
Prime Editor 3 Mediated Beta-Thalassemia Mutations of the HBB Gene in Human Erythroid Progenitor Cells


Abstract
:1. Introduction
2. Results
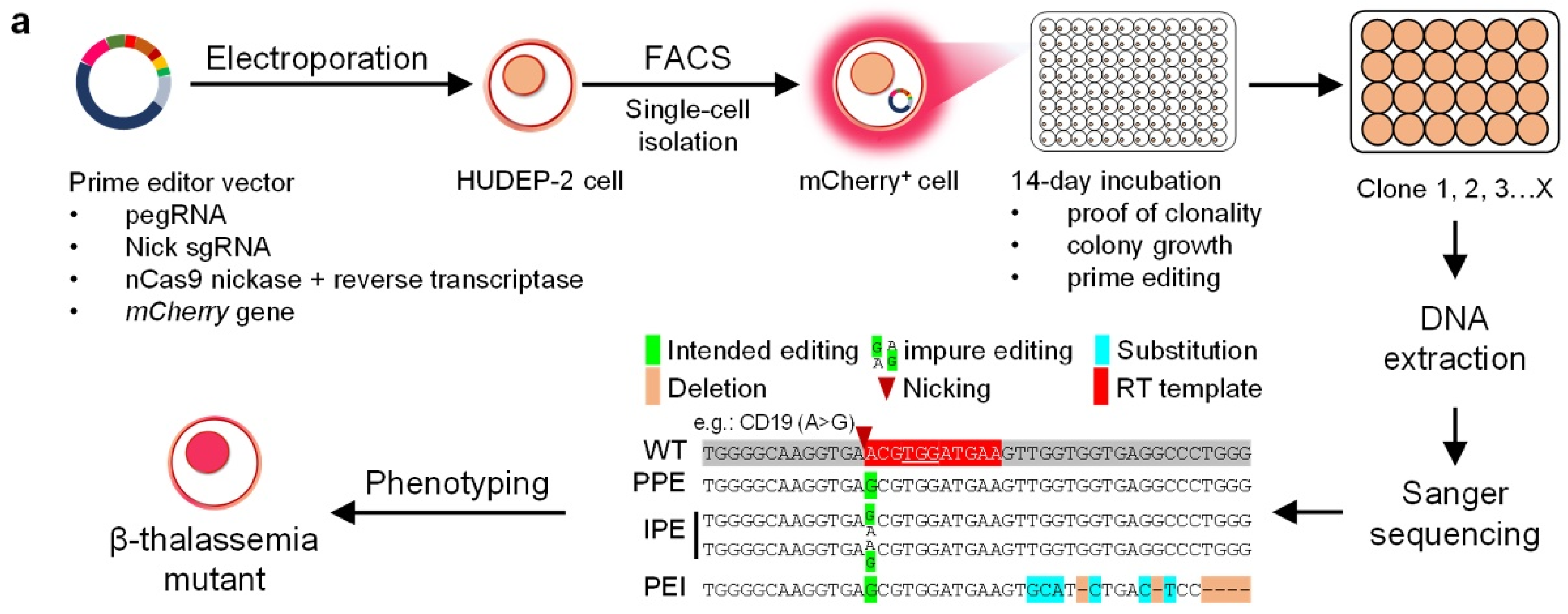
2.1. PE3 Effectively Induce Beta-Thal Mutations in HUDEP-2 Cells
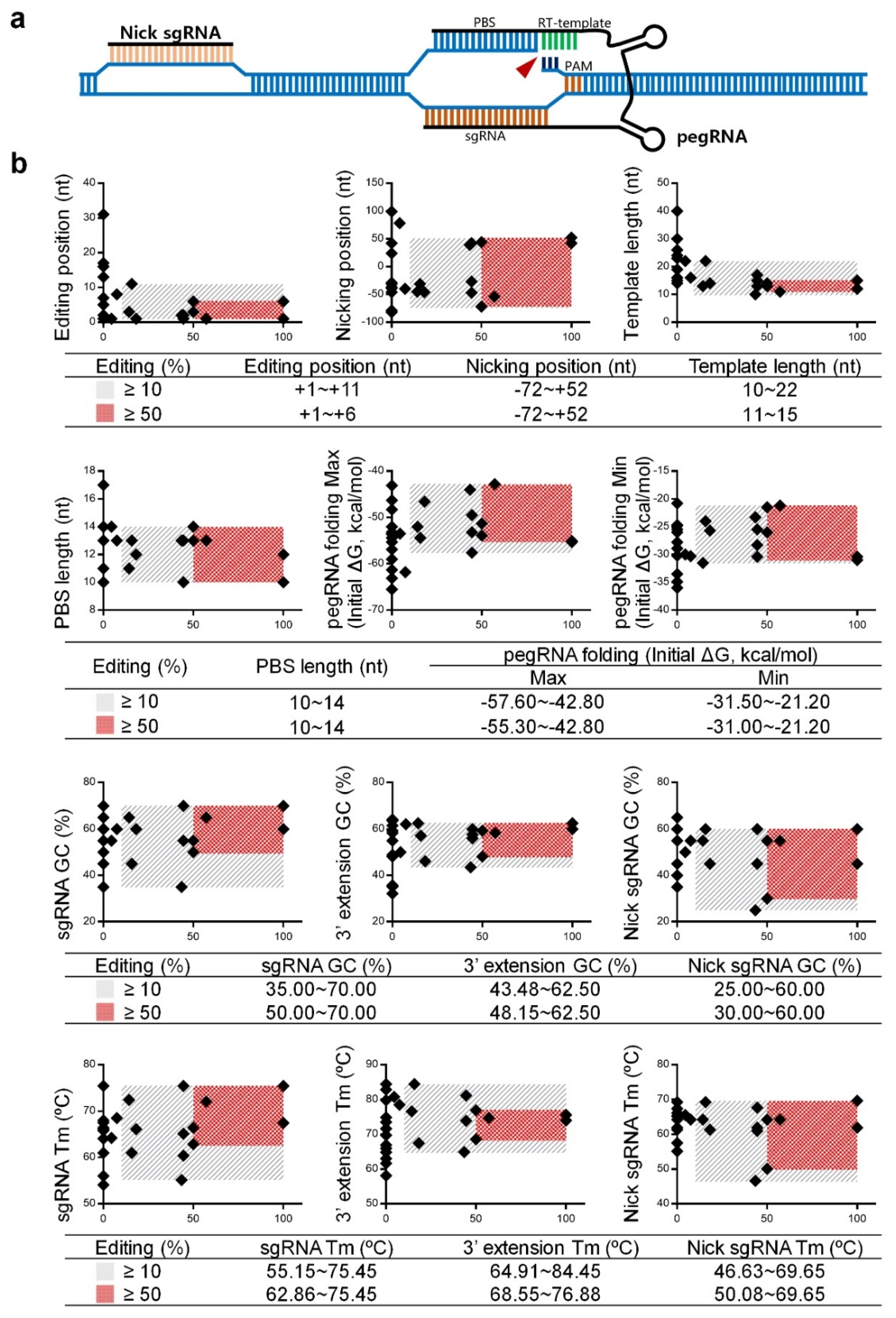
2.2. Data Analysis and Optimization of pegRNAs and Nick sgRNAs
- (a)
- pegRNA position is restricted to +1~+6 nt away from the nCas9 nicking site;
- (b)
- template length is restricted to 11~15 nt;
- (c)
- pegRNA folding requires less energy, with the upper and lower limits from −55.30~−42.80 and −31.00~−21.20 kcal/mol, respectively;
- (d)
- GC content and Tm value of the sgRNA is restricted to 50.00~70.00% and 62.86~75.45 °C, respectively;
- (e)
- GC content and Tm value of the 3′ extension is restricted to 48.15~62.50% and 68.55~76.88 °C, respectively;
- (f)
- GC content and Tm value of the nicking sgRNA is restricted to 30.00~60.00% and 50.08~69.65 °C, respectively.
2.3. Off-Target Analysis
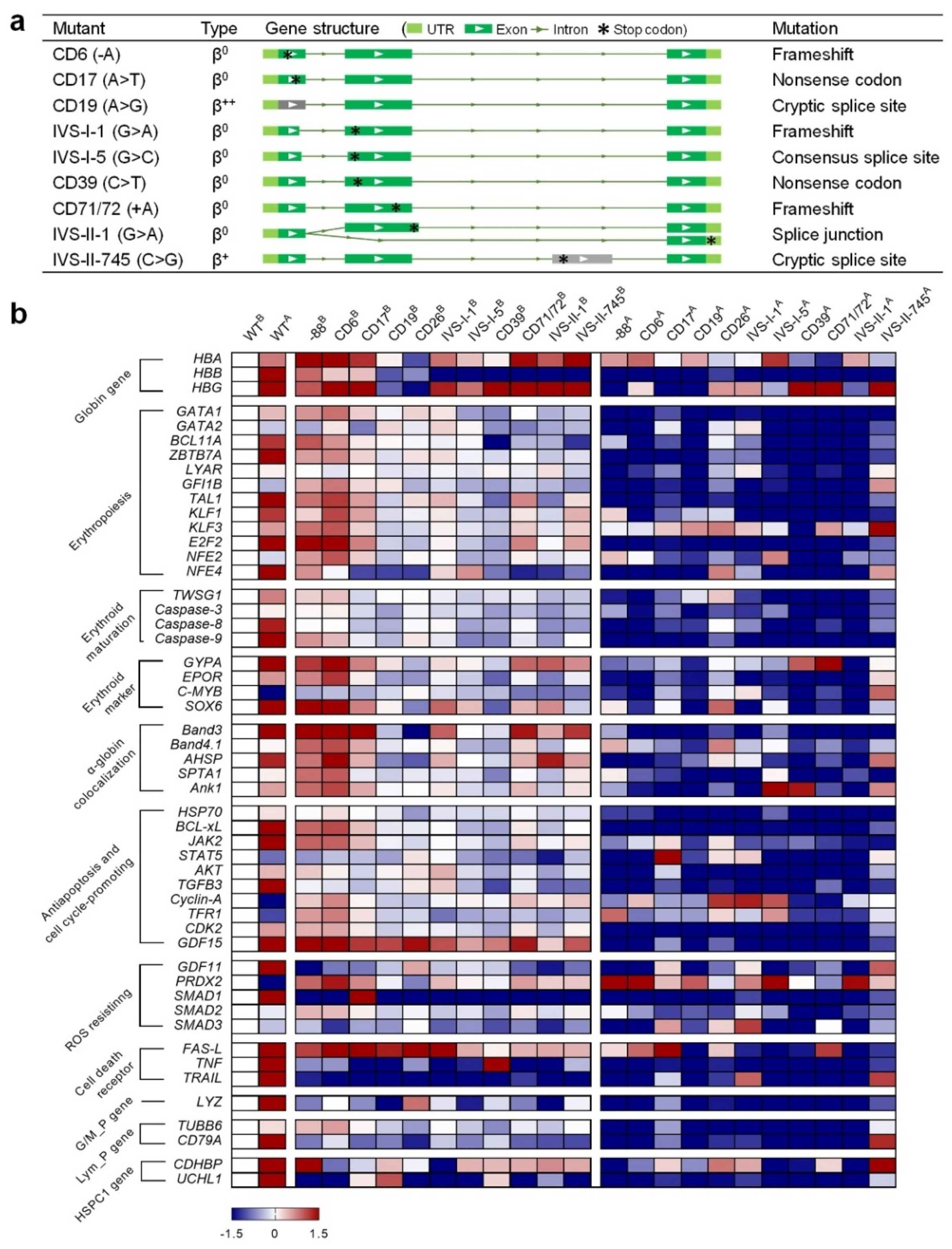
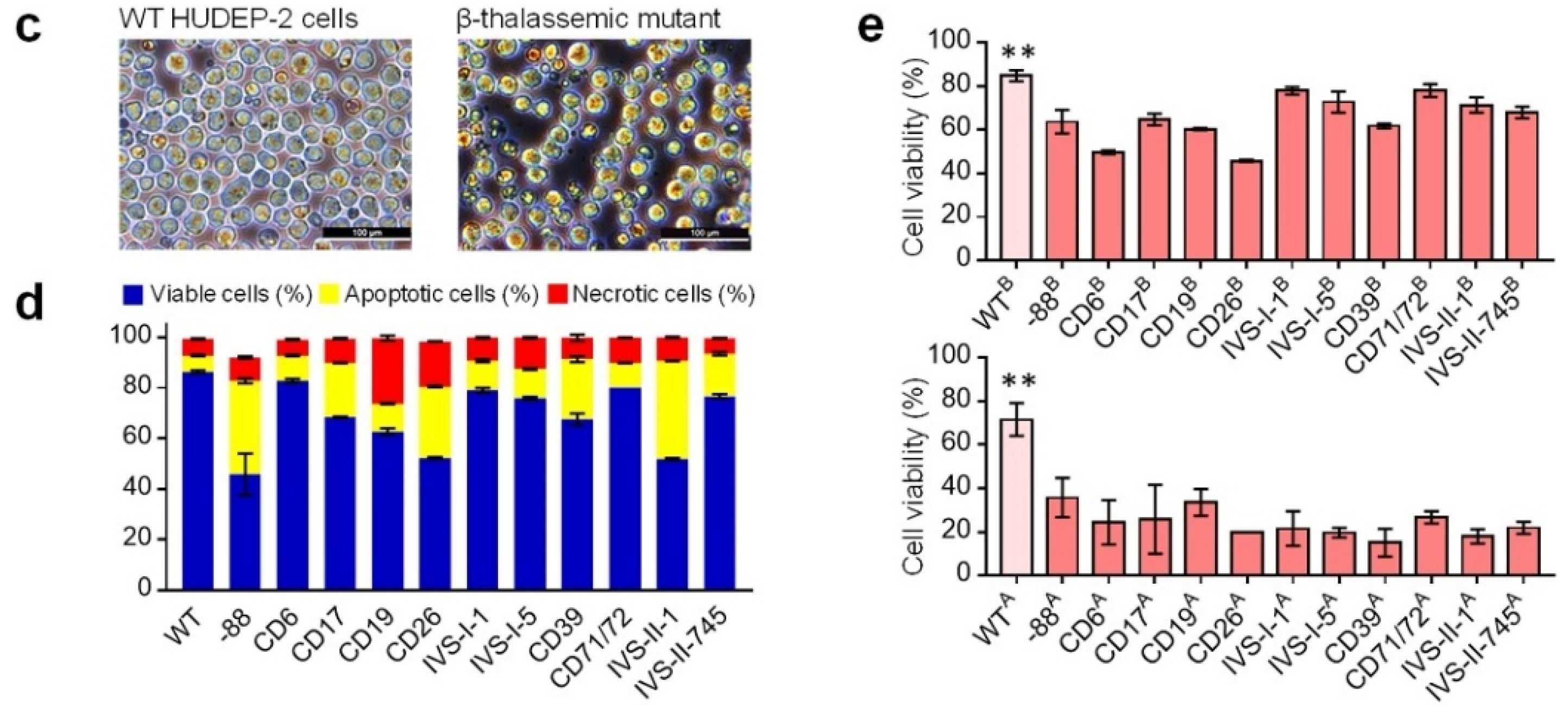
2.4. Characterization of the HUDEP-2 Beta-Thal Cells
- WT vs. beta-thal before ED: value = ln (beta-thalB/WTB);
- WT vs. WT after ED: value = ln (WTA/WTB);
- WT vs. beta-thal after ED: value = ln (beta-thalA/WTA).
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. PE3 Plasmid Construction and Purification
4.3. Establishment of HUDEP-2 Beta-Thal Mutant Clones
4.4. Identification of Desired Edits of Interest
4.5. Off-Target Analysis
4.6. RT-PCR Assays
4.7. Cell Apoptosis and Viability Assays
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Thein, S.L.; Hesketh, C.; Taylor, P.; Temperley, I.J.; Hutchinson, R.M.; Old, J.M.; Wood, W.G.; Clegg, J.B.; Weatherall, D.J. Molecular basis for dominantly inherited inclusion body beta-thalassemia. Proc. Natl. Acad. Sci. USA 1990, 87, 3924–3928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rund, D.; Rachmilewitz, E. β-thalassemia. N. Engl. J. Med. 2005, 353, 1135–1146. [Google Scholar] [CrossRef] [PubMed]
- Thein, S.L. The molecular basis of β-thalassemia. Cold Spring Harb. Perspect. Med. 2013, 3, a011700. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.H.; Fang, J.P. The current status of β-thalassemia major in Mainland China. Hemoglobin 2013, 37, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Malik, P.; Tisdale, J. Gene and Cell Therapies for Beta-Globinopathies. In American Society of Gene & Cell Therapy, 1st ed.; Springer: New York, NY, USA, 2017; p. 1. [Google Scholar]
- Thein, S.L. Genetic association studies in β-hemoglobinopathies. Hematol. Am. Soc. Hematol. Educ. Program 2013, 2013, 354–361. [Google Scholar] [CrossRef] [Green Version]
- Thein, S.L. Genetic modifiers of β-thalassemia. Haematologica 2005, 90, 649–660. [Google Scholar]
- Giardine, B.; Borg, J.; Higgs, D.R.; Peterson, K.R.; Philipsen, S.; Maglott, D.; Singleton, B.K.; Anstee, D.J.; Basak, A.N.; Clark, B.; et al. Systematic documentation and analysis of human genetic variation in hemoglobinopathies using the microattribution approach. Nat. Genet. 2011, 43, 295–301. [Google Scholar] [CrossRef]
- Hardison, R.C.; Chui, D.H.; Riemer, C.R.; Miller, W.; Carver, M.F.; Molchanova, T.P.; Efremov, G.D.; Huisman, T.H. Access to a syllabus of human hemoglobin variants (1996) via the World Wide Web. Hemoglobin 1998, 22, 113–127. [Google Scholar] [CrossRef]
- Sadelain, M.; Lisowski, L.; Samakoglu, S.; Rivella, S.; May, C.; Riviere, I. Progress toward the genetic treatment of the β-thalassemias. Ann. N. Y. Acad. Sci. 2005, 1054, 78–91. [Google Scholar] [CrossRef]
- Malik, P.; Arumugam, P.I. Gene Therapy for β-thalassemia. Hematol. Am. Soc. Hematol. Educ. Program 2005, 2005, 45–50. [Google Scholar] [CrossRef] [Green Version]
- Quek, L.; Thein, S.L. Molecular therapies in β-thalassaemia. Br. J. Haematol. 2007, 136, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Mansilla-Soto, J.; Riviere, I.; Boulad, F.; Sadelain, M. Cell and Gene Therapy for the Βeta-thalassemias: Advances and Prospects. Hum. Gene Ther. 2016, 27, 295–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thein, S.L. Molecular basis of beta thalassemia and potential therapeutic targets. Blood Cells Mol. Dis. 2018, 70, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Wu, Y.; Ren, C.; Bonanno, J.; Shen, A.H.; Shea, D.; Gehrke, J.M.; Clement, K.; Luk, K.; Yao, Q.; et al. Therapeutic base editing of human hematopoietic stem cells. Nat. Med. 2020, 26, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Anzalone, A.V.; Koblan, L.W.; Liu, D.R. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 2020, 38, 824–844. [Google Scholar] [CrossRef]
- Yang, L.; Tang, J.; Ma, X.; Lin, Y.; Ma, G.; Shan, M.; Wang, L.; Yang, Y. Progression and application of CRISPR-Cas genomic editors. Methods 2021, 194, 65–74. [Google Scholar] [CrossRef]
- Tang, L. Prime editing progress. Nat. Methods 2021, 18, 592. [Google Scholar] [CrossRef]
- Daniels, D.E.; Downes, D.J.; Ferrer-Vicens, I.; Ferguson, D.C.J.; Singleton, B.K.; Wilson, M.C.; Trakarnsanga, K.; Kurita, R.; Nakamura, Y.; Anstee, D.J.; et al. Comparing the two leading erythroid lines BEL-A and HUDEP-2. Haematologica 2020, 105, e389–e394. [Google Scholar] [CrossRef] [Green Version]
- Kurita, R.; Suda, N.; Sudo, K.; Miharada, K.; Hiroyama, T.; Miyoshi, H.; Tani, K.; Nakamura, Y. Establishment of immortalized human erythroid progenitor cell lines able to produce enucleated red blood cells. PLoS ONE 2013, 8, e59890. [Google Scholar]
- Oikonomidou, P.R.; Rivella, S. What can we learn from ineffective erythropoiesis in thalassemia? Blood Rev. 2018, 32, 130–143. [Google Scholar] [CrossRef] [PubMed]
- Chow, R.D.; Chen, J.S.; Shen, J.; Chen, S. A web tool for the design of prime-editing guide RNAs. Nat. Biomed. Eng. 2021, 5, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Petri, K.; Zhang, W.; Ma, J.; Schmidts, A.; Lee, H.; Horng, J.E.; Kim, D.Y.; Kurt, I.C.; Clement, K.; Hsu, J.Y.; et al. CRISPR prime editing with ribonucleoprotein complexes in zebrafish and primary human cells. Nat. Biotechnol. 2022, 40, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.; Park, J.; Kim, J.S. Cas-OFFinder: A fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 2014, 30, 1473–1475. [Google Scholar] [CrossRef] [Green Version]
- Trakarnsanga, K.; Wilson, M.C.; Lau, W.; Singleton, B.K.; Parsons, S.F.; Sakuntanaga, P.; Kurita, R.; Nakamura, Y.; Anstee, D.J.; Frayne, J. Induction of adult levels of beta-globin in human erythroid cells that intrinsically express embryonic or fetal globin by transduction with KLF1 and BCL11A-XL. Haematologica 2014, 99, 1677–1685. [Google Scholar] [CrossRef]
- Canver, M.C.; Smith, E.C.; Sher, F.; Pinello, L.; Sanjana, N.E.; Shalem, O.; Chen, D.D.; Schupp, P.G.; Vinjamur, D.S.; Garcia, S.P.; et al. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature 2015, 527, 192–197. [Google Scholar] [CrossRef] [Green Version]
- Wienert, B.; Martyn, G.E.; Kurita, R.; Nakamura, Y.; Quinlan, K.G.R.; Crossley, M. KLF1 drives the expression of fetal hemoglobin in British HPFH. Blood 2017, 130, 803–807. [Google Scholar] [CrossRef]
- Liu, N.; Hargreaves, V.V.; Zhu, Q.; Kurland, J.V.; Hong, J.; Kim, W.; Sher, F.; Macias-Trevino, C.; Rogers, J.M.; Kurita, R.; et al. Direct Promoter Repression by BCL11A Controls the Fetal to Adult Hemoglobin Switch. Cell 2018, 173, 430–442.e17. [Google Scholar] [CrossRef] [Green Version]
- Martyn, G.E.; Wienert, B.; Kurita, R.; Nakamura, Y.; Quinlan, K.G.R.; Crossley, M. A natural regulatory mutation in the proximal promoter elevates fetal globin expression by creating a de novo GATA1 site. Blood 2019, 133, 852–856. [Google Scholar] [CrossRef] [Green Version]
- Gardenghi, S.; Grady, R.W.; Rivella, S. Anemia, ineffective erythropoiesis, and hepcidin: Interacting factors in abnormal iron metabolism leading to iron overload in β-thalassemia. Hematol. Oncol. Clin. 2010, 24, 1089–1107. [Google Scholar] [CrossRef] [Green Version]
- Ribeil, J.A.; Arlet, J.B.; Dussiot, M.; Moura, I.C.; Courtois, G.; Hermine, O. Ineffective erythropoiesis in β-thalassemia. Sci. World J. 2013, 2013, 394295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arlet, J.B.; Dussiot, M.; Moura, I.C.; Hermine, O.; Courtois, G. Novel players in β-thalassemia dyserythropoiesis and new therapeutic strategies. Curr. Opin. Hematol. 2016, 23, 181–188. [Google Scholar] [CrossRef] [PubMed]
- El Nemer, W.; Godard, A.; El Hoss, S. Ineffective erythropoiesis in sickle cell disease: New insights and future implications. Curr. Opin. Hematol. 2021, 28, 171–176. [Google Scholar] [CrossRef] [PubMed]
- El Hoss, S.; Cochet, S.; Godard, A.; Yan, H.; Dussiot, M.; Frati, G.; Boutonnat-Faucher, B.; Laurance, S.; Renaud, O.; Joseph, L.; et al. Fetal hemoglobin rescues ineffective erythropoiesis in sickle cell disease. Haematologica 2021, 106, 2707–2719. [Google Scholar] [CrossRef] [PubMed]
- Longo, F.; Piolatto, A.; Ferrero, G.B.; Piga, A. Ineffective Erythropoiesis in β-Thalassaemia: Key Steps and Therapeutic Options by Drugs. Int. J. Mol. Sci. 2021, 22, 7229. [Google Scholar] [CrossRef]
- Hua, P.; Roy, N.; de la Fuente, J.; Wang, G.; Thongjuea, S.; Clark, K.; Roy, A.; Psaila, B.; Ashley, N.; Harrington, Y.; et al. Single-cell analysis of bone marrow-derived CD34+ cells from children with sickle cell disease and thalassemia. Blood 2019, 134, 2111–2115. [Google Scholar] [CrossRef]
- Mao, B.; Huang, S.; Lu, X.; Sun, W.; Zhou, Y.; Pan, X.; Yu, J.; Lai, M.; Chen, B.; Zhou, Q.; et al. Early Development of Definitive Erythroblasts from Human Pluripotent Stem Cells Defined by Expression of Glycophorin A/CD235a, CD34, and CD36. Stem Cell Rep. 2016, 7, 869–883. [Google Scholar] [CrossRef] [Green Version]
- Tanno, T.; Bhanu, N.V.; Oneal, P.A.; Goh, S.H.; Staker, P.; Lee, Y.T.; Moroney, J.W.; Reed, C.H.; Luban, N.L.; Wang, R.H.; et al. High levels of GDF15 in thalassemia suppress expression of the iron regulatory protein hepcidin. Nat. Med. 2007, 13, 1096–1101. [Google Scholar] [CrossRef]
- Forster, L.; Cornwall, S.; Finlayson, J.; Ghassemifar, R. Cell cycle, proliferation and apoptosis in erythroblasts cultured from patients with beta-thalassaemia major. Br. J. Haematol. 2016, 175, 539–542. [Google Scholar] [CrossRef] [Green Version]
- Forster, L.; McCooke, J.; Bellgard, M.; Joske, D.; Finlayson, J.; Ghassemifar, R. Differential gene expression analysis in early and late erythroid progenitor cells in beta-thalassaemia. Br. J. Haematol. 2015, 170, 257–267. [Google Scholar] [CrossRef]
- Libani, I.V.; Guy, E.C.; Melchiori, L.; Schiro, R.; Ramos, P.; Breda, L.; Scholzen, T.; Chadburn, A.; Liu, Y.; Kernbach, M.; et al. Decreased differentiation of erythroid cells exacerbates ineffective erythropoiesis in β-thalassemia. Blood 2008, 112, 875–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surun, D.; Schneider, A.; Mircetic, J.; Neumann, K.; Lansing, F.; Paszkowski-Rogacz, M.; Hanchen, V.; Lee-Kirsch, M.A.; Buchholz, F. Efficient Generation and Correction of Mutations in Human iPS Cells Utilizing mRNAs of CRISPR Base Editors and Prime Editors. Genes 2020, 11, 511. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, X.; He, S.; Huang, S.; Li, C.; Chen, Y.; Liu, Z.; Huang, X.; Wang, X. Efficient generation of mouse models with the prime editing system. Cell Discov. 2020, 6, 27. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Li, J.; Chen, J.; Yan, L.; Xia, L. Precise Modifications of Both Exogenous and Endogenous Genes in Rice by Prime Editing. Mol. Plant 2020, 13, 671–674. [Google Scholar] [CrossRef] [PubMed]
- Schene, I.F.; Joore, I.P.; Oka, R.; Mokry, M.; van Vugt, A.H.M.; van Boxtel, R.; van der Doef, H.P.J.; van der Laan, L.J.W.; Verstegen, M.M.A.; van Hasselt, P.M.; et al. Prime editing for functional repair in patient-derived disease models. Nat. Commun. 2020, 11, 5352. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Zong, Y.; Xue, C.; Wang, S.; Jin, S.; Zhu, Z.; Wang, Y.; Anzalone, A.V.; Raguram, A.; Doman, J.L.; et al. Prime genome editing in rice and wheat. Nat. Biotechnol. 2020, 38, 582–585. [Google Scholar] [CrossRef] [PubMed]
- Bosch, J.A.; Birchak, G.; Perrimon, N. Precise genome engineering in Drosophila using prime editing. Proc. Natl. Acad. Sci. USA 2021, 118, e2021996118. [Google Scholar] [CrossRef]
- Caso, F.; Davies, B. Base editing and prime editing in laboratory animals. Lab. Anim. 2021, 56, 35–49. [Google Scholar] [CrossRef]
- Park, S.J.; Jeong, T.Y.; Shin, S.K.; Yoon, D.E.; Lim, S.Y.; Kim, S.P.; Choi, J.; Lee, H.; Hong, J.I.; Ahn, J.; et al. Targeted mutagenesis in mouse cells and embryos using an enhanced prime editor. Genome Biol. 2021, 22, 170. [Google Scholar] [CrossRef]
- Kweon, J.; Yoon, J.K.; Jang, A.H.; Shin, H.R.; See, J.E.; Jang, G.; Kim, J.I.; Kim, Y. Engineered prime editors with PAM flexibility. Mol. Ther. 2021, 29, 2001–2007. [Google Scholar] [CrossRef]
- Concordet, J.P.; Haeussler, M. CRISPOR: Intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 2018, 46, W242–W245. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Zhang, H.; Lin, A.; Wu, Z.; Li, T.; Zhang, X.; Chen, H.; Lu, D. Multi-Omics Analysis in β-Thalassemia Using an HBB Gene-Knockout Human Erythroid Progenitor Cell Model. Int. J. Mol. Sci. 2022, 23, 2807. [Google Scholar] [CrossRef]
- Truett, G.E.; Heeger, P.; Mynatt, R.L.; Truett, A.A.; Walker, J.A.; Warman, M.L. Preparation of PCR-quality mouse genomic DNA with hot sodium hydroxide and tris (HotSHOT). Biotechniques 2000, 29, 52–54. [Google Scholar] [CrossRef] [PubMed]
- David, F.P.; Rougemont, J.; Deplancke, B. GETPrime 2.0: Gene- and transcript-specific qPCR primers for 13 species including polymorphisms. Nucleic Acids Res. 2017, 45, D56–D60. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutant | Distribution | Type |
|---|---|---|
| -88 (C>T) | African-American, Asian Indian | beta++ |
| -28 (A>G) | African, Southeast Asian | beta+ |
| CD1 (-G) | Mediterranean | beta0 |
| CD6 (-A) | North African | beta0 |
| CD14/15 (+G) | Chinese | beta0 |
| CD17 (A>T) | Chinese, Japanese | beta0 |
| CD19 (A>G) | Southeast Asian | beta++ |
| CD26 (G>A) | Southeast Asian, European | beta+ |
| CD27/28 (+C) | Chinese, Thai | beta0 |
| IVS-I-1 (G>A) | North African, Italian, Greek, Balkan, West European | beta0 |
| IVS-I-5 (G>C) | Asian Indian, Southeast Asian, Melanesian, Middle East | beta0 |
| IVS-I-6 (T>C) | Italian, Greek, Balkan, West European (Portugal) | beta+ |
| IVS-I-110 (G>A) | Italian, Greek, Cypriot, Balkan, Israeli, Lebanese, North African, West European | beta+ |
| CD39 (C>T) | Italian, Greek, Balkan, North African, Israeli, West European, Middle East | beta0 |
| CD41/42 (-TTCT) | Chinese, Southeast Asian, Indian | beta++ |
| CD44 (-C) | Israeli, Middle East | beta++ |
| CD71/72 (+A) | Chinese | beta0 |
| IVS-II-1 (G>A) | Israeli, Middle East, Japanese, Turkish | beta0 |
| IVS-II-654 (C>T) | Chinese, Southeast Asian, Japanese | beta0/beta+ |
| IVS-II-745 (C>G) | Italian, Greek, Turkish | beta+ |
| Mutation | Formula | Total Samples | Type of Edits | ||
|---|---|---|---|---|---|
| PPE | IPE | PEI | |||
| -88 | 1 | 1 | |||
| -28 | 26 | 1 | |||
| CD1 | 22 | 1 | 4 | ||
| CD6 | 9 | 4 | 5 | ||
| CD14/15 | 3 | ||||
| CD17 | F1 | 67 | 5 | 1 | |
| F2 | 14 | 1 | 1 | ||
| CD19 | 7 | 2 | 2 | 2 | |
| CD26 | F1 | 19 | 3 | ||
| F2 | 2 | 1 | |||
| CD27/28 | F1 | 43 | |||
| F2 | 21 | 5 | |||
| IVS-I-1 | 9 | 1 | 3 | 2 | |
| IVS-I-5 | 2 | 1 | 1 | ||
| IVS-I-6 | 3 | ||||
| IVS-I-110 | F1 | 77 | |||
| F2 | 3 | ||||
| CD39 | 11 | 1 | 1 | 4 | |
| CD41/42 | F1 | 24 | 1 | ||
| F2 | 38 | ||||
| CD44 | 5 | ||||
| CD71/72 | 18 | 2 | 6 | ||
| IVS-II-1 | 2 | 1 | |||
| IVS-II-654 | F1 | 12 | |||
| F2 | 20 | ||||
| F3 | 1 | ||||
| IVS-II-745 | 23 | 3 | 7 | 4 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.; Zhou, Q.; Chen, H.; Lu, D. Prime Editor 3 Mediated Beta-Thalassemia Mutations of the HBB Gene in Human Erythroid Progenitor Cells. Int. J. Mol. Sci. 2022, 23, 5002. https://doi.org/10.3390/ijms23095002
Zhang H, Zhou Q, Chen H, Lu D. Prime Editor 3 Mediated Beta-Thalassemia Mutations of the HBB Gene in Human Erythroid Progenitor Cells. International Journal of Molecular Sciences. 2022; 23(9):5002. https://doi.org/10.3390/ijms23095002
Chicago/Turabian StyleZhang, Haokun, Qinlinglan Zhou, Hongyan Chen, and Daru Lu. 2022. "Prime Editor 3 Mediated Beta-Thalassemia Mutations of the HBB Gene in Human Erythroid Progenitor Cells" International Journal of Molecular Sciences 23, no. 9: 5002. https://doi.org/10.3390/ijms23095002
APA StyleZhang, H., Zhou, Q., Chen, H., & Lu, D. (2022). Prime Editor 3 Mediated Beta-Thalassemia Mutations of the HBB Gene in Human Erythroid Progenitor Cells. International Journal of Molecular Sciences, 23(9), 5002. https://doi.org/10.3390/ijms23095002