
Bivalent EGFR-Targeting DARPin-MMAE Conjugates

 , , , and
, , , and
Abstract
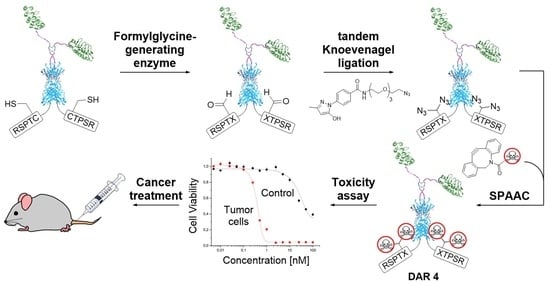
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Chemical Synthesis and Protein Conjugation
2.1.1. Synthesis of PEGylated DBCO-MMAE Linker
2.1.2. FGly Formation in Proteins
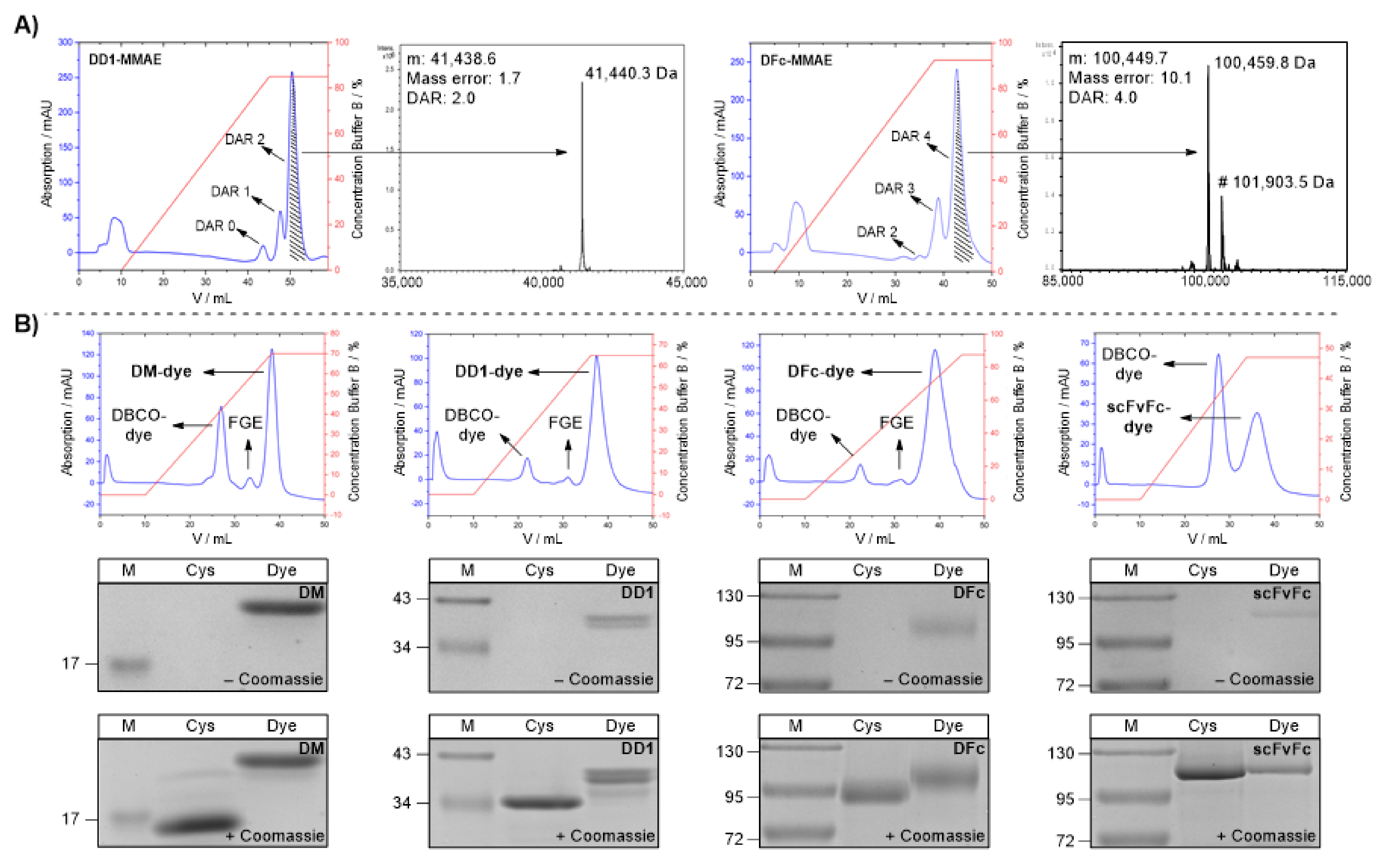
2.1.3. Protein-MMAE Conjugates
2.1.4. Protein-Alexa Fluor 647 Conjugates
2.2. In Vitro Studies
2.2.1. EGFR-Mediated Endocytosis of Alexa Fluor 647 Conjugates
2.2.2. MMAE Conjugates with Sub-Nanomolar Cytotoxicity In Vitro
2.3. In Vivo Studies
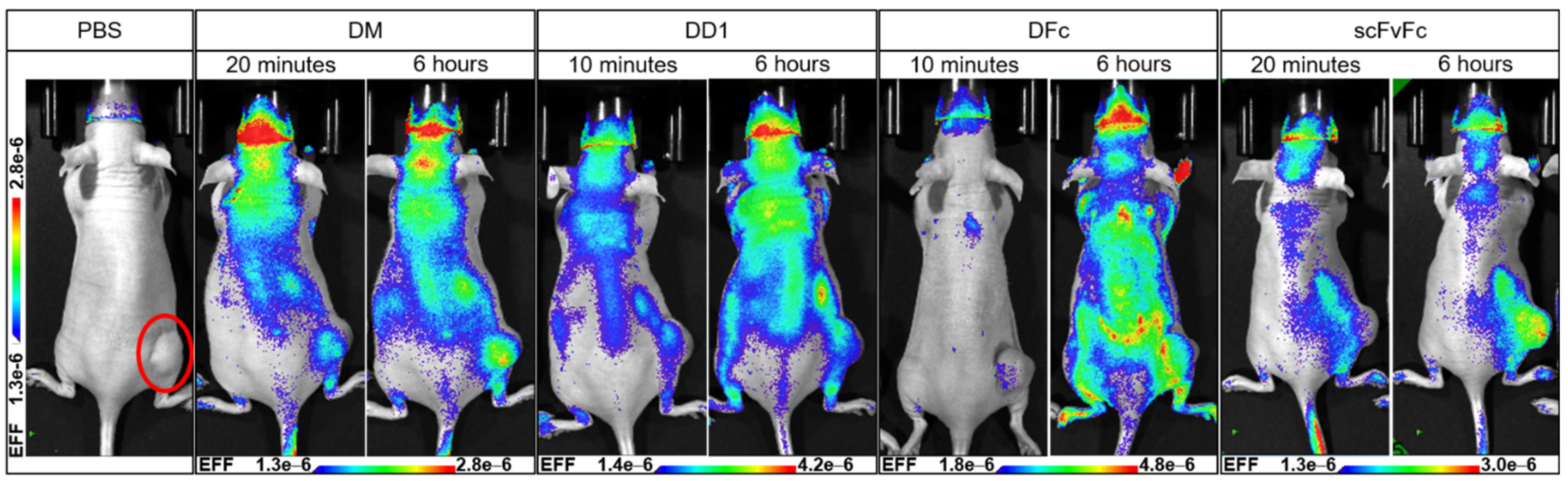
2.3.1. In Vivo Imaging of Anti-EGFR Dye Conjugates
2.3.2. Ex Vivo Imaging of Anti-EGFR Dye Conjugates
2.3.3. In Vivo Efficacy of MMAE Conjugates
3. Materials and Methods
3.1. Organic Synthesis
3.2. Biochemical/Biological Experiments
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Feiner, R.C.; Müller, K.M. Recent progress in protein-protein interaction study for EGFR-targeted therapeutics. Expert Rev. Proteom. 2016, 13, 817–832. [Google Scholar] [CrossRef] [PubMed]
- Heukers, R.; Vermeulen, J.F.; Fereidouni, F.; Bader, A.N.; Voortman, J.; Roovers, R.C.; Gerritsen, H.C.; Van Bergen En Henegouwen, P.M.P. Endocytosis of EGFR requires its kinase activity and N-terminal transmembrane dimerization motif. J. Cell Sci. 2013, 126, 4900–4912. [Google Scholar] [CrossRef] [Green Version]
- Ogiso, H.; Ishitani, R.; Nureki, O.; Fukai, S.; Yamanaka, M.; Kim, J.H.; Saito, K.; Sakamoto, A.; Inoue, M.; Shirouzu, M.; et al. Crystal structure of the complex of human epidermal growth factor and receptor extracellular domains. Cell 2002, 110, 775–787. [Google Scholar] [CrossRef] [Green Version]
- Tomas, A.; Futter, C.E.; Eden, E.R. EGF receptor trafficking: Consequences for signaling and cancer. Trends Cell Biol. 2014, 24, 26–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roskoski, R. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol. Res. 2014, 79, 34–74. [Google Scholar] [CrossRef]
- Purba, E.; Saita, E.; Maruyama, I. Activation of the EGF Receptor by Ligand Binding and Oncogenic Mutations: The Rotation Model. Cells 2017, 6, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, Y.; Kimura, T.; Nakagawa, T.; Okamoto, K.; Fukuya, A.; Goji, T.; Fujimoto, S.; Sogabe, M.; Miyamoto, H.; Muguruma, N.; et al. EGFR Downregulation after Anti-EGFR Therapy Predicts the Antitumor Effect in Colorectal Cancer. Mol. Cancer Res. 2017, 15, 1445–1454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Interlandi, G.; Wetzel, S.K.; Settanni, G.; Plückthun, A.; Caflisch, A. Characterization and further stabilization of designed ankyrin repeat proteins by combining molecular dynamics simulations and experiments. J. Mol. Biol. 2008, 375, 837–854. [Google Scholar] [CrossRef] [Green Version]
- Steiner, D.; Forrer, P.; Plückthun, A. Efficient selection of DARPins with sub-nanomolar affinities using SRP phage display. J. Mol. Biol. 2008, 382, 1211–1227. [Google Scholar] [CrossRef] [Green Version]
- Boersma, Y.L.; Chao, G.; Steiner, D.; Wittrup, K.D.; Plückthun, A. Bispecific designed ankyrin repeat proteins (DARPins) targeting epidermal growth factor receptor inhibit A431 cell proliferation and receptor recycling. J. Biol. Chem. 2011, 286, 41273–41285. [Google Scholar] [CrossRef] [Green Version]
- Müller, K.M.; Arndt, K.M.; Strittmatter, W.; Plückthun, A. The first constant domain (C(H)1 and C(L)) of an antibody used as heterodimerization domain for bispecific miniantibodies. FEBS Lett. 1998, 422, 259–264. [Google Scholar] [CrossRef] [Green Version]
- Müller, K.M.; Arndt, K.M.; Plückthun, A. A dimeric bispecific miniantibody combines two specificities with avidity. FEBS Lett. 1998, 432, 45–49. [Google Scholar] [CrossRef] [Green Version]
- Murthy, U.; Basu, A.; Rodeck, U.; Herlyn, M.; Ross, A.H.; Das, M. Binding of an antagonistic monoclonal antibody to an intact and fragmented EGF-receptor polypeptide. Arch. Biochem. Biophys. 1987, 252, 549–560. [Google Scholar] [CrossRef]
- Schmiedel, J.; Blaukat, A.; Li, S.; Knöchel, T.; Ferguson, K.M.; Knoechel, T.; Ferguson, K.M. Matuzumab binding to EGFR prevents the conformational rearrangement required for dimerization. Cancer Cell 2008, 13, 365–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, W.-Q.; Zeng, L.-S.; Wang, L.-F.; Wang, Y.-Y.; Cheng, J.-T.; Zhang, Y.; Han, Z.-W.; Zhou, Y.; Huang, S.-L.; Wang, X.-W.; et al. The Latest Battles Between EGFR Monoclonal Antibodies and Resistant Tumor Cells. Front. Oncol. 2020, 10, 1249. [Google Scholar] [CrossRef]
- Joubert, N.; Beck, A.; Dumontet, C.; Denevault-Sabourin, C. Antibody-Drug Conjugates: The Last Decade. Pharmaceuticals 2020, 13, 245. [Google Scholar] [CrossRef] [PubMed]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef] [PubMed]
- Junutula, J.R.; Raab, H.; Clark, S.; Bhakta, S.; Leipold, D.D.; Weir, S.; Chen, Y.; Simpson, M.; Tsai, S.P.; Dennis, M.S.; et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat. Biotechnol. 2008, 26, 925–932. [Google Scholar] [CrossRef] [PubMed]
- Strijbis, K.; Spooner, E.; Ploegh, H.L. Protein ligation in living cells using sortase. Traffic 2012, 13, 780–789. [Google Scholar] [CrossRef] [Green Version]
- Beerli, R.R.; Hell, T.; Merkel, A.S.; Grawunder, U. Sortase Enzyme-Mediated Generation of Site-Specifically Conjugated Antibody Drug Conjugates with High In Vitro and In Vivo Potency. PLoS ONE 2015, 10, e0131177. [Google Scholar] [CrossRef] [Green Version]
- Spycher, P.R.; Amann, C.A.; Wehrmüller, J.E.; Hurwitz, D.R.; Kreis, O.; Messmer, D.; Ritler, A.; Küchler, A.; Blanc, A.; Béhé, M.; et al. Dual, Site-Specific Modification of Antibodies by Using Solid-Phase Immobilized Microbial Transglutaminase. Chembiochem 2017, 18, 1923–1927. [Google Scholar] [CrossRef] [PubMed]
- Dennler, P.; Chiotellis, A.; Fischer, E.; Brégeon, D.; Belmant, C.; Gauthier, L.; Lhospice, F.; Romagne, F.; Schibli, R. Transglutaminase-based chemo-enzymatic conjugation approach yields homogeneous antibody-drug conjugates. Bioconjug. Chem. 2014, 25, 569–578. [Google Scholar] [CrossRef]
- Siegmund, V.; Schmelz, S.; Dickgiesser, S.; Beck, J.; Ebenig, A.; Fittler, H.; Frauendorf, H.; Piater, B.; Betz, U.A.K.; Avrutina, O.; et al. Locked by Design: A Conformationally Constrained Transglutaminase Tag Enables Efficient Site-Specific Conjugation. Angew. Chem. Int. Ed. Engl. 2015, 54, 13420–13424. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, C.M.; Yamaguchi, A.; Anami, Y.; Xiong, W.; Otani, Y.; Lee, J.; Ueno, N.T.; Zhang, N.; An, Z.; Tsuchikama, K. Antibody-drug conjugates with dual payloads for combating breast tumor heterogeneity and drug resistance. Nat. Commun. 2021, 12, 3528. [Google Scholar] [CrossRef] [PubMed]
- Anami, Y.; Yamazaki, C.M.; Xiong, W.; Gui, X.; Zhang, N.; An, Z.; Tsuchikama, K. Glutamic acid-valine-citrulline linkers ensure stability and efficacy of antibody-drug conjugates in mice. Nat. Commun. 2018, 9, 2512. [Google Scholar] [CrossRef]
- Schneider, H.; Deweid, L.; Avrutina, O.; Kolmar, H. Recent progress in transglutaminase-mediated assembly of antibody-drug conjugates. Anal. Biochem. 2020, 595, 113615. [Google Scholar] [CrossRef]
- Rashidian, M.; Kumarapperuma, S.C.; Gabrielse, K.; Fegan, A.; Wagner, C.R.; Distefano, M.D. Simultaneous dual protein labeling using a triorthogonal reagent. J. Am. Chem. Soc. 2013, 135, 16388–16396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landgrebe, J.; Dierks, T.; Schmidt, B.; von Figura, K. The human SUMF1 gene, required for posttranslational sulfatase modification, defines a new gene family which is conserved from pro- to eukaryotes. Gene 2003, 316, 47–56. [Google Scholar] [CrossRef]
- Peng, J.; Alam, S.; Radhakrishnan, K.; Mariappan, M.; Rudolph, M.G.; May, C.; Dierks, T.; von Figura, K.; Schmidt, B. Eukaryotic formylglycine-generating enzyme catalyses a monooxygenase type of reaction. FEBS J. 2015, 282, 3262–3274. [Google Scholar] [CrossRef] [Green Version]
- Von Figura, K.; Schmidt, B.; Selmer, T.; Dierks, T. A novel protein modification generating an aldehyde group in sulfatases: Its role in catalysis and disease. Bioessays 1998, 20, 505–510. [Google Scholar] [CrossRef]
- Knop, M.; Dang, T.Q.; Jeschke, G.; Seebeck, F.P. Copper is a Cofactor of the Formylglycine-Generating Enzyme. Chembiochem 2017, 18, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Holder, P.G.; Jones, L.C.; Drake, P.M.; Barfield, R.M.; Bañas, S.; De Hart, G.W.; Baker, J.; Rabuka, D. Reconstitution of formylglycine-generating enzyme with copper(II) for aldehyde tag conversion. J. Biol. Chem. 2015, 290, 15730–15745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dierks, T.; Schmidt, B.; Borissenko, L.V.; Peng, J.; Preusser, A.; Mariappan, M.; von Figura, K. Multiple sulfatase deficiency is caused by mutations in the gene encoding the human C(alpha)-formylglycine generating enzyme. Cell 2003, 113, 435–444. [Google Scholar] [CrossRef] [Green Version]
- Dierks, T.; Lecca, M.R.; Schlotterhose, P.; Schmidt, B.; von Figura, K. Sequence determinants directing conversion of cysteine to formylglycine in eukaryotic sulfatases. EMBO J. 1999, 18, 2084–2091. [Google Scholar] [CrossRef] [Green Version]
- Krüger, T.; Weiland, S.; Falck, G.; Gerlach, M.; Boschanski, M.; Alam, S.; Müller, K.M.; Dierks, T.; Sewald, N. Two-fold Bioorthogonal Derivatization by Different Formylglycine Generating Enzymes. Angew. Chem. Int. Ed. 2018, 57, 7245–7249. [Google Scholar] [CrossRef]
- Krüger, T.; Weiland, S.; Boschanski, M.; Sinha, P.K.; Falck, G.; Müller, K.M.; Dierks, T.; Sewald, N. Conversion of Serine-Type Aldehyde Tags by the Radical SAM Protein AtsB from Methanosarcina mazei. Chembiochem 2019, 20, 2074–2078. [Google Scholar] [CrossRef]
- Agarwal, P.; Kudirka, R.; Albers, A.E.; Barfield, R.M.; de Hart, G.W.; Drake, P.M.; Jones, L.C.; Rabuka, D. Hydrazino-Pictet-Spengler ligation as a biocompatible method for the generation of stable protein conjugates. Bioconjug. Chem. 2013, 24, 846–851. [Google Scholar] [CrossRef]
- Drake, P.M.; Albers, A.E.; Baker, J.; Banas, S.; Barfield, R.M.; Bhat, A.S.; de Hart, G.W.; Garofalo, A.W.; Holder, P.; Jones, L.C.; et al. Aldehyde tag coupled with HIPS chemistry enables the production of ADCs conjugated site-specifically to different antibody regions with distinct in vivo efficacy and PK outcomes. Bioconjug. Chem. 2014, 25, 1331–1341. [Google Scholar] [CrossRef]
- Boschanski, M.; Krüger, T.; Karsten, L.; Falck, G.; Alam, S.; Gerlach, M.; Müller, B.; Müller, K.M.; Sewald, N.; Dierks, T. Site-Specific Conjugation Strategy for Dual Antibody-Drug Conjugates Using Aerobic Formylglycine-Generating Enzymes. Bioconjug. Chem. 2021, 32, 1167–1174. [Google Scholar] [CrossRef]
- Janson, N.; Krüger, T.; Karsten, L.; Boschanski, M.; Dierks, T.; Müller, K.M.; Sewald, N. Bifunctional Reagents for Formylglycine Conjugation: Pitfalls and Breakthroughs. Chembiochem 2020, 21, 3580–3593. [Google Scholar] [CrossRef]
- Kudirka, R.; Barfield, R.M.; McFarland, J.; Albers, A.E.; de Hart, G.W.; Drake, P.M.; Holder, P.G.; Banas, S.; Jones, L.C.; Garofalo, A.W.; et al. Generating site-specifically modified proteins via a versatile and stable nucleophilic carbon ligation. Chem. Biol. 2015, 22, 293–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudirka, R.A.; Barfield, R.M.; McFarland, J.M.; Drake, P.M.; Carlson, A.; Bañas, S.; Zmolek, W.; Garofalo, A.W.; Rabuka, D. Site-Specific Tandem Knoevenagel Condensation-Michael Addition To Generate Antibody-Drug Conjugates. ACS Med. Chem. Lett. 2016, 7, 994–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Shen, D.; Zhang, H.; Yin, Z. Rapid, Stoichiometric, Site-Specific Modification of Aldehyde-Containing Proteins Using a Tandem Knoevenagel-Intra Michael Addition Reaction. Bioconjug. Chem. 2018, 29, 1016–1020. [Google Scholar] [CrossRef] [PubMed]
- Burke, P.J.; Hamilton, J.Z.; Jeffrey, S.C.; Hunter, J.H.; Doronina, S.O.; Okeley, N.M.; Miyamoto, J.B.; Anderson, M.E.; Stone, I.J.; Ulrich, M.L.; et al. Optimization of a PEGylated Glucuronide-Monomethylauristatin E Linker for Antibody-Drug Conjugates. Mol. Cancer Ther. 2017, 16, 116–123. [Google Scholar] [CrossRef] [Green Version]
- Zahnd, C.; Kawe, M.; Stumpp, M.T.; De Pasquale, C.; Tamaskovic, R.; Nagy-Davidescu, G.; Dreier, B.; Schibli, R.; Binz, H.K.; Waibel, R.; et al. Efficient tumor targeting with high-affinity designed ankyrin repeat proteins: Effects of affinity and molecular size. Cancer Res. 2010, 70, 1595–1605. [Google Scholar] [CrossRef] [Green Version]
- Janson, N.; Heinks, T.; Beuel, T.; Alam, S.; Höhne, M.; Bornscheuer, U.T.; Fischer von Mollard, G.; Sewald, N. Efficient site-selective immobilization of aldehyde-tagged peptides and proteins by Knoevenagel ligation. ChemCatChem 2021, 14, e202101485. [Google Scholar] [CrossRef]
- Padlan, E.A. Anatomy of the antibody molecule. Mol. Immunol. 1994, 31, 169–217. [Google Scholar] [CrossRef] [Green Version]
- Davidson, N.E.; Gelmann, E.P.; Lippman, M.E.; Dickson, R.B. Epidermal growth factor receptor gene expression in estrogen receptor-positive and negative human breast cancer cell lines. Mol. Endocrinol. 1987, 1, 216–223. [Google Scholar] [CrossRef] [Green Version]
- Giard, D.J.; Aaronson, S.A.; Todaro, G.J.; Arnstein, P.; Kersey, J.H.; Dosik, H.; Parks, W.P. In vitro cultivation of human tumors: Establishment of cell lines derived from a series of solid tumors. J. Natl. Cancer Inst. 1973, 51, 1417–1423. [Google Scholar] [CrossRef]
- Lakadamyali, M.; Rust, M.J.; Zhuang, X. Ligands for Clathrin-Mediated Endocytosis Are Differentially Sorted into Distinct Populations of Early Endosomes. Cell 2006, 124, 997–1009. [Google Scholar] [CrossRef] [Green Version]
- Björkelund, H.; Gedda, L.; Malmqvist, M.; Andersson, K. Resolving the EGF-EGFR interaction characteristics through a multiple-temperature, multiple-inhibitor, real-time interaction analysis approach. Mol. Clin. Oncol. 2013, 1, 343–352. [Google Scholar] [CrossRef] [Green Version]
- Le Joncour, V.; Filppu, P.; Hyvönen, M.; Holopainen, M.; Turunen, S.P.; Sihto, H.; Burghardt, I.; Joensuu, H.; Tynninen, O.; Jääskeläinen, J.; et al. Vulnerability of invasive glioblastoma cells to lysosomal membrane destabilization. EMBO Mol. Med. 2019, 11, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Feiner, R.C.; Pennè, I.; Müller, B.; Müller, K.M. EGF-mCherry Fusion Protein Expressed in E. coli Shows Product Heterogeneity but a High Biological Activity. Biochemistry 2019, 58, 1043–1047. [Google Scholar] [CrossRef]
- Hu, X.-Y.; Wang, R.; Jin, J.; Liu, X.-J.; Cui, A.-L.; Sun, L.-Q.; Li, Y.-P.; Li, Y.; Wang, Y.-C.; Zhen, Y.-S.; et al. An EGFR-targeting antibody-drug conjugate LR004-VC-MMAE: Potential in esophageal squamous cell carcinoma and other malignancies. Mol. Oncol. 2019, 13, 246–263. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Wang, S.; Yin, L.; Yang, Y.; Guan, Y.; Wang, W.; Xu, H.; Tao, N. Quantification of epidermal growth factor receptor expression level and binding kinetics on cell surfaces by surface plasmon resonance imaging. Anal. Chem. 2015, 87, 9960–9965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamat, V.; Donaldson, J.M.; Kari, C.; Quadros, M.R.D.; Lelkes, P.I.; Chaiken, I.; Cocklin, S.; Williams, J.C.; Papazoglou, E.; Rodeck, U. Enhanced EGFR inhibition and distinct epitope recognition by EGFR antagonistic mAbs C225 and 425. Cancer Biol. Ther. 2008, 7, 726–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van den Brand, D.; van Lith, S.A.M.; de Jong, J.M.; Gorris, M.A.J.; Palacio-Castañeda, V.; Couwenbergh, S.T.; Goldman, M.R.G.; Ebisch, I.; Massuger, L.F.; Leenders, W.P.J.; et al. EpCAM-Binding DARPins for Targeted Photodynamic Therapy of Ovarian Cancer. Cancers 2020, 12, 1762. [Google Scholar] [CrossRef]
- Wen, X.; Wu, Q.P.; Ke, S.; Ellis, L.; Charnsangavej, C.; Delpassand, A.S.; Wallace, S.; Li, C. Conjugation with (111)In-DTPA-poly(ethylene glycol) improves imaging of anti-EGF receptor antibody C225. J. Nucl. Med. 2001, 42, 1530–1537. [Google Scholar]
- Lyon, R.P.; Bovee, T.D.; Doronina, S.O.; Burke, P.J.; Hunter, J.H.; Neff-LaFord, H.D.; Jonas, M.; Anderson, M.E.; Setter, J.R.; Senter, P.D. Reducing hydrophobicity of homogeneous antibody-drug conjugates improves pharmacokinetics and therapeutic index. Nat. Biotechnol. 2015, 33, 733–735. [Google Scholar] [CrossRef]
- Yang, Y.-P.; Ma, H.; Starchenko, A.; Huh, W.J.; Li, W.; Hickman, F.E.; Zhang, Q.; Franklin, J.L.; Mortlock, D.P.; Fuhrmann, S.; et al. A Chimeric Egfr Protein Reporter Mouse Reveals Egfr Localization and Trafficking In Vivo. Cell Rep. 2017, 19, 1257–1267. [Google Scholar] [CrossRef] [Green Version]
- Bult, C.J.; Blake, J.A.; Smith, C.L.; Kadin, J.A.; Richardson, J.E.; Anagnostopoulos, A.; Asabor, R.; Baldarelli, R.M.; Beal, J.S.; Bello, S.M.; et al. Mouse Genome Database (MGD). Nucleic Acids Res. 2019, 47, D801–D806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorovits, B.; Krinos-Fiorotti, C. Proposed mechanism of off-target toxicity for antibody-drug conjugates driven by mannose receptor uptake. Cancer Immunol. Immunother. 2013, 62, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Swinson, D.E.B.; Jones, J.L.; Richardson, D.; Cox, G.; Edwards, J.G.; O’Byrne, K.J. Tumour necrosis is an independent prognostic marker in non-small cell lung cancer: Correlation with biological variables. Lung Cancer 2002, 37, 235–240. [Google Scholar] [CrossRef]
- Yang, X.D.; Jia, X.C.; Corvalan, J.R.F.; Wang, P.; Davis, C.G.; Jakobovits, A. Eradication of established tumors by a fully human monoclonal antibody to the epidermal growth factor receptor without concomitant chemotherapy. Cancer Res. 1999, 59, 1236–1243. [Google Scholar] [PubMed]
- Kitamura, S.; Maeda, T.; Yanagi, T. Vandetanib inhibits cell growth in EGFR-expressing cutaneous squamous cell carcinoma. Biochem. Biophys. Res. Commun. 2020, 531, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Kochneva, G.; Zonov, E.; Grazhdantseva, A.; Yunusova, A.; Sibolobova, G.; Popov, E.; Taranov, O.; Netesov, S.; Chumakov, P.; Ryabchikova, E. Apoptin enhances the oncolytic properties of vaccinia virus and modifies mechanisms of tumor regression. Oncotarget 2014, 5, 11269–11282. [Google Scholar] [CrossRef] [PubMed]
- Wiig, H.; Tenstad, O.; Iversen, P.O.; Kalluri, R.; Bjerkvig, R. Interstitial fluid: The overlooked component of the tumor microenvironment? Fibrogenesis Tissue Repair 2010, 3, 12. [Google Scholar] [CrossRef] [Green Version]
- Leu, A.J.; Berk, D.A.; Lymboussaki, A.; Alitalo, K.; Jain, R.K. Absence of functional lymphatics within a murine sarcoma: A molecular and functional evaluation. Cancer Res. 2000, 60, 4324–4327. [Google Scholar]
- Pflanzer, R.; Hofmann, M.; Shelke, A.; Habib, A.; Derwich, W.; Schmitz-Rixen, T.; Bernd, A.; Kaufmann, R.; Bereiter-Hahn, J. Advanced 3D-Sonographic Imaging as a Precise Technique to Evaluate Tumor Volume. Transl. Oncol. 2014, 7, 681–686. [Google Scholar] [CrossRef] [Green Version]
- Pflanzer, R.; Shelke, A.; Bereiter-Hahn, J.; Hofmann, M. Ultrasonic Quantification of Tumor Interstitial Fluid Pressure through Scanning Acoustic Microscopy; Springer: Berlin/Heidelberg, Germany, 2012; pp. 291–298. [Google Scholar] [CrossRef]
- Singh, A.P.; Shah, D.K. A “Dual” Cell-Level Systems PK-PD Model to Characterize the Bystander Effect of ADC. J. Pharm. Sci. 2019, 108, 2465–2475. [Google Scholar] [CrossRef]
- Martin-Killias, P.; Stefan, N.; Rothschild, S.; Plückthun, A.; Zangemeister-Wittke, U. A novel fusion toxin derived from an EpCAM-specific designed ankyrin repeat protein has potent antitumor activity. Clin. Cancer Res. 2011, 17, 100–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, M.; Frey, R.; Zangemeister-Wittke, U.; Plückthun, A. Orthogonal assembly of a designed ankyrin repeat protein-cytotoxin conjugate with a clickable serum albumin module for half-life extension. Bioconjug. Chem. 2013, 24, 1955–1966. [Google Scholar] [CrossRef] [PubMed]
- Brandl, F.; Busslinger, S.; Zangemeister-Wittke, U.; Plückthun, A. Optimizing the anti-tumor efficacy of protein-drug conjugates by engineering the molecular size and half-life. J. Control. Release 2020, 327, 186–197. [Google Scholar] [CrossRef]
- Lu, C.; Mi, L.-Z.; Grey, M.J.; Zhu, J.; Graef, E.; Yokoyama, S.; Springer, T.A. Structural Evidence for Loose Linkage between Ligand Binding and Kinase Activation in the Epidermal Growth Factor Receptor. Mol. Cell. Biol. 2010, 30, 5432–5443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karsten, L.; Janson, N.; Le Joncour, V.; Alam, S.; Müller, B.; Tanjore Ramanathan, J.; Laakkonen, P.; Sewald, N.; Müller, K.M. Bivalent EGFR-Targeting DARPin-MMAE Conjugates. Int. J. Mol. Sci. 2022, 23, 2468. https://doi.org/10.3390/ijms23052468
Karsten L, Janson N, Le Joncour V, Alam S, Müller B, Tanjore Ramanathan J, Laakkonen P, Sewald N, Müller KM. Bivalent EGFR-Targeting DARPin-MMAE Conjugates. International Journal of Molecular Sciences. 2022; 23(5):2468. https://doi.org/10.3390/ijms23052468
Chicago/Turabian StyleKarsten, Lennard, Nils Janson, Vadim Le Joncour, Sarfaraz Alam, Benjamin Müller, Jayendrakishore Tanjore Ramanathan, Pirjo Laakkonen, Norbert Sewald, and Kristian M. Müller. 2022. "Bivalent EGFR-Targeting DARPin-MMAE Conjugates" International Journal of Molecular Sciences 23, no. 5: 2468. https://doi.org/10.3390/ijms23052468
APA StyleKarsten, L., Janson, N., Le Joncour, V., Alam, S., Müller, B., Tanjore Ramanathan, J., Laakkonen, P., Sewald, N., & Müller, K. M. (2022). Bivalent EGFR-Targeting DARPin-MMAE Conjugates. International Journal of Molecular Sciences, 23(5), 2468. https://doi.org/10.3390/ijms23052468