
A Novel, Apparently Silent Variant in MFSD8 Causes Neuronal Ceroid Lipofuscinosis with Marked Intrafamilial Variability

 ,
,  and
and
Abstract
:1. Introduction
2. Results
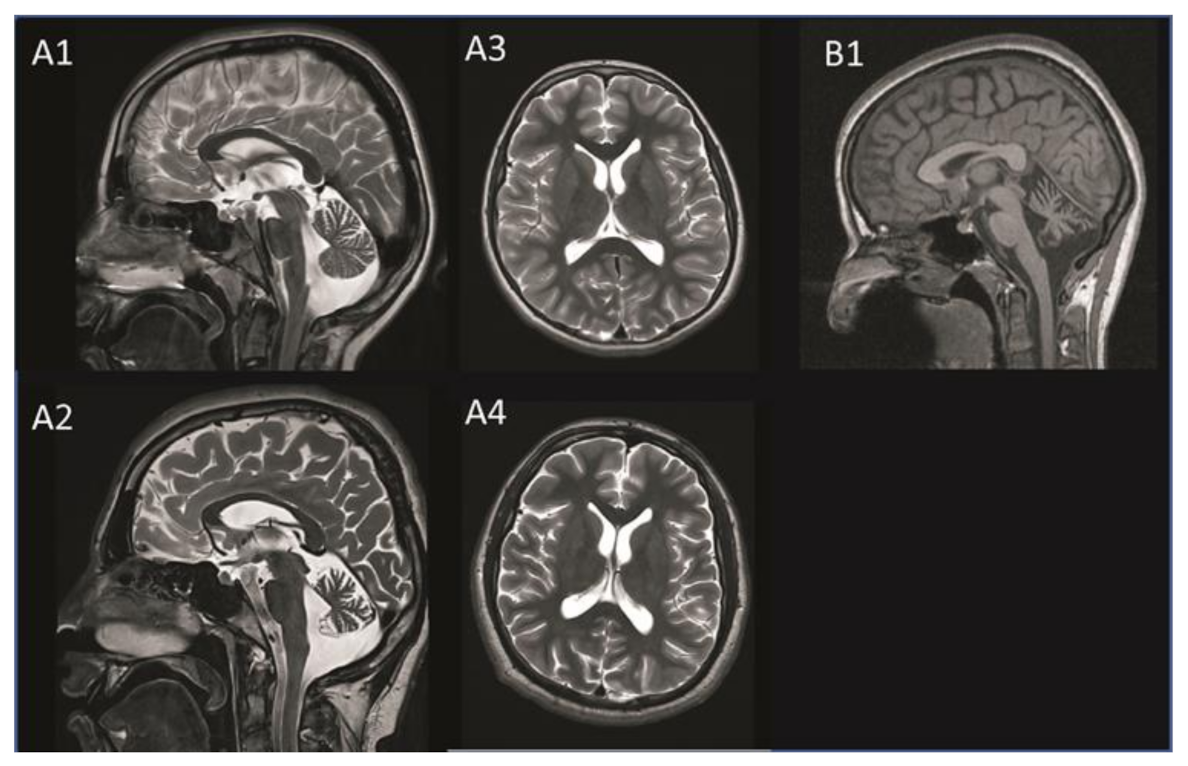
2.1. Clinical Phenotypes
2.1.1. Patient A
2.1.2. Patient B
2.2. Genetic Testing and Bioinformatic Analysis
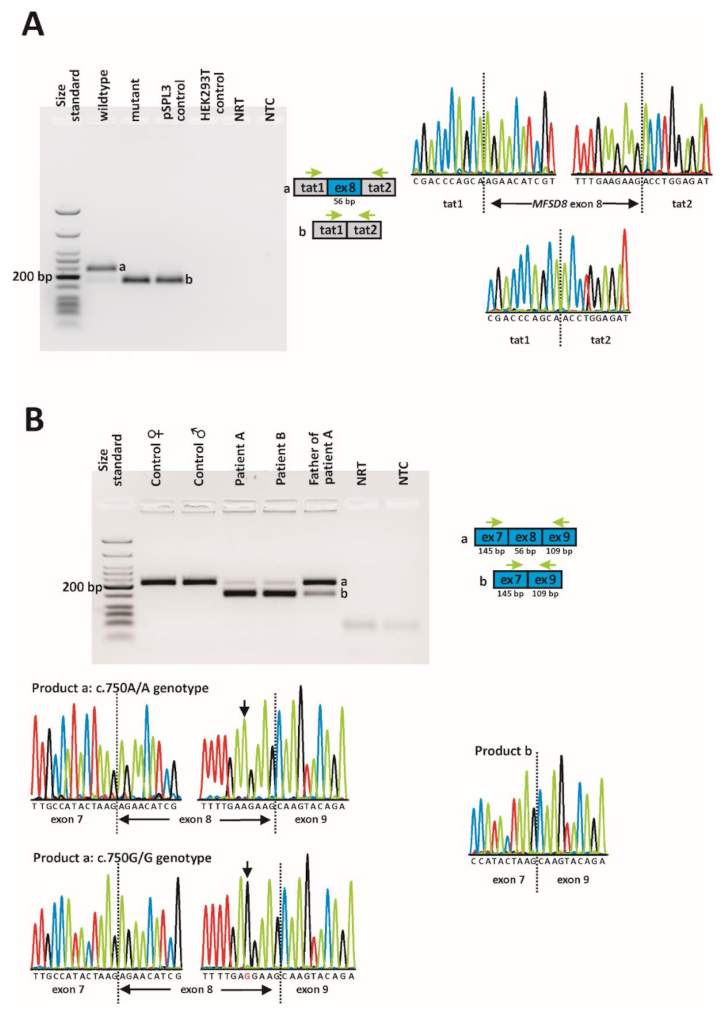
2.3. In Vitro and In Vivo Splicing Assessments
3. Discussion
4. Materials and Methods
4.1. Subjects
4.2. Clinical Assessment
4.3. Genetic Diagnostic Testing
4.4. Bioinformatic Analysis
4.5. Minigene Assays
4.6. Direct mRNA Analysis from Blood Cells
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Anderson, G.W.; Goebel, H.H.; Simonati, A. Human pathology in NCL. Biochim. Biophys. Acta 2013, 1832, 1807–1826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, R.E.; Mole, S.E. New nomenclature and classification scheme for the neuronal ceroid lipofuscinoses. Neurology 2012, 79, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Kousi, M.; Lehesjoki, A.E.; Mole, S.E. Update of the mutation spectrum and clinical correlations of over 360 mutations in eight genes that underlie the neuronal ceroid lipofuscinoses. Hum. Mutat. 2012, 33, 42–63. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.D.; Tarczyluk, M.A.; Nelvagal, H.R. Towards a new understanding of NCL pathogenesis. Biochim. Biophys. Acta 2015, 1852, 2256–2261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, A.B.; Appu, A.P.; Sadhukhan, T.; Casey, S.; Mondal, A.; Zhang, Z.; Bagh, M.B. Emerging new roles of the lysosome and neuronal ceroid lipofuscinoses. Mol. Neurodegener. 2019, 14, 4. [Google Scholar] [CrossRef] [Green Version]
- Nelvagal, H.R.; Lange, J.; Takahashi, K.; Tarczyluk-Wells, M.A.; Cooper, J.D. Pathomechanisms in the neuronal ceroid lipofuscinoses. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165570. [Google Scholar] [CrossRef]
- Palmer, D.N.; Barry, L.A.; Tyynelä, J.; Cooper, J.D. NCL disease mechanisms. Biochim. Biophys. Acta 2013, 1832, 1882–1893. [Google Scholar] [CrossRef]
- Siintola, E.; Topcu, M.; Aula, N.; Lohi, H.; Minassian, B.A.; Paterson, A.D.; Liu, X.Q.; Wilson, C.; Lahtinen, U.; Anttonen, A.K.; et al. The novel neuronal ceroid lipofuscinosis gene MFSD8 encodes a putative lysosomal transporter. Am. J. Hum. Genet. 2007, 81, 136–146. [Google Scholar] [CrossRef] [Green Version]
- Pao, S.S.; Paulsen, I.T.; Saier, M.H., Jr. Major facilitator superfamily. Microbiol. Mol. Biol. Rev. 1998, 62, 1–34. [Google Scholar] [CrossRef] [Green Version]
- Sharifi, A.; Kousi, M.; Sagné, C.; Bellenchi, G.C.; Morel, L.; Darmon, M.; Hulková, H.; Ruivo, R.; Debacker, C.; El Mestikawy, S.; et al. Expression and lysosomal targeting of CLN7, a major facilitator superfamily transporter associated with variant late-infantile neuronal ceroid lipofuscinosis. Hum. Mol. Genet. 2010, 19, 4497–4514. [Google Scholar] [CrossRef] [Green Version]
- Damme, M.; Brandenstein, L.; Fehr, S.; Jankowiak, W.; Bartsch, U.; Schweizer, M.; Hermans-Borgmeyer, I.; Storch, S. Gene disruption of Mfsd8 in mice provides the first animal model for CLN7 disease. Neurobiol. Dis. 2014, 65, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Brandenstein, L.; Schweizer, M.; Sedlacik, J.; Fiehler, J.; Storch, S. Lysosomal dysfunction and impaired autophagy in a novel mouse model deficient for the lysosomal membrane protein Cln7. Hum. Mol. Genet. 2016, 25, 777–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kousi, M.; Siintola, E.; Dvorakova, L.; Vlaskova, H.; Turnbull, J.; Topcu, M.; Yuksel, D.; Gokben, S.; Minassian, B.A.; Elleder, M.; et al. Mutations in CLN7/MFSD8 are a common cause of variant late-infantile neuronal ceroid lipofuscinosis. Brain 2009, 132, 810–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aldahmesh, M.A.; Al-Hassnan, Z.N.; Aldosari, M.; Alkuraya, F.S. Neuronal ceroid lipofuscinosis caused by MFSD8 mutations: A common theme emerging. Neurogenetics 2009, 10, 307–311. [Google Scholar] [CrossRef] [PubMed]
- Mandel, H.; Cohen Katsanelson, K.; Khayat, M.; Chervinsky, I.; Vladovski, E.; Iancu, T.C.; Indelman, M.; Horovitz, Y.; Sprecher, E.; Shalev, S.A.; et al. Clinico-pathological manifestations of variant late infantile neuronal ceroid lipofuscinosis (vLINCL) caused by a novel mutation in MFSD8 gene. Eur. J. Med. Genet. 2014, 57, 607–612. [Google Scholar] [CrossRef] [PubMed]
- Kozina, A.A.; Okuneva, E.G.; Baryshnikova, N.V.; Krasnenko, A.Y.; Tsukanov, K.Y.; Klimchuk, O.I.; Kondakova, O.B.; Larionova, A.N.; Batysheva, T.T.; Surkova, E.I.; et al. A novel MFSD8 mutation in a Russian patient with neuronal ceroid lipofuscinosis type 7: A case report. BMC Med. Genet. 2018, 19, 151. [Google Scholar] [CrossRef]
- Hosseini Bereshneh, A.; Garshasbi, M. Novel in-frame deletion in MFSD8 gene revealed by trio whole exome sequencing in an Iranian affected with neuronal ceroid lipofuscinosis type 7: A case report. J. Med. Case Rep. 2018, 12, 281. [Google Scholar] [CrossRef]
- Roosing, S.; van den Born, L.I.; Sangermano, R.; Banfi, S.; Koenekoop, R.K.; Zonneveld-Vrieling, M.N.; Klaver, C.C.; van Lith-Verhoeven, J.J.; Cremers, F.P.; den Hollander, A.I.; et al. Mutations in MFSD8, encoding a lysosomal membrane protein, are associated with nonsyndromic autosomal recessive macular dystrophy. Ophthalmology 2015, 122, 170–179. [Google Scholar] [CrossRef]
- Khan, K.N.; El-Asrag, M.E.; Ku, C.A.; Holder, G.E.; McKibbin, M.; Arno, G.; Poulter, J.A.; Carss, K.; Bommireddy, T.; Bagheri, S.; et al. Specific Alleles of CLN7/MFSD8, a Protein That Localizes to Photoreceptor Synaptic Terminals, Cause a Spectrum of Nonsyndromic Retinal Dystrophy. Investig. Ophthalmol. Vis. Sci. 2017, 58, 2906–2914. [Google Scholar] [CrossRef] [Green Version]
- Stone, E.M.; Andorf, J.L.; Whitmore, S.S.; DeLuca, A.P.; Giacalone, J.C.; Streb, L.M.; Braun, T.A.; Mullins, R.F.; Scheetz, T.E.; Sheffield, V.C.; et al. Clinically Focused Molecular Investigation of 1000 Consecutive Families with Inherited Retinal Disease. Ophthalmology 2017, 124, 1314–1331. [Google Scholar] [CrossRef]
- Birtel, J.; Gliem, M.; Mangold, E.; Müller, P.L.; Holz, F.G.; Neuhaus, C.; Lenzner, S.; Zahnleiter, D.; Betz, C.; Eisenberger, T.; et al. Next-generation sequencing identifies unexpected genotype-phenotype correlations in patients with retinitis pigmentosa. PLoS ONE 2018, 13, e0207958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zare-Abdollahi, D.; Bushehri, A.; Alavi, A.; Dehghani, A.; Mousavi-Mirkala, M.; Effati, J.; Miratashi, S.A.M.; Dehani, M.; Jamali, P.; Khorram Khorshid, H.R. MFSD8 gene mutations; evidence for phenotypic heterogeneity. Ophthalmic Genet. 2019, 40, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Q.; Cao, Y.; Xu, H.; Yang, Z.; Tang, L.; Xiang, J.; Li, J.; Deng, H.; Yuan, L. Novel MFSD8 Variants in a Chinese Family with Nonsyndromic Macular Dystrophy. J. Ophthalmol. 2021, 2021, 6684045. [Google Scholar] [CrossRef] [PubMed]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Evans, K.; Hayden, M.; Heywood, S.; Hussain, M.; Phillips, A.D.; Cooper, D.N. The Human Gene Mutation Database: Towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum. Genet. 2017, 136, 665–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cartegni, L.; Chew, S.L.; Krainer, A.R. Listening to silence and understanding nonsense: Exonic mutations that affect splicing. Nat. Rev. Genet. 2002, 3, 285–298. [Google Scholar] [CrossRef]
- Sherry, S.T.; Ward, M.H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [Green Version]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- 1000 Genomes Project Consortium; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Sharma, Y.; Miladi, M.; Dukare, S.; Boulay, K.; Caudron-Herger, M.; Groß, M.; Backofen, R.; Diederichs, S. A pan-cancer analysis of synonymous mutations. Nat. Commun. 2019, 10, 2569. [Google Scholar] [CrossRef] [Green Version]
- Jaganathan, K.; Panagiotopoulou, S.K.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019, 176, 535–548.e24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blencowe, B.J. Exonic splicing enhancers: Mechanism of action, diversity and role in human genetic diseases. Trends Biochem. Sci. 2000, 25, 106–110. [Google Scholar] [CrossRef]
- Wang, Z.; Xiao, X.; Van Nostrand, E.; Burge, C.B. General and specific functions of exonic splicing silencers in splicing control. Mol. Cell. 2006, 23, 61–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raponi, M.; Kralovicova, J.; Copson, E.; Divina, P.; Eccles, D.; Johnson, P.; Baralle, D.; Vorechovsky, I. Prediction of single-nucleotide substitutions that result in exon skipping: Identification of a splicing silencer in BRCA1 exon 6. Hum. Mutat. 2011, 32, 436–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piva, F.; Giulietti, M.; Nocchi, L.; Principato, G. SpliceAid: A database of experimental RNA target motifs bound by splicing proteins in humans. Bioinformatics 2009, 25, 1211–1213. [Google Scholar] [CrossRef]
- Graveley, B.R. Sorting out the complexity of SR protein functions. RNA 2000, 6, 1197–1211. [Google Scholar] [CrossRef] [Green Version]
- Erkelenz, S.; Mueller, W.F.; Evans, M.S.; Busch, A.; Schöneweis, K.; Hertel, K.J.; Schaal, H. Position-dependent splicing activation and repression by SR and hnRNP proteins rely on common mechanisms. RNA 2013, 19, 96–102. [Google Scholar] [CrossRef] [Green Version]
- Fairbrother, W.G.; Yeh, R.F.; Sharp, P.A.; Burge, C.B. Predictive identification of exonic splicing enhancers in human genes. Science 2002, 297, 1007–1013. [Google Scholar] [CrossRef] [Green Version]
- Giulietti, M.; Piva, F.; D’Antonio, M.; D’Onorio De Meo, P.; Paoletti, D.; Castrignanò, T.; D’Erchia, A.M.; Picardi, E.; Zambelli, F.; Principato, G.; et al. SpliceAid-F: A database of human splicing factors and their RNA-binding sites. Nucleic Acids Res. 2013, 41, D125–D131. [Google Scholar] [CrossRef]
- Kohlschütter, A.; Laabs, R.; Albani, M. Juvenile neuronal ceroid lipofuscinosis (JNCL): Quantitative description of its clinical variability. Acta Paediatr. Scand. 1988, 77, 867–872. [Google Scholar] [CrossRef]
- Lebrun, A.H.; Moll-Khosrawi, P.; Pohl, S.; Makrypidi, G.; Storch, S.; Kilian, D.; Streichert, T.; Otto, B.; Mole, S.E.; Ullrich, K.; et al. Analysis of potential biomarkers and modifier genes affecting the clinical course of CLN3 disease. Mol. Med. 2011, 17, 1253–1261. [Google Scholar] [CrossRef] [PubMed]
- Gilani, N.; Razmara, E.; Ozaslan, M.; Abdulzahra, I.K.; Arzhang, S.; Tavasoli, A.R.; Garshasbi, M. A novel deletion variant in CLN3 with highly variable expressivity is responsible for juvenile neuronal ceroid lipofuscinoses. Acta Neurol. Belg. 2021, 121, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Cannelli, N.; Garavaglia, B.; Simonati, A.; Aiello, C.; Barzaghi, C.; Pezzini, F.; Cilio, M.R.; Biancheri, R.; Morbin, M.; Dalla Bernardina, B.; et al. Variant late infantile ceroid lipofuscinoses associated with novel mutations in CLN6. Biochem. Biophys. Res. Commun. 2009, 379, 892–897. [Google Scholar] [CrossRef] [PubMed]
- Järvelä, I.; Autti, T.; Lamminranta, S.; Aberg, L.; Raininko, R.; Santavuori, P. Clinical and magnetic resonance imaging findings in Batten disease: Analysis of the major mutation (1.02-kb deletion). Ann. Neurol. 1997, 42, 799–802. [Google Scholar] [CrossRef]
- Wisniewski, K.E.; Kida, E.; Connell, F.; Zhong, N. Neuronal ceroid lipofuscinoses: Research update. Neurol. Sci. 2000, 21, S49–S56. [Google Scholar] [CrossRef]
- Pasquinelli, G.; Cenacchi, G.; Piane, E.L.; Russo, C.; Aguglia, U. The problematic issue of Kufs disease diagnosis as performed on rectal biopsies: A case report. Ultrastruct. Pathol. 2004, 28, 43–48. [Google Scholar] [CrossRef]
- Goebel, H.H.; Braak, H. Adult neuronal ceroid-lipofuscinosis. Clin Neuropathol. 1989, 8, 109–119. [Google Scholar]
- Capalbo, A.; Valero, R.A.; Jimenez-Almazan, J.; Pardo, P.M.; Fabiani, M.; Jiménez, D.; Simon, C.; Rodriguez, J.M. Optimizing clinical exome design and parallel gene-testing for recessive genetic conditions in preconception carrier screening: Translational research genomic data from 14,125 exomes. PLoS Genet. 2019, 15, e1008409. [Google Scholar] [CrossRef]
- Frischmeyer, P.A.; Dietz, H.C. Nonsense-mediated mRNA decay in health and disease. Hum. Mol. Genet. 1999, 8, 1893–1900. [Google Scholar] [CrossRef] [Green Version]
- Schmitz-Hübsch, T.; du Montcel, S.T.; Baliko, L.; Berciano, J.; Boesch, S.; Depondt, C.; Giunti, P.; Globas, C.; Infante, J.; Kang, J.S.; et al. Scale for the assessment and rating of ataxia: Development of a new clinical scale. Neurology 2006, 66, 1717–1720. [Google Scholar] [CrossRef]
- Weisschuh, N.; Mazzola, P.; Heinrich, T.; Haack, T.; Wissinger, B.; Tonagel, F.; Kelbsch, C. First submicroscopic inversion of the OPA1 gene identified in dominant optic atrophy—A case report. BMC Med. Genet. 2020, 21, 236. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [Green Version]
- Den Dunnen, J.T.; Dalgleish, R.; Maglott, D.R.; Hart, R.K.; Greenblatt, M.S.; McGowan-Jordan, J.; Roux, A.F.; Smith, T.; Antonarakis, S.E.; Taschner, P.E. HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Hum. Mutat. 2016, 37, 564–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weisschuh, N.; Wissinger, B.; Gramer, E. A splice site mutation in the PAX6 gene which induces exon skipping causes autosomal dominant inherited aniridia. Mol. Vis. 2012, 18, 751–757. [Google Scholar] [PubMed]
- Weisschuh, N.; Marino, V.; Schäferhoff, K.; Richter, P.; Park, J.; Haack, T.B.; Dell’Orco, D. Mutations at a split codon in the GTPase-encoding domain of OPA1 cause dominant optic atrophy through different molecular mechanisms. Hum. Mol. Genet. 2021, 24, ddab286. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Main Feature | Detailed Characteristics | Patient A | Patient B |
|---|---|---|---|
| Medical history | Gender | Male | Female |
| First neurological symptom | Speech delay | Epileptic seizure | |
| First ophthalmological symptom | Blurred vision | Blurred vision | |
| Age at the first neurological symptom | 4 years | 19 years | |
| Age at the first ophthalmological symptom | 10 years | 22 years | |
| Age at the last visit | 20 years | 26 years | |
| Neurological symptoms | Delayed speech | + | - |
| Intellectual disability | + | - | |
| Psychomotor degeneration | + | - | |
| Visual hallucinations | - | - | |
| Aphasia | - | - | |
| Dysarthria | + | - | |
| Seizures | + | + | |
| Ataxia of extremities | + | + | |
| Gait ataxia | + | + | |
| Saccadic gaze | + | + | |
| Myoclonus | - | + | |
| Vision problems | Photophobia | + | + |
| Nystagmus | + | - | |
| Loss of visual acuity | + | + | |
| Foveal thinning of the retinal layer | + | + | |
| Diminished or absent ERG responses | n/a | + | |
| cMRI | Cerebellar atrophy | + | + |
| Cerebral atrophy | + | - | |
| White matter changes | - | - | |
| Other abnormalities | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reith, M.; Zeltner, L.; Schäferhoff, K.; Witt, D.; Zuleger, T.; Haack, T.B.; Bornemann, A.; Alber, M.; Ruf, S.; Schoels, L.; et al. A Novel, Apparently Silent Variant in MFSD8 Causes Neuronal Ceroid Lipofuscinosis with Marked Intrafamilial Variability. Int. J. Mol. Sci. 2022, 23, 2271. https://doi.org/10.3390/ijms23042271
Reith M, Zeltner L, Schäferhoff K, Witt D, Zuleger T, Haack TB, Bornemann A, Alber M, Ruf S, Schoels L, et al. A Novel, Apparently Silent Variant in MFSD8 Causes Neuronal Ceroid Lipofuscinosis with Marked Intrafamilial Variability. International Journal of Molecular Sciences. 2022; 23(4):2271. https://doi.org/10.3390/ijms23042271
Chicago/Turabian StyleReith, Milda, Lena Zeltner, Karin Schäferhoff, Dennis Witt, Theresia Zuleger, Tobias B. Haack, Antje Bornemann, Michael Alber, Susanne Ruf, Ludger Schoels, and et al. 2022. "A Novel, Apparently Silent Variant in MFSD8 Causes Neuronal Ceroid Lipofuscinosis with Marked Intrafamilial Variability" International Journal of Molecular Sciences 23, no. 4: 2271. https://doi.org/10.3390/ijms23042271
APA StyleReith, M., Zeltner, L., Schäferhoff, K., Witt, D., Zuleger, T., Haack, T. B., Bornemann, A., Alber, M., Ruf, S., Schoels, L., Stingl, K., & Weisschuh, N. (2022). A Novel, Apparently Silent Variant in MFSD8 Causes Neuronal Ceroid Lipofuscinosis with Marked Intrafamilial Variability. International Journal of Molecular Sciences, 23(4), 2271. https://doi.org/10.3390/ijms23042271