B.; Mkumba, L.; Wagner, M.B.; Jobe, S.M. Mitochondrial calcium and reactive oxygen species regulate agonist-initiated platelet phosphatidylserine exposure. Arter. Thromb. Vasc. Biol. 2012, 32, 2946–2955. [Google Scholar] [CrossRef] [Green Version]
- Jobe, S.M.; Wilson, K.M.; Leo, L.; Raimondi, A.; Molkentin, J.D.; Lentz, S.R.; Di Paola, J. Critical role for the mitochondrial permeability transition pore and cyclophilin D in platelet activation and thrombosis. Blood 2008, 111, 1257–1265. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Gamez, G.; Myers, D.R.; Clemmons, W.; Lam, W.A.; Jobe, S.M. Mitochondrially mediated integrin alphaIIbbeta3 protein inactivation limits thrombus growth. J. Biol. Chem. 2013, 288, 30672–30681. [Google Scholar] [CrossRef] [Green Version]
- Reddy, E.C.; Rand, M.L. Procoagulant Phosphatidylserine-Exposing Platelets in vitro and in vivo. Front. Cardiovasc. Med. 2020, 7, 15. [Google Scholar] [CrossRef] [Green Version]
- Kunzelmann, K.; Nilius, B.; Owsianik, G.; Schreiber, R.; Ousingsawat, J.; Sirianant, L.; Wanitchakool, P.; Bevers, E.M.; Heemskerk, J.W.M. Molecular functions of anoctamin 6 (TMEM16F): A chloride channel, cation channel, or phospholipid scramblase? Pflügers Arch.-Eur. J. Physiol. 2014, 466, 407–414. [Google Scholar] [CrossRef]
- Hynes, R.O. Integrins. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef] [Green Version]
- Durrant, T.N.; Van Den Bosch, M.T.; Hers, I. Integrin αIIbβ3 outside-in signaling. Blood 2017, 130, 1607–1619. [Google Scholar] [CrossRef] [Green Version]
- Rosado, J.A.; Meijer, E.M.Y.; Hamulyak, K.; Novakova, I.; Heemskerk, J.W.M.; Sage, S.O. Fibrinogen binding to the integrin αIIbβ3 modulates store-mediated calcium entry in human platelets. Blood 2001, 97, 2648–2656. [Google Scholar] [CrossRef]
- Morgenstern, E. Human Platelet Morphology/Ultrastructure. In Platelets and Their Factors; von Bruchhausen, F., Walter, U., Eds.; Springer: Berlin/Heidelberg, Germany, 1997; pp. 27–60. [Google Scholar]
- Penington, D.G.; Streatfield, K.; Roxburgh, A.E. Megakaryocytes and the Heterogeneity of Circulating Platelets. Br. J. Haematol. 1976, 34, 639–653. [Google Scholar] [CrossRef]
- Rand, M.; Greenberg, J.; Packham, M.; Mustard, J. Density subpopulations of rabbit platelets: Size, protein, and sialic acid content, and specific radioactivity changes following labeling with 35S-sulfate in vivo. Blood 1981, 57, 741–746. [Google Scholar] [CrossRef] [Green Version]
- Opper, C.; Fett, C.; Capito, B.; Raha, S.; Wesemann, W. Plasma membrane properties in heterogeneous human blood platelet subfractions modulate the cellular response at the second messenger level. Thromb. Res. 1993, 72, 39–47. [Google Scholar] [CrossRef]
- Opper, C.; Schrumpf, E.; Gear, A.R.L.; Wesemann, W. Involvement of guanylate cyclase and phosphodiesterases in the functional heterogeneity of human blood platelet subpopulations. Thromb. Res. 1995, 80, 461–470. [Google Scholar] [CrossRef]
- Handtke, S.; Wesche, J.; Palankar, R.; Greinacher, A.; Thiele, T. Function of Large and Small Platelets Differs, Depending on Extracellular Calcium Availability and Type of Inductor. Thromb. Haemost. 2020, 120, 1075–1086. [Google Scholar] [CrossRef]
- Handtke, S.; Thiele, T. Large and small platelets—(When) do they differ? J. Thromb. Haemost. 2020, 18, 1256–1267. [Google Scholar] [CrossRef]
- Handtke, S.; Steil, L.; Palankar, R.; Conrad, J.; Cauhan, S.; Kraus, L.; Ferrara, M.; Dhople, V.; Wesche, J.; Völker, U.; et al. Role of Platelet Size Revisited—Function and Protein Composition of Large and Small Platelets. Thromb. Haemost. 2019, 119, 407–420. [Google Scholar] [CrossRef]
- Thompson, C.; Love, D.; Quinn, P.; Valeri, C. Platelet size does not correlate with platelet age. Blood 1983, 62, 487–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braekkan, S.K.; Mathiesen, E.B.; Njølstad, I.; Wilsgaard, T.; Størmer, J.; Hansen, J.B. Mean platelet volume is a risk factor for venous thromboembolism: The Tromsø study. J. Thromb. Haemost. 2010, 8, 157–162. [Google Scholar] [CrossRef]
- Chu, S.G.; Becker, R.C.; Berger, P.B.; Bhatt, D.L.; Eikelboom, J.W.; Konkle, B.; Mohler, E.R.; Reilly, M.P.; Berger, J.S. Mean platelet volume as a predictor of cardiovascular risk: A systematic review and meta-analysis. J. Thromb. Haemost. 2010, 8, 148–156. [Google Scholar] [CrossRef]
- Opper, C.; Schuessler, G.; Kuschel, M.; Clement, H.-W.; Gear, A.R.L.; Hinsch, E.; Hinsch, K.; Wesemann, W. Analysis of GTP-binding proteins, phosphoproteins, and cytosolic calcium in functional heterogeneous human blood platelet subpopulations. Biochem. Pharmacol. 1997, 54, 1027–1035. [Google Scholar] [CrossRef]
- Lesyk, G.; Jurasz, P. Advances in Platelet Subpopulation Research. Front. Cardiovasc. Med. 2019, 6, 138. [Google Scholar] [CrossRef] [Green Version]
- Eckly, A.; Rinckel, J.-Y.; Proamer, F.; Ulas, N.; Joshi, S.; Whiteheart, S.W.; Gachet, C. Respective contributions of single and compound granule fusion to secretion by activated platelets. Blood 2016, 128, 2538–2549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonnalagadda, D.; Izu, L.T.; Whiteheart, S.W. Platelet secretion is kinetically heterogeneous in an agonist-responsive manner. Blood 2012, 120, 5209–5216. [Google Scholar] [CrossRef] [PubMed]
- Obydennyi, S.I.; Artemenko, E.O.; Sveshnikova, A.N.; Ignatova, A.A.; Varlamova, T.V.; Gambaryan, S.; Lomakina, G.Y.; Ugarova, N.N.; Kireev, I.I.; Ataullakhanov, F.I.; et al. Mechanisms of increased mitochondria-dependent necrosis in Wiskott-Aldrich syndrome platelets. Haematologica 2020, 105, 1095–1106. [Google Scholar] [CrossRef]
- Nechipurenko, D.Y.; Shibeko, A.M.; Sveshnikova, A.N.; Panteleev, M.A. In Silico Hemostasis Modeling and Prediction. Hämostaseologie 2020, 40, 524–535. [Google Scholar] [CrossRef]
- Ramström, S.; Öberg, K.V.; Åkerström, F.; Enström, C.; Lindahl, T.L. Platelet PAR1 receptor density—Correlation to platelet activation response and changes in exposure after platelet activation. Thromb. Res. 2008, 121, 681–688. [Google Scholar] [CrossRef] [Green Version]
- Furihata, K.; Clemetson, K.J.; Deguchi, H.; Kunicki, T.J. Variation in Human Platelet Glycoprotein VI Content Modulates Glycoprotein VI–Specific Prothrombinase Activity. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1857–1863. [Google Scholar] [CrossRef] [Green Version]
- Tohidi-Esfahani, I.; Tan, S.; Tan, C.W.; Johnson, L.; Marks, D.C.; Chen, V.M. Platelet procoagulant potential is reduced in platelet concentrates ex vivo but appears restored following transfusion. Transfusion 2021, 61, 3420–3431. [Google Scholar] [CrossRef]
- Kholmukhamedov, A.; Jobe, S.M. Necrotic but Not Apoptotic Platelets Are Functionally Procoagulant. Blood 2018, 132, 2420. [Google Scholar] [CrossRef]
- Grozovsky, R.; Hoffmeister, K.M.; Falet, H. Novel clearance mechanisms of platelets. Curr. Opin. Hematol. 2010, 17, 585–589. [Google Scholar] [CrossRef] [Green Version]
- Jain, K.; Tyagi, T.; Patell, K.; Xie, Y.; Kadado, A.J.; Lee, S.H.; Yarovinsky, T.; Du, J.; Hwang, J.; Martin, K.A.; et al. Age associated non-linear regulation of redox homeostasis in the anucleate platelet: Implications for CVD risk patients. EBioMedicine 2019, 44, 28–40. [Google Scholar] [CrossRef] [Green Version]
- KrȯTz, F.; Sohn, H.-Y.; Pohl, U. Reactive Oxygen Species. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1988–1996. [Google Scholar]
- Dayal, S.; Wilson, K.M.; Motto, D.G.; Miller, F.J.; Chauhan, A.K.; Lentz, S.R. Hydrogen Peroxide Promotes Aging-Related Platelet Hyperactivation and Thrombosis. Circulation 2013, 127, 1308–1316. [Google Scholar] [CrossRef] [Green Version]
- Masselli, E.; Pozzi, G.; Vaccarezza, M.; Mirandola, P.; Galli, D.; Vitale, M.; Carubbi, C.; Gobbi, G. ROS in Platelet Biology: Functional Aspects and Methodological Insights. Int. J. Mol. Sci. 2020, 21, 4866. [Google Scholar] [CrossRef] [PubMed]
- Andrews, R.K.; Gardiner, E.E. Metalloproteolytic receptor shedding…platelets “acting their age”. Platelets 2016, 27, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Bergmeier, W.; Piffath, C.L.; Cheng, G.; Dole, V.S.; Zhang, Y.; Von Andrian, U.H.; Wagner, D.D. Tumor Necrosis Factor-α–Converting Enzyme (ADAM17) Mediates GPIbα Shedding from Platelets In Vitro and In Vivo. Circ. Res. 2004, 95, 677–683. [Google Scholar] [CrossRef] [Green Version]
- Alberio, L.; Friese, P.; Clemetson, K.J.; Dale, G.L. Collagen response and glycoprotein VI function decline progressively as canine platelets age in vivo. Thromb. Haemost. 2002, 88, 510–516. [Google Scholar] [CrossRef]
- Ng, M.S.Y.; Tung, J.-P.; Fraser, J.F. Platelet Storage Lesions: What More Do We Know Now? Transfus. Med. Rev. 2018, 32, 144–154. [Google Scholar] [CrossRef] [Green Version]
- Hosseini, E.; Ghasemzadeh, M.; Azizvakili, E.; Beshkar, P. Platelet spreading on fibrinogen matrix, a reliable and sensitive marker of platelet functional activity during storage. J. Thromb. Thrombolysis 2019, 48, 430–438. [Google Scholar] [CrossRef]
- Baaten, C.C.F.M.J.; Swieringa, F.; Misztal, T.; Mastenbroek, T.G.; Feijge, M.A.H.; Bock, P.E.; Donners, M.M.P.C.; Collins, P.W.; Li, R.; van der Meijden, P.E.J.; et al. Platelet heterogeneity in activation-induced glycoprotein shedding: Functional effects. Blood Adv. 2018, 2, 2320–2331. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Shimizu, S.; Watanabe, T.; Yamaguchi, O.; Otsu, K.; Yamagata, H.; Inohara, H.; Kubo, T.; Tsujimoto, Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 2005, 434, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Hurst, S.; Gonnot, F.; Dia, M.; Crola Da Silva, C.; Gomez, L.; Sheu, S.-S. Phosphorylation of cyclophilin D at serine 191 regulates mitochondrial permeability transition pore opening and cell death after ischemia-reperfusion. Cell Death Dis. 2020, 11, 661. [Google Scholar] [CrossRef] [PubMed]
- Topalov, N.N.; Yakimenko, A.O.; Canault, M.; Artemenko, E.O.; Zakharova, N.V.; Abaeva, A.A.; Loosveld, M.; Ataullakhanov, F.I.; Nurden, A.T.; Alessi, M.-C.; et al. Two Types of Procoagulant Platelets Are Formed Upon Physiological Activation and Are Controlled by Integrin α IIb β 3. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2475–2483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandyopadhyay, S.K.; Azharuddin, M.; Dasgupta, A.K.; Ganguli, B.; SenRoy, S.; Patra, H.K.; Deb, S. Probing ADP Induced Aggregation Kinetics During Platelet-Nanoparticle Interactions: Functional Dynamics Analysis to Rationalize Safety and Benefits. Front. Bioeng. Biotechnol. 2019, 7, 163. [Google Scholar] [CrossRef] [Green Version]
- Löf, A.; Müller, J.P.; Brehm, M.A. A biophysical view on von Willebrand factor activation. J. Cell. Physiol. 2018, 233, 799–810. [Google Scholar] [CrossRef]
- Stalker, T.J.; Welsh, J.D.; Tomaiuolo, M.; Wu, J.; Colace, T.V.; Diamond, S.L.; Brass, L.F. A systems approach to hemostasis: 3. Thrombus consolidation regulates intrathrombus solute transport and local thrombin activity. Blood 2014, 124, 1824–1831. [Google Scholar] [CrossRef]
- Tomaiuolo, M.; Stalker, T.J.; Welsh, J.D.; Diamond, S.L.; Sinno, T.; Brass, L.F. A systems approach to hemostasis: 2. Computational analysis of molecular transport in the thrombus microenvironment. Blood 2014, 124, 1816–1823. [Google Scholar] [CrossRef] [Green Version]
- Kholmukhamedov, A.; Jobe, S. Procoagulant Platelets Get Squeezed to Define the Boundaries of the Hemostatic Plug. Arter. Thromb. Vasc. Biol. 2019, 39, 5–6. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Veuthey, L.; Aliotta, A.; Bertaggia Calderara, D.; Pereira Portela, C.; Alberio, L. Mechanisms Underlying Dichotomous Procoagulant COAT Platelet Generation—A Conceptual Review Summarizing Current Knowledge. Int. J. Mol. Sci. 2022, 23, 2536. https://doi.org/10.3390/ijms23052536
Veuthey L, Aliotta A, Bertaggia Calderara D, Pereira Portela C, Alberio L. Mechanisms Underlying Dichotomous Procoagulant COAT Platelet Generation—A Conceptual Review Summarizing Current Knowledge. International Journal of Molecular Sciences. 2022; 23(5):2536. https://doi.org/10.3390/ijms23052536
Chicago/Turabian StyleVeuthey, Lucas, Alessandro Aliotta, Debora Bertaggia Calderara, Cindy Pereira Portela, and Lorenzo Alberio. 2022. "Mechanisms Underlying Dichotomous Procoagulant COAT Platelet Generation—A Conceptual Review Summarizing Current Knowledge" International Journal of Molecular Sciences 23, no. 5: 2536. https://doi.org/10.3390/ijms23052536
APA StyleVeuthey, L., Aliotta, A., Bertaggia Calderara, D., Pereira Portela, C., & Alberio, L. (2022). Mechanisms Underlying Dichotomous Procoagulant COAT Platelet Generation—A Conceptual Review Summarizing Current Knowledge. International Journal of Molecular Sciences, 23(5), 2536. https://doi.org/10.3390/ijms23052536