
Employing CRISPR-Cas9 to Generate CD133 Synthetic Lethal Melanoma Stem Cells

 ,
,  , , , , and
, , , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
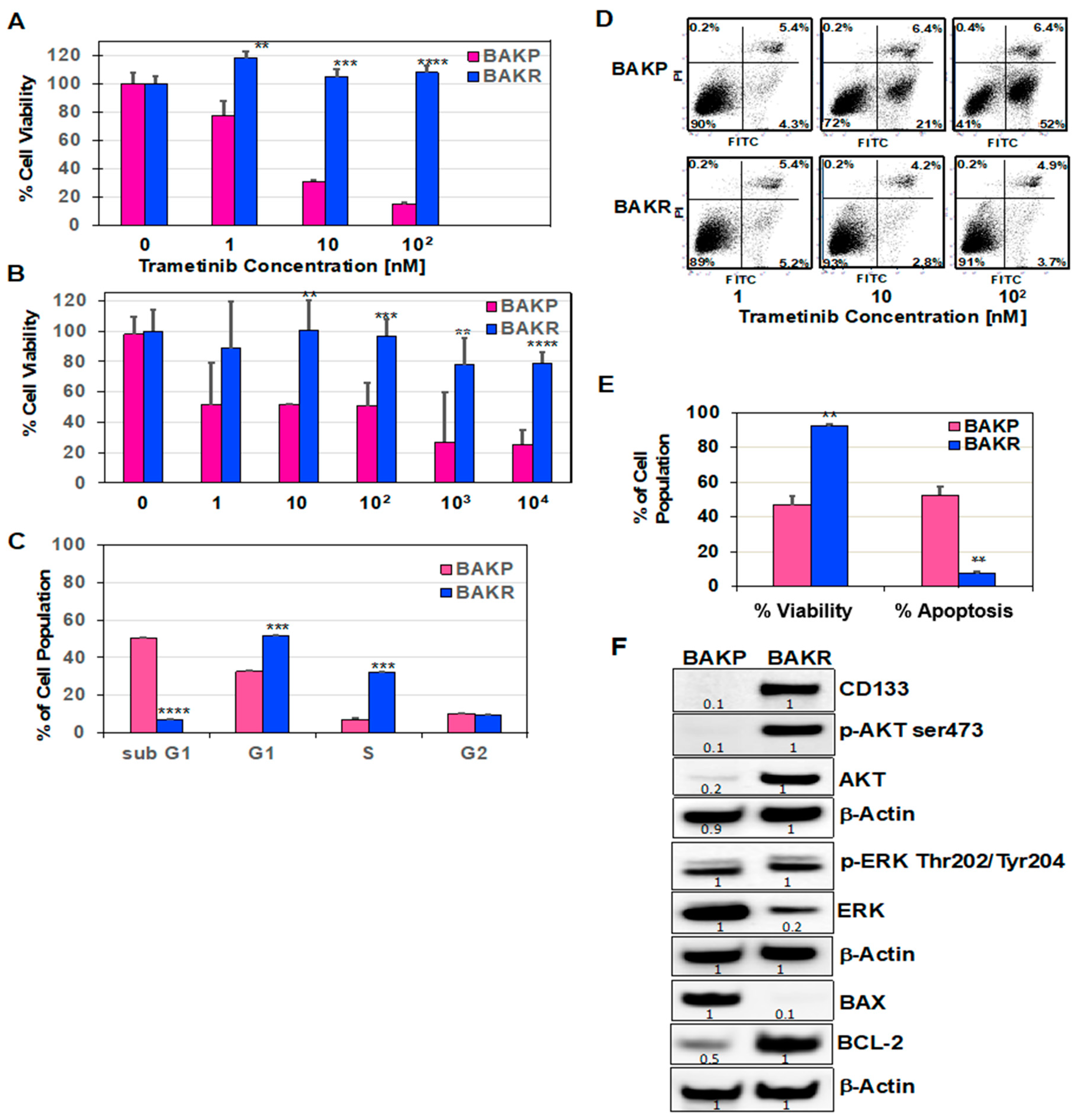
2.1. CD133 Overexpression in Conditionally Reprogrammed BAKP Melanoma Cells (BAKR) Increases Cell Viability and Inhibits Apoptosis in Response to Trametinib or DTIC
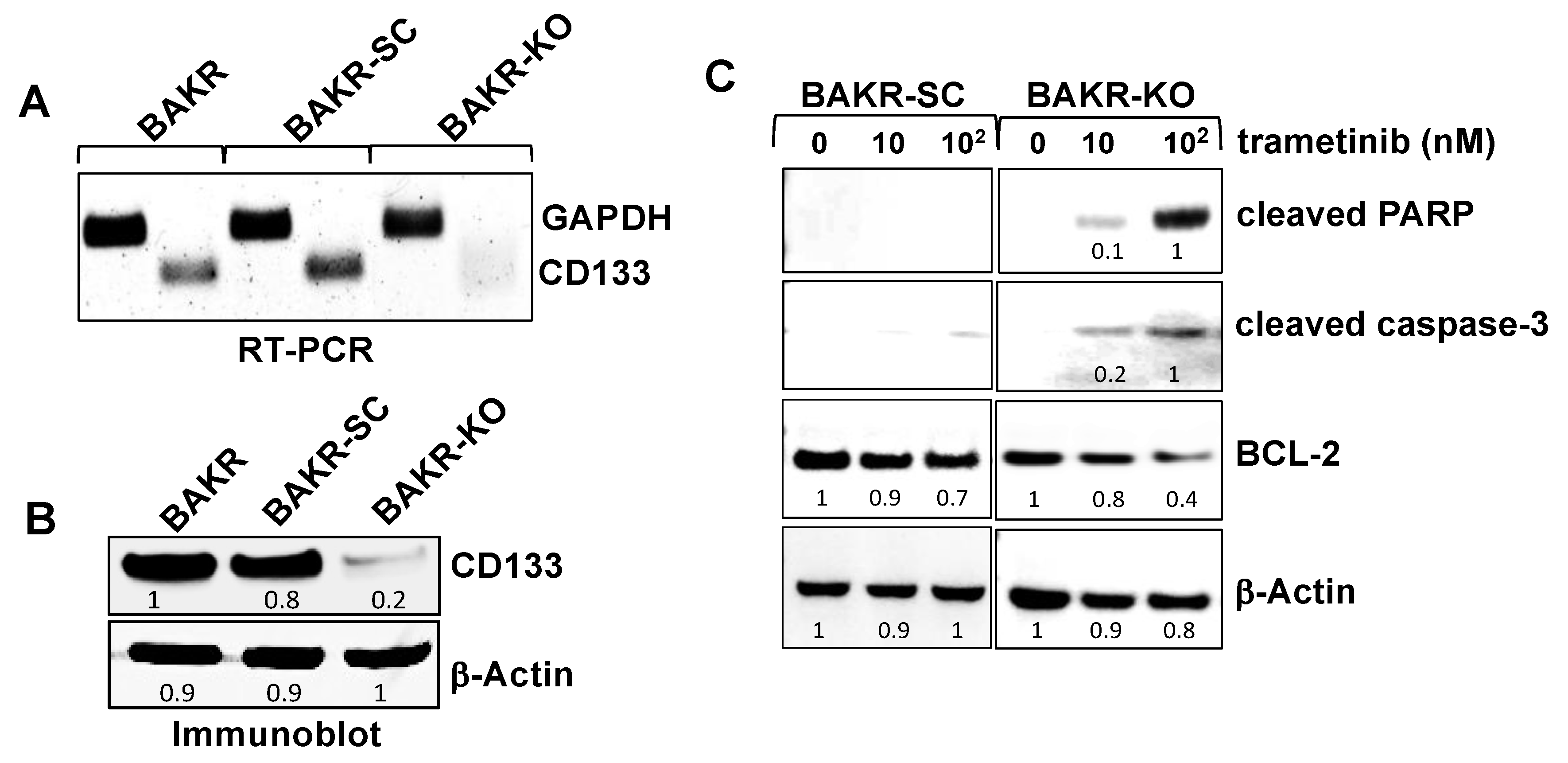
2.2. CRISPR-Cas9 Knockout of CD133 in Conditionally Reprogrammed BAKR Cells Increases Apoptotic Caspase-3 Activation in Response to Trametinib via Bcl2 Downregulation
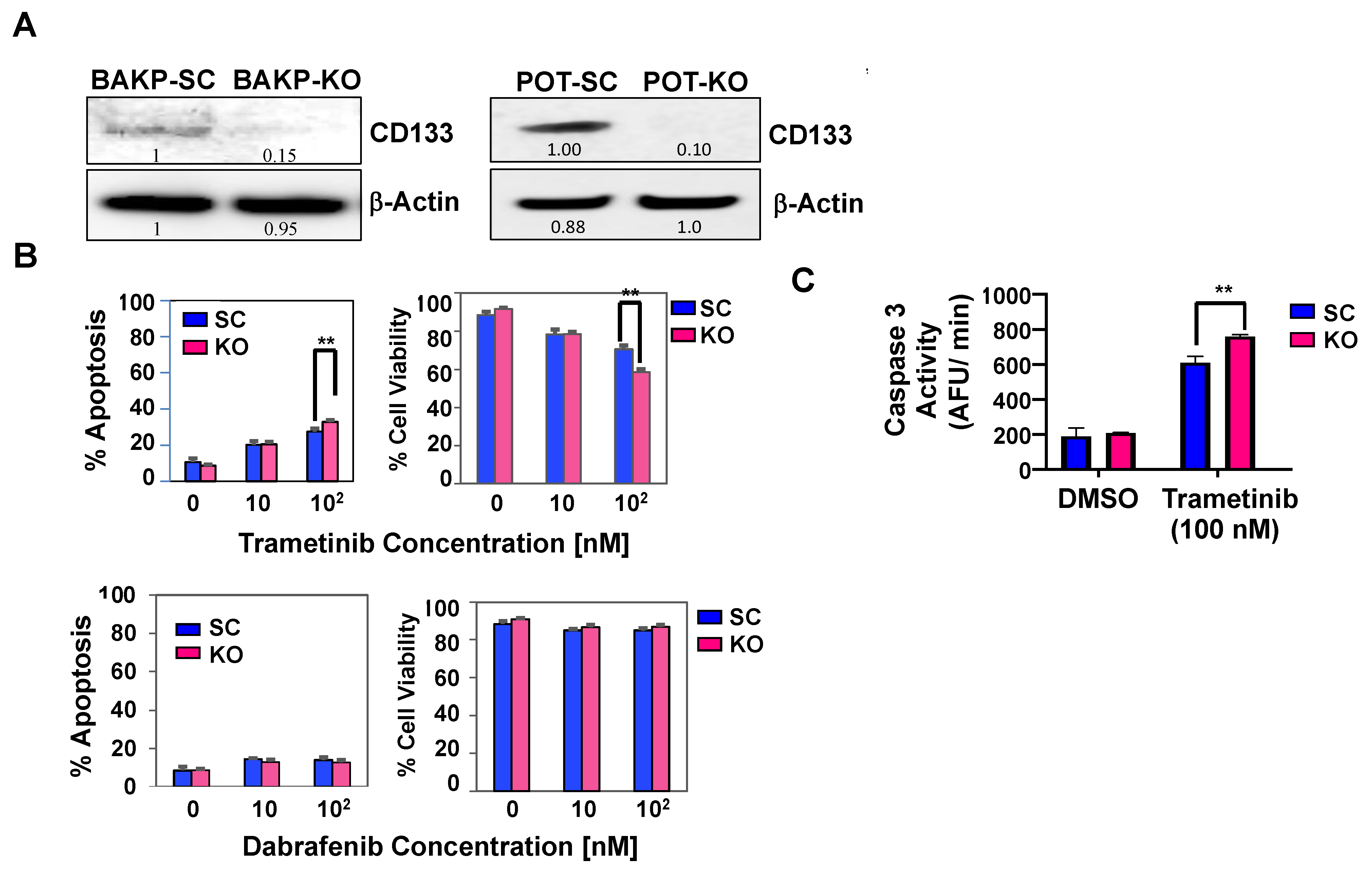
2.3. CRISPR-Cas9 Knockout of CD133 Expression in BAKP Melanoma Cells Increases Caspase 3-Mediated Apoptosis in Response to Trametinib
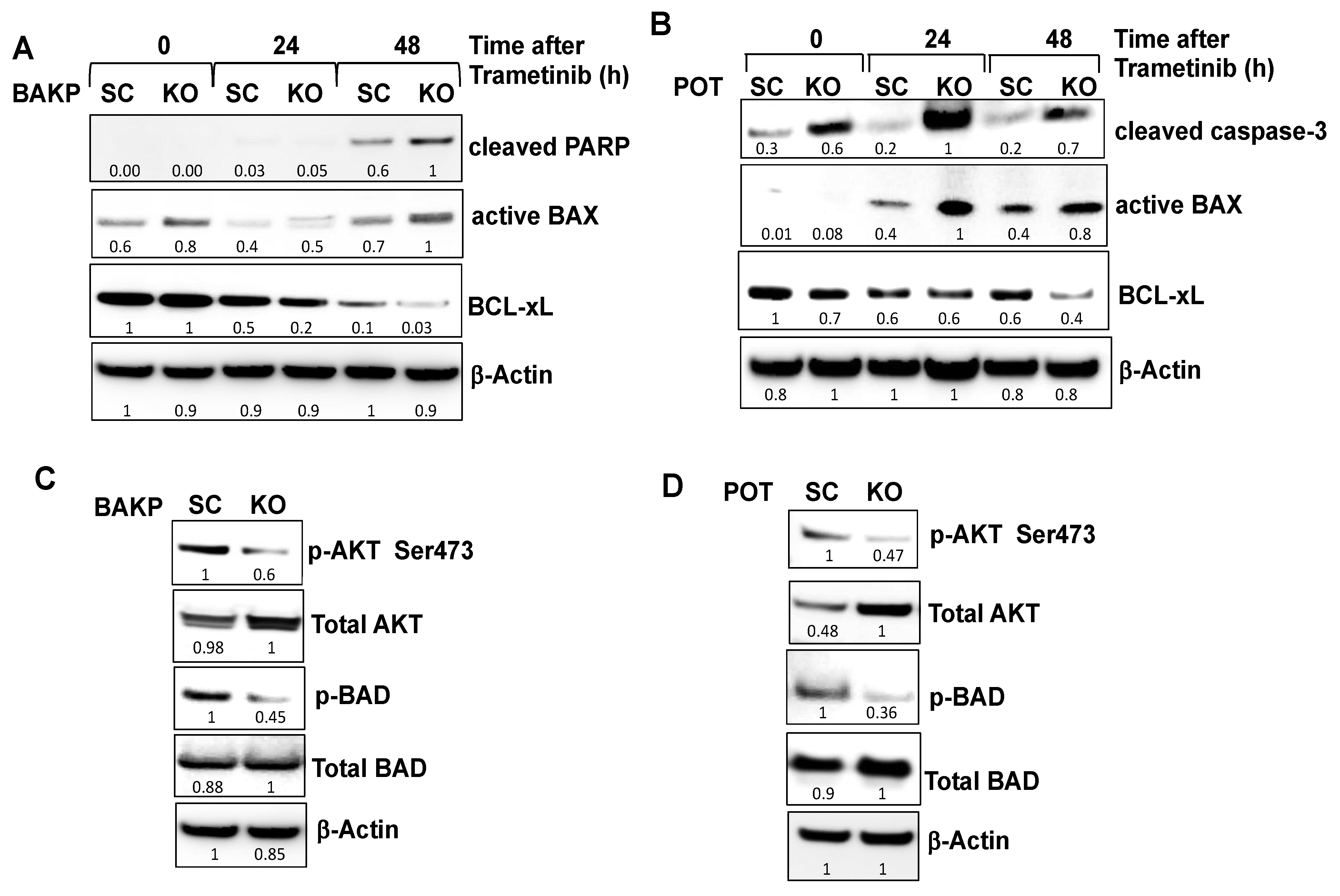
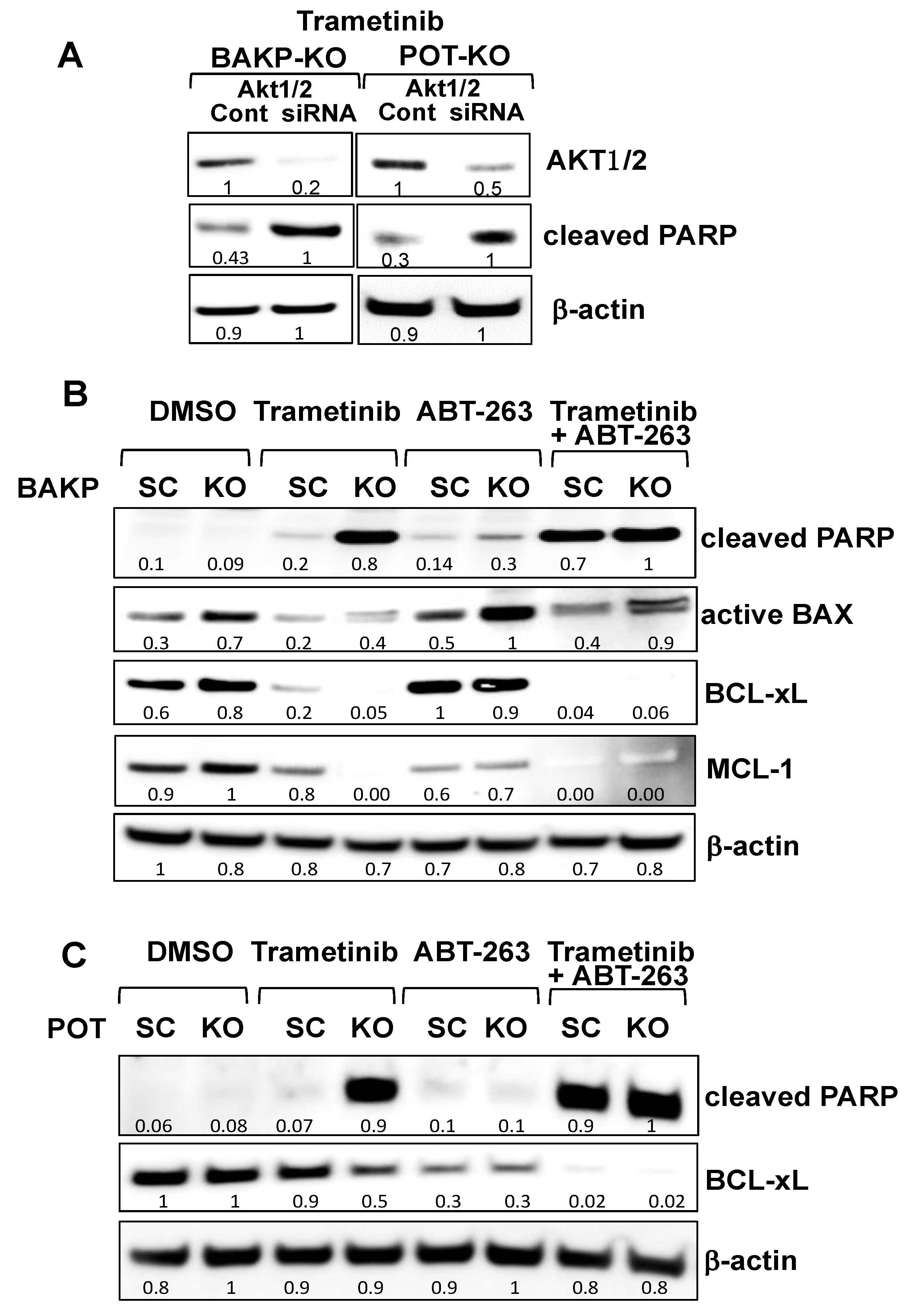
2.4. CRISPR-Cas9 Knockout of CD133 in BAKP and POT Cells Enhances Trametinib-Induced Apoptosis via Downregulation of Pro-Survival BCL-2 Family Members and AKT
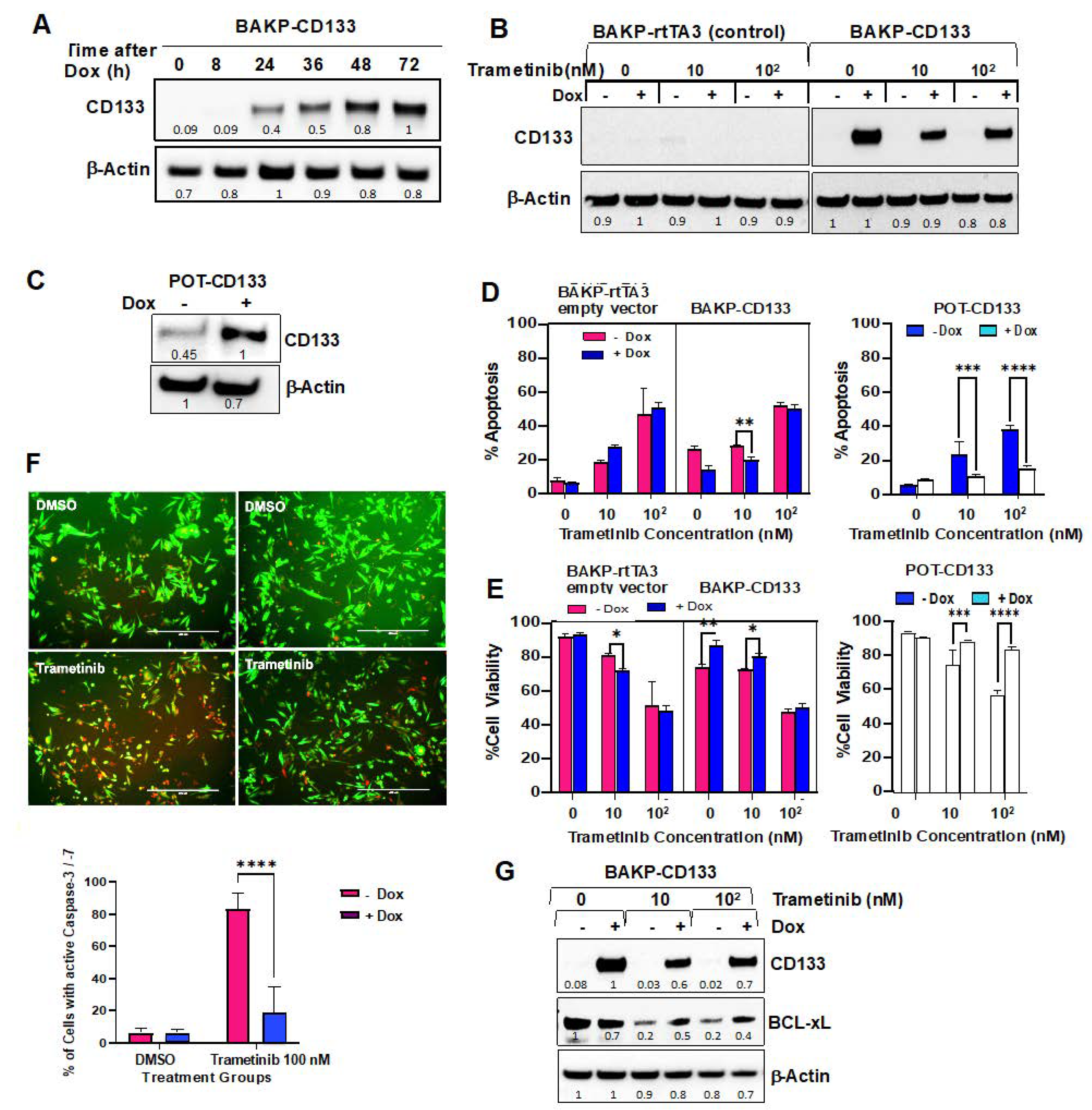
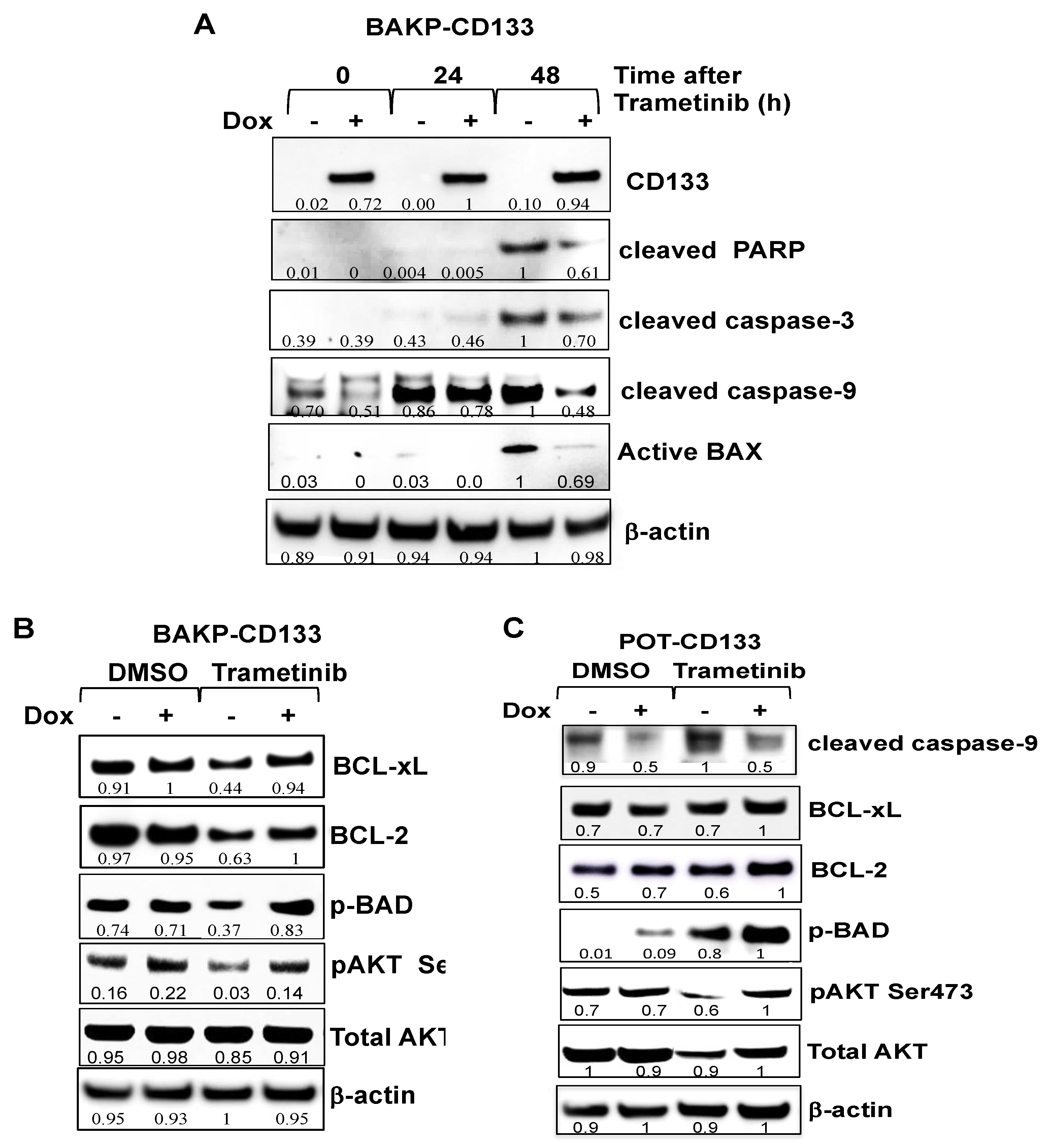
2.5. Dox-Inducible Expression of CD133 Has Opposite Effects of CRISPR-cas9 CD133 Knockout, Suppressing Trametinib-Induced Apoptosis via Modulation of BCL-2 Family Members Mediated by AKT in BAKP and POT Cells
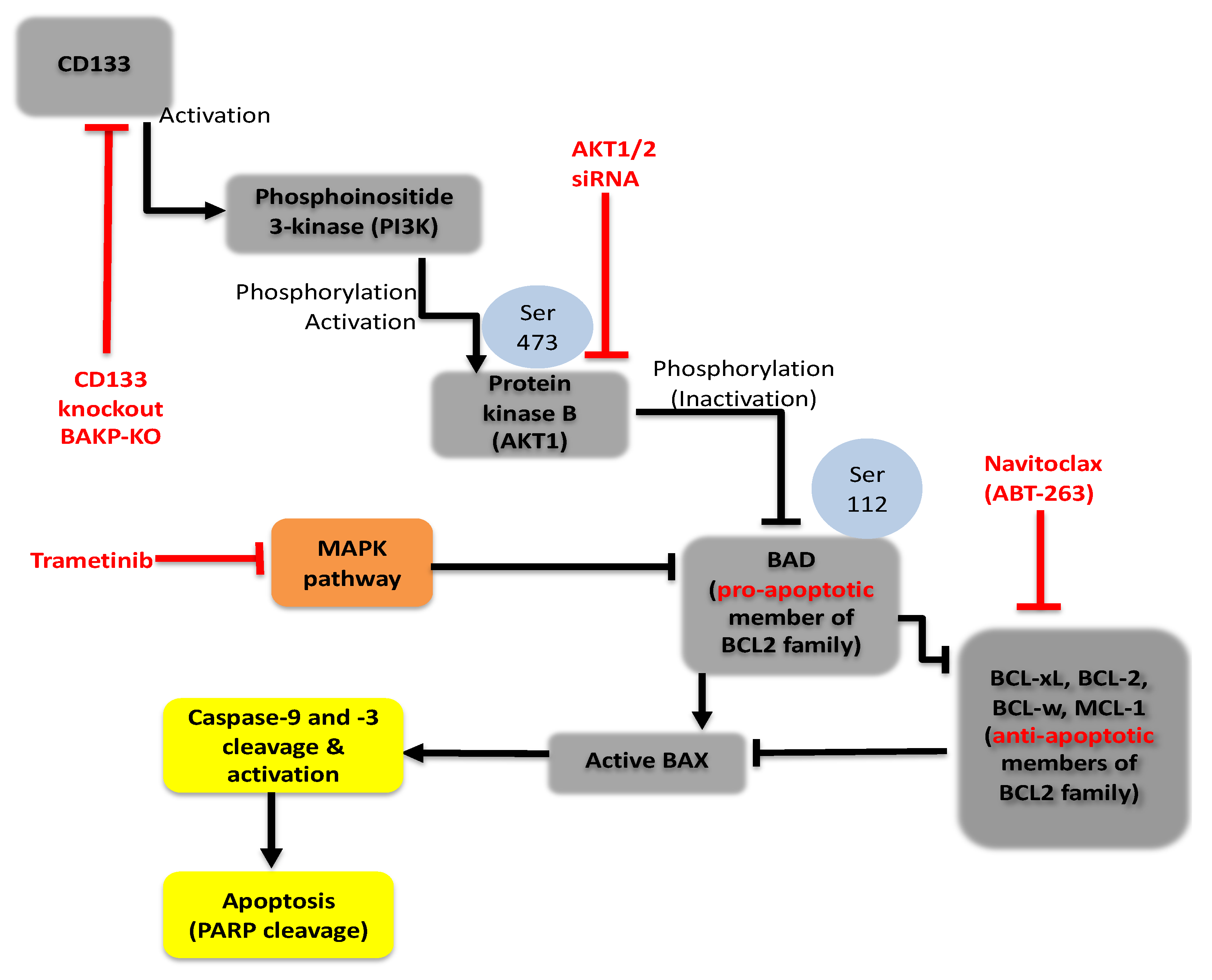
2.6. Targeting Nodes of the CD133, AKT, and BCL-2 Family Survival Pathway, and/or Trametini, Reveals the Potential for Combination Therapies for NRAS Mutant Melanoma
3. Discussion
4. Materials and Methods
4.1. Cells
4.2. Magnetic Sorting, Pre- and Post-Staining for CD133-Positivity and Conditional Reprogramming of CD133(+) Cells
4.3. AKT Knockdown by siRNA
4.4. CRISPR-Cas9 Deletion of CD133
- sgRNA1: CAACAGGGAGCCGAGTACGA (complement of Target 1 underlined below)
- sgRNA2: TTCATCCACAGATGCTCCTA (complement of Target 2 underlined below)
- sgRNA3: TTACCTTCTGGGAAATCACGC (complement of Target 3 underlined below)
- ATG GCC CTC GTA CTC GGC TCC CTG TTG CTG CTG GGG CTG TGC GGG AAC T1
- TCC TTT TCA GGA GGG CAG CCT TCA TCC ACA GAT GCT CCT AAG GCT TGG T2
- AAT TAT GAA TTG CCT GCA ACA AAT TAT GAG ACC CAA GAC TCC CAT AAA GCT GGA CCC ATT GGC ATT CTC TTT GAA CTA GTG CAT ATC TTT CTT ATG TGG TAC AGC CGC GTG ATT TCC CAG AAG GTA A T3
- Target 1-forward (F1a) TTCCCCAAGGCTTCCAGAAG
- Target 1-reverse (R1a) GCCCTCCTGAAAAGGAGTTC
- Target 2-forward (F2a) GAACTCCTTTTCAGGAGGGC
- Target 2-reverse (R2a) GAGAATGCCAATGGGTCCAG
- Target 3-forward (F3a) CTGGACCCATTGGCATTCTC
- Target 3-reverse (R3a) CATTCT1TCCCTGCCATCAGC
4.5. Quantitative Reverse-Transcription PCR (qRT-PCR)
- CD133 forward-5ʹ-CCC GGG GCT GCT GTT TAT A
- CD133 reverse-5ʹ-ATC ACC AAC AGG GAG ATT G
4.6. Generation of Dox-Inducible Cells
4.7. Drug Treatment and Cell Viability Assays
4.8. Annexin V/PI Staining and Flow Cytometry
4.9. Fluorometric Caspase-3 Activity Assays
4.10. Live FLICA Caspase-3/7 Activity Assays
4.11. Immunoblot Analysis
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Surveillance, Epidemiology, and End Results Program. In Cancer Stat Facts: Melanoma of the Skin; Bethesda: Rockville, MD, USA, 2021. Available online: https://seer.cancer.gov/statfacts/html/melan.html (accessed on 18 May 2021).
- Paluncic, J.; Kovacevic, Z.; Jansson, P.J.; Kalinowski, D.; Merlot, A.M.; Huang, M.L.-H.; Lok, H.C.; Sahni, S.; Lane, D.J.R.; Richardson, D.R. Roads to melanoma: Key pathways and emerging players in melanoma progression and oncogenic signaling. Biochim. Biophys. Acta 2016, 1863, 770–784. [Google Scholar] [CrossRef] [PubMed]
- Spagnolo, F.; Picasso, V.; Lambertini, M.; Ottaviano, V.; Dozin, B.; Queirolo, P. Survival of patients with metastatic melanoma and brain metastases in the era of MAP-kinase inhibitors and immunologic checkpoint blockade antibodies: A systematic review. Cancer Treat. Rev. 2016, 45, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Ugurel, S.; Röhmel, J.; Ascierto, P.A.; Flaherty, K.T.; Grob, J.J.; Hauschild, A.; Larkin, J.; Long, G.V.; Lorigan, P.; McArthur, G.A.; et al. Survival of patients with advanced metastatic melanoma: The impact of novel therapies-update 2017. Eur. J. Cancer 2017, 83, 247–257. [Google Scholar] [CrossRef] [Green Version]
- Sandri, S.; Faião-Flores, F.; Tiago, M.; Pennacchi, P.C.; Massaro, R.R.; Alves-Fernandes, D.K.; Berardinelli, G.N.; Evangelista, A.F.; Vazquez, V.D.; Reis, R.M.; et al. Vemurafenib resistance increases melanoma invasiveness and modulates the tumor microenvironment by MMP-2 upregulation. Pharmacol. Res. 2016, 111, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Kalal, B.S.; Upadhya, D.; Pai, V.R. Chemotherapy Resistance Mechanisms in Advanced Skin Cancer. Oncol. Rev. 2017, 11, 326. [Google Scholar] [CrossRef]
- Berger, M.F.; Hodis, E.; Heffernan, T.P.; Deribe, Y.L.; Lawrence, M.S.; Protopopov, A.; Ivanova, E.; Watson, I.R.; Nickerson, E.; Ghosh, P.; et al. Melanoma genome sequencing reveals frequent PREX2 mutations. Nature 2012, 485, 502. [Google Scholar] [CrossRef]
- Pleasance, E.D.; Cheetham, R.K.; Stephens, P.J.; McBride, D.J.; Humphray, S.J.; Greenman, C.D.; Varela, I.; Lin, M.-L.; Ordóñez, G.R.; Bignell, G.R.; et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature 2009, 463, 191. [Google Scholar] [CrossRef]
- Tian, Y.; Guo, W. A review of the molecular pathways involved in resistance to BRAF inhibitors in patients with advanced-stage melanoma. Med. Sci. Monit. 2020, 26, e920957. [Google Scholar] [CrossRef]
- Bhatia, S.; Tykodi, S.S.; Thompson, J. Treatment of metastatic melanoma: An overview. Oncology 2009, 23, 488–496. [Google Scholar]
- Flaherty, K.T.; Infante, J.R.; Daud, A.; Gonzalez, R.; Kefford, R.F.; Sosman, J.; Hamid, O.; Schuchter, L.; Cebon, J.; Ibrahim, N.; et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N. Engl. J. Med. 2012, 367, 1694–1703. [Google Scholar] [CrossRef] [Green Version]
- Ascierto, P.A.; Schadendorf, D.; Berking, C.; Agarwala, S.S.; van Herpen, C.M.; Queirolo, P.; Blank, C.U.; Hauschild, A.; Beck, J.T.; St-Pierre, A.; et al. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: A non-randomised, open-label phase 2 study. Lancet Oncol. 2013, 14, 249–256. [Google Scholar] [CrossRef]
- Falchook, G.S.; Lewis, K.D.; Infante, J.R.; Gordon, M.S.; Vogelzang, N.J.; DeMarini, D.J.; Sun, P.; Moy, C.; Szabo, S.A.; Roadcap, L.T.; et al. Activity of the oral MEK inhibitor trametinib in patients with advanced melanoma: A phase 1 dose-escalation trial. Lancet Oncol. 2012, 13, 782–789. [Google Scholar] [CrossRef] [Green Version]
- Thota, R.; Johnson, D.B.; Sosman, J.A. Trametinib in the treatment of melanoma. Expert Opin. Biol. Ther. 2015, 15, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Bedard, P.L.; Tabernero, J.; Janku, F.; Wainberg, Z.A.; Paz-Ares, L.; Vansteenkiste, J.; van Cutsem, E.; Pérez-García, J.; Stathis, A.; Britten, C.D.; et al. A phase Ib dose-escalation study of the oral pan-PI3K inhibitor buparlisib (BKM120) in combination with the oral MEK1/2 inhibitor trametinib (GSK1120212) in patients with selected advanced solid tumors. Clin. Cancer Res. 2015, 21, 730–738. [Google Scholar] [CrossRef] [Green Version]
- Grilley-Olson, J.E.; Bedard, P.L.; Fasolo, A.; Cornfeld, M.; Cartee, L.; Razak, A.R.A.; Stayner, L.-A.; Wu, Y.; Greenwood, R.; Singh, R.; et al. A phase Ib dose-escalation study of the MEK inhibitor trametinib in combination with the PI3K/mTOR inhibitor GSK2126458 in patients with advanced solid tumors. Investig. New Drugs 2016, 34, 740–749. [Google Scholar] [CrossRef]
- Posch, C.; Moslehi, H.; Feeney, L.; Green, G.A.; Ebaee, A.; Feichtenschlager, V.; Chong, K.; Peng, L.; Dimon, M.T.; Phillips, T.; et al. Combined targeting of MEK and PI3K/mTOR effector pathways is necessary to effectively inhibit NRAS mutant melanoma in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2013, 110, 4015–4020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagler, A.; Vredevoogd, D.W.; Alon, M.; Cheng, P.F.; Trabish, S.; Kalaora, S.; Arafeh, R.; Goldin, V.; Levesque, M.P.; Peeper, D.S.; et al. A genome-wide CRISPR screen identifies FBXO42 involvement in resistance toward MEK inhibition in NRAS-mutant melanoma. Pigment. Cell Melanoma Res. 2020, 33, 334–344. [Google Scholar] [CrossRef]
- Vujic, I.; Sanlorenzo, M.; Posch, C.; Esteve-Puig, R.; Yen, A.J.; Kwong, A.; Tsumura, A.; Murphy, R.; Rappersberger, K.; Ortiz-Urda, S. Metformin and trametinib have synergistic effects on cell viability and tumor growth in NRAS mutant cancer. Oncotarget 2015, 6, 969–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vu, H.L.; Aplin, A.E. Targeting TBK1 inhibits migration and resistance to MEK inhibitors in mutant NRAS melanoma. Mol. Cancer Res. 2014, 12, 1509–1519. [Google Scholar] [CrossRef] [Green Version]
- Vogel, C.J.; Smit, M.A.; Maddalo, G.; Possik, P.A.; Sparidans, R.W.; van der Burg, S.H.; Verdegaal, E.M.; Heck, A.J.R.; Samatar, A.A.; Beijnen, J.H.; et al. Cooperative induction of apoptosis in NRAS mutant melanoma by inhibition of MEK and ROCK. Pigment Cell Melanoma Res. 2015, 28, 307–317. [Google Scholar] [CrossRef]
- Marzagalli, M.; Marelli, M.M.; Casati, L.; Fontana, F.; Moretti, R.M.; Limonta, P. Estrogen receptor beta in melanoma: From molecular insights to potential clinical utility. Front. Endocrinol. 2016, 7, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Posch, C.; Cholewa, B.D.; Vujic, I.; Sanlorenzo, M.; Ma, J.; Kim, S.T.; Kleffel, S.; Schatton, T.; Rappersberger, K.; Gutteridge, R.; et al. Combined inhibition of MEK and Plk1 has synergistic antitumor activity in NRAS mutant melanoma. J. Investig. Derm. 2015, 135, 2475–2483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Mayes, P.A.; Eastman, S.; Shi, H.; Yadavilli, S.; Zhang, T.; Yang, J.; Seestaller-Wehr, L.; Zhang, S.-Y.; Hopson, C.; et al. The BRAF and MEK inhibitors dabrafenib and trametinib: Effects on immune function and in combination with immunomodulatory antibodies targeting PD-1, PD-L1, and CTLA-4. Clin. Cancer Res. 2015, 21, 1639–1651. [Google Scholar] [CrossRef] [Green Version]
- Giampietri, C.; Petrungaro, S.; Cordella, M.; Tabolacci, C.; Tomaipitinca, L.; Facchiano, A.; Eramo, A.; Filippini, A.; Facchiano, F.; Ziparo, E. Lipid storage and autophagy in melanoma cancer cells. Int. J. Mol. Sci. 2017, 18, 1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schatton, T.; Murphy, G.F.; Frank, N.Y.; Yamaura, K.; Waaga-Gasser, A.M.; Gasser, M.; Zhan, Q.; Jordan, S.; Duncan, L.M.; Weishaupt, C.; et al. Identification of cells initiating human melanomas. Nature 2008, 451, 345–349. [Google Scholar] [CrossRef] [Green Version]
- Sharma, B.K.; Manglik, V.; O’Connell, M.; Weeraratna, A.; McCarron, E.C.; Broussard, J.N.; Divito, K.A.; Simbulan-Rosenthal, C.M.; Rosenthal, D.S.; Zapas, J.L. Clonal dominance of CD133+ subset population as risk factor in tumor progression and disease recurrence of human cutaneous melanoma. Int. J. Oncol. 2012, 41, 1570–1576. [Google Scholar] [CrossRef] [Green Version]
- Fang, D.; Nguyen, T.K.; Leishear, K.; Finko, R.; Kulp, A.N.; Hotz, S.; van Belle, P.A.; Xu, X.; Elder, D.E.; Herlyn, M. A tumorigenic subpopulation with stem cell properties in melanomas. Cancer Res. 2005, 65, 9328–9337. [Google Scholar] [CrossRef] [Green Version]
- Thapa, R.; Wilson, G.D. The importance of CD44 as a stem cell biomarker and therapeutic target in cancer. Stem Cells Int. 2016, 2016, 2087204. [Google Scholar] [CrossRef] [Green Version]
- Hendrix, M.J.C.; Seftor, E.A.; Hess, A.R.; Seftor, R.E.B. Molecular plasticity of human melanoma cells. Oncogene 2003, 22, 3070–3075. [Google Scholar] [CrossRef] [Green Version]
- Klein, W.M.; Wu, B.P.; Zhao, S.; Wu, H.; Klein-Szanto, A.J.P.; Tahan, S.R. Increased expression of stem cell markers in malignant melanoma. Mod. Pathol. 2007, 20, 102–107. [Google Scholar] [CrossRef]
- Civenni, G.; Walter, A.; Kobert, N.; Mihic-Probst, D.; Zipser, M.; Belloni, B.; Seifert, B.; Moch, H.; Dummer, R.; van den Broek, M.; et al. Human CD271-positive melanoma stem cells associated with metastasis establish tumor heterogeneity and long-term growth. Cancer Res. 2011, 71, 3098–3109. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Dallaglio, K.; Chen, Y.; Robinson, W.A.; Robinson, S.E.; McCarter, M.D.; Wang, J.; Gonzalez, R.; Thompson, D.C.; Norris, D.A.; et al. ALDH1A isozymes are markers of human melanoma stem cells and potential therapeutic targets. Stem Cells 2012, 30, 2100–2113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akiyama, M.; Matsuda, Y.; Ishiwata, T.; Naito, Z.; Kawana, S. Inhibition of the stem cell marker nestin reduces tumor growth and invasion of malignant melanoma. J. Investig. Derm. 2013, 133, 1384–1387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, N.Y.; Schatton, T.; Kim, S.; Zhan, Q.; Wilson, B.J.; Ma, J.; Saab, K.R.; Osherov, V.; Widlund, H.R.; Gasser, M.; et al. VEGFR-1 expressed by malignant melanoma-initiating cells is required for tumor growth. Cancer Res. 2011, 71, 1474–1485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roesch, A.; Fukunaga-Kalabis, M.; Schmidt, E.C.; Zabierowski, S.E.; Brafford, P.A.; Vultur, A.; Basu, D.; Gimotty, P.; Vogt, T.; Herlyn, M. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell 2010, 141, 583–594. [Google Scholar] [CrossRef] [Green Version]
- Sanai, N.; Alvarez-Buylla, A.; Berger, M.S. Neural stem cells and the origin of gliomas. N. Engl. J. Med. 2005, 353, 811–822. [Google Scholar] [CrossRef]
- Yin, A.H.; Miraglia, S.; Zanjani, E.D.; Almeida-Porada, G.; Ogawa, M.; Leary, A.G.; Olweus, J.; Kearney, J.; Buck, D.W. AC133, a novel marker for human hematopoietic stem and progenitor cells. Blood 1997, 90, 5002–5012. [Google Scholar] [CrossRef] [Green Version]
- Lai, G.-M.; Yao, C.-J.; Yeh, C.-T.; Yeh, C.-F.; Chang, C.-K.; Yan, J.-L.; Li, C.-H.; Chuang, S.-E. Active fraction of Taiwanofungus camphoratus (HS7) exerted anticancer effects through multiple molecule targeting and elimination of cancer stem-like cells in lung cancer and hepatoma cells. Cancer Res. 2009, 69, 5475. [Google Scholar]
- Mizrak, D.; Brittan, M.; Alison, M.R. CD133: Molecule of the moment. J. Pathol. 2008, 214, 3–9. [Google Scholar] [CrossRef]
- Shmelkov, S.V.; Clair, R.S.; Lyden, D.; Rafii, S. AC133/CD133/Prominin-1. Int. J. Biochem. Cell Biol. 2005, 37, 715–719. [Google Scholar] [CrossRef]
- Dowland, S.N.; Madawala, R.J.; Poon, C.E.; Lindsay, L.A.; Murphy, C.R. Prominin-1 glycosylation changes throughout early pregnancy in uterine epithelial cells under the influence of maternal ovarian hormones. Reprod. Fertil. Dev. 2017, 29, 1194–1208. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.T.; Berry, P.A.; Hyde, C.; Stower, M.J.; Maitland, N.J. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005, 65, 10946–10951. [Google Scholar] [CrossRef] [Green Version]
- Ferrandina, G.; Bonanno, G.; Pierelli, L.; Perillo, A.; Procoli, A.; Mariotti, A.; Corallo, M.; Martinelli, E.; Rutella, S.; Paglia, A.; et al. Expression of CD133-1 and CD133-2 in ovarian cancer. Int. J. Gynecol. Cancer 2008, 18, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 2007, 1, 313–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.S.; Gisina, A.M.; Chiang, J.-H.; Yarygin, K.N.; Lupatov, A.Y. Cancer Stem Cell Molecular Markers Verified in vivo. Biochem. Suppl. Ser. B Biomed. Chem. 2017, 11, 43–54. [Google Scholar] [CrossRef]
- O’Brien, C.A.; Pollett, A.; Gallinger, S.; Dick, J.E. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 2007, 445, 106–110. [Google Scholar] [CrossRef]
- Ricci-Vitiani, L.; Lombardi, D.G.; Pilozzi, E.; Biffoni, M.; Todaro, M.; Peschle, C.; de Maria, R. Identification and expansion of human colon-cancer-initiating cells. Nature 2007, 445, 111–115. [Google Scholar] [CrossRef]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003, 63, 5821–5828. [Google Scholar]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Suetsugu, A.; Nagaki, M.; Aoki, H.; Motohashi, T.; Kunisada, T.; Moriwaki, H. Characterization of CD133+ hepatocellular carcinoma cells as cancer stem/progenitor cells. Biochem. Biophys. Res. Commun. 2006, 351, 820–824. [Google Scholar] [CrossRef]
- Galli, R.; Binda, E.; Orfanelli, U.; Cipelletti, B.; Gritti, A.; de Vitis, S.; Fiocco, R.; Foroni, C.; Dimeco, F.; Vescovi, A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004, 64, 7011–7021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintana, E.; Shackleton, M.; Sabel, M.S.; Fullen, D.R.; Johnson, T.M.; Morrison, S.J. Efficient tumour formation by single human melanoma cells. Nature 2008, 456, 593–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendrix, M.J.C.; Seftor, E.A.; Seftor, R.E.B.; Kasemeier-Kulesa, J.; Kulesa, P.M.; Postovit, L.-M. Reprogramming metastatic tumour cells with embryonic microenvironments. Nat. Rev. Cancer 2007, 7, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Kirschmann, D.A.; Seftor, E.A.; Hardy, K.M.; Seftor, R.E.B.; Hendrix, M.J.C. Molecular pathways: Vasculogenic mimicry in tumor cells: Diagnostic and therapeutic implications. Clin. Cancer Res. 2012, 18, 2726–2732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mihic-Probst, D.; Ikenberg, K.; Tinguely, M.; Schraml, P.; Behnke, S.; Seifert, B.; Civenni, G.; Sommer, L.; Moch, H.; Dummer, R. Tumor cell plasticity and angiogenesis in human melanomas. PLoS ONE 2012, 7, e33571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simbulan-Rosenthal, C.M.; Gaur, A.; Zhou, H.; AbdusSamad, M.; Qin, Q.; Dougherty, R.; Aljehane, L.; Kuo, L.-W.; Vakili, S.; Karna, K.; et al. CD133 is associated with increased melanoma cell survival after multikinase inhibition. J. Oncol. 2019, 2019, 19. [Google Scholar] [CrossRef] [Green Version]
- Simbulan-Rosenthal, C.M.; Dougherty, R.; Vakili, S.; Ferraro, A.M.; Kuo, L.-W.; Alobaidi, R.; Aljehane, L.; Gaur, A.; Sykora, P.; Glasgow, E.; et al. CRISPR-Cas9 knockdown and induced expression of CD133 reveal essential roles in melanoma invasion and metastasis. Cancers 2019, 11, 1490. [Google Scholar] [CrossRef] [Green Version]
- Angelastro, J.M.; Lamé, M.W. Overexpression of CD133 promotes drug resistance in C6 glioma cells. Mol. Cancer Res. 2010, 8, 1105–1115. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Jiang, Y.; Zou, F.; Liu, Y.; Wang, S.; Xu, N.; Xu, W.; Cui, C.; Xing, Y.; Liu, Y.; et al. Activation of PI3K/Akt pathway by CD133-p85 interaction promotes tumorigenic capacity of glioma stem cells. Proc. Natl. Acad. Sci. USA 2013, 110, 6829–6834. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.; Lee, T.K.; Zheng, B.-J.; Chan, K.W.; Guan, X.-Y. CD133+ HCC cancer stem cells confer chemoresistance by preferential expression of the Akt/PKB survival pathway. Oncogene 2008, 27, 1749–1758. [Google Scholar] [CrossRef] [Green Version]
- Song, S.; Pei, G.; Du, Y.; Wu, J.; Ni, X.; Wang, S.; Jiang, B.; Luo, M.; Yu, J. Interaction between CD133 and PI3K-p85 promotes chemoresistance in gastric cancer cells. Am. J. Transl. Res. 2018, 10, 304–314. [Google Scholar]
- Simbulan-Rosenthal, C.M.; Dakshanamurthy, S.; Gaur, A.; Chen, Y.-S.; Fang, H.-B.; Abdussamad, M.; Zhou, H.; Zapas, J.; Calvert, V.; Petricoin, E.F.; et al. The repurposed anthelmintic mebendazole in combination with trametinib suppresses refractory NRASQ61K melanoma. Oncotarget 2017, 8, 12576–12595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tse, C.; Shoemaker, A.R.; Adickes, J.; Anderson, M.G.; Chen, J.; Jin, S.; Johnson, E.F.; Marsh, K.C.; Mitten, M.J.; Nimmer, P.; et al. ABT-263: A potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008, 68, 3421–3428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frederick, D.T.; Fragomeni, R.A.S.; Schalck, A.; Ferreiro-Neira, I.; Hoff, T.; Cooper, Z.A.; Haq, R.; Panka, D.J.; Kwong, L.N.; Davies, M.A.; et al. Clinical profiling of BCL-2 family members in the setting of BRAF inhibition offers a rationale for targeting de novo resistance using BH3 mimetics. PLoS ONE 2014, 9, e101286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmohl, J.U.; Gleason, M.K.; Dougherty, P.R.; Miller, J.S.; Vallera, D.A. Heterodimeric bispecific single chain variable fragments (scFv) killer engagers (BiKEs) enhance NK-cell activity against CD133+ colorectal cancer cells. Target. Oncol. 2016, 11, 353–361. [Google Scholar] [CrossRef] [Green Version]
- Waldron, N.N.; Kaufman, D.S.; Oh, S.; Inde, Z.; Hexum, M.K.; Ohlfest, J.R.; Vallera, D.A. Targeting tumor-initiating cancer cells with dCD133KDEL shows impressive tumor reductions in a xenotransplant model of human head and neck cancer. Mol. Cancer Ther. 2011, 10, 1829–1838. [Google Scholar] [CrossRef] [Green Version]
- Alibolandi, M.; Abnous, K.; Anvari, S.; Mohammadi, M.; Ramezani, M.; Taghdisi, S.M. CD133-targeted delivery of self-assembled PEGylated carboxymethylcellulose-SN38 nanoparticles to colorectal cancer. Artif. Cells Nanomed. Biotechnol. 2018, 46, 1159–1169. [Google Scholar] [CrossRef] [Green Version]
- DiVito, K.A.; Simbulan-Rosenthal, C.M.; Chen, Y.-S.; Trabosh, V.A.; Rosenthal, D.S. Id2, Id3 and Id4 overcome a Smad7-mediated block in tumorigenesis, generating TGF-beta-independent melanoma. Carcinogenesis 2014, 35, 951–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiVito, K.A.; Trabosh, V.A.; Chen, Y.-S.; Chen, Y.; Albanese, C.; Javelaud, D.; Mauviel, A.; Simbulan-Rosenthal, C.M.; Rosenthal, D.S. Smad7 restricts melanoma invasion by restoring N-cadherin expression and establishing heterotypic cell-cell interactions in vivo. Pigment Cell Melanoma Res. 2010, 23, 795–808. [Google Scholar] [CrossRef] [Green Version]
- Falzone, L.; Candido, S.; Salemi, R.; Basile, M.S.; Scalisi, A.; McCubrey, J.A.; Torino, F.; Signorelli, S.S.; Montella, M.; Libra, M. Computational identification of microRNAs associated to both epithelial to mesenchymal transition and NGAL/MMP-9 pathways in bladder cancer. Oncotarget 2016, 7, 72758–72766. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Song, X.; Zhu, J.; Li, M.; Ji, Y.; Wu, F.; Chen, Y.; Cui, X.; Hu, J.; Wang, L.; et al. Tumor suppressor microRNA-34a inhibits cell migration and invasion by targeting MMP-2/MMP-9/FNDC3B in esophageal squamous cell carcinoma. Int. J. Oncol. 2017, 51, 378–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiVito, K.A.; Trabosh, V.A.; Chen, Y.-S.; Simbulan-Rosenthal, C.M.; Rosenthal, D.S. Inhibitor of differentiation-4 (Id4) stimulates pigmentation in melanoma leading to histiocyte infiltration. Exp. Dermatol. 2015, 24, 101–107. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simbulan-Rosenthal, C.M.; Haribabu, Y.; Vakili, S.; Kuo, L.-W.; Clark, H.; Dougherty, R.; Alobaidi, R.; Carney, B.; Sykora, P.; Rosenthal, D.S. Employing CRISPR-Cas9 to Generate CD133 Synthetic Lethal Melanoma Stem Cells. Int. J. Mol. Sci. 2022, 23, 2333. https://doi.org/10.3390/ijms23042333
Simbulan-Rosenthal CM, Haribabu Y, Vakili S, Kuo L-W, Clark H, Dougherty R, Alobaidi R, Carney B, Sykora P, Rosenthal DS. Employing CRISPR-Cas9 to Generate CD133 Synthetic Lethal Melanoma Stem Cells. International Journal of Molecular Sciences. 2022; 23(4):2333. https://doi.org/10.3390/ijms23042333
Chicago/Turabian StyleSimbulan-Rosenthal, Cynthia M., Yogameenakshi Haribabu, Sahar Vakili, Li-Wei Kuo, Havens Clark, Ryan Dougherty, Ryyan Alobaidi, Bonnie Carney, Peter Sykora, and Dean S. Rosenthal. 2022. "Employing CRISPR-Cas9 to Generate CD133 Synthetic Lethal Melanoma Stem Cells" International Journal of Molecular Sciences 23, no. 4: 2333. https://doi.org/10.3390/ijms23042333
APA StyleSimbulan-Rosenthal, C. M., Haribabu, Y., Vakili, S., Kuo, L.-W., Clark, H., Dougherty, R., Alobaidi, R., Carney, B., Sykora, P., & Rosenthal, D. S. (2022). Employing CRISPR-Cas9 to Generate CD133 Synthetic Lethal Melanoma Stem Cells. International Journal of Molecular Sciences, 23(4), 2333. https://doi.org/10.3390/ijms23042333