
A Review of Alpha-1 Antitrypsin Binding Partners for Immune Regulation and Potential Therapeutic Application

 , ,
, ,
Abstract
:1. An Introduction to Alpha-1 Antitrypsin
Control of Alpha-1 Antitrypsin Production
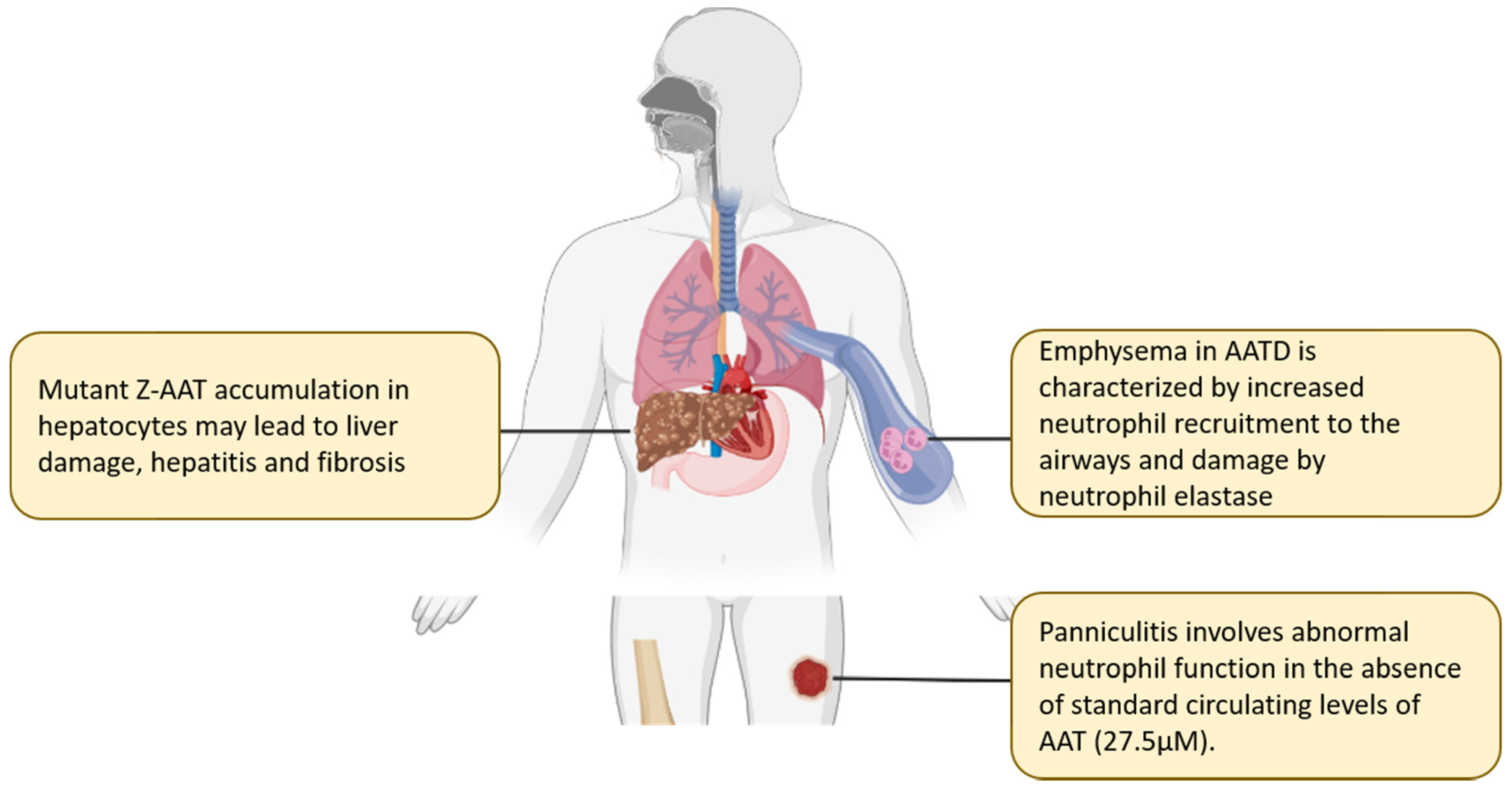
2. Alpha-1 Antitrypsin Deficiency States
3. Anti-Inflammatory Effects of Alpha-1 Antitrypsin beyond Protease Inhibition
4. Alpha-1 Antitrypsin Molecular Interactions
4.1. Mechanisms of Binding
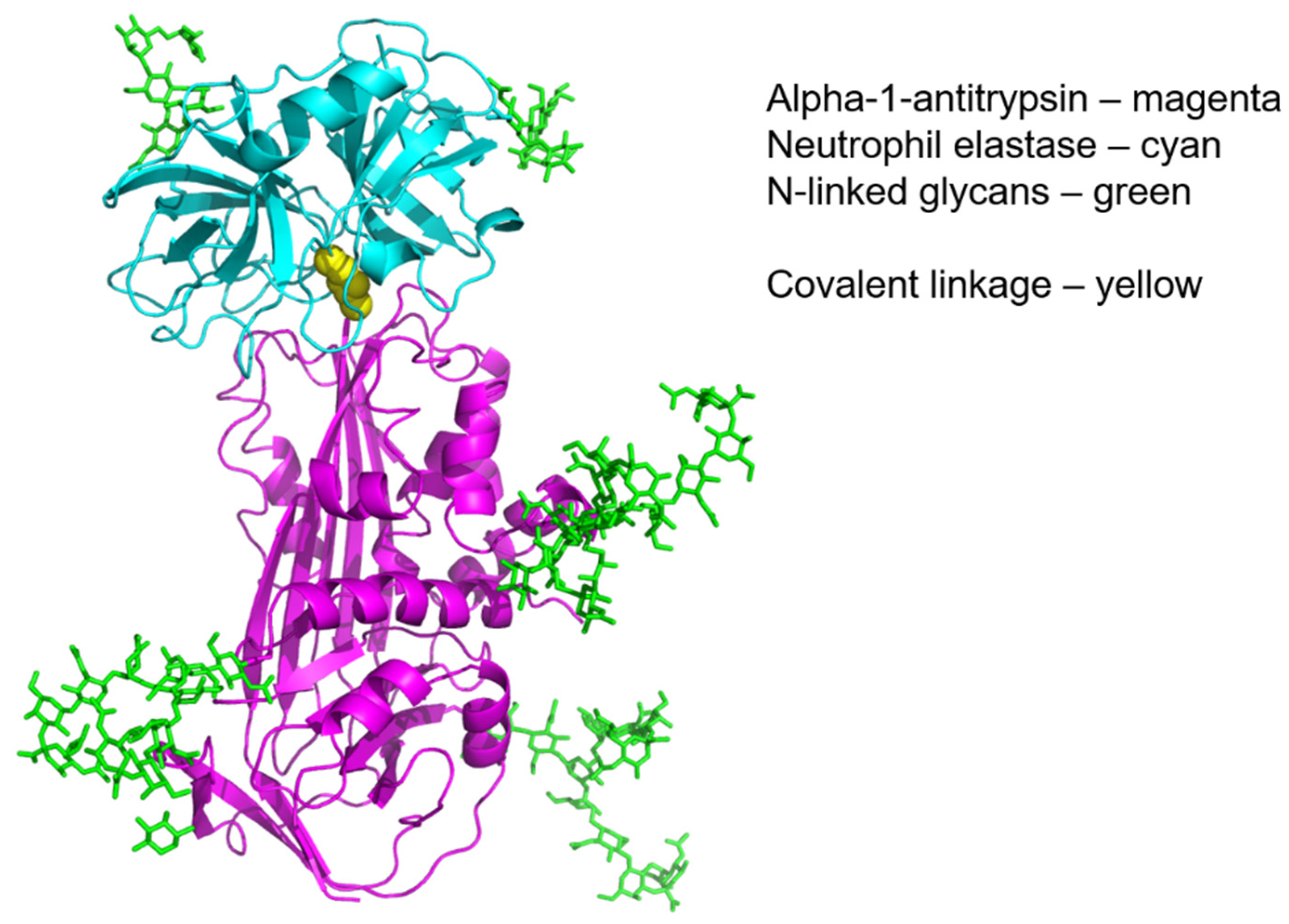
4.2. Alpha-1 Antitrypsin-Specific RCL Protease Binding
4.3. Alpha-1 Antitrypsin RCL Self-Binding Leading to Polymer Formation
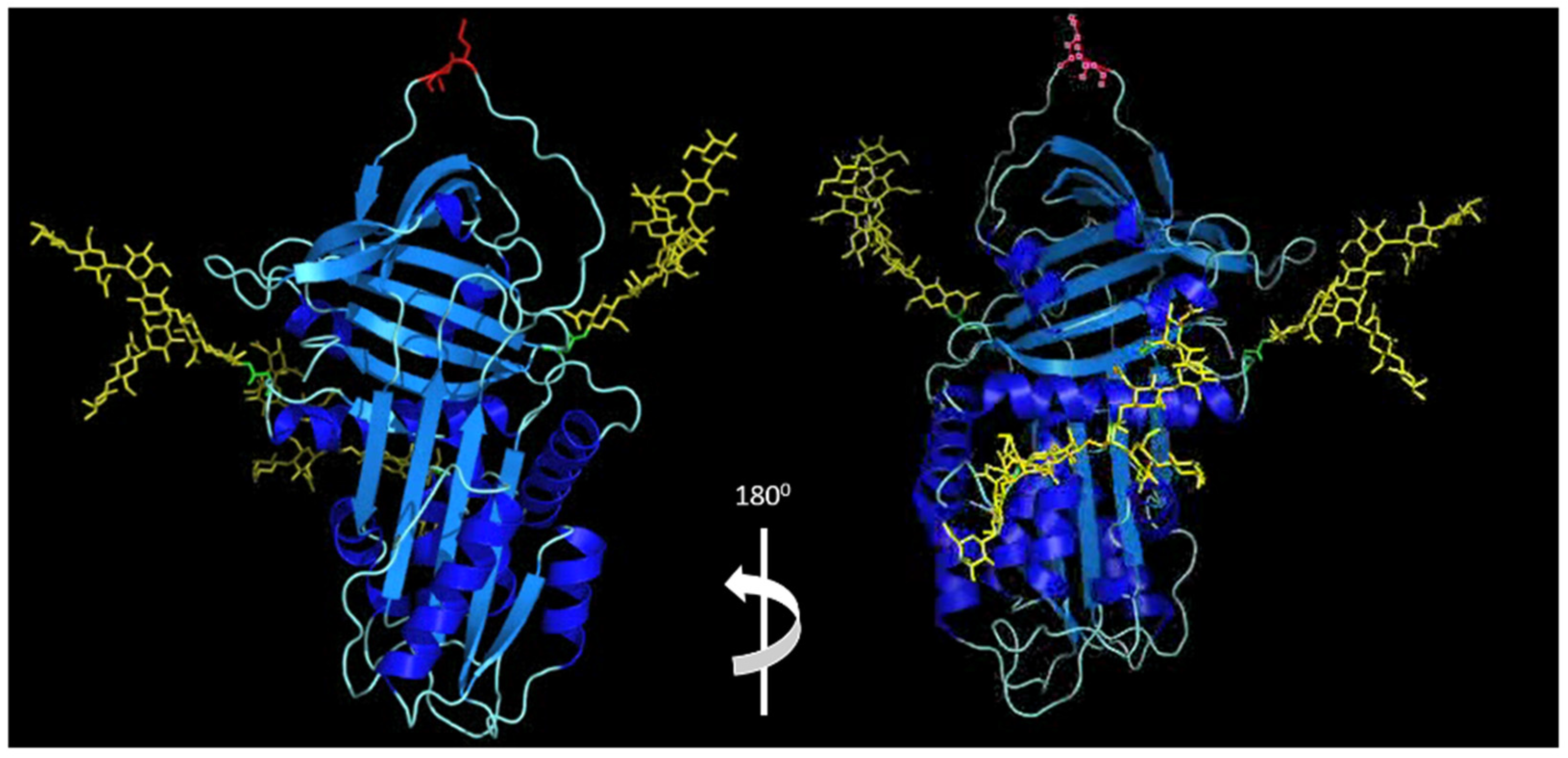
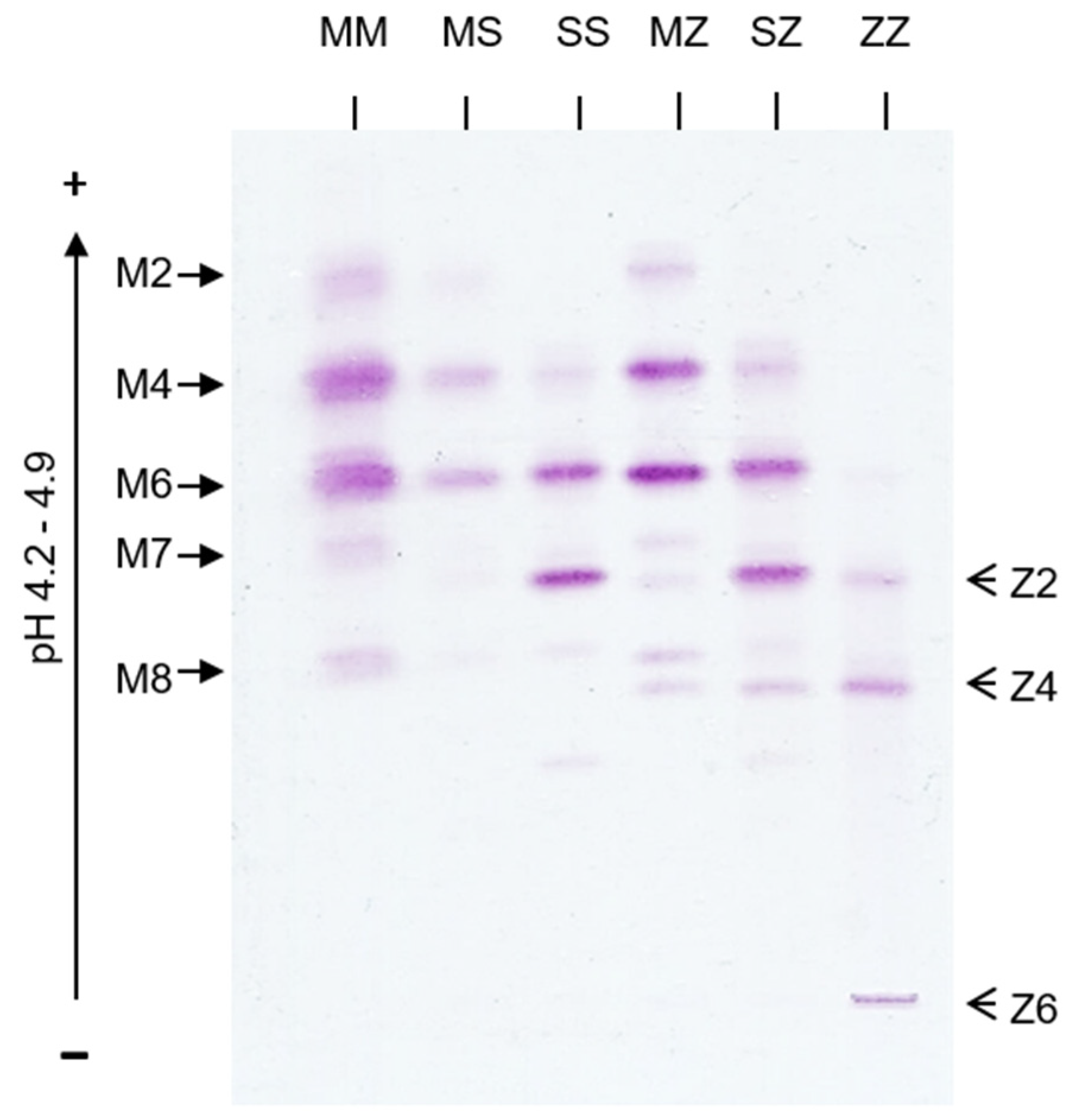
4.4. Alpha-1 Antitrypsin Electrostatic Interactions and Post-Translational Glycosylation Effects
4.5. Hydrophobic Binding of Alpha-1 Antitrypsin with the Lipoprotein System
4.6. Alpha-1 Antitrypsin Cysteine Binding Potential
4.7. The Heparin Binding Motif of Alpha-1 Antitrypsin
5. The Impact of Alpha-1 Antitrypsin Binding in Health and Disease
5.1. Alpha-1 Antitrypsin Protease Binding and the Coagulation System
5.2. Alpha-1 Antitrypsin Protein Complexes and Tissue Inflammation
5.3. Alpha-1 Antitrypsin Binding Partners and the Complement System
5.4. Alpha-1 Antitrypsin Protease Binding and COVID-19
6. AAT Augmentation Therapy
AAT Replacement Therapy in Acute and Chronic Disease
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Schroeder, W.T.; Miller, M.F.; Woo, S.L.; Saunders, G.F. Chromosomal localization of the human alpha 1-antitrypsin gene (PI) to 14q31–32. Am. J. Hum. Genet. 1985, 37, 868–872. [Google Scholar] [PubMed]
- Hunt, L.T.; Dayhoff, M.O. A surprising new protein superfamily containing ovalbumin, antithrombin-III, and alpha1-proteinase inhibitor. Biochem. Biophys. Res. Commun. 1980, 95, 864–871. [Google Scholar] [CrossRef]
- Van Gent, D.; Sharp, P.; Morgan, K.; Kalsheker, N. Serpins: Structure, function and molecular evolution. Int. J. Biochem. Cell Biol. 2003, 35, 1536–1547. [Google Scholar] [CrossRef]
- Hammond, G.L.; Smith, C.L.; Paterson, N.A.M.; Sibbald, W.J. A role for corticosteroid-binding globulin in delivery of cortisol to activated neutrophils. J. Clin. Endocrinol. Metab. 1990, 71, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Huber, R.; Carrell, R.W. Implications of the three-dimensional structure of alpha.1-antitrypsin for structure and function of serpins. Biochemistry 1989, 28, 8951–8966. [Google Scholar] [CrossRef]
- O’Dwyer, C.A.; O’Brien, M.E.; Wormald, M.R.; White, M.M.; Banville, N.; Hurley, K.; McCarthy, C.; McElvaney, N.G.; Reeves, E.P. The BLT1 inhibitory function of α-1 antitrypsin augmentation therapy disrupts leukotriene B4 neutrophil signaling. J. Immunol. 2015, 195, 3628–3641. [Google Scholar] [CrossRef] [Green Version]
- Bergin, D.A.; Reeves, E.P.; Meleady, P.; Henry, M.; McElvaney, O.J.; Carroll, T.P.; Condron, C.; Chotirmall, S.H.; Clynes, M.; O’Neill, S.J.; et al. α-1 antitrypsin regulates human neutrophil chemotaxis induced by soluble immune complexes and IL-8. J. Clin. Investig. 2010, 120, 4236–4250. [Google Scholar] [CrossRef] [Green Version]
- Mashiba, S.; Wada, Y.; Takeya, M.; Sugiyama, A.; Hamakubo, T.; Nakamura, A.; Noguchi, N.; Niki, E.; Izumi, A.; Kobayashi, M.; et al. In Vivo complex formation of oxidized α1-antitrypsin and LDL. Arter. Thromb. Vasc. Biol. 2001, 21, 1801–1808. [Google Scholar] [CrossRef] [Green Version]
- Tomasi, T.B., Jr.; Hauptman, S.P. The binding of alpha-1 antitrypsin to human IgA. J. Immunol. 1974, 112, 2274–2277. [Google Scholar]
- Laurell, C.B.; Thulin, E. Complexes in human plasma between α1-antitrypsin and IgA, and α1-antitrypsin and fibrinogen. Scand. J. Immunol. 1975, 4, 7–12. [Google Scholar] [CrossRef]
- Laurell, C.B.; Thulin, E. Complexes in plasma between light chain κ immunoglobulins and α1-antitrypsin respectively prealbumin. Immunochemistry 1974, 11, 703–709. [Google Scholar] [CrossRef]
- Finotti, P.; Pagetta, A. A heat shock protein70 fusion protein with α1-antitrypsin in plasma of Type 1 diabetic subjects. Biochem. Biophys. Res. Commun. 2004, 315, 297–305. [Google Scholar] [CrossRef]
- Pagetta, A.; Folda, A.; Brunati, A.M.; Finotti, P. Identification and purification from the plasma of Type 1 diabetic subjects of a proteolytically active Grp94: Evidence that Grp94 is entirely responsible for plasma proteolytic activity. Diabetologia 2003, 46, 996–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.-M.; Finne, P.; Leinonen, J.; Vesalainen, S.; Nordling, S.; Stenman, U.-H. Measurement of the complex between prostate-specific antigen and α1-protease inhibitor in serum. Clin. Chem. 1999, 45, 814–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janciauskiene, S.; Eriksson, S. In vitro complex formation between cholesterol and α1-proteinase inhibitor. FEBS Lett. 1993, 316, 269–272. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, M.E.; Fee, L.; Browne, N.; Carroll, T.P.; Meleady, P.; Henry, M.; McQuillan, K.; Murphy, M.P.; Logan, M.; McCarthy, C.; et al. Activation of complement component 3 is associated with airways disease and pulmonary emphysema in alpha-1 antitrypsin deficiency. Thorax 2020, 75, 321–330. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, C.; Dunlea, D.M.; Saldova, R.; Henry, M.; Meleady, P.; McElvaney, O.J.; Marsh, B.; Rudd, P.M.; Reeves, E.P.; McElvaney, N.G. Glycosylation repurposes alpha-1 antitrypsin for resolution of community-acquired pneumonia. Am. J. Respir. Crit. Care Med. 2018, 197, 1346–1349. [Google Scholar] [CrossRef]
- Bergin, D.A.; Reeves, E.P.; Hurley, K.; Wolfe, R.; Jameel, R.; Fitzgerald, S.; McElvaney, N.G. The circulating proteinase inhibitor α-1 antitrypsin regulates neutrophil degranulation and autoimmunity. Sci. Transl. Med. 2014, 6, 217. [Google Scholar] [CrossRef]
- Reeves, E.P.; Bergin, D.A.; Fitzgerald, S.; Hayes, E.; Keenan, J.; Henry, M.; Meleady, P.; Vega-Carrascal, I.; Murray, M.A.; Low, T.B.; et al. A novel neutrophil derived inflammatory biomarker of pulmonary exacerbation in cystic fibrosis. J. Cyst. Fibros. 2012, 11, 100–107. [Google Scholar] [CrossRef] [Green Version]
- Karnaukhova, E.; Krupnikova, S.S.; Rajabi, M.; Alayash, A.I. Heme binding to human alpha-1 proteinase inhibitor. Biochim. Biophys. Acta Gen. Subj. 2012, 1820, 2020–2029. [Google Scholar] [CrossRef]
- Pandur, E.; Nagy, J.; Poór, V.S.; Sarnyai, Á.; Huszár, A.; Miseta, A.; Sipos, K. α-1 antitrypsin binds preprohepcidin intracellularly and prohepcidin in the serum. FEBS J. 2009, 276, 2012–2021. [Google Scholar] [CrossRef] [PubMed]
- Karnaukhova, E. Interactions of α1–proteinase inhibitor with small ligands of therapeutic potential: Binding with retinoic acid. Amino Acids 2010, 38, 1011–1020. [Google Scholar] [CrossRef] [PubMed]
- Petrache, I.; Fijalkowska, I.; Medler, T.R.; Skirball, J.; Cruz, P.; Zhen, L.; Petrache, H.I.; Flotte, T.R.; Tuder, R.M. Alpha-1 antitrypsin inhibits caspase-3 activity, preventing lung endothelial cell apoptosis. Am. J. Pathol. 2006, 169, 1155–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; He, Y.; Abraham, B.; Rouhani, F.N.; Brantly, M.L.; Scott, D.E.; Reed, J.L. Cytosolic, autocrine alpha-1 proteinase inhibitor (A1PI) inhibits caspase-1 and blocks IL-1β dependent cytokine release in monocytes. PLoS ONE 2012, 7, e51078. [Google Scholar] [CrossRef]
- Toldo, S.; Seropian, I.M.; Mezzaroma, E.; Van Tassell, B.W.; Salloum, F.N.; Lewis, E.C.; Voelkel, N.; Dinarello, C.A.; Abbate, A. Alpha-1 antitrypsin inhibits caspase-1 and protects from acute myocardial ischemia–reperfusion injury. J. Mol. Cell. Cardiol. 2011, 51, 244–251. [Google Scholar] [CrossRef]
- Al-Omari, M.; Korenbaum, E.; Ballmaier, M.; Lehmann, U.; Jonigk, D.; Manstein, D.J.; Welte, T.; Mahadeva, R.; Janciauskiene, S. Acute-phase protein α1-antitrypsin inhibits neutrophil calpain I and induces random migration. Mol. Med. 2011, 17, 865–874. [Google Scholar] [CrossRef]
- Gorrini, M.; Lupi, A.; Iadarola, P.; Dos Santos, C.; Rognoni, P.; Dalzoppo, D.; Carrabino, N.; Pozzi, E.; Baritussio, A.; Luisetti, M. SP-A binds alpha1-antitrypsin in vitro and reduces the association rate constant for neutrophil elastase. Respir. Res. 2005, 6, 146. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, K.; Suzuki, Y.; Saito, A.; Fukuda, K.; Hamanishi, C.; Munakata, H. Aggrecanase-1 (ADAMTS-4) interacts with α1-antitrypsin. Biochim. Biophys. Acta Gen. Subj. 2005, 1725, 152–159. [Google Scholar] [CrossRef]
- Janciauskiene, S.; Eriksson, S. The interaction of hydrophobic bile acids with the α1-proteinase inhibitor. FEBS Lett. 1994, 343, 141–145. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, Y.; Akaike, T.; Maeda, H. S-Nitrosylated human α1-protease inhibitor. Biochim. Biophys. Acta Protein Struct. Mol. Enzym. 2000, 1477, 90–97. [Google Scholar] [CrossRef]
- Perlmutter, D.H.; Cole, F.S.; Kilbridge, P.; Rossing, T.H.; Colten, H.R. Expression of the alpha 1-proteinase inhibitor gene in human monocytes and macrophages. Proc. Natl. Acad. Sci. USA 1985, 82, 795–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulgrew, A.T.; Taggart, C.; Lawless, M.W.; Greene, C.; Brantly, M.L.; O’Neill, S.J.; McElvaney, N.G. Z α1-antitrypsin polymerizes in the lung and acts as a neutrophil chemoattractant. Chest 2004, 125, 1952–1957. [Google Scholar] [CrossRef] [PubMed]
- Molmenti, E.P.; Perlmutter, D.H.; Rubin, D.C. Cell-specific expression of alpha 1-antitrypsin in human intestinal epithelium. J. Clin. Investig. 1993, 92, 2022–2034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perlino, E.; Cortese, R.; Ciliberto, G. The human alpha 1-antitrypsin gene is transcribed from two different promoters in macrophages and hepatocytes. EMBO J. 1987, 6, 2767–2771. [Google Scholar] [CrossRef]
- Kalsheker, N.; Morley, S.; Morgan, K. Gene regulation of the serine proteinase inhibitors alpha1-antitrypsin and alpha1-antichymotrypsin. Biochem. Soc. Trans. 2002, 30, 93–98. [Google Scholar] [CrossRef]
- Yuan, Y.; Diciaccio, B.; Li, Y.; Elshikha, A.S.; Titov, D.; Brenner, B.; Seifer, L.; Pan, H.; Karic, N.; Akbar, M.A.; et al. Anti-inflammaging effects of human alpha-1 antitrypsin. Aging Cell 2018, 17, e12694. [Google Scholar] [CrossRef]
- Knoell, D.L.; Ralston, D.R.; Coulter, K.R.; Wewers, M.D. Alpha 1-antitrypsin and protease complexation is induced by lipopolysaccharide, interleukin-1β, and tumor necrosis factor-α in monocytes. Am. J. Respir. Crit. Care Med. 1998, 157, 246–255. [Google Scholar] [CrossRef]
- Rotondo, J.C.; Oton-Gonzalez, L.; Selvatici, R.; Rizzo, P.; Pavasini, R.; Campo, G.C.; Lanzillotti, C.; Mazziotta, C.; De Mattei, M.; Tognon, M.; et al. SERPINA1 gene promoter is differentially methylated in peripheral blood mononuclear cells of pregnant women. Front. Cell Dev. Biol. 2020, 8, 550543. [Google Scholar] [CrossRef]
- Beckmeyer-Borowko, A.; Imboden, M.; Rezwan, F.I.; Wielscher, M.; Amaral, A.F.S.; Jeong, A.; Schaffner, E.; Auvinen, J.; Sebert, S.; Karhunen, V.; et al. SERPINA1 methylation and lung function in tobacco-smoke exposed European children and adults: A meta-analysis of ALEC population-based cohorts. Respir. Res. 2018, 19, 156. [Google Scholar] [CrossRef]
- Loebermann, H.; Tokuoka, R.; Deisenhofer, J.; Huber, R. Human α1-proteinase inhibitor: Crystal structure analysis of two crystal modifications, molecular model and preliminary analysis of the implications for function. J. Mol. Biol. 1984, 177, 531–557. [Google Scholar] [CrossRef]
- Ryu, S.-E.; Choi, H.-J.; Kwon, K.-S.; Lee, K.N.; Yu, M.-H. The native strains in the hydrophobic core and flexible reactive loop of a serine protease inhibitor: Crystal structure of an uncleaved α1-antitrypsin at 2.7 Å. Structure 1996, 4, 1181–1192. [Google Scholar] [CrossRef] [Green Version]
- Ogushi, F.; Fells, G.A.; Hubbard, R.C.; Straus, S.D.; Crystal, R.G. Z-type alpha 1-antitrypsin is less competent than M1-type alpha 1-antitrypsin as an inhibitor of neutrophil elastase. J. Clin. Investig. 1987, 80, 1366–1374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrodinger, L. The PyMOL Molecular Graphics System; Version 1.3r1; Schrodinger, LLC: New York, NY, USA, 2010. [Google Scholar]
- McCarthy, C.; Saldova, R.; Wormald, M.R.; Rudd, P.M.; McElvaney, N.G.; Reeves, E.P. The role and importance of glycosylation of acute phase proteins with focus on alpha-1 antitrypsin in acute and chronic inflammatory conditions. J. Proteome Res. 2014, 13, 3131–3143. [Google Scholar] [CrossRef] [PubMed]
- Gabay, C.; Kushner, I. Acute-phase proteins and other systemic responses to inflammation. N. Engl. J. Med. 1999, 340, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Karadagi, A.; Johansson, H.; Zemack, H.; Salipalli, S.; Mörk, L.-M.; Kannisto, K.; Jorns, C.; Gramignoli, R.; Strom, S.; Stokkeland, K.; et al. Exogenous alpha 1-antitrypsin down-regulates SERPINA1 expression. PLoS ONE 2017, 12, e0177279. [Google Scholar] [CrossRef]
- Gadek, J.E.; Fells, G.A.; Zimmerman, R.L.; Rennard, S.I.; Crystal, R.G. Antielastases of the human alveolar structures. Implications for the protease-antiprotease theory of emphysema. J. Clin. Investig. 1981, 68, 889–898. [Google Scholar] [CrossRef] [Green Version]
- Ferrarotti, I.; Thun, G.A.; Zorzetto, M.; Ottaviani, S.; Imboden, M.; Schindler, C.; Von Eckardstein, A.; Rohrer, L.; Rochat, T.; Russi, E.W.; et al. Serum levels and genotype distribution of α1-antitrypsin in the general population. Thorax 2012, 67, 669–674. [Google Scholar] [CrossRef] [Green Version]
- Corley, M.; Solem, A.; Phillips, G.; Lackey, L.; Ziehr, B.; Vincent, H.A.; Mustoe, A.M.; Ramos, S.B.V.; Weeks, K.M.; Moorman, N.J.; et al. An RNA structure-mediated, posttranscriptional model of human α-1-antitrypsin expression. Proc. Natl. Acad. Sci. USA 2017, 114, E10244–E10253. [Google Scholar] [CrossRef] [Green Version]
- Chappell, S.; Daly, L.; Morgan, K.; Guetta Baranes, T.; Roca, J.; Rabinovich, R.; Millar, A.; Donnelly, S.C.; Keatings, V.; MacNee, W.; et al. Cryptic haplotypes of SERPINA 1 confer susceptibility to chronic obstructive pulmonary disease. Hum. Mutat. 2005, 27, 103–109. [Google Scholar] [CrossRef]
- American Thoracic Society; European Respiratory Society. American Thoracic Society/European Respiratory Society statement: Standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Am. J. Respir. Crit. Care Med. 2003, 168, 818–900. [Google Scholar] [CrossRef]
- Abboud, R.T.; Nelson, T.N.; Jung, B.; Mattman, A. Alpha1-antitrypsin deficiency: A clinical-genetic overview. Appl. Clin. Genet. 2011, 4, 55–65. [Google Scholar] [CrossRef] [Green Version]
- Pannell, R.; Johnson, D.; Travis, J. Isolation and properties of human plasma α-1-proteinase inhibitor. Biochemistry 1974, 13, 5439–5445. [Google Scholar] [CrossRef] [PubMed]
- Fagerhol, M.K.; Laurell, C.-B. The polymorphism of “prealbumins” and α1-antitrypsin in human sera. Clin. Chim. Acta 1967, 16, 199–203. [Google Scholar] [CrossRef]
- Greene, C.M.; Marciniak, S.J.; Teckman, J.; Ferrarotti, I.; Brantly, M.L.; Lomas, D.A.; Stoller, J.K.; McElvaney, N.G. α1-antitrypsin deficiency. Nat. Rev. Dis. Prim. 2016, 2, 16051. [Google Scholar] [CrossRef] [PubMed]
- Sveger, T. Liver disease in alpha1-antitrypsin deficiency detected by screening of 200,000 infants. N. Engl. J. Med. 1976, 294, 1316–1321. [Google Scholar] [CrossRef]
- Carroll, T.P.; O’Connor, C.A.; Floyd, O.; McPartlin, J.; Kelleher, D.P.; O’Brien, G.; Dimitrov, B.D.; Morris, V.B.; Taggart, C.C.; McElvaney, N.G. The prevalence of alpha-1 antitrypsin deficiency in Ireland. Respir. Res. 2011, 12, 91. [Google Scholar] [CrossRef]
- Lomas, D.A.; Evans, D.L.; Stone, S.R.; Chang, W.S.W.; Carrell, R.W. Effect of the Z mutation on the physical and inhibitory properties of alpha1-antitrypsin. Biochemistry 1993, 32, 500–508. [Google Scholar] [CrossRef]
- Taggart, C.C.; Greene, C.M.; Carroll, T.P.; O’Neill, S.J.; McElvaney, N.G. Elastolytic proteases: Inflammation resolution and dysregulation in chronic infective lung disease. Am. J. Respir. Crit. Care Med. 2005, 171, 1070–1076. [Google Scholar] [CrossRef]
- Franciosi, A.N.; Hobbs, B.D.; McElvaney, O.J.; Molloy, K.; Hersh, C.; Clarke, L.; Gunaratnam, C.; Silverman, E.K.; Carroll, T.P.; McElvaney, N.G. Clarifying the risk of lung disease in SZ alpha-1 antitrypsin deficiency. Am. J. Respir. Crit. Care Med. 2020, 202, 73–82. [Google Scholar] [CrossRef]
- Lockett, A.D.; Kimani, S.; Ddungu, G.; Wrenger, S.; Tuder, R.M.; Janciauskiene, S.M.; Petrache, I. α1-antitrypsin modulates lung endothelial cell inflammatory responses to TNF-α. Am. J. Respir. Cell Mol. Biol. 2013, 49, 143–150. [Google Scholar] [CrossRef] [Green Version]
- Lewis, E.C. Expanding the clinical indications for α1-antitrypsin therapy. Mol. Med. 2012, 18, 957–970. [Google Scholar] [CrossRef] [PubMed]
- Geraghty, P.; Eden, E.; Pillai, M.; Campos, M.; McElvaney, N.G.; Foronjy, R.F. α1-antitrypsin activates protein phosphatase 2A to counter lung inflammatory responses. Am. J. Respir. Crit. Care Med. 2014, 190, 1229–1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nita, I.M.; Serapinas, D.; Janciauskiene, S.M. α1-antitrypsin regulates CD14 expression and soluble CD14 levels in human monocytes in vitro. Int. J. Biochem. Cell Biol. 2007, 39, 1165–1176. [Google Scholar] [CrossRef]
- Hurley, K.; Lacey, N.; O’Dwyer, C.A.; Bergin, D.A.; McElvaney, O.J.; O’Brien, M.E.; McElvaney, O.F.; Reeves, E.P.; McElvaney, N.G. Alpha-1 antitrypsin augmentation therapy corrects accelerated neutrophil apoptosis in deficient individuals. J. Immunol. 2014, 193, 3978–3991. [Google Scholar] [CrossRef] [Green Version]
- Petrache, I.; Fijalkowska, I.; Zhen, L.; Medler, T.R.; Brown, E.; Cruz, P.; Choe, K.-H.; Taraseviciene-Stewart, L.; Scerbavicius, R.; Shapiro, L.; et al. A novel antiapoptotic role for α1-antitrypsin in the prevention of pulmonary emphysema. Am. J. Respir. Crit. Care Med. 2006, 173, 1222–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janciauskiene, S. The beneficial effects of antioxidants in health and diseases. Chronic Obstr. Pulm. Dis. J. COPD Found. 2020, 7, 182–202. [Google Scholar] [CrossRef] [PubMed]
- Dabbagh, K.; Laurent, G.J.; Shock, A.; Leoni, P.; Papakrivopoulou, J.; Chambers, R.C. Alpha-1-antitrypsin stimulates fibroblast proliferation and procollagen production and activates classical MAP kinase signalling pathways. J. Cell. Physiol. 2001, 186, 73–81. [Google Scholar] [CrossRef]
- Liu, Z.; Zhou, X.; Shapiro, S.D.; Shipley, J.M.; Twining, S.S.; Diaz, L.A.; Senior, R.M.; Werb, Z. The serpin α1-proteinase inhibitor is a critical substrate for gelatinase B/MMP-9 in vivo. Cell 2000, 102, 647–655. [Google Scholar] [CrossRef] [Green Version]
- Janciauskiene, S.; Nita, I.; Subramaniyam, D.; Li, Q.; Lancaster, J.R., Jr.; Matalon, S. α1-antitrypsin inhibits the activity of the matriptase catalytic domain in vitro. Am. J. Respir. Cell Mol. Biol. 2008, 39, 631–637. [Google Scholar] [CrossRef] [Green Version]
- Hada, K.; Isshiki, K.; Matsuda, S.; Yuasa, K.; Tsuji, A. Engineering of alpha1-antitrypsin variants with improved specificity for the proprotein convertase furin using site-directed random mutagenesis. Protein Eng. Des. Sel. 2013, 26, 123–131. [Google Scholar] [CrossRef]
- Münch, J.; Ständker, L.; Adermann, K.; Schulz, A.; Schindler, M.; Chinnadurai, R.; Pöhlmann, S.; Chaipan, C.; Biet, T.; Peters, T.; et al. Discovery and optimization of a natural HIV-1 entry inhibitor targeting the gp41 fusion peptide. Cell 2007, 129, 263–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wettstein, L.; Weil, T.; Conzelmann, C.; Müller, J.A.; Groß, R.; Hirschenberger, M.; Seidel, A.; Klute, S.; Zech, F.; Prelli Bozzo, C.; et al. Alpha-1 antitrypsin inhibits TMPRSS2 protease activity and SARS-CoV-2 infection. Nat. Commun. 2021, 12, 1726. [Google Scholar] [CrossRef] [PubMed]
- Azouz, N.P.; Klingler, A.M.; Callahan, V.; Akhrymuk, I.V.; Elez, K.; Raich, L.; Henry, B.M.; Benoit, J.L.; Benoit, S.W.; Noé, F.; et al. Alpha 1 antitrypsin is an inhibitor of the SARS-CoV-2–priming protease TMPRSS2. Pathog. Immun. 2021, 6, 55–74. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.; Thornton, J.M. Principles of protein-protein interactions. Proc. Natl. Acad. Sci. USA 1996, 93, 13–20. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Woodbury, N.W. Selective protein–peptide interactions at surfaces. Acta Biomater. 2014, 10, 761–768. [Google Scholar] [CrossRef]
- Beatty, K.; Bieth, J.; Travis, J. Kinetics of association of serine proteinases with native and oxidized alpha-1-proteinase inhibitor and alpha-1-antichymotrypsin. J. Biol. Chem. 1980, 255, 3931–3934. [Google Scholar] [CrossRef]
- Duranton, J.; Bieth, J.G. Inhibition of proteinase 3 by α1-antitrypsin in vitro predicts very fast inhibition in vivo. Am. J. Respir. Cell Mol. Biol. 2003, 29, 57–61. [Google Scholar] [CrossRef]
- Wuillemin, W.A.; Minnema, M.; Meijers, J.C.; Roem, D.; Eerenberg, A.J.; Nuijens, J.H.; ten Cate, H.; Hack, C.E. Inactivation of factor XIa in human plasma assessed by measuring factor XIa-protease inhibitor complexes: Major role for C1-inhibitor. Blood 1995, 85, 1517–1526. [Google Scholar] [CrossRef] [Green Version]
- Scott, C.F.; Schapira, M.; James, H.L.; Cohen, A.B.; Colman, R.W. Inactivation of factor XIa by plasma protease inhibitors: Predominant role of alpha 1-protease inhibitor and protective effect of high molecular weight kininogen. J. Clin. Investig. 1982, 69, 844–852. [Google Scholar] [CrossRef] [Green Version]
- Rao, N.V.; Wehner, N.G.; Marshall, B.C.; Gray, W.R.; Gray, B.H.; Hoidal, J.R. Characterization of proteinase-3 (PR-3), a neutrophil serine proteinase. Structural and functional properties. J. Biol. Chem. 1991, 266, 9540–9548. [Google Scholar] [CrossRef]
- Huntington, J.A.; Read, R.J.; Carrell, R.W. Structure of a serpin–protease complex shows inhibition by deformation. Nature 2000, 407, 923–926. [Google Scholar] [CrossRef] [PubMed]
- Lomas, D.A.; Parfrey, H. α1-antitrypsin deficiency •4: Molecular pathophysiology. Thorax 2004, 59, 529–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perlmutter, D.H.; Joslin, G.; Nelson, P.; Schasteen, C.; Adams, S.P.; Fallon, R.J. Endocytosis and degradation of alpha 1-antitrypsin-protease complexes is mediated by the serpin-enzyme complex (SEC) receptor. J. Biol. Chem. 1990, 265, 16713–16716. [Google Scholar] [CrossRef]
- Joslin, G.; Wittwer, A.; Adams, S.; Tollefsen, D.M.; August, A.; Perlmutter, D.H. Cross-competition for binding of alpha 1-antitrypsin (alpha 1 AT)-elastase complexes to the serpin-enzyme complex receptor by other serpin-enzyme complexes and by proteolytically modified alpha 1 AT. J. Biol. Chem. 1993, 268, 1886–1893. [Google Scholar] [CrossRef]
- Joslin, G.; Fallon, R.J.; Bullock, J.; Adams, S.P.; Perlmutter, D.H. The SEC receptor recognizes a pentapeptide neodomain of alpha 1-antitrypsin-protease complexes. J. Biol. Chem. 1991, 266, 11282–11288. [Google Scholar] [CrossRef]
- Owen, M.C.; Brennan, S.O.; Lewis, J.H.; Carrell, R.W. Mutation of antitrypsin to antithrombin. Alpha 1-antitrypsin Pittsburgh (358 Met leads to Arg), a fatal bleeding disorder. N. Engl. J. Med. 1983, 309, 694–698. [Google Scholar] [CrossRef] [PubMed]
- Carp, H.; Miller, F.; Hoidal, J.R.; Janoff, A. Potential mechanism of emphysema: Alpha 1-proteinase inhibitor recovered from lungs of cigarette smokers contains oxidized methionine and has decreased elastase inhibitory capacity. Proc. Natl. Acad. Sci. USA 1982, 79, 2041–2045. [Google Scholar] [CrossRef] [Green Version]
- Taggart, C.; Cervantes-Laurean, D.; Kim, G.; McElvaney, N.G.; Wehr, N.; Moss, J.; Levine, R.L. Oxidation of either methionine 351 or methionine 358 in alpha 1-antitrypsin causes loss of anti-neutrophil elastase activity. J. Biol. Chem. 2000, 275, 27258–27265. [Google Scholar] [CrossRef]
- Griffiths, S.W.; King, J.; Cooney, C.L. The reactivity and oxidation pathway of cysteine 232 in recombinant human α1-antitrypsin. J. Biol. Chem. 2002, 277, 25486–25492. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.-D.; Gan, J.C. The role of sialic acid and galactose residues in determining the survival of human plasma α1-antitrypsin in the blood circulation. Arch. Biochem. Biophys. 1977, 179, 477–485. [Google Scholar] [CrossRef]
- Churg, A.; Wang, R.D.; Xie, C.; Wright, J.L. α-1-antitrypsin ameliorates cigarette smoke–induced emphysema in the mouse. Am. J. Respir. Crit. Care Med. 2003, 168, 199–207. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, M.E.; Pennycooke, K.; Carroll, T.; Shum, J.; Fee, L.T.; O’Connor, C.; Logan, P.M.; Reeves, E.P.; McElvaney, N.G. The impact of smoke exposure on the clinical phenotype of alpha-1 antitrypsin deficiency in Ireland: Exploiting a National Registry to understand a rare disease. COPD J. Chronic Obstr. Pulm. Dis. 2015, 12, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Alam, S.; Li, Z.; Janciauskiene, S.; Mahadeva, R. Oxidation of Z α1-antitrypsin by cigarette smoke induces polymerization: A novel mechanism of early-onset emphysema. Am. J. Respir. Cell Mol. Biol. 2011, 45, 261–269. [Google Scholar] [CrossRef]
- Levi, M.; Roem, D.; Kamp, A.M.; De Boer, J.P.; Hack, C.E.; Cate, J.W.T. Assessment of the relative contribution of different protease inhibitors to the inhibition of plasmin in vivo. Thromb. Haemost. 1993, 69, 141–146. [Google Scholar] [CrossRef]
- Fredenburgh, J.C.; Stafford, A.R.; Weitz, J.I. Conformational changes in thrombin when complexed by serpins. J. Biol. Chem. 2001, 276, 44828–44834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heeb, M.J.; Bischoff, R.; Courtney, M.; Griffin, J. Inhibition of activated protein C by recombinant alpha 1-antitrypsin variants with substitution of arginine or leucine for methionine358. J. Biol. Chem. 1990, 265, 2365–2369. [Google Scholar] [CrossRef]
- Gooptu, B.; Hazes, B.; Chang, W.-S.W.; Dafforn, T.R.; Carrell, R.W.; Read, R.J.; Lomas, D.A. Inactive conformation of the serpin alpha 1-antichymotrypsin indicates two-stage insertion of the reactive loop: Implications for inhibitory function and conformational disease. Proc. Natl. Acad. Sci. USA 2000, 97, 67–72. [Google Scholar] [CrossRef] [Green Version]
- Yamasaki, M.; Li, W.; Johnson, D.J.D.; Huntington, J.A. Crystal structure of a stable dimer reveals the molecular basis of serpin polymerization. Nature 2008, 455, 1255–1258. [Google Scholar] [CrossRef]
- Carrell, R.W.; Lomas, D.A. Alpha1-antitrypsin deficiency—A model for conformational diseases. N. Engl. J. Med. 2002, 346, 45–53. [Google Scholar] [CrossRef]
- Lawless, M.W.; Greene, C.M.; Mulgrew, A.; Taggart, C.C.; O’Neill, S.J.; McElvaney, N.G. Activation of endoplasmic reticulum-specific stress responses associated with the conformational disease Z α1-antitrypsin deficiency. J. Immunol. 2004, 172, 5722–5726. [Google Scholar] [CrossRef] [Green Version]
- Carroll, T.P.; Greene, C.M.; O’Connor, C.A.; Nolan, A.M.; O’Neill, S.J.; McElvaney, N.G. Evidence for unfolded protein response activation in monocytes from individuals with α-1 antitrypsin deficiency. J. Immunol. 2010, 184, 4538–4546. [Google Scholar] [CrossRef] [Green Version]
- Mahadeva, R.; Chang, W.-S.W.; Dafforn, T.R.; Oakley, D.J.; Foreman, R.C.; Calvin, J.; Wight, D.G.; Lomas, D.A. Heteropolymerization of S, I, and Z α1-antitrypsin and liver cirrhosis. J. Clin. Investig. 1999, 103, 999–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahadeva, R.; Atkinson, C.; Li, Z.; Stewart, S.; Janciauskiene, S.; Kelley, D.G.; Parmar, J.; Pitman, R.; Shapiro, S.D.; Lomas, D.A. Polymers of Z α1-antitrypsin co-localize with neutrophils in emphysematous alveoli and are chemotactic in vivo. Am. J. Pathol. 2005, 166, 377–386. [Google Scholar] [CrossRef]
- Pini, L.; Tiberio, L.; Venkatesan, N.; Bezzi, M.; Corda, L.; Luisetti, M.; Ferrarotti, I.; Malerba, M.; Lomas, D.A.; Janciauskiene, S.; et al. The role of bronchial epithelial cells in the pathogenesis of COPD in Z-alpha-1 antitrypsin deficiency. Respir. Res. 2014, 15, 112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Pak, S.C.; O’Reilly, L.P.; Benson, J.A.; Wang, Y.; Hidvegi, T.; Hale, P.; Dippold, C.; Ewing, M.; Silverman, G.A.; et al. Fluphenazine reduces proteotoxicity in C. elegans and mammalian models of alpha-1-antitrypsin deficiency. PLoS ONE 2014, 9, e87260. [Google Scholar] [CrossRef] [Green Version]
- Hidvegi, T.; Ewing, M.; Hale, P.; Dippold, C.; Beckett, C.; Kemp, C.; Maurice, N.; Mukherjee, A.; Goldbach, C.; Watkins, S.; et al. An autophagy-enhancing drug promotes degradation of mutant α1-antitrypsin Z and reduces hepatic fibrosis. Science 2010, 329, 229–232. [Google Scholar] [CrossRef]
- Washington University School of Medicine. Carbamazepine in Severe Liver Disease Due to Alpha-1 Antitrypsin Deficiency (CBZ). 2021. Available online: https://clinicaltrials.gov/ct2/show/results/NCT01379469 (accessed on 1 July 2021).
- Wooddell, C.I.; Blomenkamp, K.; Peterson, R.M.; Subbotin, V.M.; Schwabe, C.; Hamilton, J.; Chu, Q.; Christianson, D.R.; Hegge, J.O.; Kolbe, J.; et al. Development of an RNAi therapeutic for alpha-1-antitrypsin liver disease. JCI Insight 2020, 5, e135348. [Google Scholar] [CrossRef]
- Santos, G.; Turner, A.M. Alpha-1 antitrypsin deficiency: An update on clinical aspects of diagnosis and management. Fac. Rev. 2020, 9, 1. [Google Scholar] [CrossRef]
- McCarthy, C.; Saldova, R.; O’Brien, M.E.; Bergin, D.A.; Carroll, T.P.; Keenan, J.; Meleady, P.; Henry, M.; Clynes, M.; Rudd, P.M.; et al. Increased outer arm and core fucose residues on the N-glycans of mutated alpha-1 antitrypsin protein from alpha-1 antitrypsin deficient individuals. J. Proteome Res. 2014, 13, 596–605. [Google Scholar] [CrossRef]
- Dudev, T.; Lim, C. Principles governing Mg, Ca, and Zn binding and selectivity in proteins. Chem. Rev. 2003, 103, 773–788. [Google Scholar] [CrossRef]
- Kolarich, D.; Weber, A.; Turecek, P.L.; Schwarz, H.-P.; Altmann, F. Comprehensive glyco-proteomic analysis of human α1-antitrypsin and its charge isoforms. Proteomics 2006, 6, 3369–3380. [Google Scholar] [CrossRef] [PubMed]
- Rudd, P.M.; Dwek, R.A. Glycosylation: Heterogeneity and the 3D structure of proteins. Crit. Rev. Biochem. Mol. Biol. 1997, 32, 1–100. [Google Scholar] [CrossRef] [PubMed]
- Kelleher, D.J.; Gilmore, R. An evolving view of the eukaryotic oligosaccharyltransferase. Glycobiology 2006, 16, 47R–62R. [Google Scholar] [CrossRef] [PubMed]
- Trombetta, E.S.; Helenius, A. Lectins as chaperones in glycoprotein folding. Curr. Opin. Struct. Biol. 1998, 8, 587–592. [Google Scholar] [CrossRef]
- Schachter, H. Biosynthetic controls that determine the branching and microheterogeneity of protein-bound oligosaccharides. Biochem. Cell Biol. 1986, 64, 163–181. [Google Scholar] [CrossRef]
- Kuhn, B.; Benz, J.; Greif, M.; Engel, A.M.; Sobek, H.; Rudolph, M.G. The structure of human α-2,6-sialyltransferase reveals the binding mode of complex glycans. Acta Crystallogr. Sect. D Biol. Crystallogr. 2013, 69, 1826–1838. [Google Scholar] [CrossRef]
- Casolaro, M.A.; Fells, G.; Wewers, M.; Pierce, J.E.; Ogushi, F.; Hubbard, R.; Sellers, S.; Forstrom, J.; Lyons, D.; Kawasaki, G.; et al. Augmentation of lung antineutrophil elastase capacity with recombinant human alpha-1-antitrypsin. J. Appl. Physiol. 1987, 63, 2015–2023. [Google Scholar] [CrossRef]
- Karnaukhova, E.; Ophir, Y.; Golding, B. Recombinant human alpha-1 proteinase inhibitor: Towards therapeutic use. Amino Acids 2006, 30, 317–332. [Google Scholar] [CrossRef]
- Mast, A.E.; Enghild, J.J.; Pizzo, S.V.; Salvesen, G. Analysis of the plasma elimination kinetics and conformational stabilities of native, proteinase-complexed and reactive site cleaved serpins: Comparison of alpha-1-proteinase inhibitor, alpha-1-antichymotrypsin, antithrombin III, alpha-2-antiplasmin, angiotensinogen, and ovalbumin. Biochemistry 1991, 30, 1723–1730. [Google Scholar] [CrossRef]
- Lusch, A.; Kaup, M.; Marx, U.; Tauber, R.; Blanchard, V.; Berger, M. Development and analysis of alpha 1-antitrypsin neoglycoproteins: The impact of additional N-glycosylation sites on serum half-life. Mol. Pharm. 2013, 10, 2616–2629. [Google Scholar] [CrossRef]
- Zerimech, F.; Jourdain, M.; Onraed, B.; Bouchecareilh, M.; Sendid, B.; Duhamel, A.; Balduyck, M.; Pigny, P. LICORNE Study Groupa Protease-antiprotease imbalance in patients with severe COVID-19. Clin. Chem. Lab. Med. 2021, 59, e330–e334. [Google Scholar] [CrossRef] [PubMed]
- Ferrarotti, I.; Carroll, T.P.; Ottaviani, S.; Fra, A.M.; O’Brien, G.; Molloy, K.; Corda, L.; Medicina, D.; Curran, D.R.; McElvaney, N.G.; et al. Identification and characterisation of eight novel SERPINA1 Null mutations. Orphanet J. Rare Dis. 2014, 9, 172. [Google Scholar] [CrossRef] [Green Version]
- Reeves, E.P.; O’Dwyer, C.A.; Dunlea, D.M.; Wormald, M.R.; Hawkins, P.; Alfares, M.; Kotton, D.N.; Rowe, S.M.; Wilson, A.A.; McElvaney, N.G. Ataluren, a new therapeutic for alpha-1 antitrypsin–deficient individuals with nonsense mutations. Am. J. Respir. Crit. Care Med. 2018, 198, 1099–1102. [Google Scholar] [CrossRef]
- Reeves, E.P.; Dunlea, D.M.; McQuillan, K.; O’Dwyer, C.A.; Carroll, T.P.; Saldova, R.; Akepati, P.R.; Wormald, M.R.; McElvaney, O.J.; Shutchaidat, V.; et al. Circulating truncated alpha-1 antitrypsin glycoprotein in patient plasma retains anti-inflammatory capacity. J. Immunol. 2019, 202, 2240–2253. [Google Scholar] [CrossRef]
- Goodarzi, M.T.; Turner, G.A. Reproducible and sensitive determination of charged oligosaccharides from haptoglobin by PNGase F digestion and HPAEC/PAD analysis: Glycan composition varies with disease. Glycoconj. J. 1998, 15, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Patschull, A.O.M.; Gooptu, B.; Ashford, P.; Daviter, T.; Nobeli, I. In silico assessment of potential druggable pockets on the surface of α1-antitrypsin conformers. PLoS ONE 2012, 7, e36612. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.-Y.; Chen, S.-F.; Lai, M.-D.; Chang, T.-T.; Chen, T.-L.; Li, P.-Y.; Shieh, D.-B.; Young, K.-C. Comparative proteomic profiling of plasma very-low-density and low-density lipoproteins. Clin. Chim. Acta 2010, 411, 336–344. [Google Scholar] [CrossRef]
- Kumaraswamy, S.B.; Linder, A.; Akesson, P.; Dahlback, B. Decreased plasma concentrations of apolipoprotein M in sepsis and systemic inflammatory response syndromes. Crit. Care 2012, 16, R60. [Google Scholar] [CrossRef] [Green Version]
- Ortiz-Muñoz, G.; Houard, X.; Martín-Ventura, J.-L.; Ishida, B.Y.; Loyau, S.; Rossignol, P.; Moreno, J.-A.; Kane, J.P.; Chalkley, R.J.; Burlingame, A.L.; et al. HDL antielastase activity prevents smooth muscle cell anoikis, a potential new antiatherogenic property. FASEB J. 2009, 23, 3129–3139. [Google Scholar] [CrossRef] [Green Version]
- Moreno, J.-A.; Ortega-Gomez, A.; Rubio-Navarro, A.; Louedec, L.; Ho-Tin-Noé, B.; Caligiuri, G.; Nicoletti, A.; Levoye, A.; Plantier, L.; Meilhac, O. High-density lipoproteins potentiate α1-antitrypsin therapy in elastase-induced pulmonary emphysema. Am. J. Respir. Cell Mol. Biol. 2014, 51, 536–549. [Google Scholar] [CrossRef]
- Vaerman, J.P.; Hagiwara, K.; Kobayashi, K.; Rits, M. Complexes of albumin and α1-antitrypsin with Fc-fragment of IgA monomer are disulfide-bound to penultimate C-terminal cysteine in the Cα3-domain. Immunol. Lett. 1987, 15, 67–72. [Google Scholar] [CrossRef]
- Musiani, P.; Lauriola, L.; Piantelli, M. Inhibitory activity of alpha-1-antitrypsin bound to human IgA. Clin. Chim. Acta 1978, 85, 61–66. [Google Scholar] [CrossRef]
- Moldthan, H.L.; Hirko, A.C.; Thinschmidt, J.S.; Grant, M.B.; Li, Z.; Peris, J.; Lu, Y.; Elshikha, A.S.; King, M.A.; Hughes, J.A.; et al. Alpha 1-antitrypsin therapy mitigated ischemic stroke damage in rats. J. Stroke Cerebrovasc. Dis. 2014, 23, e355–e363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, W.; Zhao, J.; Kim, H.; Xu, S.; Chen, M.; Bai, X.; Toba, H.; Cho, H.-R.; Zhang, H.; Keshavjeel, S.; et al. α1-antitrypsin inhibits ischemia reperfusion-induced lung injury by reducing inflammatory response and cell death. J. Hear. Lung Transplant. 2014, 33, 309–315. [Google Scholar] [CrossRef]
- Abbate, A.; Van Tassell, B.W.; Christopher, S.; Abouzaki, N.A.; Sonnino, C.; Oddi, C.; Carbone, S.; Melchior, R.D.; Gambill, M.L.; Roberts, C.S.; et al. Effects of prolastin C (plasma-derived alpha-1 antitrypsin) on the acute inflammatory response in patients with ST-segment elevation myocardial infarction (from the VCU-alpha 1-RT pilot study). Am. J. Cardiol. 2015, 115, 8–12. [Google Scholar] [CrossRef]
- Rosenberg, R.D.; Damus, P.S. The purification and mechanism of action of human antithrombin-heparin cofactor. J. Biol. Chem. 1973, 248, 6490–6505. [Google Scholar] [CrossRef]
- Frommherz, K.J.; Faller, B.; Bieth, J.G. Heparin strongly decreases the rate of inhibition of neutrophil elastase by alpha 1-proteinase inhibitor. J. Biol. Chem. 1991, 266, 15356–15362. [Google Scholar] [CrossRef]
- Talens, S.; Malfliet, J.J.M.C.; van Hal, P.T.W.; Leebeek, F.W.G.; Rijken, D.C. Identification and characterization of α1-antitrypsin in fibrin clots. J. Thromb. Haemost. 2013, 11, 1319–1328. [Google Scholar] [CrossRef]
- Massberg, S.; Grahl, L.; von Bruehl, M.-L.; Manukyan, D.; Pfeiler, S.; Goosmann, C.; Brinkmann, V.; Lorenz, M.; Bidzhekov, K.; Khandagale, A.B.; et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat. Med. 2010, 16, 887–896. [Google Scholar] [CrossRef]
- Opal, S.M. Phylogenetic and functional relationships between coagulation and the innate immune response. Crit. Care Med. 2000, 28, S77–S80. [Google Scholar] [CrossRef]
- Diamond, M.S.; Springer, T.A. A subpopulation of Mac-1 (CD11b/CD18) molecules mediates neutrophil adhesion to ICAM-1 and fibrinogen. J. Cell Biol. 1993, 120, 545–556. [Google Scholar] [CrossRef] [Green Version]
- Weitz, J.I.; Huang, A.J.; Landman, S.L.; Nicholson, S.C.; Silverstein, S.C. Elastase-mediated fibrinogenolysis by chemoattractant-stimulated neutrophils occurs in the presence of physiologic concentrations of antiproteinases. J. Exp. Med. 1987, 166, 1836–1850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, R.; Mumford, R.A.; Treonze, K.M.; Finke, P.E.; Davies, P.; Si, Q.; Humes, J.L.; Dirksen, A.; Piitulainen, E.; Ahmad, A.; et al. The fibrinogen cleavage product alpha-Val360, a specific marker of neutrophil elastase activity in vivo. Thorax 2011, 66, 686–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, R.I.; Ungurs, M.J.; Pillai, A.; Mumford, R.A.; Stockley, R.A. The relationship of the fibrinogen cleavage biomarker Aa-Val360 with disease severity and activity in α1-antitrypsin deficiency. Chest 2015, 148, 382–388. [Google Scholar] [CrossRef] [PubMed]
- Rao, C.N.; Ladin, D.A.; Liu, Y.Y.; Chilukuri, K.; Hou, Z.Z.; Woodley, D.T. α1-antitrypsin is degraded and non-functional in chronic wounds but intact and functional in acute wounds: The inhibitor protects fibronectin from degradation by chronic wound fluid enzymes. J. Investig. Dermatol. 1995, 105, 572–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grinnell, F.; Zhu, M. Fibronectin degradation in chronic wounds depends on the relative levels of elastase, α1-proteinase inhibitor, and α2-macroglobulin. J. Investig. Dermatol. 1996, 106, 335–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, L.J.; Evans, E.L.; Dawes, P.T.; Russell, G.I.; Mattey, D.L. Comparison of IgA-alpha1-antitrypsin levels in rheumatoid arthritis and seronegative oligoarthritis: Complex formation is not associated with inflammation per se. Br. J. Rheumatol. 1998, 37, 398–404. [Google Scholar] [CrossRef] [Green Version]
- Swedlund, H.A.; Hunder, G.G.; Gleich, G.J. Alpha 1-antitrypsin in serum and synovial fluid in rheumatoid arthritis. Ann. Rheum. Dis. 1974, 33, 162–164. [Google Scholar] [CrossRef] [Green Version]
- Chrostek, L.; Cylwik, B.; Gińdzieńska-Sieśkiewicz, E.; Gruszewska, E.; Szmitkowski, M.; Sierakowski, S. Sialic acid level reflects the disturbances of glycosylation and acute-phase reaction in rheumatic diseases. Rheumatol. Int. 2014, 34, 393–399. [Google Scholar] [CrossRef] [Green Version]
- Cylwik, B.; Chrostek, L.; Gindzienska-Sieskiewicz, E.; Sierakowski, S.; Szmitkowski, M. Relationship between serum acute-phase proteins and high disease activity in patients with rheumatoid arthritis. Adv. Med. Sci. 2010, 55, 80–85. [Google Scholar] [CrossRef]
- McCarthy, C.; Orr, C.; Fee, L.T.; Carroll, T.; Dunlea, D.M.; Hunt, D.; Dunne, E.; O’Connell, P.; McCarthy, G.; Kenny, D.; et al. Brief report: Genetic variation of the α1-antitrypsin gene is associated with increased autoantibody production in rheumatoid arthritis. Arthritis Rheumatol. 2017, 69, 1576–1579. [Google Scholar] [CrossRef] [PubMed]
- Marc, M.M.; Korosec, P.; Kosnik, M.; Kern, I.; Flezar, M.; Suskovic, S.; Sorli, J. Complement factors C3a, C4a, and C5a in chronic obstructive pulmonary disease and asthma. Am. J. Respir. Cell Mol. Biol. 2004, 31, 216–219. [Google Scholar] [CrossRef] [PubMed]
- Ramadass, M.; Ghebrehiwet, B.; Smith, R.J.; Kew, R.R. Generation of multiple fluid-phase C3b: Plasma protein complexes during complement activation: Possible implications in C3 glomerulopathies. J. Immunol. 2013, 192, 1220–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fee, L.T.; Gogoi, D.; O’Brien, M.E.; McHugh, E.; Casey, M.; Gough, C.; Murphy, M.; Hopkins, A.M.; Carroll, T.P.; McElvaney, N.G.; et al. C3d elicits neutrophil degranulation and decreases endothelial cell migration, with implications for patients with alpha-1 antitrypsin deficiency. Biomedicines 2021, 9, 1925. [Google Scholar] [CrossRef]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Shapira, G.; Shomron, N.; Gurwitz, D. Ethnic differences in alpha--1 antitrypsin deficiency allele frequencies may partially explain national differences in COVID--19 fatality rates. FASEB J. 2020, 34, 14160–14165. [Google Scholar] [CrossRef]
- Massi, G.; Cotumaccio, R.; Auconi, P. Alpha-1-antitrypsin (α1AT) phenotypes and PiM subtypes in Italy. Evidence of considerable geographic variability. Hum. Genet. 1982, 61, 76–77. [Google Scholar] [CrossRef]
- De Loyola, M.B.; dos Reis, T.T.A.; de Oliveira, G.X.L.M.; Palmeira, J.; Argañaraz, G.A.; Argañaraz, E.R. Alpha--1--antitrypsin: A possible host protective factor against COVID--19. Rev. Med. Virol. 2021, 31, e2157. [Google Scholar] [CrossRef]
- Vianello, A.; Braccioni, F. Geographical overlap between alpha-1 antitrypsin deficiency and COVID-19 infection in Italy: Casual or causal? Arch. Bronconeumol. 2020, 56, 609–610. [Google Scholar] [CrossRef]
- McElvaney, O.J.; McEvoy, N.L.; McElvaney, O.F.; Carroll, T.P.; Murphy, M.P.; Dunlea, D.M.; Ni Choileain, O.; Clarke, J.; O’Connor, E.; Hogan, G.; et al. Characterization of the inflammatory response to severe COVID-19 illness. Am. J. Respir. Crit. Care Med. 2020, 202, 812–821. [Google Scholar] [CrossRef]
- Bhattacharyya, C.; Das, C.; Ghosh, A.; Singh, A.K.; Mukherjee, S.; Majumder, P.P.; Basu, A.; Biswas, N.K. SARS-CoV-2 mutation 614G creates an elastase cleavage site enhancing its spread in high AAT-deficient regions. Infect. Genet. Evol. 2021, 90, 104760. [Google Scholar] [CrossRef] [PubMed]
- Wewers, M.D.; Casolaro, M.A.; Sellers, S.E.; Swayze, S.C.; McPhaul, K.M.; Wittes, J.T.; Crystal, R.G. Replacement therapy for alpha 1-antitrypsin deficiency associated with emphysema. N. Engl. J. Med. 1987, 316, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Crystal, R.G. Alpha 1-antitrypsin deficiency, emphysema, and liver disease. Genetic basis and strategies for therapy. J. Clin. Investig. 1990, 85, 1343–1352. [Google Scholar] [CrossRef] [PubMed]
- Chapman, K.R.; Burdon, J.G.W.; Piitulainen, E.; Sandhaus, R.A.; Seersholm, N.; Stocks, J.M.; Stoel, B.C.; Huang, L.; Yao, Z.; Edelman, J.M.; et al. Intravenous augmentation treatment and lung density in severe α1 antitrypsin deficiency (RAPID): A randomised, double-blind, placebo-controlled trial. Lancet 2015, 386, 360–368. [Google Scholar] [CrossRef]
- Sandhaus, R.A.; Turino, G.; Brantly, M.L.; Campos, M.; Cross, C.E.; Goodman, K.; Hogarth, D.K.; Knight, S.L.; Stocks, J.M.; Stoller, J.K.; et al. The diagnosis and management of alpha-1 antitrypsin deficiency in the adult. Chronic Obstr. Pulm. Dis. J. COPD Found. 2016, 3, 668–682. [Google Scholar] [CrossRef] [Green Version]
- Campos, M.A.; Kueppers, F.; Stocks, J.M.; Strange, C.; Chen, J.; Griffin, R.; Wang-Smith, L.; Brantly, M.L. Safety and pharmacokinetics of 120 mg/kg versus 60 mg/kg weekly intravenous infusions of alpha-1 proteinase inhibitor in alpha-1 antitrypsin deficiency: A multicenter, randomized, double-blind, crossover study (SPARK). COPD J. Chronic Obstr. Pulm. Dis. 2013, 10, 687–695. [Google Scholar] [CrossRef]
- Campos, M.A.; Geraghty, P.; Holt, G.; Mendes, E.; Newby, P.R.; Ma, S.; Luna-Diaz, L.V.; Turino, G.M.; Stockley, R.A. The biological effects of double-dose alpha-1 antitrypsin augmentation therapy. A pilot clinical trial. Am. J. Respir. Crit. Care Med. 2019, 200, 318–326. [Google Scholar] [CrossRef]
- Greulich, T.; Chlumsky, J.; Wencker, M.; Vit, O.; Fries, M.; Chung, T.; Shebl, A.; Vogelmeier, C.; Chapman, K.R.; McElvaney, N.G. Safety of biweekly α1-antitrypsin treatment in the RAPID programme. Eur. Respir. J. 2018, 52, 1800897. [Google Scholar] [CrossRef] [Green Version]
- McElvaney, N.G.; Hubbard, R.C.; Birrer, P.; Chernick, M.S.; Caplan, D.B.; Frank, M.M.; Crystal, R.G. Aerosol α1-antitrypsin treatment for cystic fibrosis. Lancet 1991, 337, 392–394. [Google Scholar] [CrossRef]
- Martin, S.L.; Downey, D.; Bilton, D.; Keogan, M.T.; Edgar, J.; Elborn, J.S. Safety and efficacy of recombinant alpha1-antitrypsin therapy in cystic fibrosis. Pediatr. Pulmonol. 2006, 41, 177–183. [Google Scholar] [CrossRef]
- Griese, M.; Latzin, P.; Kappler, M.; Weckerle, K.; Heinzlmaier, T.; Bernhardt, T.; Hartl, D. Alpha1-antitrypsin inhalation reduces airway inflammation in cystic fibrosis patients. Eur. Respir. J. 2006, 29, 240–250. [Google Scholar] [CrossRef] [Green Version]
- Park, S.S.; Rodriguez Ortega, R.; Agudelo, C.W.; Perez, J.P.; Gandara, B.P.; Garcia-Arcos, I.; McCarthy, C.; Geraghty, P. Therapeutic potential of alpha-1 antitrypsin in Type 1 and Type 2 diabetes mellitus. Medicina 2021, 57, 397. [Google Scholar] [CrossRef]
- Lewis, E.C.; Shapiro, L.; Bowers, O.J.; Dinarello, C.A. Alpha1-antitrypsin monotherapy prolongs islet allograft survival in mice. Proc. Natl. Acad. Sci. USA 2005, 102, 12153–12158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, E.C.; Mizrahi, M.; Toledano, M.; DeFelice, N.; Wright, J.L.; Churg, A.; Shapiro, L.; Dinarello, C.A. Alpha1-antitrypsin monotherapy induces immune tolerance during islet allograft transplantation in mice. Proc. Natl. Acad. Sci. USA 2008, 105, 16236–16241. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Yan, H.-J.; Zhou, S.-Y.; Wang, Y.-S.; Qi, H.; Deng, C.-Y.; Li, F.-R. The immunoregulation effect of alpha 1-antitrypsin prolong β-cell survival after transplantation. PLoS ONE 2014, 9, e94548. [Google Scholar] [CrossRef]
- Lagarde, W.H.; Courtney, K.L.; Reiner, B.; Steinmann, K.; Tsalikian, E.; Willi, S.M. Human plasma--derived alpha 1--proteinase inhibitor in patients with new--onset type 1 diabetes mellitus: A randomized, placebo--controlled proof--of--concept study. Pediatr. Diabetes 2021, 22, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Rachmiel, M.; Strauss, P.; Dror, N.; Benzaquen, H.; Horesh, O.; Tov, N.; Weintrob, N.; Landau, Z.; Ben-Ami, M.; Haim, A.; et al. Alpha-1 antitrypsin therapy is safe and well tolerated in children and adolescents with recent onset Type 1 diabetes mellitus. Pediatr. Diabetes 2016, 17, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Brener, A.; Lebenthal, Y.; Interator, H.; Horesh, O.; Leshem, A.; Weintrob, N.; Loewenthal, N.; Shalitin, S.; Rachmiel, M. Long-term safety of α-1 antitrypsin therapy in children and adolescents with Type 1 diabetes. Immunotherapy 2018, 10, 1137–1148. [Google Scholar] [CrossRef] [PubMed]
- Elshikha, A.S.; Lu, Y.; Chen, M.-J.; Akbar, M.; Zeumer, L.; Ritter, A.; Elghamry, H.; Mahdi, M.A.; Morel, L.; Song, S. Alpha 1 antitrypsin inhibits dendritic cell activation and attenuates nephritis in a mouse model of lupus. PLoS ONE 2016, 11, e0156583. [Google Scholar] [CrossRef] [Green Version]
- Elshikha, A.S.; Yuan, Y.; Lu, Y.; Chen, M.-J.; Abboud, G.; Akbar, M.A.; Plate, H.; Wolney, H.; Hoffmann, T.; Tagari, E.; et al. Alpha 1 antitrypsin gene therapy extends the lifespan of lupus-prone mice. Mol. Ther. Methods Clin. Dev. 2018, 11, 131–142. [Google Scholar] [CrossRef]
- Singh, B.B.; Ohm, J.; Quenum Zanbede, F.O.; Chauhan, P.; Kroese, F.G.M.; Vissink, A.; Ambrus, J.L.; Mishra, B.B. Decrease in alpha-1 antiproteinase antitrypsin is observed in primary Sjogren’s syndrome condition. Autoimmunity 2020, 53, 270–282. [Google Scholar] [CrossRef] [PubMed]
- Marcondes, A.M.; Hockenbery, D.; Lesnikova, M.; Dinarello, C.A.; Woolfrey, A.; Gernsheimer, T.; Loghman-Adham, M.; Gelmont, D.; Storer, B.; Hansen, J.A.; et al. Response of steroid-refractory acute GVHD to α1-antitrypsin. Biol. Blood Marrow Transplant. 2016, 22, 1596–1601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magenau, J.M.; Goldstein, S.C.; Peltier, D.; Soiffer, R.J.; Braun, T.; Pawarode, A.; Riwes, M.M.; Kennel, M.; Antin, J.H.; Cutler, C.S.; et al. α1-antitrypsin infusion for treatment of steroid-resistant acute graft-versus-host disease. Blood 2018, 131, 1372–1379. [Google Scholar] [CrossRef] [Green Version]
- Strassmair, M.; Stangl, M. Alpha-1 antitrypsin deficiency and COVID-19 infection. Arch. Bronconeumol. 2021, 57, 97. [Google Scholar] [CrossRef] [PubMed]
- McEvoy, N.L.; Clarke, J.L.; Mc Elvaney, O.J.; Mc Elvaney, O.F.; Boland, F.; Hyland, D.; Geoghegan, P.; Donnelly, K.; Friel, O.; Cullen, A.; et al. A randomised, double-blind, placebo-controlled, pilot trial of intravenous plasma purified alpha-1 antitrypsin for SARS-CoV-2-induced Acute Respiratory Distress Syndrome: A structured summary of a study protocol for a randomised, controlled trial. Trials 2021, 22, 288. [Google Scholar] [CrossRef]
- McElvaney, O.J.; O’Connor, E.; McEvoy, N.L.; Fraughan, D.D.; Clarke, J.; McElvaney, O.F.; Gunaratnam, C.; O’Rourke, J.; Curley, G.F.; McElvaney, N.G. Alpha-1 antitrypsin for cystic fibrosis complicated by severe cytokinemic COVID-19. J. Cyst. Fibros. 2021, 20, 31–35. [Google Scholar] [CrossRef]
- Ritzmann, F.; Chitirala, P.; Krüger, N.; Hoffmann, M.; Zuo, W.; Lammert, F.; Smola, S.; Tov, N.; Alagem, N.; Lepper, P.M.; et al. Therapeutic application of alpha-1-antitrypsin in COVID-19. Am. J. Respir. Crit. Care Med. 2021, 204, 224–227. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Binding Partner | Disease Processes | Reference | |
|---|---|---|---|
| Plasma andcirculating cells | Apolipoprotein B-100 | Atheroma | [8] |
| IgA complexes | Rheumatoid arthritis, myeloma | [9] | |
| Fibrinogen | Healthy | [10] | |
| IgK light chains | Myeloma | [11] | |
| HSP 70 | Diabetes mellitus | [12] | |
| Grp94 | Diabetes mellitus | [13] | |
| PSA/kallikrein 3 | Benin prostatic hypertrophy, prostate cancer | [14] | |
| Cholesterol | Not specified | [15] | |
| Leukotriene B4 | Inflammation | [6] | |
| Complement C3 | Complement activation | [16] | |
| Complement C4-A | Not specified | [16] | |
| Serum albumin | Not specified | [16] | |
| Apolipoprotein A-I | Not specified | [16] | |
| Prothrombin | Not specified | [16] | |
| IL-8 | Emphysema/pneumonia | [7,17] | |
| TNFR | Emphysema | [18] | |
| FcγRIIIb (CD16b) | Biomarker of pulmonary exacerbation | [19] | |
| Intracellular | Heme | Not specified | [20] |
| Prohepcidin | Iron metabolism | [21] | |
| Retinoic acid | Emphysema | [22] | |
| Caspase 3 | Apoptosis, emphysema | [23] | |
| Caspase 1 | Apoptosis | [24,25] | |
| Calpain 1 | Neutrophil activation | [26] | |
| Extracellular/Tissue | IgA complexes | Synovial fluid/rheumatoid arthritis | [9,10] |
| Surfactant protein A | Airway surface liquid | [27] | |
| Aggrecanase 1 | Synovial tissue/OA | [28] | |
| Bile acids | Bile | [29] | |
| NO | Inflammation/innate immunity | [30] |
| Role of Alpha-1 Antitrypsin | Function | Reference |
|---|---|---|
| Protease inhibitor | Anti-NE, -Cath-G and -PR3 | [47] |
| Anti-apoptosis | Inhibition of caspase-1, caspase-3, and calpain-1 | [23,25,26] |
| Antioxidant | Oxidative stress inhibition | [67] |
| Anti-inflammatory/tissue repair | Repair, fibroblast proliferation, procollagen synthesis, and activation of MAP kinase pathways | [68] |
| Modulation of ADAM-17 activity | [28] | |
| Substrate for metalloproteinase MMP-9 activity | [69] | |
| Inactivation of matriptase in vitro and inhibition of epithelial sodium transport in vitro and in vivo | [70] | |
| Antibacterial | Bacteriostasis—binding to furin (inhibits bacterial toxin activation) | [71] |
| Antiviral | Inhibition of HIV-1 viral cell entry | [72] |
| Inhibition of SARS-CoV-2 entry by inhibiting transmembrane serine protease 2 and ADAM-17 | [73,74] |
| Proteinase | AAT | Oxidised AAT | Reference |
|---|---|---|---|
| Neutrophil Elastase | 6.5 ± 4.0 × 107 | 3.1 ± 0.2 × 104 | [77] |
| Proteinase 3 | 8.1 × 106 | - | [78] |
| Cathepsin G | 4.1 ± 0.6 × 105 | 6.5 ± 0.3 × 102 | [77] |
| Chymotrypsin | 5.4 ± 0.6 × 106 | 1.0 ± 0.4 × 106 | [77] |
| Trypsin 2 (Anionic) | 7.3 ± 1.8 × 104 | 3.2 ± 0.1 × 104 | [77] |
| Trypsin 1 (Cationic) | 1.1 ± 0.2 × 104 | 3.0 ± 1.1 × 103 | [77] |
| Factor Xia | 1.3 × 104 | - | [80] |
| Matriptase | 3.1 × 102 | 0 | [70] |
| Plasmin | 1.9 ± 0.1 × 102 | 0 | [95] |
| Thrombin | 4.8 ± 0.5 × 101 | 0 | [96] |
| Activated Protein C | 1.1 × 101 | - | [97] |
| Transmembrane Serine Protease 2 | - | - | [74] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
O’Brien, M.E.; Murray, G.; Gogoi, D.; Yusuf, A.; McCarthy, C.; Wormald, M.R.; Casey, M.; Gabillard-Lefort, C.; McElvaney, N.G.; Reeves, E.P. A Review of Alpha-1 Antitrypsin Binding Partners for Immune Regulation and Potential Therapeutic Application. Int. J. Mol. Sci. 2022, 23, 2441. https://doi.org/10.3390/ijms23052441
O’Brien ME, Murray G, Gogoi D, Yusuf A, McCarthy C, Wormald MR, Casey M, Gabillard-Lefort C, McElvaney NG, Reeves EP. A Review of Alpha-1 Antitrypsin Binding Partners for Immune Regulation and Potential Therapeutic Application. International Journal of Molecular Sciences. 2022; 23(5):2441. https://doi.org/10.3390/ijms23052441
Chicago/Turabian StyleO’Brien, Michael E., Grace Murray, Debananda Gogoi, Azeez Yusuf, Cormac McCarthy, Mark R. Wormald, Michelle Casey, Claudie Gabillard-Lefort, Noel G. McElvaney, and Emer P. Reeves. 2022. "A Review of Alpha-1 Antitrypsin Binding Partners for Immune Regulation and Potential Therapeutic Application" International Journal of Molecular Sciences 23, no. 5: 2441. https://doi.org/10.3390/ijms23052441
APA StyleO’Brien, M. E., Murray, G., Gogoi, D., Yusuf, A., McCarthy, C., Wormald, M. R., Casey, M., Gabillard-Lefort, C., McElvaney, N. G., & Reeves, E. P. (2022). A Review of Alpha-1 Antitrypsin Binding Partners for Immune Regulation and Potential Therapeutic Application. International Journal of Molecular Sciences, 23(5), 2441. https://doi.org/10.3390/ijms23052441