
Recent Advances in Structural Studies of Cytochrome bd and Its Potential Application as a Drug Target



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Recent Advances in the Structural Biology of Cytochrome bd Oxidases
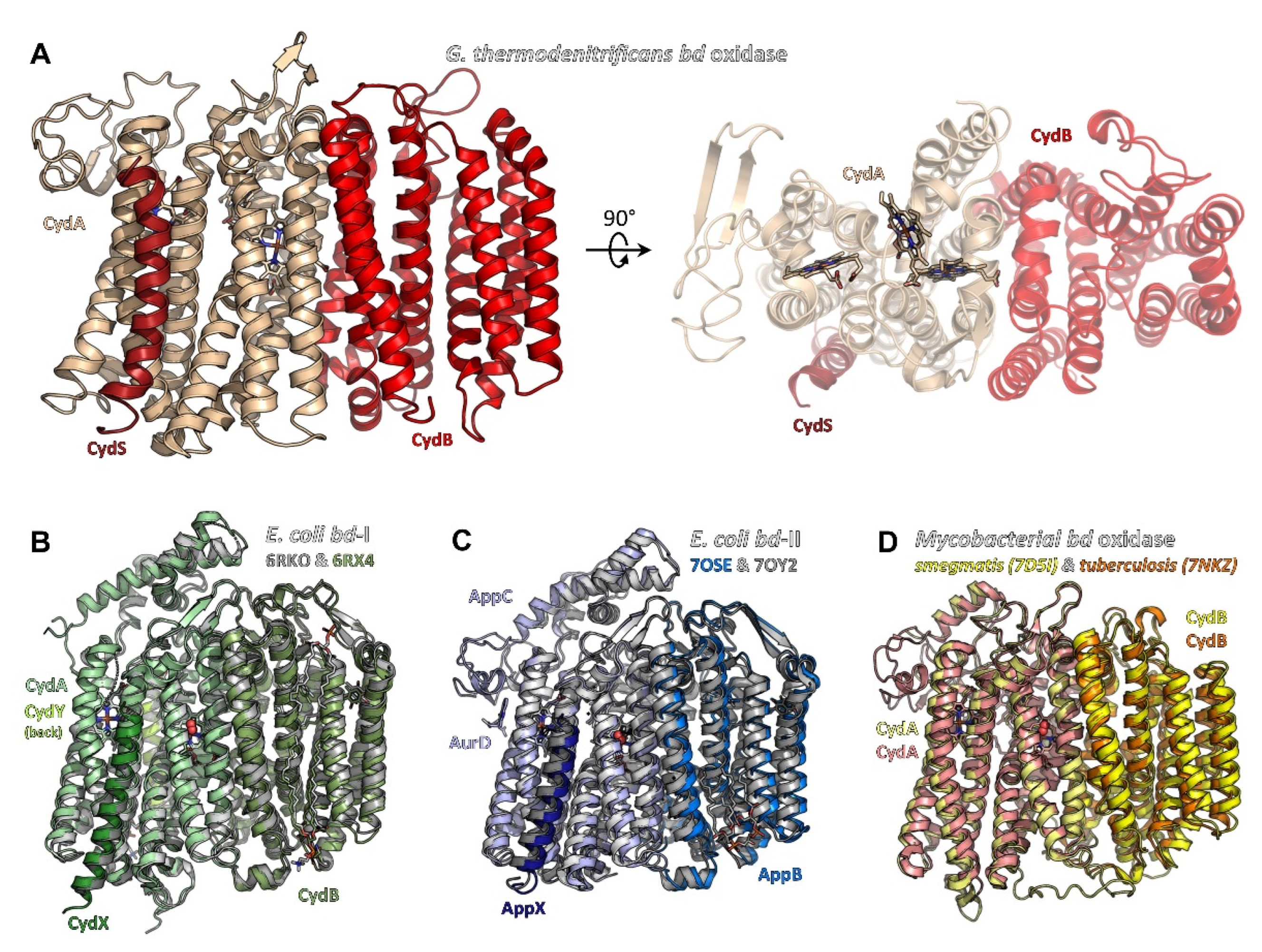
2.1. Common Architecture of Cytochrome bd Oxidases
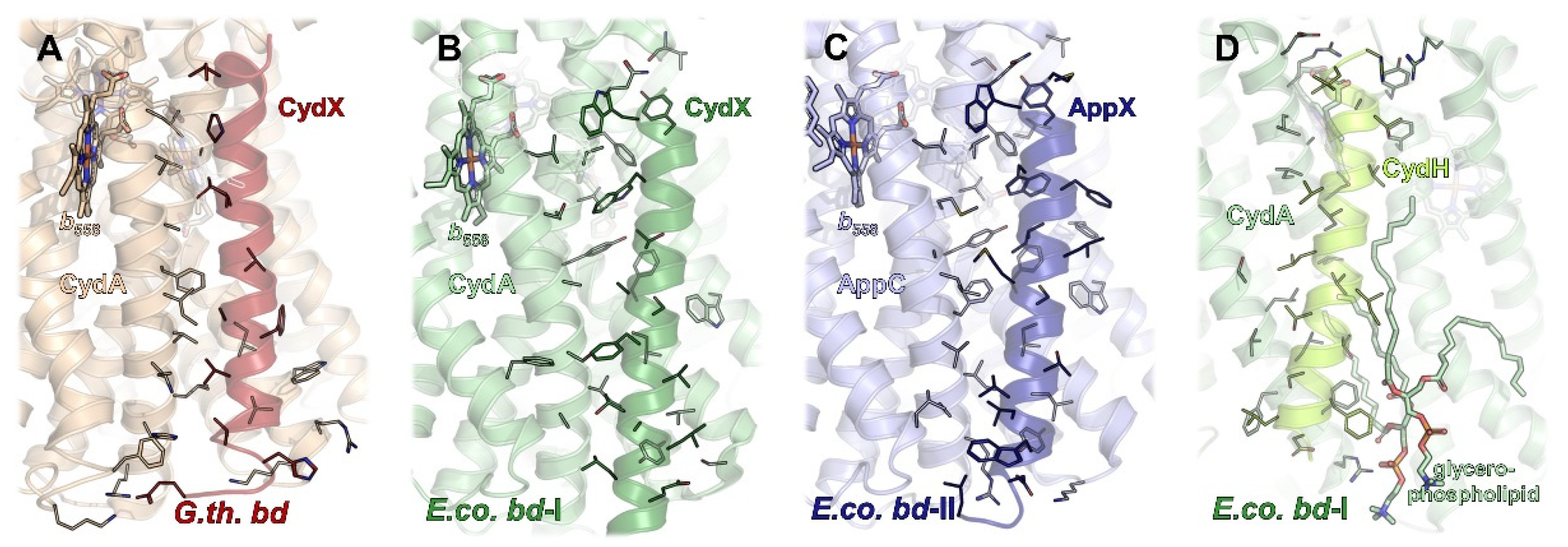
2.2. Additional Subunits
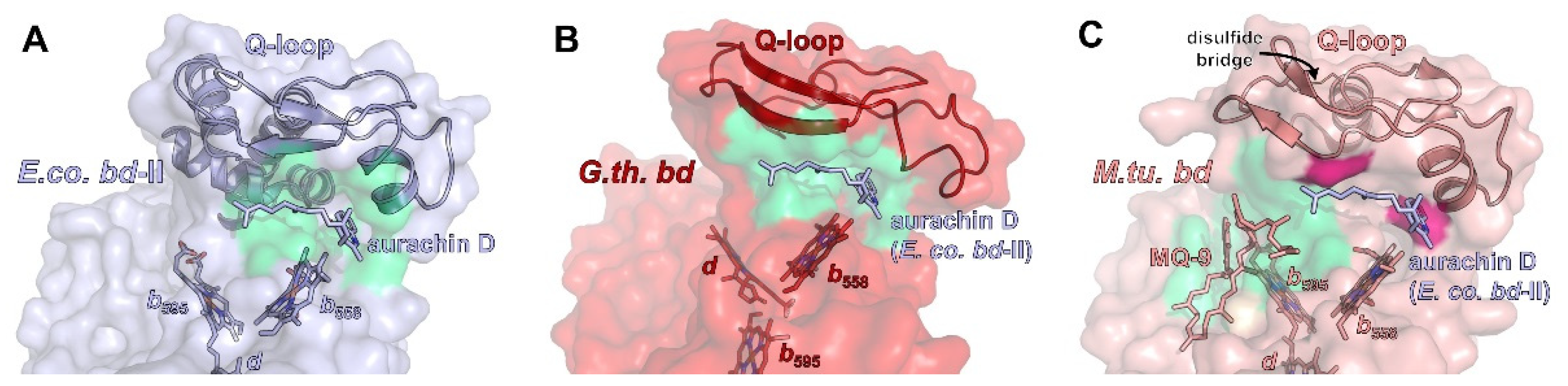
2.3. Quinol Binding Site(s)
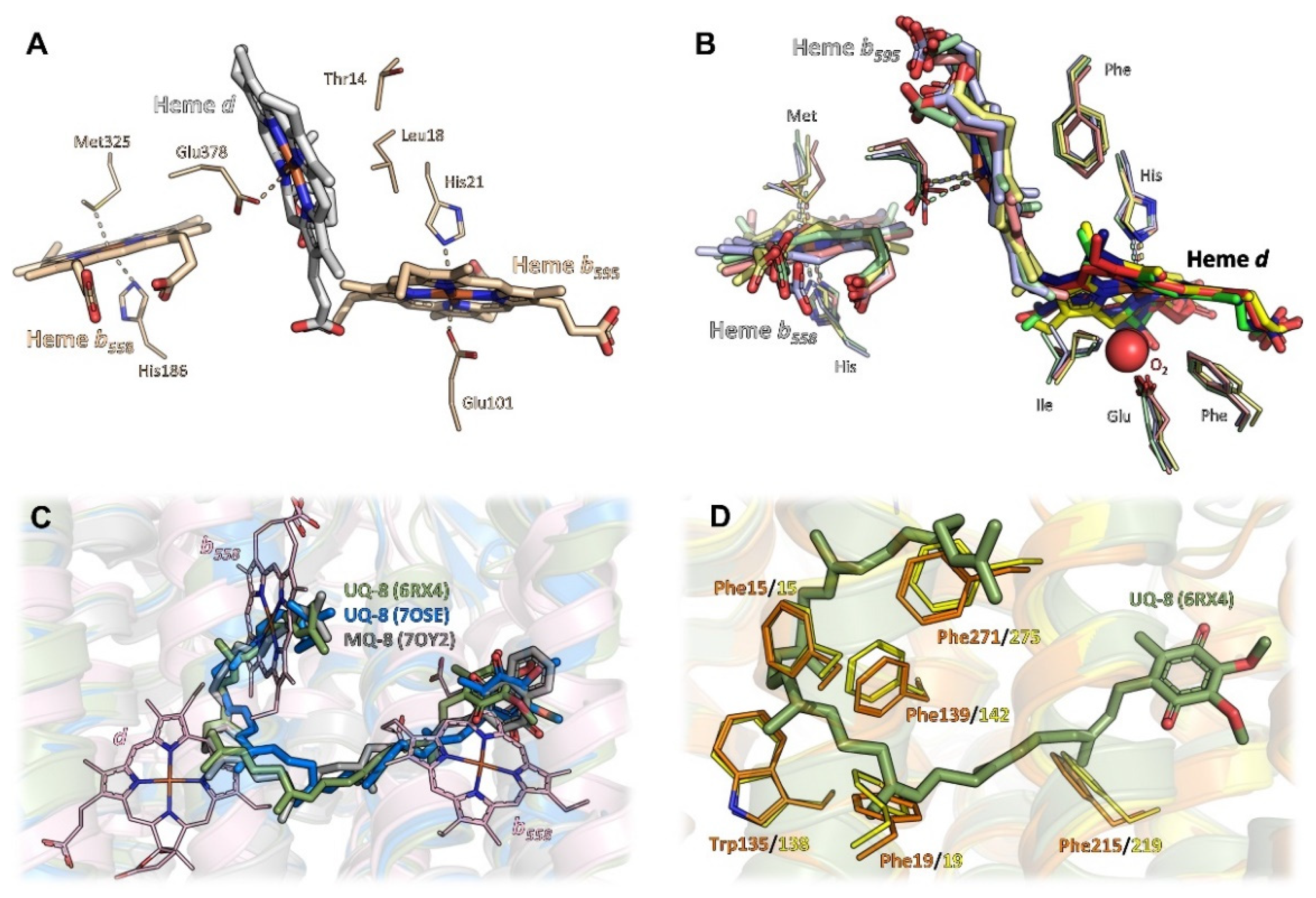
2.4. Heme Arrangement and Electron Transfer
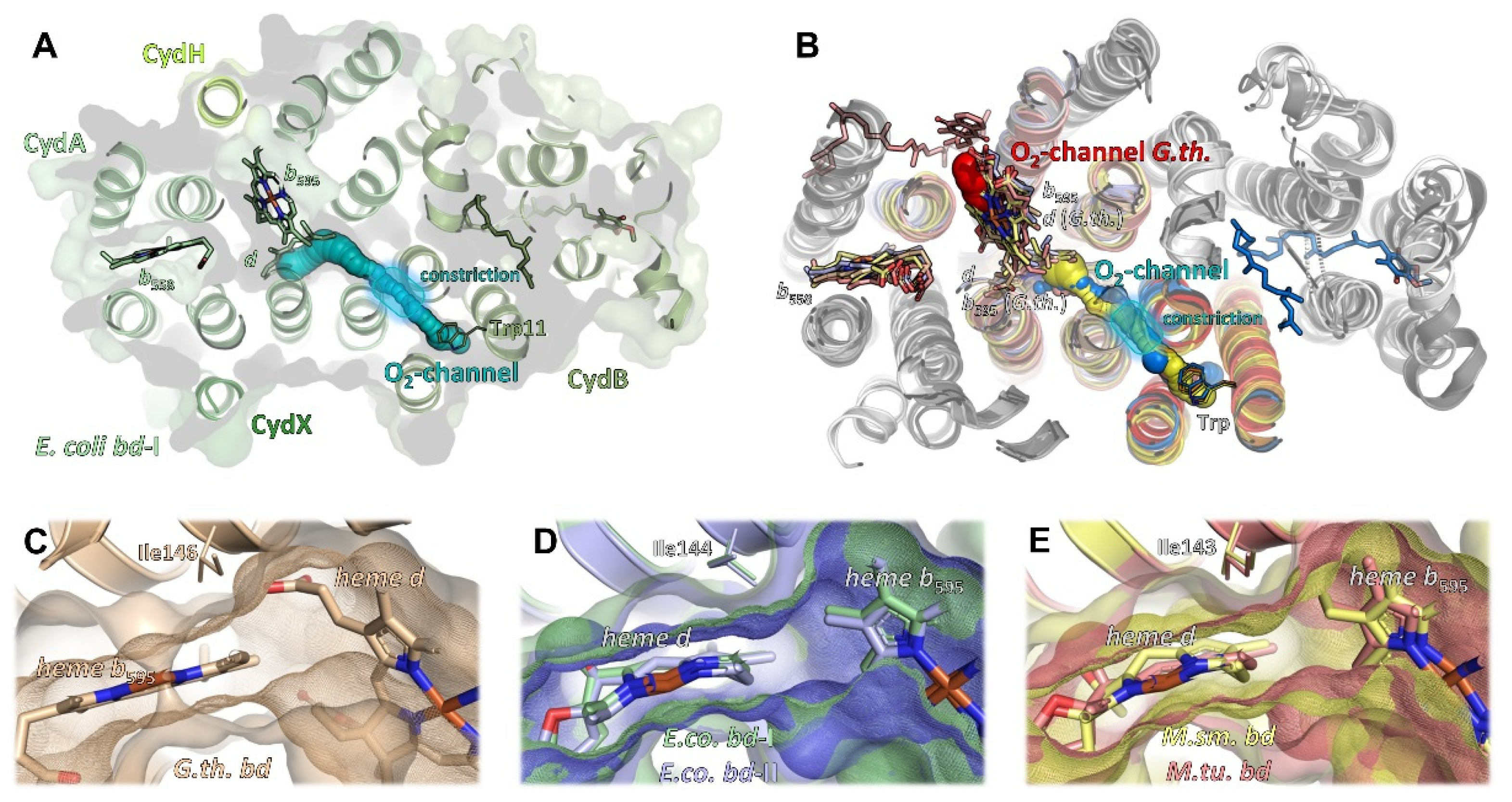
2.5. Oxygen Access
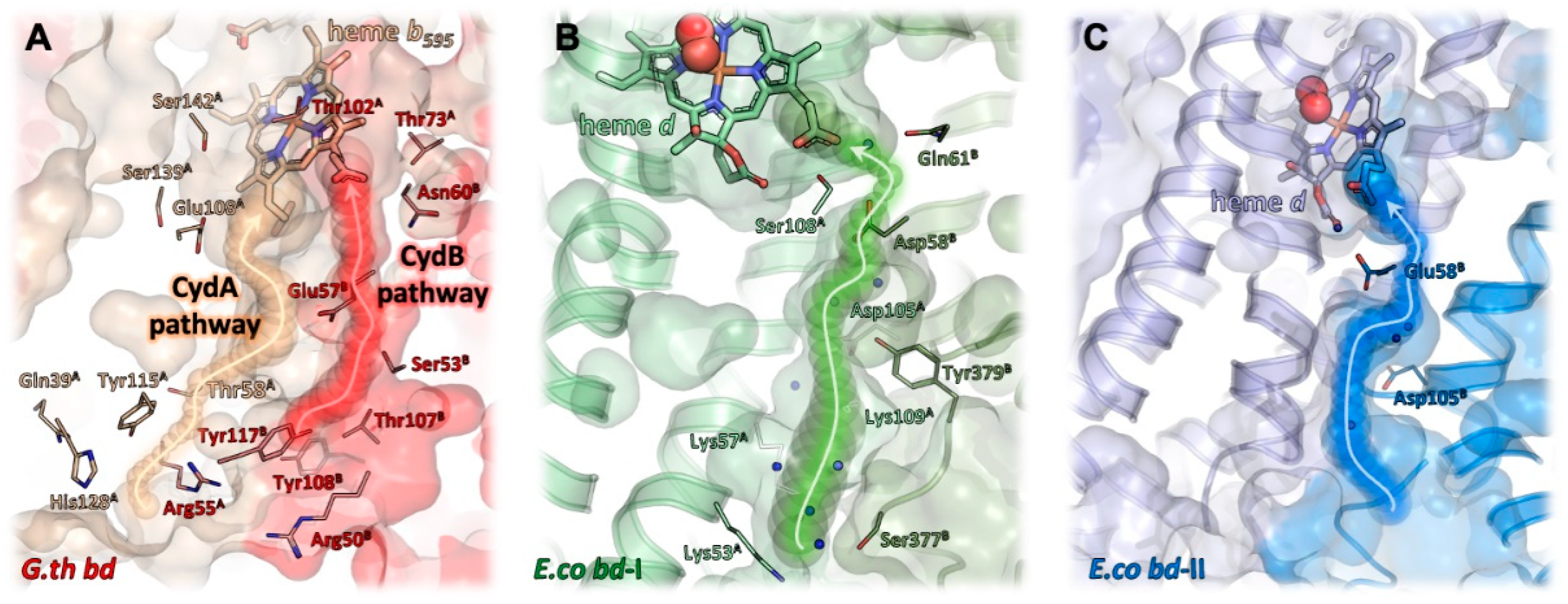
2.6. Proton Pathways
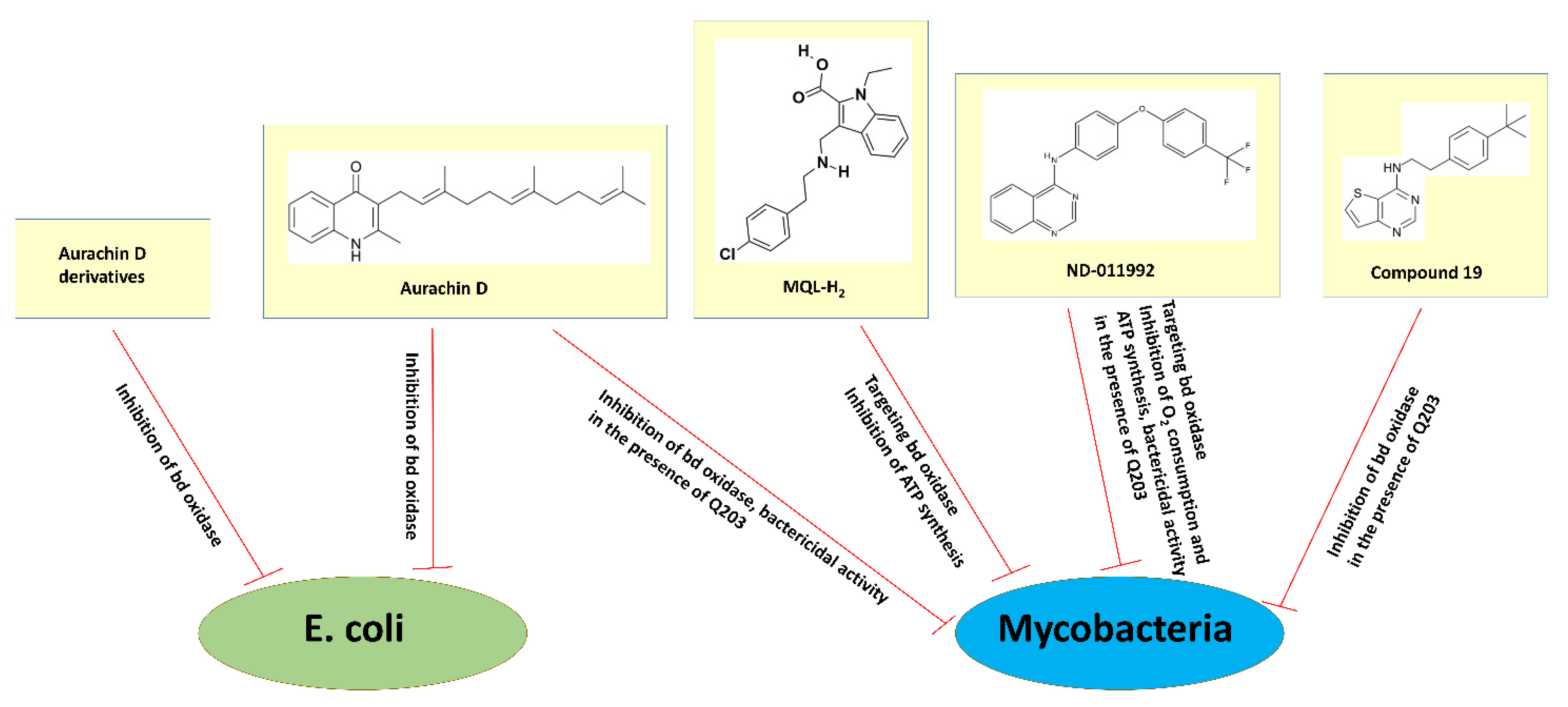
3. Cytochrome bd as a Prospective Target for the Development of New Antibiotics
4. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Murali, R.; Gennis, R.B.; Hemp, J. Evolution of the cytochrome bd oxygen reductase superfamily and the function of CydAA’ in Archaea. ISME J. 2021, 15, 3534–3548. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Bayona, L.; Coyne, M.J.; Hantman, N.; Montero-Llopis, P.; Von, S.S.; Ito, T.; Malamy, M.H.; Basler, M.; Barquera, B.; Comstock, L.E. Nanaerobic growth enables direct visualization of dynamic cellular processes in human gut symbionts. Proc. Natl. Acad. Sci. USA 2020, 117, 24484–24493. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Imlay, J.A. When anaerobes encounter oxygen: Mechanisms of oxygen toxicity, tolerance and defence. Nat. Rev. Microbiol. 2021, 19, 774–785. [Google Scholar] [CrossRef]
- Siletsky, S.A.; Borisov, V.B.; Mamedov, M.D. Photosystem II and terminal respiratory oxidases: Molecular machines operating in opposite directions. Front. Biosci. 2017, 22, 1379–1426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikolaev, A.; Safarian, S.; Thesseling, A.; Wohlwend, D.; Friedrich, T.; Michel, H.; Kusumoto, T.; Sakamoto, J.; Melin, F.; Hellwig, P. Electrocatalytic evidence of the diversity of the oxygen reaction in the bacterial bd oxidase from different organisms. Biochim. Biophys. Acta Bioenerg. 2021, 1862, 148436. [Google Scholar] [CrossRef] [PubMed]
- Belevich, I.; Borisov, V.B.; Verkhovsky, M.I. Discovery of the true peroxy intermediate in the catalytic cycle of terminal oxidases by real-time measurement. J. Biol. Chem. 2007, 282, 28514–28519. [Google Scholar] [CrossRef] [Green Version]
- Borisov, V.B.; Belevich, I.; Bloch, D.A.; Mogi, T.; Verkhovsky, M.I. Glutamate 107 in subunit I of cytochrome bd from Escherichia coli is part of a transmembrane intraprotein pathway conducting protons from the cytoplasm to the heme b595/heme d active site. Biochemistry 2008, 47, 7907–7914. [Google Scholar] [CrossRef]
- Borisov, V.B.; Murali, R.; Verkhovskaya, M.L.; Bloch, D.A.; Han, H.; Gennis, R.B.; Verkhovsky, M.I. Aerobic respiratory chain of Escherichia coli is not allowed to work in fully uncoupled mode. Proc. Natl. Acad. Sci. USA 2011, 108, 17320–17324. [Google Scholar] [CrossRef] [Green Version]
- Safarian, S.; Rajendran, C.; Muller, H.; Preu, J.; Langer, J.D.; Ovchinnikov, S.; Hirose, T.; Kusumoto, T.; Sakamoto, J.; Michel, H. Structure of a bd oxidase indicates similar mechanisms for membrane-integrated oxygen reductases. Science 2016, 352, 583–586. [Google Scholar] [CrossRef] [Green Version]
- Thesseling, A.; Rasmussen, T.; Burschel, S.; Wohlwend, D.; Kagi, J.; Muller, R.; Bottcher, B.; Friedrich, T. Homologous bd oxidases share the same architecture but differ in mechanism. Nat. Commun. 2019, 10, 5138. [Google Scholar] [CrossRef] [Green Version]
- Safarian, S.; Hahn, A.; Mills, D.J.; Radloff, M.; Eisinger, M.L.; Nikolaev, A.; Meier-Credo, J.; Melin, F.; Miyoshi, H.; Gennis, R.B.; et al. Active site rearrangement and structural divergence in prokaryotic respiratory oxidases. Science 2019, 366, 100–104. [Google Scholar] [CrossRef]
- Wang, W.; Gao, Y.; Tang, Y.; Zhou, X.; Lai, Y.; Zhou, S.; Zhang, Y.; Yang, X.; Liu, F.; Guddat, L.W.; et al. Cryo-EM structure of mycobacterial cytochrome bd reveals two oxygen access channels. Nat. Commun. 2021, 12, 4621. [Google Scholar] [CrossRef]
- Safarian, S.; Opel-Reading, H.K.; Wu, D.; Mehdipour, A.R.; Hards, K.; Harold, L.K.; Radloff, M.; Stewart, I.; Welsch, S.; Hummer, G.; et al. The cryo-EM structure of the bd oxidase from M. tuberculosis reveals a unique structural framework and enables rational drug design to combat TB. Nat. Commun. 2021, 12, 5236. [Google Scholar] [CrossRef] [PubMed]
- Grauel, A.; Kagi, J.; Rasmussen, T.; Makarchuk, I.; Oppermann, S.; Moumbock, A.F.A.; Wohlwend, D.; Muller, R.; Melin, F.; Gunther, S.; et al. Structure of Escherichia coli cytochrome bd-II type oxidase with bound aurachin D. Nat. Commun. 2021, 12, 6498. [Google Scholar] [CrossRef] [PubMed]
- Grund, T.N.; Radloff, M.; Wu, D.; Goojani, H.G.; Witte, L.F.; Josting, W.; Buschmann, S.; Muller, H.; Elamri, I.; Welsch, S.; et al. Mechanistic and structural diversity between cytochrome bd isoforms of Escherichia coli. Proc. Natl. Acad. Sci. USA 2021, 118, e2114013118. [Google Scholar] [CrossRef] [PubMed]
- Borisov, V.B.; Gennis, R.B.; Hemp, J.; Verkhovsky, M.I. The cytochrome bd respiratory oxygen reductases. Biochim. Biophys. Acta 2011, 1807, 1398–1413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arutyunyan, A.M.; Sakamoto, J.; Inadome, M.; Kabashima, Y.; Borisov, V.B. Optical and magneto-optical activity of cytochrome bd from Geobacillus thermodenitrificans. Biochim. Biophys. Acta 2012, 1817, 2087–2094. [Google Scholar] [CrossRef] [Green Version]
- Belevich, I.; Borisov, V.B.; Konstantinov, A.A.; Verkhovsky, M.I. Oxygenated complex of cytochrome bd from Escherichia coli: Stability and photolability. FEBS Lett. 2005, 579, 4567–4570. [Google Scholar] [CrossRef] [Green Version]
- Belevich, I.; Borisov, V.B.; Bloch, D.A.; Konstantinov, A.A.; Verkhovsky, M.I. Cytochrome bd from Azotobacter vinelandii: Evidence for high-affinity oxygen binding. Biochemistry 2007, 46, 11177–11184. [Google Scholar] [CrossRef]
- Vos, M.H.; Borisov, V.B.; Liebl, U.; Martin, J.L.; Konstantinov, A.A. Femtosecond resolution of ligand-heme interactions in the high-affinity quinol oxidase bd: A di-heme active site? Proc. Natl. Acad. Sci. USA 2000, 97, 1554–1559. [Google Scholar] [CrossRef] [Green Version]
- Borisov, V.B.; Sedelnikova, S.E.; Poole, R.K.; Konstantinov, A.A. Interaction of cytochrome bd with carbon monoxide at low and room temperatures: Evidence that only a small fraction of heme b595 reacts with CO. J. Biol. Chem. 2001, 276, 22095–22099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arutyunyan, A.M.; Borisov, V.B.; Novoderezhkin, V.I.; Ghaim, J.; Zhang, J.; Gennis, R.B.; Konstantinov, A.A. Strong excitonic interactions in the oxygen-reducing site of bd-type oxidase: The Fe-to-Fe distance between hemes d and b595 is 10 A. Biochemistry 2008, 47, 1752–1759. [Google Scholar] [CrossRef]
- Siletsky, S.A.; Rappaport, F.; Poole, R.K.; Borisov, V.B. Evidence for fast electron transfer between the high-spin haems in cytochrome bd-I from Escherichia coli. PLoS ONE 2016, 11, e0155186. [Google Scholar] [CrossRef] [PubMed]
- Murali, R.; Gennis, R.B. Functional importance of Glutamate-445 and Glutamate-99 in proton-coupled electron transfer during oxygen reduction by cytochrome bd from Escherichia coli. Biochim. Biophys. Acta 2018, 1859, 577–590. [Google Scholar] [CrossRef]
- Poole, R.K.; Cook, G.M. Redundancy of aerobic respiratory chains in bacteria? Routes, reasons and regulation. Adv. Microb. Physiol. 2000, 43, 165–224. [Google Scholar] [CrossRef] [PubMed]
- May, B.; Young, L.; Moore, A.L. Structural insights into the alternative oxidases: Are all oxidases made equal? Biochem. Soc. Trans. 2017, 45, 731–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melo, A.M.; Teixeira, M. Supramolecular organization of bacterial aerobic respiratory chains: From cells and back. Biochim. Biophys. Acta 2016, 1857, 190–197. [Google Scholar] [CrossRef]
- Refojo, P.N.; Sena, F.V.; Calisto, F.; Sousa, F.M.; Pereira, M.M. The plethora of membrane respiratory chains in the phyla of life. Adv. Microb. Physiol. 2019, 74, 331–414. [Google Scholar] [CrossRef]
- Kopcova, K.; Mikulova, L.; Pechova, I.; Sztachova, T.; Cizmar, E.; Jancura, D.; Fabian, M. Modulation of the electron-proton coupling at cytochrome a by the ligation of the oxidized catalytic center in bovine cytochrome c oxidase. Biochim. Biophys. Acta Bioenerg. 2020, 1861, 148237. [Google Scholar] [CrossRef]
- Forte, E.; Giuffre, A.; Huang, L.S.; Berry, E.A.; Borisov, V.B. Nitric oxide does not inhibit but is metabolized by the cytochrome bcc-aa3 supercomplex. Int. J. Mol. Sci. 2020, 21, 8521. [Google Scholar] [CrossRef]
- Giamogante, F.; Cali, T.; Malatesta, F. Physiological cyanide concentrations do not stimulate mitochondrial cytochrome c oxidase activity. Proc. Natl. Acad. Sci. USA 2021, 118, e2112373118. [Google Scholar] [CrossRef]
- Martino, P.L.; Capitanio, G.; Capitanio, N.; Papa, S. Inhibition of proton pumping in membrane reconstituted bovine heart cytochrome c oxidase by zinc binding at the inner matrix side. Biochim. Biophys. Acta 2011, 1807, 1075–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hederstedt, L. Molecular biology of Bacillus subtilis cytochromes anno 2020. Biochemistry 2021, 86, 8–21. [Google Scholar] [CrossRef] [PubMed]
- Siletsky, S.A.; Soulimane, T.; Belevich, I.; Gennis, R.B.; Wikstrom, M. Specific inhibition of proton pumping by the T315V mutation in the K channel of cytochrome ba3 from Thermus thermophilus. Biochim. Biophys. Acta Bioenerg. 2021, 1862, 148450. [Google Scholar] [CrossRef]
- Wikstrom, M.; Krab, K.; Sharma, V. Oxygen activation and energy conservation by cytochrome c oxidase. Chem. Rev. 2018, 118, 2469–2490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borisov, V.B.; Siletsky, S.A. Features of organization and mechanism of catalysis of two families of terminal oxidases: Heme-copper and bd-type. Biochemistry 2019, 84, 1390–1402. [Google Scholar] [CrossRef]
- Fedotovskaya, O.; Albertsson, I.; Nordlund, G.; Hong, S.; Gennis, R.B.; Brzezinski, P.; Adelroth, P. Identification of a cytochrome bc1-aa3 supercomplex in Rhodobacter sphaeroides. Biochim. Biophys. Acta Bioenerg. 2021, 1862, 148433. [Google Scholar] [CrossRef]
- Kaila, V.R.I.; Wikstrom, M. Architecture of bacterial respiratory chains. Nat. Rev. Microbiol. 2021, 19, 319–330. [Google Scholar] [CrossRef]
- Marechal, A.; Xu, J.Y.; Genko, N.; Hartley, A.M.; Haraux, F.; Meunier, B.; Rich, P.R. A common coupling mechanism for A-type heme-copper oxidases from bacteria to mitochondria. Proc. Natl. Acad. Sci. USA 2020, 117, 9349–9355. [Google Scholar] [CrossRef] [Green Version]
- Shimada, A.; Hara, F.; Shinzawa-Itoh, K.; Kanehisa, N.; Yamashita, E.; Muramoto, K.; Tsukihara, T.; Yoshikawa, S. Critical roles of the CuB site in efficient proton pumping as revealed by crystal structures of mammalian cytochrome c oxidase catalytic intermediates. J. Biol. Chem. 2021, 297, 100967. [Google Scholar] [CrossRef]
- Kolbe, F.; Safarian, S.; Piorek, Z.; Welsch, S.; Muller, H.; Michel, H. Cryo-EM structures of intermediates suggest an alternative catalytic reaction cycle for cytochrome c oxidase. Nat. Commun. 2021, 12, 6903. [Google Scholar] [CrossRef] [PubMed]
- Sedlak, E.; Kozar, T.; Musatov, A. The interplay among subunit composition, cardiolipin content, and aggregation state of bovine heart cytochrome c oxidase. Cells 2020, 9, 2588. [Google Scholar] [CrossRef] [PubMed]
- Ramzan, R.; Kadenbach, B.; Vogt, S. Multiple mechanisms regulate eukaryotic cytochrome c oxidase. Cells 2021, 10, 514. [Google Scholar] [CrossRef] [PubMed]
- Vogt, S.; Ramzan, R.; Grossman, L.I.; Singh, K.K.; Ferguson-Miller, S.; Yoshikawa, S.; Lee, I.; Huttemann, M. Mitochondrial respiration is controlled by allostery, subunit composition and phosphorylation sites of cytochrome c oxidase: A trailblazer’s tale—Bernhard Kadenbach. Mitochondrion 2021, 60, 228–233. [Google Scholar] [CrossRef]
- Blomberg, M.R.A. The redox-active tyrosine is essential for proton pumping in cytochrome c oxidase. Front. Chem. 2021, 9, 640155. [Google Scholar] [CrossRef]
- Chen, J.; Xie, P.; Huang, Y.; Gao, H. Complex interplay of heme-copper oxidases with nitrite and nitric oxide. Int. J. Mol. Sci. 2022, 23, 979. [Google Scholar] [CrossRef]
- Chateau, A.; Alpha-Bazin, B.; Armengaud, J.; Duport, C. Heme A synthase deficiency affects the ability of Bacillus cereus to adapt to a nutrient-limited environment. Int. J. Mol. Sci. 2022, 23, 1033. [Google Scholar] [CrossRef]
- Jasaitis, A.; Borisov, V.B.; Belevich, N.P.; Morgan, J.E.; Konstantinov, A.A.; Verkhovsky, M.I. Electrogenic reactions of cytochrome bd. Biochemistry 2000, 39, 13800–13809. [Google Scholar] [CrossRef]
- Belevich, I.; Borisov, V.B.; Zhang, J.; Yang, K.; Konstantinov, A.A.; Gennis, R.B.; Verkhovsky, M.I. Time-resolved electrometric and optical studies on cytochrome bd suggest a mechanism of electron-proton coupling in the di-heme active site. Proc. Natl. Acad. Sci. USA 2005, 102, 3657–3662. [Google Scholar] [CrossRef] [Green Version]
- Liang, R.; Swanson, J.M.J.; Wikstrom, M.; Voth, G.A. Understanding the essential proton-pumping kinetic gates and decoupling mutations in cytochrome c oxidase. Proc. Natl. Acad. Sci. USA 2017, 114, 5924–5929. [Google Scholar] [CrossRef] [Green Version]
- Siletsky, S.A.; Borisov, V.B. Proton pumping and non-pumping terminal respiratory oxidases: Active sites intermediates of these molecular machines and their derivatives. Int. J. Mol. Sci. 2021, 22, 10852. [Google Scholar] [CrossRef] [PubMed]
- Forte, E.; Borisov, V.B.; Vicente, J.B.; Giuffre, A. Cytochrome bd and gaseous ligands in bacterial physiology. Adv. Microb. Physiol. 2017, 71, 171–234. [Google Scholar] [CrossRef] [PubMed]
- Borisov, V.B.; Siletsky, S.A.; Paiardini, A.; Hoogewijs, D.; Forte, E.; Giuffre, A.; Poole, R.K. Bacterial oxidases of the cytochrome bd family: Redox enzymes of unique structure, function and utility as drug targets. Antioxid. Redox Signal. 2021, 34, 1280–1318. [Google Scholar] [CrossRef] [PubMed]
- Ramel, F.; Amrani, A.; Pieulle, L.; Lamrabet, O.; Voordouw, G.; Seddiki, N.; Brethes, D.; Company, M.; Dolla, A.; Brasseur, G. Membrane-bound oxygen reductases of the anaerobic sulfate-reducing Desulfovibrio vulgaris Hildenborough: Roles in oxygen defense and electron link with the periplasmic hydrogen oxidation. Microbiology 2013, 159, 2663–2673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramel, F.; Brasseur, G.; Pieulle, L.; Valette, O.; Hirschler-Rea, A.; Fardeau, M.L.; Dolla, A. Growth of the obligate anaerobe Desulfovibrio vulgaris Hildenborough under continuous low oxygen concentration sparging: Impact of the membrane-bound oxygen reductases. PLoS ONE 2015, 10, e0123455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beebout, C.J.; Eberly, A.R.; Werby, S.H.; Reasoner, S.A.; Brannon, J.R.; De, S.; Fitzgerald, M.J.; Huggins, M.M.; Clayton, D.B.; Cegelski, L.; et al. Respiratory heterogeneity shapes biofilm formation and host colonization in uropathogenic Escherichia coli. MBio 2019, 10, e02400-18. [Google Scholar] [CrossRef] [Green Version]
- Zhao, N.; Jiao, L.; Xu, J.; Zhang, J.; Qi, Y.; Qiu, M.; Wei, X.; Fan, M. Integrated transcriptomic and proteomic analysis reveals the response mechanisms of Alicyclobacillus acidoterrestris to heat stress. Food Res. Int. 2022, 151, 110859. [Google Scholar] [CrossRef]
- Borisov, V.B.; Forte, E.; Konstantinov, A.A.; Poole, R.K.; Sarti, P.; Giuffre, A. Interaction of the bacterial terminal oxidase cytochrome bd with nitric oxide. FEBS Lett. 2004, 576, 201–204. [Google Scholar] [CrossRef] [Green Version]
- Borisov, V.B.; Forte, E.; Sarti, P.; Brunori, M.; Konstantinov, A.A.; Giuffre, A. Redox control of fast ligand dissociation from Escherichia coli cytochrome bd. Biochem. Biophys. Res. Commun. 2007, 355, 97–102. [Google Scholar] [CrossRef]
- Mason, M.G.; Shepherd, M.; Nicholls, P.; Dobbin, P.S.; Dodsworth, K.S.; Poole, R.K.; Cooper, C.E. Cytochrome bd confers nitric oxide resistance to Escherichia coli. Nat. Chem. Biol. 2009, 5, 94–96. [Google Scholar] [CrossRef]
- Shepherd, M.; Achard, M.E.; Idris, A.; Totsika, M.; Phan, M.D.; Peters, K.M.; Sarkar, S.; Ribeiro, C.A.; Holyoake, L.V.; Ladakis, D.; et al. The cytochrome bd-I respiratory oxidase augments survival of multidrug-resistant Escherichia coli during infection. Sci. Rep. 2016, 6, 35285. [Google Scholar] [CrossRef] [PubMed]
- Holyoake, L.V.; Hunt, S.; Sanguinetti, G.; Cook, G.M.; Howard, M.J.; Rowe, M.L.; Poole, R.K.; Shepherd, M. CydDC-mediated reductant export in Escherichia coli controls the transcriptional wiring of energy metabolism and combats nitrosative stress. Biochem. J. 2016, 473, 693–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones-Carson, J.; Husain, M.; Liu, L.; Orlicky, D.J.; Vazquez-Torres, A. Cytochrome bd-dependent bioenergetics and antinitrosative defenses in Salmonella pathogenesis. MBio 2016, 7, e02052-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, Q.; Yin, J.; Jin, M.; Gao, H. Distinct nitrite and nitric oxide physiologies in Escherichia coli and Shewanella oneidensis. Appl. Environ. Microbiol. 2018, 84, e00559-18. [Google Scholar] [CrossRef] [Green Version]
- Forte, E.; Siletsky, S.A.; Borisov, V.B. In Escherichia coli ammonia inhibits cytochrome bo3 but activates cytochrome bd-I. Antioxidants 2021, 10, 13. [Google Scholar] [CrossRef]
- Fu, H.; Chen, H.; Wang, J.; Zhou, G.; Zhang, H.; Zhang, L.; Gao, H. Crp-dependent cytochrome bd oxidase confers nitrite resistance to Shewanella oneidensis. Environ. Microbiol. 2013, 15, 2198–2212. [Google Scholar] [CrossRef]
- Guo, K.; Gao, H. Physiological roles of nitrite and nitric oxide in bacteria: Similar consequences from distinct cell targets, protection, and Sensing Systems. Adv. Biol. 2021, 5, e2100773. [Google Scholar] [CrossRef]
- Forte, E.; Borisov, V.B.; Falabella, M.; Colaco, H.G.; Tinajero-Trejo, M.; Poole, R.K.; Vicente, J.B.; Sarti, P.; Giuffre, A. The terminal oxidase cytochrome bd promotes sulfide-resistant bacterial respiration and growth. Sci. Rep. 2016, 6, 23788. [Google Scholar] [CrossRef] [Green Version]
- Korshunov, S.; Imlay, K.R.; Imlay, J.A. The cytochrome bd oxidase of Escherichia coli prevents respiratory inhibition by endogenous and exogenous hydrogen sulfide. Mol. Microbiol. 2016, 101, 62–77. [Google Scholar] [CrossRef] [Green Version]
- Kunota, T.T.R.; Rahman, M.A.; Truebody, B.E.; Mackenzie, J.S.; Saini, V.; Lamprecht, D.A.; Adamson, J.H.; Sevalkar, R.R.; Lancaster, J.R., Jr.; Berney, M.; et al. Mycobacterium tuberculosis H2S functions as a sink to modulate central metabolism, bioenergetics, and drug susceptibility. Antioxidants 2021, 10, 1285. [Google Scholar] [CrossRef]
- Borisov, V.B.; Forte, E. Terminal oxidase cytochrome bd protects bacteria against hydrogen sulfide toxicity. Biochemistry 2021, 86, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Borisov, V.B.; Forte, E. Impact of hydrogen sulfide on mitochondrial and bacterial bioenergetics. Int. J. Mol. Sci. 2021, 22, 12688. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Wu, S.; Li, L.; Xu, B.; Wang, G. The cytochrome bd complex is essential for chromate and sulfide resistance and is regulated by a GbsR-type regulator, CydE, in Alishewanella sp. WH16-1. Front. Microbiol. 2018, 9, 1849. [Google Scholar] [CrossRef] [PubMed]
- Borisov, V.B.; Forte, E.; Davletshin, A.; Mastronicola, D.; Sarti, P.; Giuffre, A. Cytochrome bd oxidase from Escherichia coli displays high catalase activity: An additional defense against oxidative stress. FEBS Lett. 2013, 587, 2214–2218. [Google Scholar] [CrossRef]
- Al-Attar, S.; Yu, Y.; Pinkse, M.; Hoeser, J.; Friedrich, T.; Bald, D.; de Vries, S. Cytochrome bd displays significant quinol peroxidase activity. Sci. Rep. 2016, 6, 27631. [Google Scholar] [CrossRef] [Green Version]
- Borisov, V.B.; Siletsky, S.A.; Nastasi, M.R.; Forte, E. ROS defense systems and terminal oxidases in bacteria. Antioxidants 2021, 10, 839. [Google Scholar] [CrossRef]
- Borisov, V.B.; Forte, E.; Siletsky, S.A.; Sarti, P.; Giuffre, A. Cytochrome bd from Escherichia coli catalyzes peroxynitrite decomposition. Biochim. Biophys. Acta 2015, 1847, 182–188. [Google Scholar] [CrossRef] [Green Version]
- Kalia, N.P.; Shi Lee, B.; Ab Rahman, N.B.; Moraski, G.C.; Miller, M.J.; Pethe, K. Carbon metabolism modulates the efficacy of drugs targeting the cytochrome bc1:aa3 in Mycobacterium tuberculosis. Sci. Rep. 2019, 9, 8608. [Google Scholar] [CrossRef]
- Beebout, C.J.; Sominsky, L.A.; Eberly, A.R.; Van Horn, G.T.; Hadjifrangiskou, M. Cytochrome bd promotes Escherichia coli biofilm antibiotic tolerance by regulating accumulation of noxious chemicals. NPJ Biofilms Microbiomes 2021, 7, 35. [Google Scholar] [CrossRef]
- Schildkraut, J.A.; Coolen, J.P.M.; Burbaud, S.; Sangen, J.J.N.; Kwint, M.P.; Floto, R.A.; Op den Camp, H.J.M.; Te Brake, L.H.M.; Wertheim, H.F.L.; Neveling, K.; et al. RNA-sequencing elucidates drug-specific mechanisms of antibiotic tolerance and resistance in M. abscessus. Antimicrob. Agents Chemother. 2021, 66, e0150921. [Google Scholar] [CrossRef]
- Iwata, S.; Ostermeier, C.; Ludwig, B.; Michel, H. Structure at 2.8 A resolution of cytochrome c oxidase from Paracoccus denitrificans. Nature 1995, 376, 660–669. [Google Scholar] [CrossRef]
- Tsukihara, T.; Aoyama, H.; Yamashita, E.; Tomizaki, T.; Yamaguchi, H.; Shinzawa-Itoh, K.; Nakashima, T.; Yaono, R.; Yoshikawa, S. Structures of metal sites of oxidized bovine heart cytochrome c oxidase at 2.8 A. Science 1995, 269, 1069–1074. [Google Scholar] [CrossRef] [PubMed]
- Abramson, J.; Svensson-Ek, M.; Byrne, B.; Iwata, S. Structure of cytochrome c oxidase: A comparison of the bacterial and mitochondrial enzymes. Biochim. Biophys. Acta 2001, 1544, 1–9. [Google Scholar] [CrossRef]
- Shiba, T.; Kido, Y.; Sakamoto, K.; Inaoka, D.K.; Tsuge, C.; Tatsumi, R.; Takahashi, G.; Balogun, E.O.; Nara, T.; Aoki, T.; et al. Structure of the trypanosome cyanide-insensitive alternative oxidase. Proc. Natl. Acad. Sci. USA 2013, 110, 4580–4585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubach, V.R.A.; Guskov, A. The resolution in X-ray crystallography and single-particle cryogenic electron microscopy. Crystals 2020, 10, 580. [Google Scholar] [CrossRef]
- Dueweke, T.J.; Gennis, R.B. Epitopes of monoclonal antibodies which inhibit ubiquinol oxidase activity of Escherichia coli cytochrome d complex localize a functional domain. J. Biol. Chem. 1990, 265, 4273–4277. [Google Scholar] [CrossRef]
- Mogi, T.; Akimoto, S.; Endou, S.; Watanabe-Nakayama, T.; Mizuochi-Asai, E.; Miyoshi, H. Probing the ubiquinol-binding site in cytochrome bd by site-directed mutagenesis. Biochemistry 2006, 45, 7924–7930. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, Y.; Murai, M.; Fujita, D.; Sakamoto, K.; Miyoshi, H.; Yoshida, M.; Mogi, T. Mass spectrometric analysis of the ubiquinol-binding site in cytochrome bd from Escherichia coli. J. Biol. Chem. 2006, 281, 1905–1912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goojani, H.G.; Konings, J.; Hakvoort, H.; Hong, S.; Gennis, R.B.; Sakamoto, J.; Lill, H.; Bald, D. The carboxy-terminal insert in the Q-loop is needed for functionality of Escherichia coli cytochrome bd-I. Biochim. Biophys. Acta 2020, 1861, 148175. [Google Scholar] [CrossRef] [PubMed]
- Thesseling, A.; Burschel, S.; Wohlwend, D.; Friedrich, T. The long Q-loop of Escherichia coli cytochrome bd oxidase is required for assembly and structural integrity. FEBS Lett. 2020, 594, 1577–1585. [Google Scholar] [CrossRef] [Green Version]
- Sviriaeva, E.; Subramanian Manimekalai, M.S.; Gruber, G.; Pethe, K. Features and functional importance of key residues of the Mycobacterium tuberculosis cytochrome bd oxidase. ACS Infect. Dis. 2020, 6, 1697–1707. [Google Scholar] [CrossRef] [PubMed]
- Allen, R.J.; Brenner, E.P.; VanOrsdel, C.E.; Hobson, J.J.; Hearn, D.J.; Hemm, M.R. Conservation analysis of the CydX protein yields insights into small protein identification and evolution. BMC Genom. 2014, 15, 946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoeser, J.; Hong, S.; Gehmann, G.; Gennis, R.B.; Friedrich, T. Subunit CydX of Escherichia coli cytochrome bd ubiquinol oxidase is essential for assembly and stability of the di-heme active site. FEBS Lett. 2014, 588, 1537–1541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hummer, G.; Rasaiah, J.C.; Noworyta, J.P. Water conduction through the hydrophobic channel of a carbon nanotube. Nature 2001, 414, 188–190. [Google Scholar] [CrossRef]
- Yang, K.; Zhang, J.; Vakkasoglu, A.S.; Hielscher, R.; Osborne, J.P.; Hemp, J.; Miyoshi, H.; Hellwig, P.; Gennis, R.B. Glutamate 107 in subunit I of the cytochrome bd quinol oxidase from Escherichia coli is protonated and near the heme d/heme b595 binuclear center. Biochemistry 2007, 46, 3270–3278. [Google Scholar] [CrossRef]
- Corbett, D.; Goldrick, M.; Fernandes, V.E.; Davidge, K.; Poole, R.K.; Andrew, P.W.; Cavet, J.; Roberts, I.S. Listeria monocytogenes has both a bd-type and an aa3-type terminal oxidase which allow growth in different oxygen levels and both are important in infection. Infect. Immun. 2017, 85, e00354-17. [Google Scholar] [CrossRef] [Green Version]
- Price, E.P.; Viberg, L.T.; Kidd, T.J.; Bell, S.C.; Currie, B.J.; Sarovich, D.S. Transcriptomic analysis of longitudinal Burkholderia pseudomallei infecting the cystic fibrosis lung. Microb. Genom. 2018, 4, e000194. [Google Scholar] [CrossRef]
- WHO. Global Tuberculosis Report 2021. Global Tuberculosis Report 2021. World Health Organization, Geneva, Switzerland-Licence: CC BY-NC-SA 3.0 IGO. 2021. Available online: https://www.who.int/publications/i/item/9789240037021 (accessed on 10 March 2022).
- Cook, G.M.; Hards, K.; Dunn, E.; Heikal, A.; Nakatani, Y.; Greening, C.; Crick, D.C.; Fontes, F.L.; Pethe, K.; Hasenoehrl, E.; et al. Oxidative phosphorylation as a target space for tuberculosis: Success, caution, and future directions. Microbiol. Spectr. 2017, 5. [Google Scholar] [CrossRef] [Green Version]
- Bald, D.; Villellas, C.; Lu, P.; Koul, A. Targeting energy metabolism in Mycobacterium tuberculosis, a new paradigm in antimycobacterial drug discovery. MBio 2017, 8, e00272-17. [Google Scholar] [CrossRef] [Green Version]
- Iqbal, I.K.; Bajeli, S.; Akela, A.K.; Kumar, A. Bioenergetics of Mycobacterium: An emerging landscape for drug discovery. Pathogens 2018, 7, 24. [Google Scholar] [CrossRef] [Green Version]
- Hards, K.; Cook, G.M. Targeting bacterial energetics to produce new antimicrobials. Drug Resist. Updat. 2018, 36, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Mascolo, L.; Bald, D. Cytochrome bd in Mycobacterium tuberculosis: A respiratory chain protein involved in the defense against antibacterials. Prog. Biophys. Mol. Biol. 2020, 152, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.S.; Sviriaeva, E.; Pethe, K. Targeting the cytochrome oxidases for drug development in mycobacteria. Prog. Biophys. Mol. Biol. 2020, 152, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Bajeli, S.; Baid, N.; Kaur, M.; Pawar, G.P.; Chaudhari, V.D.; Kumar, A. Terminal respiratory oxidases: A targetables vulnerability of mycobacterial bioenergetics? Front. Cell. Infect. Microbiol. 2020, 10, 589318. [Google Scholar] [CrossRef] [PubMed]
- Appetecchia, F.; Consalvi, S.; Scarpecci, C.; Biava, M.; Poce, G. SAR analysis of small molecules interfering with energy-metabolism in Mycobacterium tuberculosis. Pharmaceuticals 2020, 13, 227. [Google Scholar] [CrossRef]
- Wani, M.A.; Dhaked, D.K. Targeting the cytochrome bc1 complex for drug development in M. tuberculosis: Review. Mol. Divers. 2021. [Google Scholar] [CrossRef]
- Sindhu, T.; Debnath, P. Cytochrome bc1-aa3 oxidase supercomplex as emerging and potential drug target against tuberculosis. Curr. Mol. Pharmacol. 2022, 15. [Google Scholar] [CrossRef]
- Urban, M.; Slachtova, V.; Brulikova, L. Small organic molecules targeting the energy metabolism of Mycobacterium tuberculosis. Eur. J. Med. Chem. 2021, 212, 113139. [Google Scholar] [CrossRef]
- Vilcheze, C.; Weinrick, B.; Leung, L.W.; Jacobs, W.R., Jr. Plasticity of Mycobacterium tuberculosis NADH dehydrogenases and their role in virulence. Proc. Natl. Acad. Sci. USA 2018, 115, 1599–1604. [Google Scholar] [CrossRef] [Green Version]
- McNeil, M.B.; Ryburn, H.W.; Tirados, J.; Cheung, C.Y.; Cook, G.M. Multiplexed transcriptional repression identifies a network of bactericidal interactions between mycobacterial respiratory complexes. iScience 2021, 25, 103573. [Google Scholar] [CrossRef]
- Hards, K.; McMillan, D.G.G.; Schurig-Briccio, L.A.; Gennis, R.B.; Lill, H.; Bald, D.; Cook, G.M. Ionophoric effects of the antitubercular drug bedaquiline. Proc. Natl. Acad. Sci. USA 2018, 115, 7326–7331. [Google Scholar] [CrossRef] [Green Version]
- de Jager, V.R.; Dawson, R.; van Niekerk, C.; Hutchings, J.; Kim, J.; Vanker, N.; van der Merwe, L.; Choi, J.; Nam, K.; Diacon, A.H. Telacebec (Q203), a new antituberculosis agent. N. Engl. J. Med. 2020, 382, 1280–1281. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.S.; Pethe, K. Telacebec: An investigational antibiotic for the treatment of tuberculosis (TB). Expert Opin. Investig. Drugs 2022, 31, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Kalia, N.P.; Hasenoehrl, E.J.; Ab Rahman, N.B.; Koh, V.H.; Ang, M.L.T.; Sajorda, D.R.; Hards, K.; Gruber, G.; Alonso, S.; Cook, G.M.; et al. Exploiting the synthetic lethality between terminal respiratory oxidases to kill Mycobacterium tuberculosis and clear host infection. Proc. Natl. Acad. Sci. USA 2017, 114, 7426–7431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, B.S.; Hards, K.; Engelhart, C.A.; Hasenoehrl, E.J.; Kalia, N.P.; Mackenzie, J.S.; Sviriaeva, E.; Chong, S.M.S.; Manimekalai, M.S.S.; Koh, V.H.; et al. Dual inhibition of the terminal oxidases eradicates antibiotic-tolerant Mycobacterium tuberculosis. EMBO Mol. Med. 2021, 13, e13207. [Google Scholar] [CrossRef]
- Meunier, B.; Madgwick, S.A.; Reil, E.; Oettmeier, W.; Rich, P.R. New inhibitors of the quinol oxidation sites of bacterial cytochromes bo and bd. Biochemistry 1995, 34, 1076–1083. [Google Scholar] [CrossRef]
- Radloff, M.; Elamri, I.; Grund, T.N.; Witte, L.F.; Hohmann, K.F.; Nakagaki, S.; Goojani, H.G.; Nasiri, H.; Hideto, M.; Bald, D.; et al. Short-chain aurachin D derivatives are selective inhibitors of E. coli cytochrome bd-I and bd-II oxidases. Sci. Rep. 2021, 11, 23852. [Google Scholar] [CrossRef]
- Miyoshi, H.; Takegami, K.; Sakamoto, K.; Mogi, T.; Iwamura, H. Characterization of the ubiquinol oxidation sites in cytochromes bo and bd from Escherichia coli using aurachin C analogues. J. Biochem. 1999, 125, 138–142. [Google Scholar] [CrossRef]
- Makarchuk, I.; Nikolaev, A.; Thesseling, A.; Dejon, L.; Lamberty, D.; Stief, L.; Speicher, A.; Friedrich, T.; Hellwig, P.; Nasiri, H.R.; et al. Identification and optimization of quinolone-based inhibitors against cytochrome bd oxidase using an electrochemical assay. Electrochim. Acta 2021, 381, 138293. [Google Scholar] [CrossRef]
- Lu, P.; Heineke, M.H.; Koul, A.; Andries, K.; Cook, G.M.; Lill, H.; van Spanning, R.; Bald, D. The cytochrome bd-type quinol oxidase is important for survival of Mycobacterium smegmatis under peroxide and antibiotic-induced stress. Sci. Rep. 2015, 5, 10333. [Google Scholar] [CrossRef] [Green Version]
- Lu, P.; Asseri, A.H.; Kremer, M.; Maaskant, J.; Ummels, R.; Lill, H.; Bald, D. The anti-mycobacterial activity of the cytochrome bcc inhibitor Q203 can be enhanced by small-molecule inhibition of cytochrome bd. Sci. Rep. 2018, 8, 2625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harikishore, A.; Chong, S.S.M.; Ragunathan, P.; Bates, R.W.; Gruber, G. Targeting the menaquinol binding loop of mycobacterial cytochrome bd oxidase. Mol. Divers. 2021, 25, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Hopfner, S.M.; Lee, B.S.; Kalia, N.P.; Miller, M.J.; Pethe, K.; Moraski, G.C. Structure guided generation of thieno[3,2-d]pyrimidin-4-amine Mycobacterium tuberculosis bd oxidase inhibitors. RSC Med. Chem. 2021, 12, 73–77. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Friedrich, T.; Wohlwend, D.; Borisov, V.B. Recent Advances in Structural Studies of Cytochrome bd and Its Potential Application as a Drug Target. Int. J. Mol. Sci. 2022, 23, 3166. https://doi.org/10.3390/ijms23063166
Friedrich T, Wohlwend D, Borisov VB. Recent Advances in Structural Studies of Cytochrome bd and Its Potential Application as a Drug Target. International Journal of Molecular Sciences. 2022; 23(6):3166. https://doi.org/10.3390/ijms23063166
Chicago/Turabian StyleFriedrich, Thorsten, Daniel Wohlwend, and Vitaliy B. Borisov. 2022. "Recent Advances in Structural Studies of Cytochrome bd and Its Potential Application as a Drug Target" International Journal of Molecular Sciences 23, no. 6: 3166. https://doi.org/10.3390/ijms23063166
APA StyleFriedrich, T., Wohlwend, D., & Borisov, V. B. (2022). Recent Advances in Structural Studies of Cytochrome bd and Its Potential Application as a Drug Target. International Journal of Molecular Sciences, 23(6), 3166. https://doi.org/10.3390/ijms23063166