
Botrytis cinerea Loss and Restoration of Virulence during In Vitro Culture Follows Flux in Global DNA Methylation


 , and
, and
Abstract
:1. Introduction
2. Results
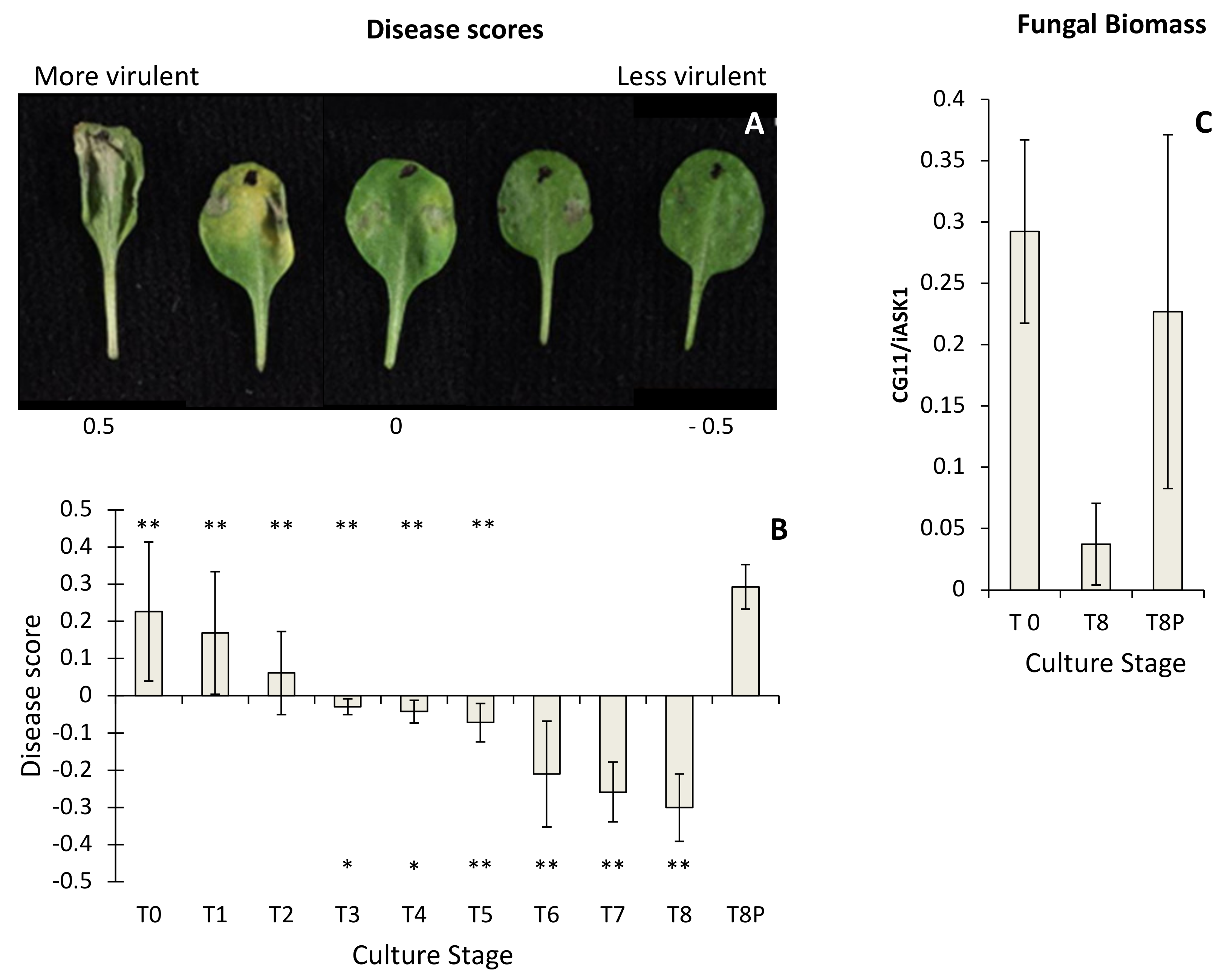
2.1. Pathogenicity Analysis of Botrytis cinerea
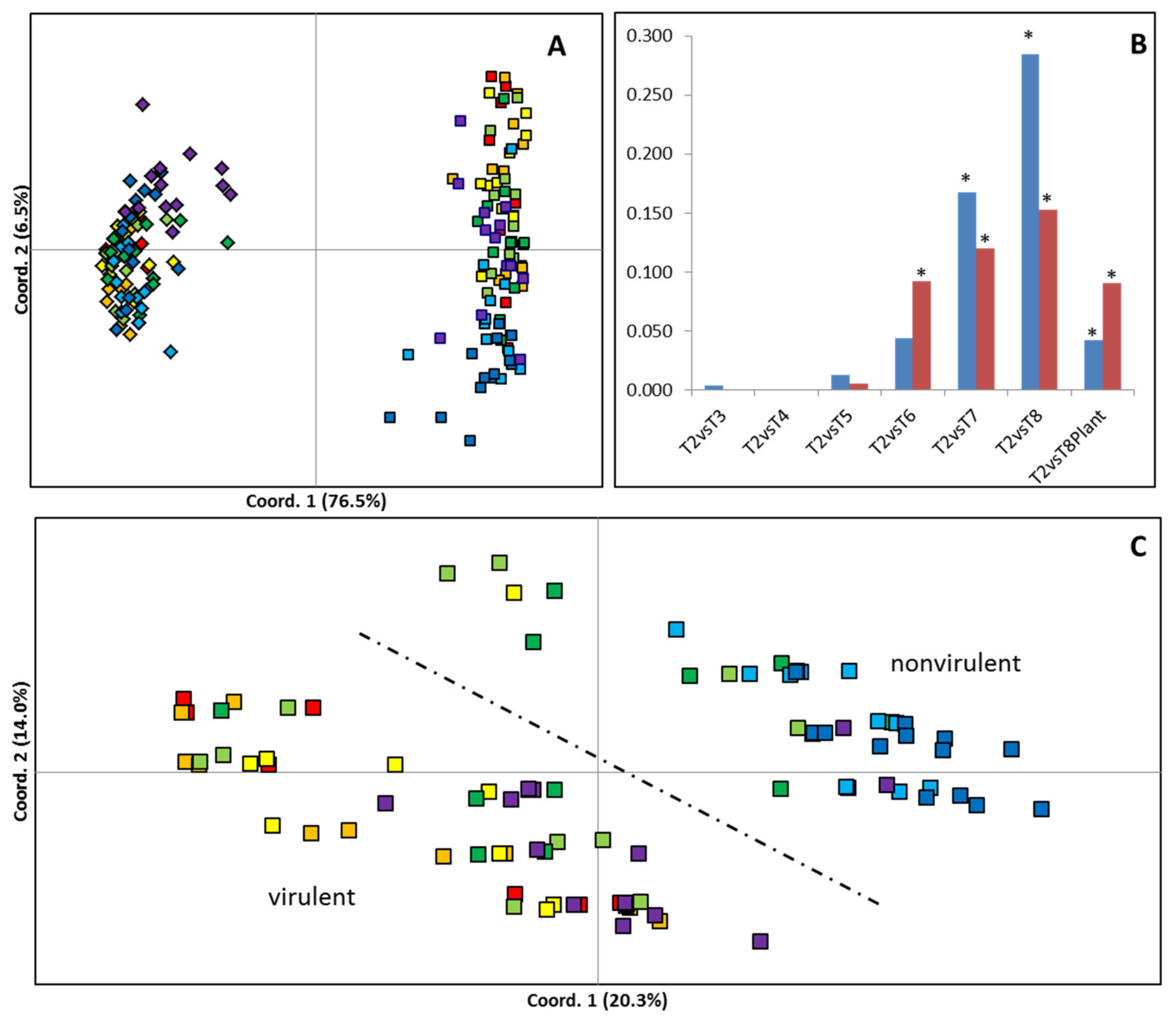
2.2. Analysis of Genetic and Epigenetic Variance during Culture Using MSAPs
2.3. B. cinerea Genome Re-sequencing
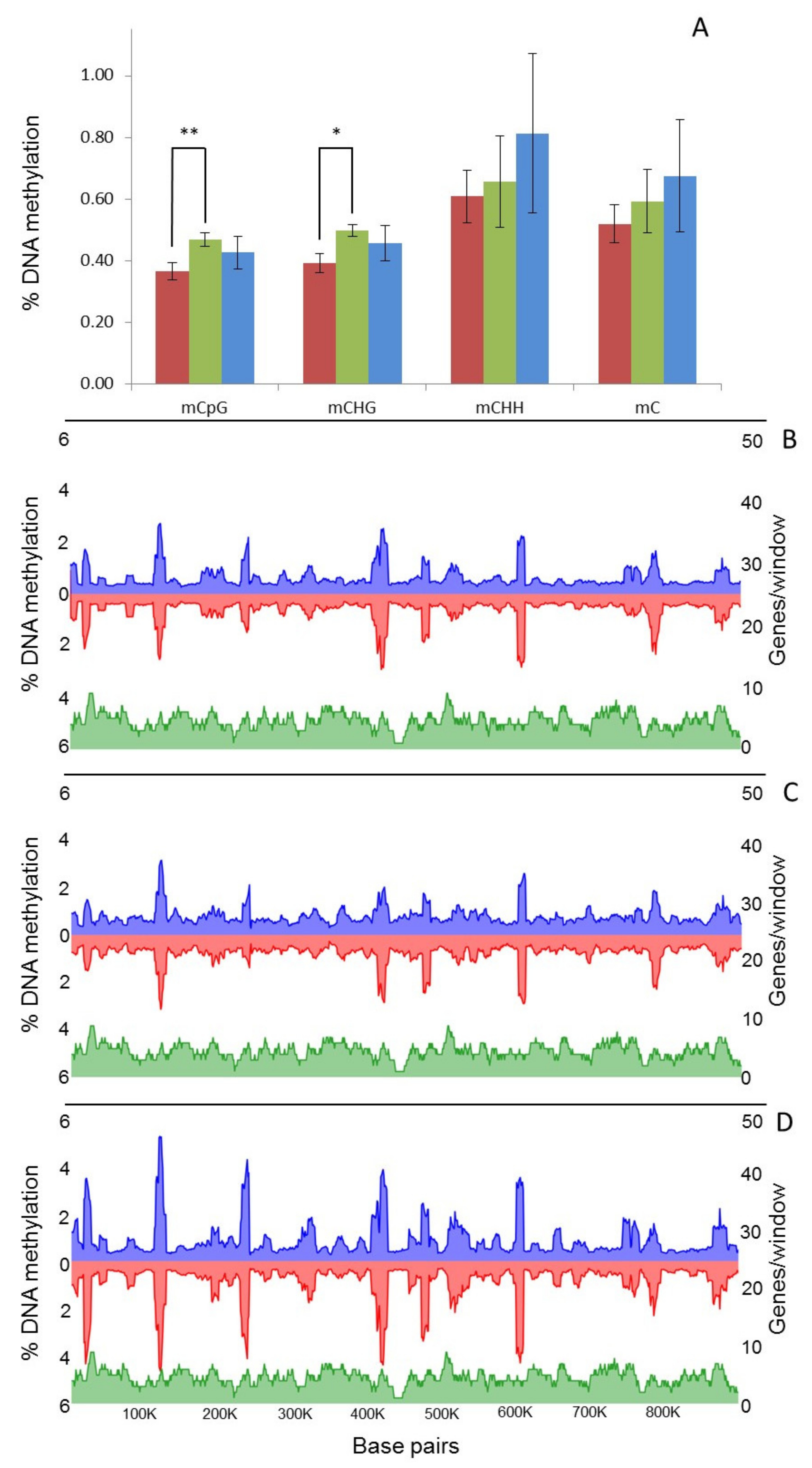
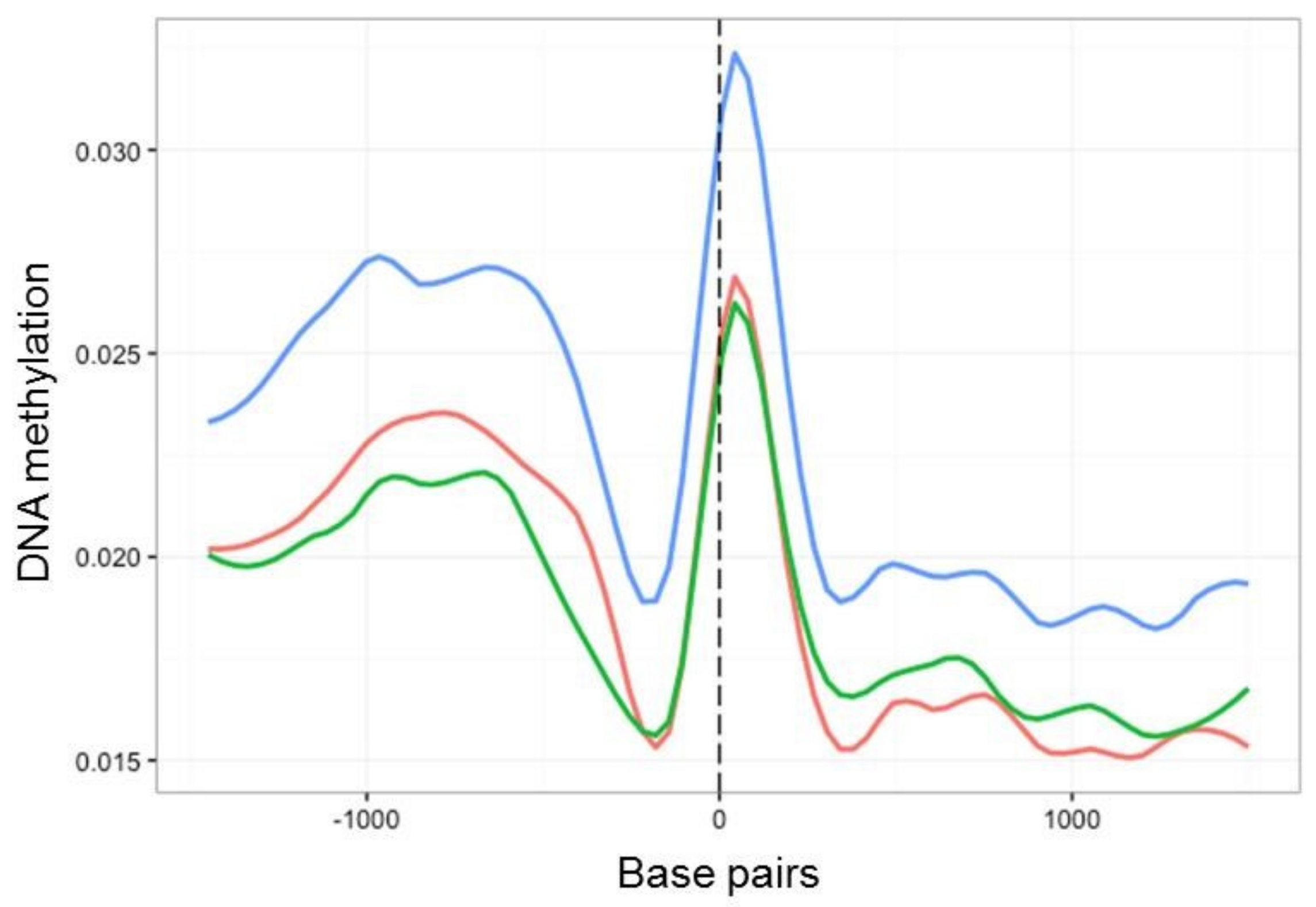
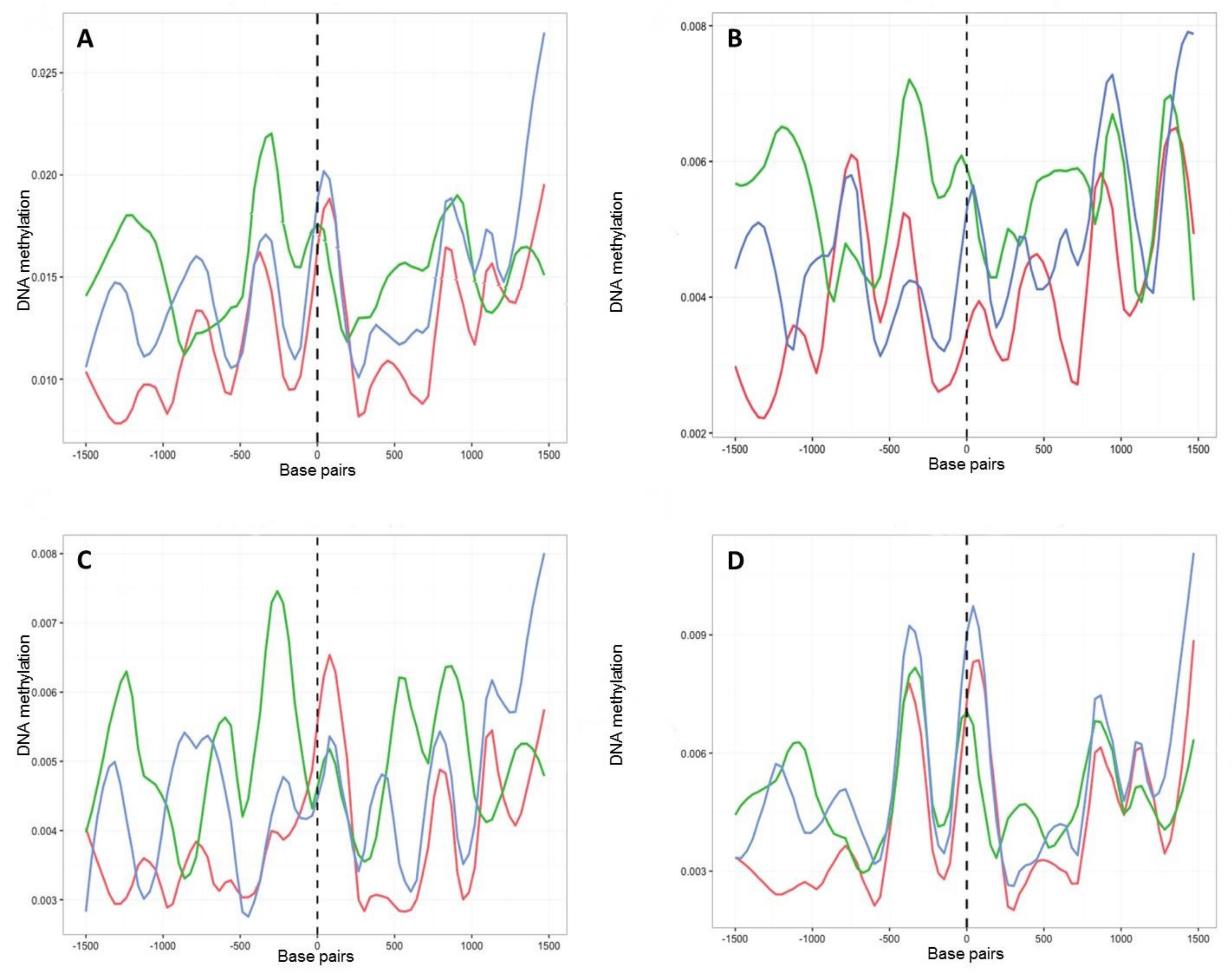
2.4. Characterisation of DNA Methylation Changes by WGBS
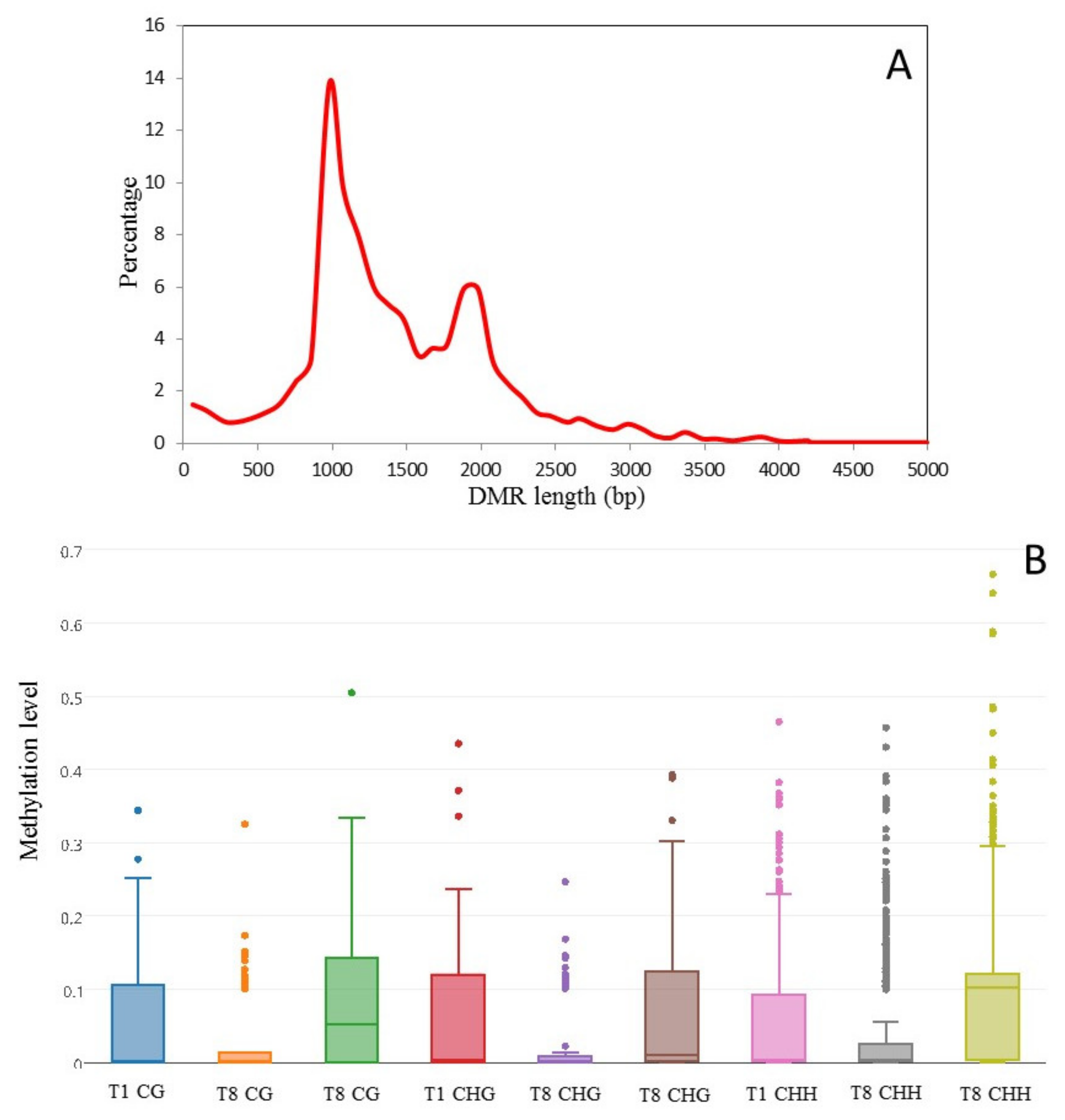
2.5. Detection of Culture-Induced DMRs
3. Discussion
3.1. Culture-Induced Changes to MSAP Profiles and Virulence Are Simultaneous and Reversible
3.2. Sequence Variants Do Not Explain Loss of Virulence during Culture
3.3. In Vitro Culture Affects Whole-Genome Methylation Patterns
3.4. Culture Induced DMRs Are Reversible and Mirror Changes in Virulence
4. Materials and Methods
4.1. Botrytis cinerea Culture and Inoculation
4.2. Plant Material
4.3. DNA Isolation
4.4. Scoring B. cinerea Lesion Phenotypes
4.5. Estimation of in Planta Fungal Growth by qPCR
4.6. MSAP Procedure
4.7. Analysis of Genetic/Epigenetic Variability during Time in Culture Using MSAP
4.8. Methylation Analysis by WGBS
4.9. Sequence Analysis and Differential Methylation Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fournier, E.; Gladieux, P.; Giraud, T. The “Dr Jekyll and Mr Hyde fungus”: Noble rot versus gray mold symptoms of Botrytis cinerea on grapes. Evol. Appl. 2013, 6, 960–969. [Google Scholar] [CrossRef] [PubMed]
- Hua, L.; Yong, C.; Zhanquan, Z.; Boqiang, L.; Guozheng, Q.; Shiping, T. Pathogenic mechanisms and control strategies of Botrytis cinerea causing post-harvest decay in fruits and vegetables. Food Qual. Saf. 2018, 2, 111–119. [Google Scholar] [CrossRef] [Green Version]
- Martinez, F.; Dubos, B.; Fermaud, M. The Role of Saprotrophy and Virulence in the Population Dynamics of Botrytis cinerea in Vineyards. Phytopathology 2005, 95, 692–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pathirana, R.; Cheah, L.H.; Carimi, F.; Carra, A. Low temperature stored in cryobank® maintains pathogenicity in grapevine. Cryoletters 2009, 30, 84. [Google Scholar]
- Butt, T.; Wang, C.; Shah, F.; Hall, R. Degneration of Entomogenous Fungi. In An Ecological and Societal Approach to Biological Control; Eilenberg, J., Hokkanen, H., Eds.; Springer: Dordrecht, The Netherlands, 2006; pp. 213–226. [Google Scholar]
- Naiki, T.; Cook, R.J. Factors in loss of pathogenicity in Gaeumannomyces graminis var. tritici. Phytopathology 1983, 73, 1652–1656. [Google Scholar] [CrossRef] [Green Version]
- Chang, T.-H.; Lin, Y.-H.; Wan, Y.-L.; Chen, K.-S.; Huang, J.-W.; Chang, P.-F.L. Degenerated Virulence and Irregular Development of Fusarium oxysporum f. sp. niveum Induced by Successive Subculture. J. Fungi 2020, 6, 382. [Google Scholar] [CrossRef]
- Chu, Y.-M.; Jeon, J.-J.; Yea, S.-J.; Kim, Y.-H.; Yun, S.-H.; Lee, Y.-W.; Kim, K.-H. Double-Stranded RNA Mycovirus from Fusarium graminearum. Appl. Environ. Microbiol. 2002, 68, 2529–2534. [Google Scholar] [CrossRef] [Green Version]
- Castro, M.; Kramer, K.; Valdivia, L.; Ortiz, S.; Castillo, A. A double-stranded RNA mycovirus confers hypovirulence-associated traits to Botrytis cinerea. FEMS Microbiol. Lett. 2003, 228, 87–91. [Google Scholar] [CrossRef]
- Akamatsu, H.; Taga, M.; Kodama, M.; Johnson, R.; Otani, H.; Kohmoto, K. Molecular karyotypes for Alternaria plant pathogens known to produce host-specific toxins. Curr. Genet. 1999, 35, 647–656. [Google Scholar] [CrossRef]
- Hatta, R.; Ito, K.; Hosaki, Y.; Tanaka, T.; Tanaka, A.; Yamamoto, M.; Akimitsu, K.; Tsuge, T. A Conditionally Dispensable Chromosome Controls Host-Specific Pathogenicity in the Fungal Plant Pathogen Alternaria alternata. Genetics 2002, 161, 59–70. [Google Scholar] [CrossRef]
- Kelly, S.A.; Panhuis, T.M.; Stoehr, A.M. Phenotypic Plasticity: Molecular Mechanisms and Adaptive Significance. Compr. Physiol. 2012, 2, 1417–1439. [Google Scholar] [CrossRef]
- Waddington, C.H. Canalization of development and the inheritance of acquired characters. Nature 1942, 150, 563–565. [Google Scholar] [CrossRef]
- Deans, C.; Maggert, K.A. What Do You Mean, “Epigenetic”? Genetics 2015, 199, 887–896. [Google Scholar] [CrossRef] [Green Version]
- Allis, C.D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef]
- Su, Z.; Han, L.; Zhao, Z. Conservation and divergence of DNA methylation in eukaryotes: New insights from single base-resolution DNA methylomes. Epigenetics 2011, 6, 134–140. [Google Scholar] [CrossRef] [Green Version]
- He, X.-J.; Chen, T.; Zhu, J.-K. Regulation and function of DNA methylation in plants and animals. Cell Res. 2011, 21, 442–465. [Google Scholar] [CrossRef]
- Jupe, E.R.; Magill, J.M.; Magill, C.W. Stage-specific DNA methylation in a fungal plant pathogen. J. Bacteriol. 1986, 165, 420–423. [Google Scholar] [CrossRef] [Green Version]
- Jeon, J.; Choi, J.; Lee, G.-W.; Park, S.-Y.; Huh, A.; Dean, R.A.; Lee, Y.-H. Genome-wide profiling of DNA methylation provides insights into epigenetic regulation of fungal development in a plant pathogenic fungus, Magnaporthe oryzae. Sci. Rep. 2015, 5, 8567. [Google Scholar] [CrossRef] [Green Version]
- Armaleo, D.; Miao, V. Symbiosis and DNA methylation in the Cladonia lichen fungus. Symbiosis 1999, 26, 143–163. [Google Scholar]
- Zemach, A.; McDaniel, I.E.; Silva, P.; Zilberman, D. Genome-Wide Evolutionary Analysis of Eukaryotic DNA Methylation. Science 2010, 328, 916–919. [Google Scholar] [CrossRef] [Green Version]
- Alonso, C.; Ramos-Cruz, D.; Becker, C. The role of plant epigenetics in biotic interactions. New Phytol. 2019, 221, 731–737. [Google Scholar] [CrossRef]
- Reyna-López, G.E.; Simpson, J.; Ruiz-Herrera, J. Differences in DNA methylation patterns are detectable during the dimorphic transition of fungi by amplification of restriction polymorphisms. Mol. Gen Genet. 1997, 253, 703–710. [Google Scholar] [CrossRef]
- Gachon, C.; Saindrenan, P. Real-time PCR monitoring of fungal development in Arabidopsis thaliana infected by Alternaria brassicicola and Botrytis cinerea. Plant Physiol. Biochem. 2004, 42, 367–371. [Google Scholar] [CrossRef]
- Amselem, J.; Cuomo, C.A.; van Kan, J.A.; Viaud, M.; Benito, E.P.; Couloux, A.; Coutinho, P.M.; de Vries, R.P.; Dyer, P.S.; Fillinger, S.; et al. Genomic Analysis of the Necrotrophic Fungal Pathogens Sclerotinia sclerotiorum and Botrytis cinerea. PLoS Genet. 2011, 7, e1002230. [Google Scholar] [CrossRef] [Green Version]
- Choquer, M.; Fournier, E.; Kunz, C.; Levis, C.; Pradier, J.M.; Simon, A.; Viaud, M. Botrytis cinerea virulence factors: New insights into a necrotrophic and polyphageous pathogen. FEMS Microbiol. Lett. 2007, 277, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Blanco-Ulate, B.; Morales-Cruz, A.; Amrine, K.C.H.; Labavitch, J.M.; Powell, A.L.T.; Cantu, D. Genome-wide transcriptional profiling of Botrytis cinerea genes targeting plant cell walls during infections of different hosts. Front. Plant Sci. 2014, 5, 435. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez López, C.M.; Wetten, A.C.; Wilkinson, M.J. Progressive erosion of genetic and epigenetic variation in callus-derived cocoa (Theobroma cacao) plants. New Phytol. 2010, 186, 856–868. [Google Scholar] [CrossRef] [PubMed]
- Dahmen, H.; Staub, T.; Schwinn, F.J. Technique for Long-Term Preservation of Phytopathogenic Fungi in Liquid Nitrogen. Phytopathology 1983, 73, 241. [Google Scholar] [CrossRef]
- Schumacher, J.; Pradier, J.M.; Simon, A.; Traeger, S.; Moraga, J.; Collado, I.G.; Viaud, M.; Tudzybski, B. Natural variation in the VELVET gene bvcel1 affects virulence and light-dependent differentiation in Botrytis cinerea. PLoS ONE 2012, 7, e47840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hevia, M.A.; Canessa, P.; Muller-Esparza, H.; Larrondo, L.F. A circadian oscillator in the fungus Botrytis cinereal regulates virulence when infecting Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2015, 112, 8744–8749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.Q.; Qin, G.Z.; Li, B.Q.; Tian, S.P. Knocking our Bcsas1 in Botrytis cinereal impacts growth, development, and secretion of extracellular proteins, which decrease virulence. Mol. Plant-Microbe Interact. 2014, 27, 590–600. [Google Scholar] [CrossRef]
- Kohn, L.M.; Schaffer, M.R.; Anderson, J.B.; Grünwald, N.J. Marker stability throughout 400 days of in vitro hyphal growth in the filamentous ascomycete, Sclerotinia sclerotiorum. Fungal Genet. Biol. 2008, 45, 613–617. [Google Scholar] [CrossRef]
- Lombard, V.; Golaconda Ramulu, H.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, D490–D495. [Google Scholar] [CrossRef] [Green Version]
- Atwell, S.; Corwin, J.; Soltis, N.E.; Subedy, A.; Denby, K.J.; Kliebenstein, D.J. Whole genome resequencing of Botrytis cinerea isolates identifies high levels of standing diversity. Front. Microbiol. 2015, 6, 996. [Google Scholar] [CrossRef] [Green Version]
- Jeon, J.; Choi, J.; Lee, G.-W.; Dean, R.A.; Lee, Y.-H. Experimental Evolution Reveals Genome-Wide Spectrum and Dynamics of Mutations in the Rice Blast Fungus, Magnaporthe oryzae. PLoS ONE 2013, 8, e65416. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; He, C.; Chen, Y.; Li, B.; Tian, S. DNA Methyltransferases Regulate Pathogenicity of Botrytis cinerea to Horticultural Crops. J. Fungi 2021, 7, 659. [Google Scholar] [CrossRef]
- Jung, M.; Pfeifer, G.P. Aging and DNA methylation. BMC Biol. 2015, 13, 7. [Google Scholar] [CrossRef] [Green Version]
- Belden, W.J.; Lewis, Z.A.; Selker, E.U.; Loros, J.J.; Dunlap, J.C. CHD1 Remodels Chromatin and Influences Transient DNA Methylation at the Clock Gene frequency. PLoS Genet. 2011, 7, e1002166. [Google Scholar] [CrossRef] [Green Version]
- Hervouet, E.; Cheray, M.; Vallette, F.M.; Cartron, P.-F. DNA Methylation and Apoptosis Resistance in Cancer Cells. Cells 2013, 2, 545–573. [Google Scholar] [CrossRef]
- Gómez-Díaz, E.; Jordà, M.; Peinado, M.A.; Rivero, A. Epigenetics of Host–Pathogen Interactions: The Road Ahead and the Road Behind. PLoS Pathog. 2012, 8, e1003007. [Google Scholar] [CrossRef] [Green Version]
- Cheeseman, K.; Weitzman, J.B. Host–parasite interactions: An intimate epigenetic relationship. Cell. Microbiol. 2015, 17, 1121–1132. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.K.; Baum, M.; Carbon, J. DNA methylation regulates phenotype-dependent transcriptional activity in Candida albicans. Proc. Natl. Acad. Sci. USA 2011, 108, 11965–11970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Consuegra, S.; López, C.M.R. Epigenetic-induced alterations in sex-ratios in response to climate change: An epigenetic trap? BioEssays 2016, 38, 950–958. [Google Scholar] [CrossRef] [PubMed]
- Thomma, B.P.; Eggermont, K.; Tierens, K.F.-J.; Broekaert, W.F. Requirement of Functional Ethylene-Insensitive 2Gene for Efficient Resistance of Arabidopsis to Infection by Botrytis cinerea. Plant Physiol. 1999, 121, 1093–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, H.E.; Lloyd, A.J.; Mur, L.A.J.; Smith, A.R.; Causton, D.R. The application of MANOVA to analyse Arabidopsis thaliana metabolomic data from factorially designed experiments. Metabolomics 2007, 3, 517–530. [Google Scholar] [CrossRef]
- Johnson, H.E.; Broadhurst, D.; Goodacre, R.; Smith, A.R. Metabolic fingerprinting of salt-stressed tomatoes. Phytochemistry 2003, 62, 919–928. [Google Scholar] [CrossRef]
- Boyes, D.C.; Zayed, A.M.; Ascenzi, R.; McCaskill, A.J.; Hoffman, N.E.; Davis, K.R.; Gorlach, J. Growth stage-based phenotypic analysis of Arabidopsis: A model for high throughput functional genomics in plants. Plant Cell 2001, 13, 1499–1510. [Google Scholar] [CrossRef] [Green Version]
- Lloyd, A.J.; Allwood, J.W.; Winder, C.L.; Dunn, W.B.; Heald, J.K.; Cristescu, S.M.; Sivakumaran, A.; Harren, F.J.M.; Mulema, J.; Denby, K.; et al. Metabolomic approaches reveal that cell wall modifications play a major role in ethylene-mediated resistance against Botrytis cinerea. Plant J. 2011, 67, 852–868. [Google Scholar] [CrossRef]
- Rodríguez López, C.; Morán, P.; Lago, F.; Espiñeira, M.; Beckmann, M.; Consuegra, S. Detection and quantification of tissue of origin in salmon and veal products using methylation sensitive AFLPs. Food Chem. 2012, 131, 1493–1498. [Google Scholar] [CrossRef]
- Caballero, A.; Quesada, H.; Rolán-Alvarez, E. Impact of Amplified Fragment Length Polymorphism Size Homoplasy on the Estimation of Population Genetic Diversity and the Detection of Selective Loci. Genetics 2008, 179, 539–554. [Google Scholar] [CrossRef] [Green Version]
- Gower, J.C. Some Distance Properties of Latent Root and Vector Methods Used in Multivariate Analysis. Biometrika 1966, 53, 325–338. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research—An update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [Green Version]
- Excoffier, L.; Smouse, P.E.; Quattro, J.M. Analysis of molecular variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction data. Genetics 1992, 131, 479–491. [Google Scholar] [CrossRef]
- Michalakis, Y.; Excoffier, L. A Generic Estimation of Population Subdivision Using Distances Between Alleles with Special Reference for Microsatellite Loci. Genetics 1996, 142, 1061–1064. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Garrison, E.; Marth, G. Haplotype-based variant detection from short-read sequencing. arXiv 2012, arXiv:1207.3907. [Google Scholar]
- Vergara, I.A.; Frech, C.; Chen, N. CooVar: Co-occurring variant analyzer. BMC Res. Notes 2012, 5, 615. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [Green Version]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Function | Total | Gene Variant | Gene/DMR Overlap | DMR Type | Reference | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| #G/D | %G/D | #G/D | %G/D | Recovery | Non- Recovery | ||||||
| 0 | 2 | 1a | 1b | ||||||||
| Housekeeping | 5 | - | - | - | - | - | - | - | - | [25] | |
| Apoptosis | 10 | 2 | 20 | 4 | 40.0 | 3 | 1 | - | - | [25] | |
| Conidiation | 16 | - | - | 6 | 37.5 | 1 | 2 | - | 3 | [25] | |
| Mating and fruit body development | 32 | 6 | 18.8 | 6 | 18.7 | 1 | 4 | 1 | 1 | [25] | |
| Secondary metabolism | 51 | 4 | 7.8 | 14 | 27.5 | 9 | 3 | 2 | 3 | [25] | |
| Signalling pathways | 176 | 6 | 3.4 | 54 | 30.7 | 12 | 17 | 7 | 20 | [25] | |
| Sclerotium formation | 249 | 7 | 2.8 | 67 | 26.9 | 26 | 31 | 10 | 13 | [25] | |
| Virul. s.l. | Appressorium | 12 | 2 | 16.7 | 3 | 25.0 | 1 | - | - | 3 | [25] |
| Virulence s.s. | 17 | 1 | 5.9 | 4 | 23.5 | 1 | 1 | 1 | 2 | [26] | |
| CAZyme genes | 1155 | 50 | 4.3 | 320 | 27.7 | 112 | 132 | 45 | 99 | [27] | |
| Total | 1577 | 68 | 4.3 | 478 | 30.3 | 166 | 191 | 66 | 144 | ||
| Total | Recovery (%) | Total Recovery (%) | No Recovery | |||
|---|---|---|---|---|---|---|
| Type 0 | Type 2 | In Vitro Induced (Type 1a) | Plant Induced (Type 1b) | |||
| mC | 2822 | 757 (26.82) | 860 (30.47) | 1617 (57.30) | 452 (16.02) | 753 (26.68) |
| CG | 70 | 17 (24.29) | 15 (21.43) | 32 (45.71) | 14 (20.00) | 24 (34.29) |
| CHG | 82 | 14 (17.07) | 30 (36.59) | 44 (53.66) | 15 (18.29) | 23 (28.05) |
| CHH | 1248 | 303 (24.28) | 490 (39.26) | 793 (63.54) | 137 (10.98)) | 318 (25.48) |
| Total | 4222 | 1395 (33.04) | 1091 (25.84) | 2486 (58.88) | 618 (14.64) | 1118 (26.48) |
| Total Genes | Promoter, Gene Body and 3’UTR (*) | Promoter Only (*) | Promoter and Gene Body (*) | Gene Body and 3’UTR (*) | Gene Body (*) | |
|---|---|---|---|---|---|---|
| mC | 3055 | 626 (20.49) | 61 (2.00) | 1022 (33.45) | 923 (30.21) | 423 (13.85) |
| CG | 68 | 3 (4.41) | 0 (0.00) | 24 (35.29) | 21 (30.88) | 20 (29.41) |
| CHG | 84 | 8 (9.52) | 0 (0.00) | 29 (34.52) | 27 (32.14) | 20 (23.81) |
| CHH | 1339 | 248 (18.52) | 32 (2.39) | 443 (33.08) | 413 (30.84) | 203 (15.16) |
| Recovery genes (**) | ||||||
| mC | 1713 (56.07) | 345 (20.14) | 43 (2.51) | 545 (31.82) | 537 (31.35) | 243 (14.19) |
| CG | 32 (47.06) | 2 (6.25) | 0 (0.00) | 11 (34.38) | 9 (28.13) | 10 (31.25) |
| CHG | 46 (54.76) | 4 (8.70) | 1 (2.17) | 13 (28.26) | 16 (34.78) | 12 (26.09) |
| CHH | 863 (64.45) | 175 (20.28) | 21 (2.43) | 286 (33.14) | 252 (29.20) | 129 (14.95) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Breen, J.; Mur, L.A.J.; Sivakumaran, A.; Akinyemi, A.; Wilkinson, M.J.; Rodriguez Lopez, C.M. Botrytis cinerea Loss and Restoration of Virulence during In Vitro Culture Follows Flux in Global DNA Methylation. Int. J. Mol. Sci. 2022, 23, 3034. https://doi.org/10.3390/ijms23063034
Breen J, Mur LAJ, Sivakumaran A, Akinyemi A, Wilkinson MJ, Rodriguez Lopez CM. Botrytis cinerea Loss and Restoration of Virulence during In Vitro Culture Follows Flux in Global DNA Methylation. International Journal of Molecular Sciences. 2022; 23(6):3034. https://doi.org/10.3390/ijms23063034
Chicago/Turabian StyleBreen, James, Luis Alejandro Jose Mur, Anushen Sivakumaran, Aderemi Akinyemi, Michael James Wilkinson, and Carlos Marcelino Rodriguez Lopez. 2022. "Botrytis cinerea Loss and Restoration of Virulence during In Vitro Culture Follows Flux in Global DNA Methylation" International Journal of Molecular Sciences 23, no. 6: 3034. https://doi.org/10.3390/ijms23063034
APA StyleBreen, J., Mur, L. A. J., Sivakumaran, A., Akinyemi, A., Wilkinson, M. J., & Rodriguez Lopez, C. M. (2022). Botrytis cinerea Loss and Restoration of Virulence during In Vitro Culture Follows Flux in Global DNA Methylation. International Journal of Molecular Sciences, 23(6), 3034. https://doi.org/10.3390/ijms23063034