
Inducible Systemic Gcn1 Deletion in Mice Leads to Transient Body Weight Loss upon Tamoxifen Treatment Associated with Decrease of Fat and Liver Glycogen Storage

 ,
,  , ,
, ,  , , ,
, , ,  , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
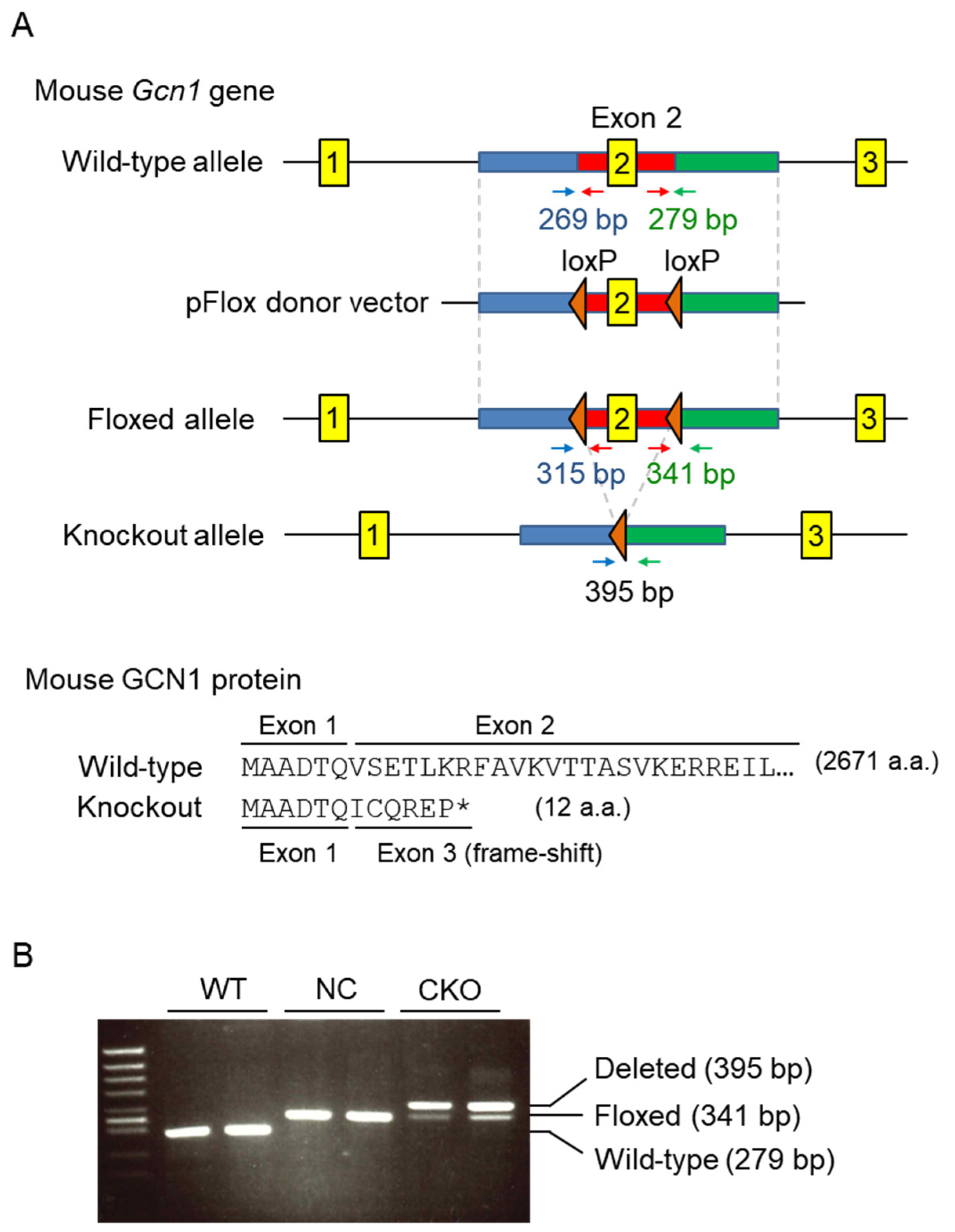
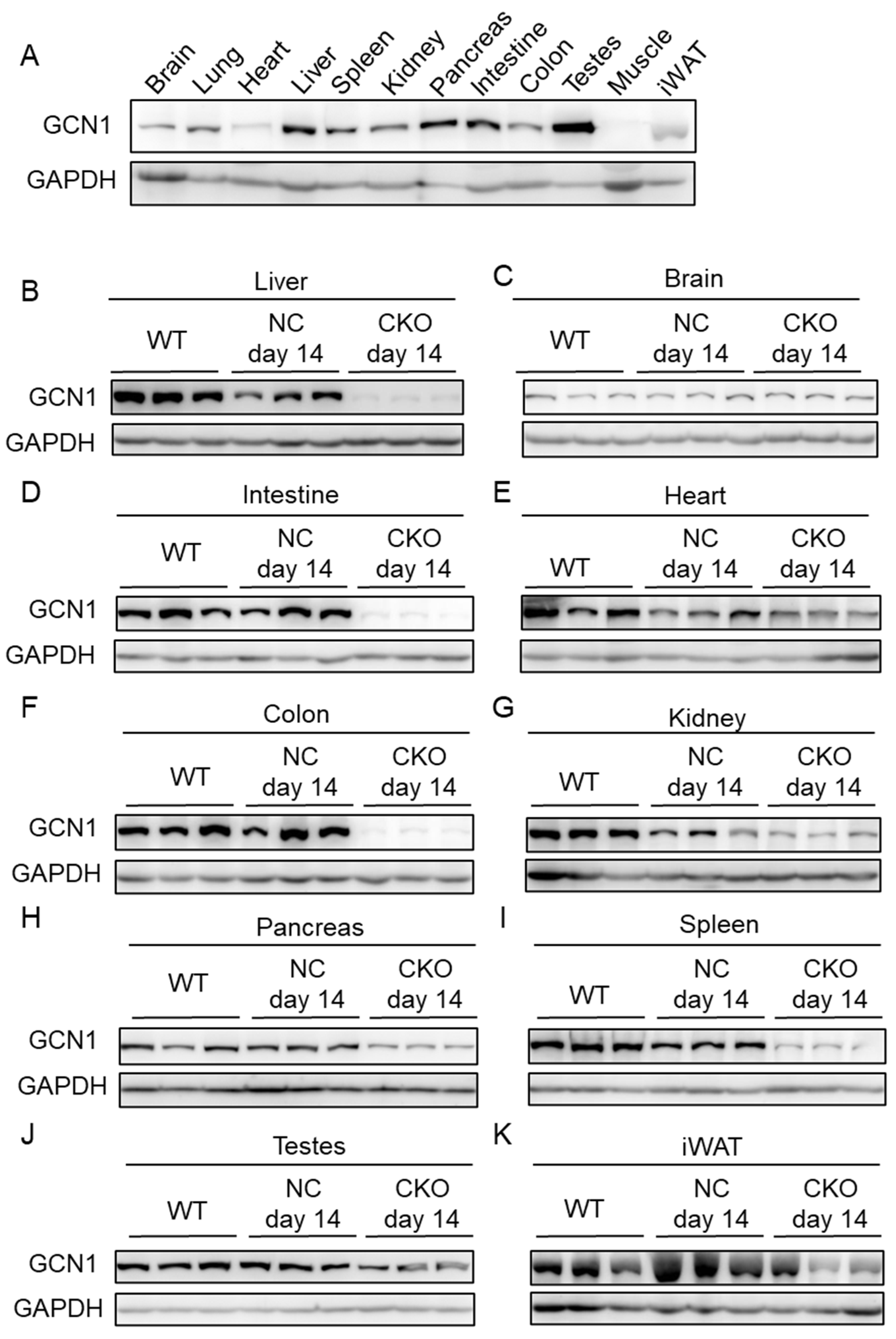
2.1. Establishment of Gcn1 CKO Mice
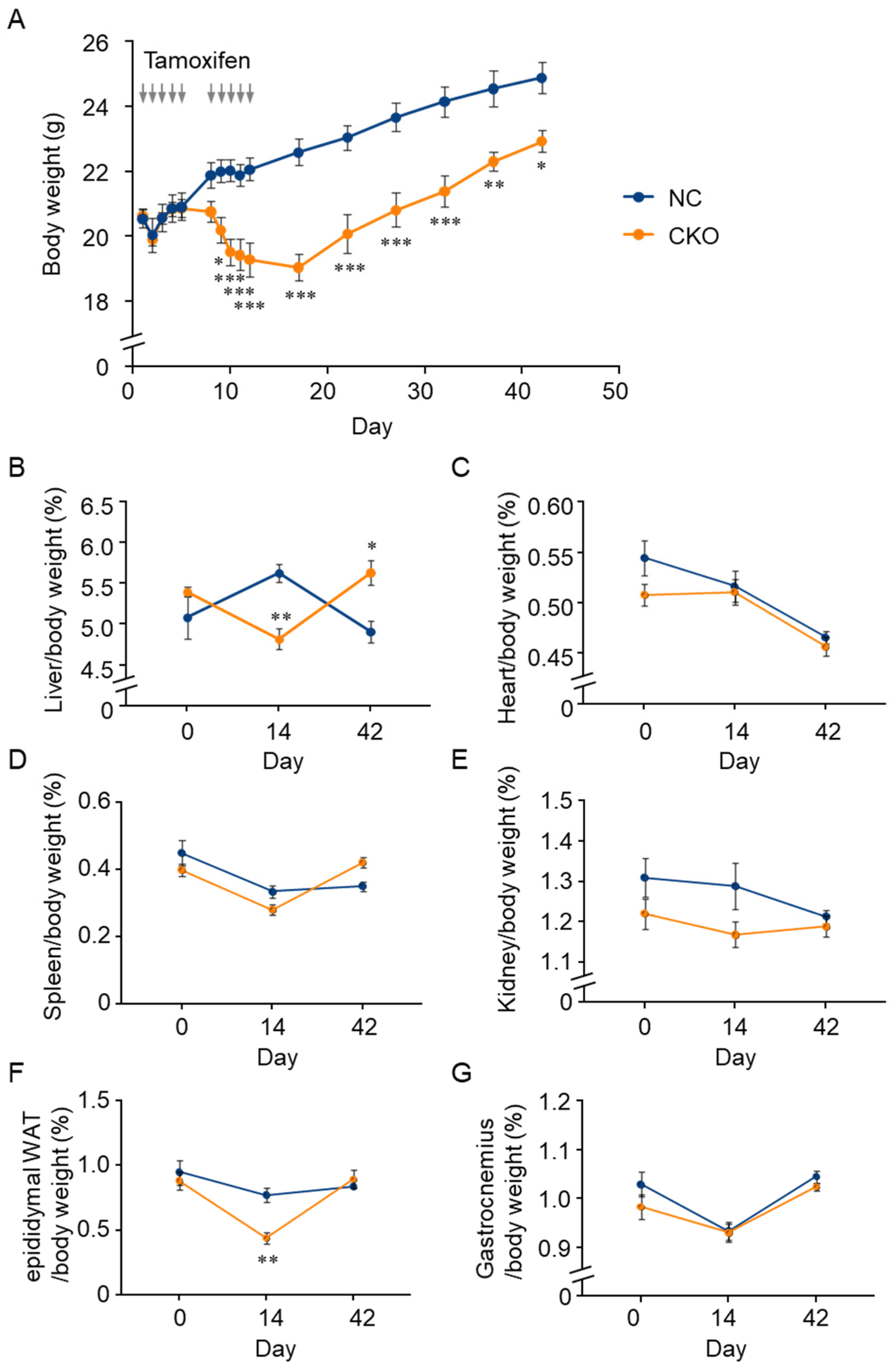
2.2. Tamoxifen-Induced Body Weight Loss in Gcn1 CKO Mice
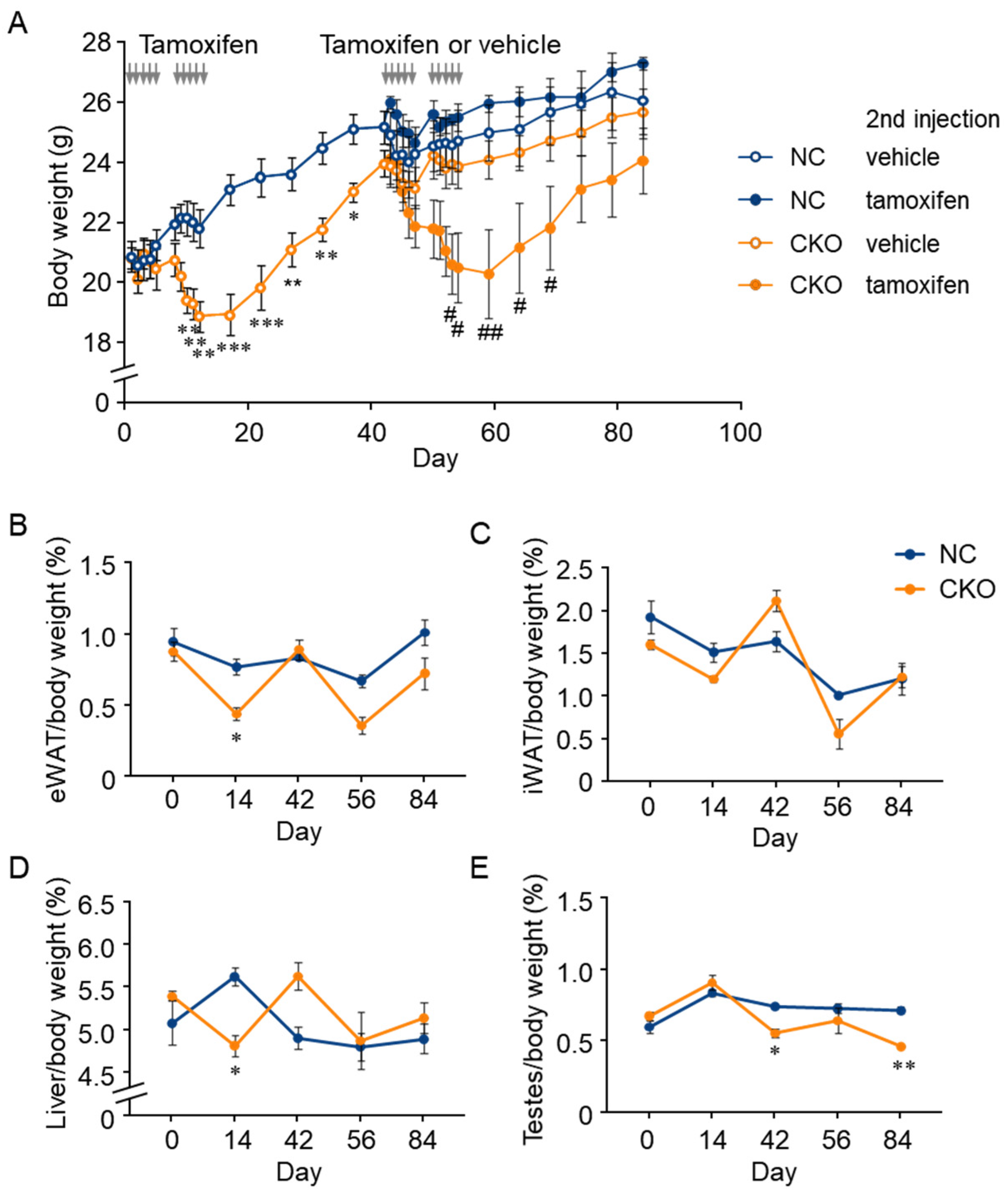
2.3. Combination of GCN1-Deletion and Tamoxifen-Treatment Induced the Rapid Loss of Body Weight Gain
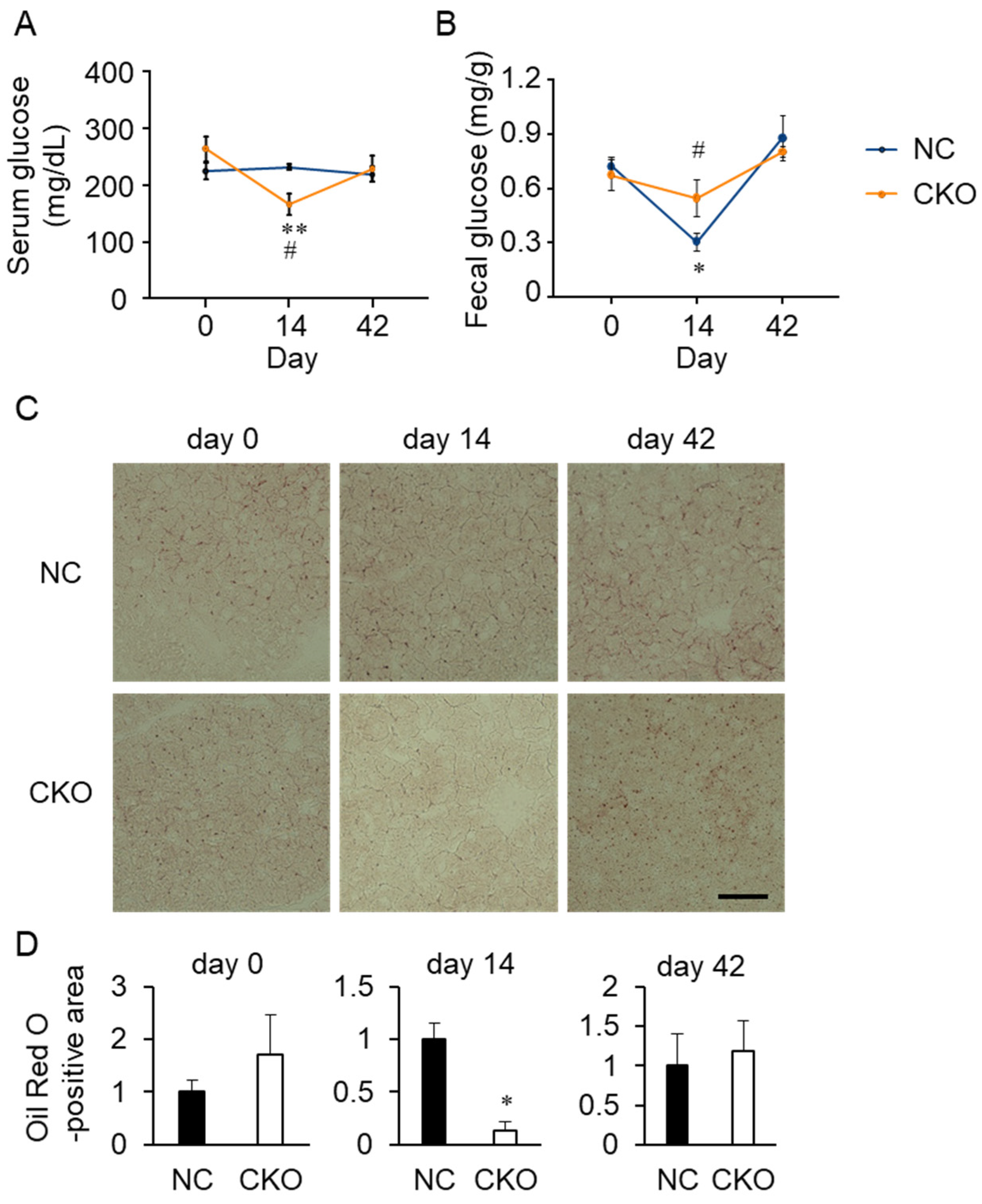
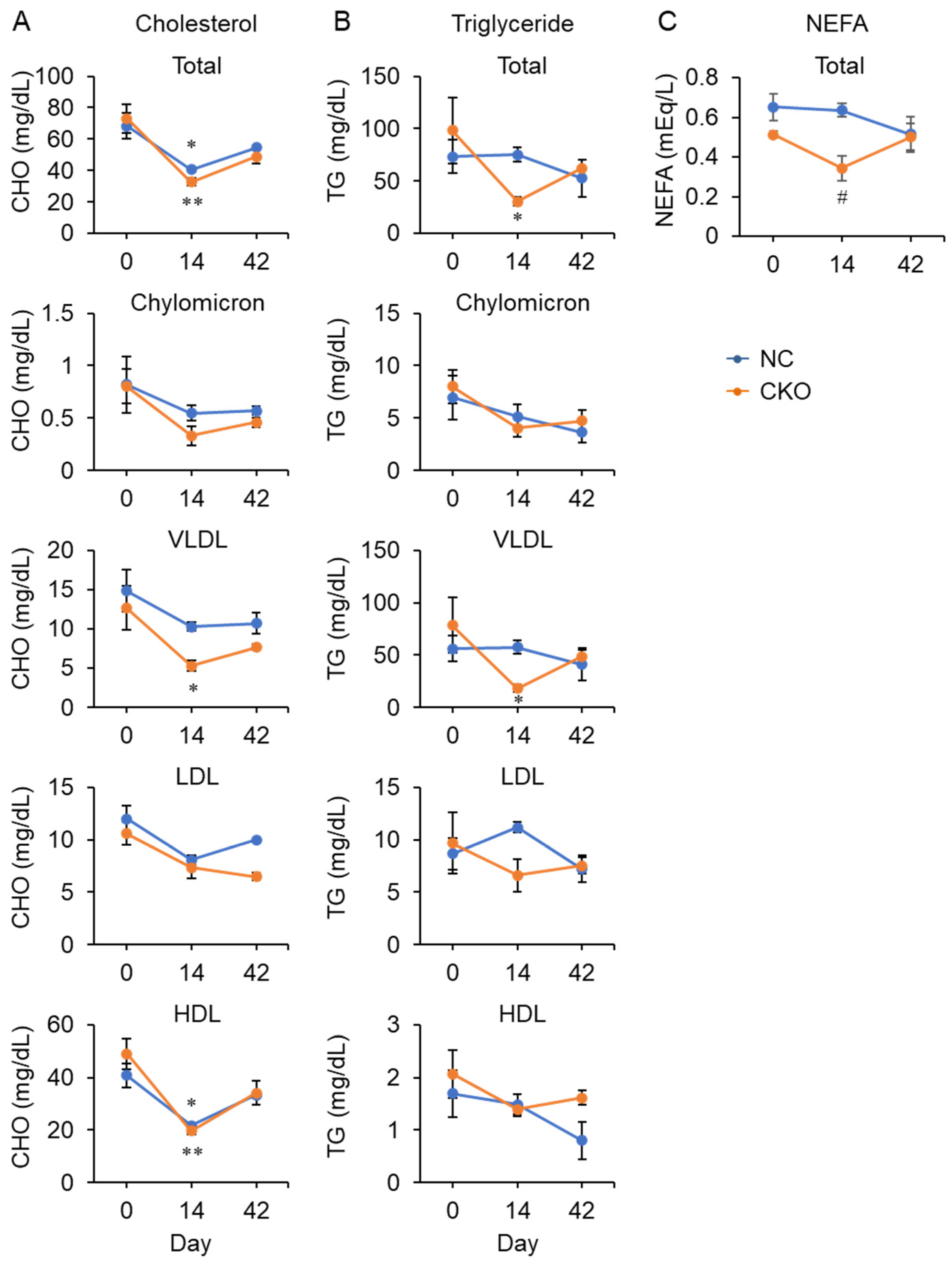
2.4. Tamoxifen Administration Drastically Impaired Glucose and Fat Metabolism in Gcn1 CKO Mice
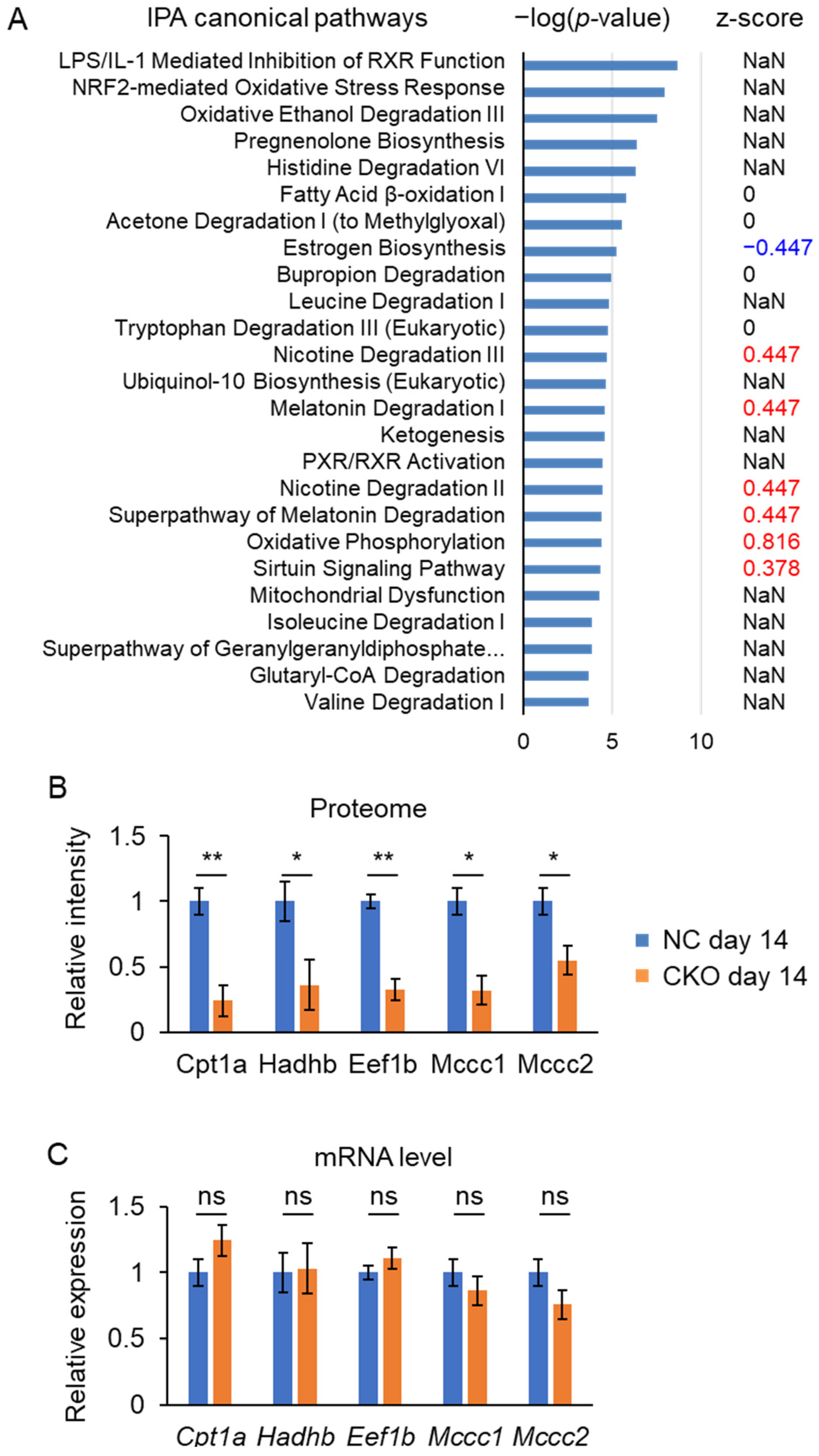
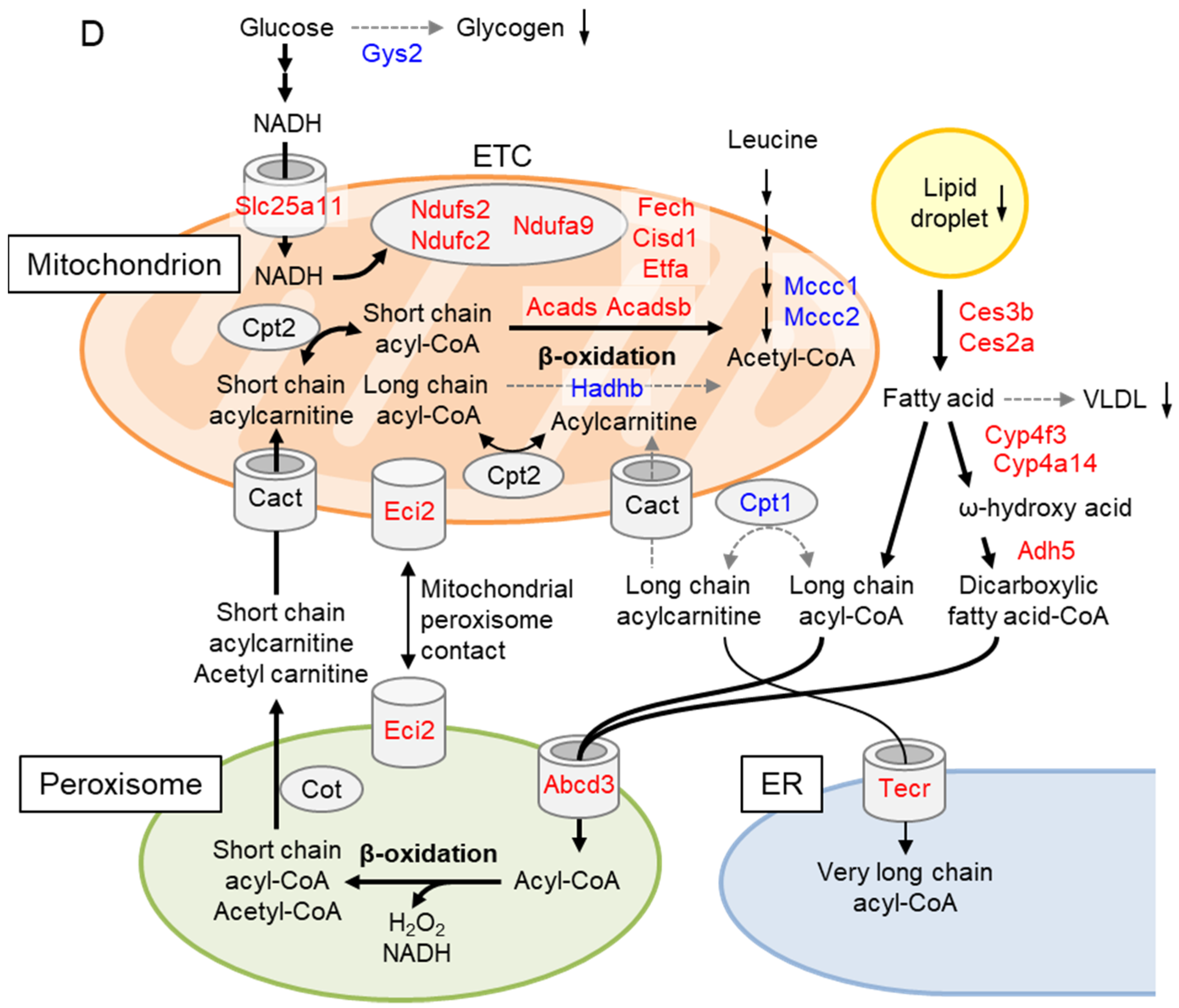
2.5. Liver Proteome Analysis Revealed Drastic Metabolic Remodeling Leading to the Inhibition of Mitochondrial FAO and Enhanced Peroxisomal FAO in Gcn1 CKO Mice
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Generation of Gcn1 CKO Mice
4.3. Tamoxifen Metabolite Quantification
4.4. Histological Analysis
4.5. Mouse Embryonic Fibroblast Preparation
4.6. Mouse Primary Hepatocyte Preparation
4.7. Reverse Transcriptase-Quantitative PCR (RT-qPCR)
4.8. Western Blot Analysis
4.9. Biochemical Marker Measurements
4.10. Liver Glycogen Measurement
4.11. Fecal Glucose Measurement
4.12. ATP Measurement
4.13. Oxygen Consumption Rate (OCR) Measurement
4.14. Proteome Analysis
4.15. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Castilho, B.A.; Shanmugam, R.; Silva, R.C.; Ramesh, R.; Himme, B.M.; Sattlegger, E. Keeping the eIF2 alpha kinase Gcn2 in check. Biochim. Biophys. Acta 2014, 1843, 1948–1968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamazaki, H.; Kasai, S.; Mimura, J.; Ye, P.; Inose-Maruyama, A.; Tanji, K.; Wakabayashi, K.; Mizuno, S.; Sugiyama, F.; Takahashi, S.; et al. Ribosome binding protein GCN1 regulates the cell cycle and cell proliferation and is essential for the embryonic development of mice. PLoS Genet. 2020, 16, e1008693. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Zhang, P.; McGrath, B.C.; Reinert, J.; Olsen, D.S.; Lei, L.; Gill, S.; Wek, S.A.; Vattem, K.M.; Wek, R.C.; Kimball, S.R.; et al. The GCN2 eIF2alpha kinase is required for adaptation to amino acid deprivation in mice. Mol. Cell Biol. 2002, 22, 6681–6688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, S.H.; Kim, A.R.; Park, N.H.; Park, J.W.; Han, I.S. DRG2 Regulates G2/M Progression via the Cyclin B1-Cdk1 Complex. Mol. Cells 2016, 39, 699–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, H.R.; Vo, M.T.; Kim, D.J.; Lee, U.H.; Yoon, J.H.; Kim, H.J.; Kim, J.; Kim, S.R.; Lee, J.Y.; Yang, C.H.; et al. DRG2 Deficient Mice Exhibit Impaired Motor Behaviors with Reduced Striatal Dopamine Release. Int. J. Mol. Sci. 2019, 21, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirose, T.; Horvitz, H.R. The translational regulators GCN-1 and ABCF-3 act together to promote apoptosis in C. elegans. PLoS Genet. 2014, 10, e1004512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izquierdo, Y.; Kulasekaran, S.; Benito, P.; Lopez, B.; Marcos, R.; Cascon, T.; Hamberg, M.; Castresana, C. Arabidopsis nonresponding to oxylipins locus NOXY7 encodes a yeast GCN1 homolog that mediates noncanonical translation regulation and stress adaptation. Plant Cell Environ. 2018, 41, 1438–1452. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Sundrud, M.S.; Zhou, C.; Edenius, M.; Zocco, D.; Powers, K.; Zhang, M.; Mazitschek, R.; Rao, A.; Yeo, C.Y.; et al. Aminoacyl-tRNA synthetase inhibition activates a pathway that branches from the canonical amino acid response in mammalian cells. Proc. Natl. Acad. Sci. USA 2020, 117, 8900–8911. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.C.; Peterson, A.; Zinshteyn, B.; Regot, S.; Green, R. Ribosome Collisions Trigger General Stress Responses to Regulate Cell Fate. Cell 2020, 182, 404–416.e14. [Google Scholar] [CrossRef] [PubMed]
- Pochopien, A.A.; Beckert, B.; Kasvandik, S.; Berninghausen, O.; Beckmann, R.; Tenson, T.; Wilson, D.N. Structure of Gcn1 bound to stalled and colliding 80S ribosomes. Proc. Natl. Acad. Sci. USA 2021, 118, e2022756118. [Google Scholar] [CrossRef] [PubMed]
- Houston, L.; Platten, E.M.; Connelly, S.M.; Wang, J.; Grayhack, E.J. Frameshifting at collided ribosomes is modulated by elongation factor eEF3 and by Integrated Stress Response regulators Gcn1 and Gcn20. RNA 2021, 28, 320–339. [Google Scholar] [CrossRef] [PubMed]
- Terrey, M.; Adamson, S.I.; Gibson, A.L.; Deng, T.; Ishimura, R.; Chuang, J.H.; Ackerman, S.L. GTPBP1 resolves paused ribosomes to maintain neuronal homeostasis. Elife 2020, 9, e62731. [Google Scholar] [CrossRef] [PubMed]
- Lyu, X.; Yang, Q.; Zhao, F.; Liu, Y. Codon usage and protein length-dependent feedback from translation elongation regulates translation initiation and elongation speed. Nucleic Acids Res. 2021, 49, 9404–9423. [Google Scholar] [CrossRef]
- Ventura, A.; Kirsch, D.G.; McLaughlin, M.E.; Tuveson, D.A.; Grimm, J.; Lintault, L.; Newman, J.; Reczek, E.E.; Weissleder, R.; Jacks, T. Restoration of p53 function leads to tumour regression in vivo. Nature 2007, 445, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Hameyer, D.; Loonstra, A.; Eshkind, L.; Schmitt, S.; Antunes, C.; Groen, A.; Bindels, E.; Jonkers, J.; Krimpenfort, P.; Meuwissen, R.; et al. Toxicity of ligand-dependent Cre recombinases and generation of a conditional Cre deleter mouse allowing mosaic recombination in peripheral tissues. Physiol. Genom. 2007, 31, 32–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohin, N.; Carlson, E.A.; Samuelson, L.C. Genome Toxicity and Impaired Stem Cell Function after Conditional Activation of CreER(T2) in the Intestine. Stem Cell Rep. 2018, 11, 1337–1346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higashi, A.Y.; Ikawa, T.; Muramatsu, M.; Economides, A.N.; Niwa, A.; Okuda, T.; Murphy, A.J.; Rojas, J.; Heike, T.; Nakahata, T.; et al. Direct hematological toxicity and illegitimate chromosomal recombination caused by the systemic activation of CreERT2. J. Immunol. 2009, 182, 5633–5640. [Google Scholar] [CrossRef] [Green Version]
- Ceasrine, A.M.; Ruiz-Otero, N.; Lin, E.E.; Lumelsky, D.N.; Boehm, E.D.; Kuruvilla, R. Tamoxifen Improves Glucose Tolerance in a Delivery-, Sex-, and Strain-Dependent Manner in Mice. Endocrinology 2019, 160, 782–790. [Google Scholar] [CrossRef] [Green Version]
- Cole, L.K.; Jacobs, R.L.; Vance, D.E. Tamoxifen induces triacylglycerol accumulation in the mouse liver by activation of fatty acid synthesis. Hepatology 2010, 52, 1258–1265. [Google Scholar] [CrossRef]
- Marek, C.B.; Peralta, R.M.; Itinose, A.M.; Bracht, A. Influence of tamoxifen on gluconeogenesis and glycolysis in the perfused rat liver. Chem. Biol. Interact. 2011, 193, 22–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birzniece, V.; Barrett, P.H.R.; Ho, K.K.Y. Tamoxifen reduces hepatic VLDL production and GH secretion in women: A possible mechanism for steatosis development. Eur. J. Endocrinol. 2017, 177, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Eechoute, K.; Mathijssen, R.H.J.; van Gelder, T. Tamoxifen-induced fatty liver disease in a Caucasian patient. Breast Cancer Res. Treat. 2018, 171, 243–244. [Google Scholar] [CrossRef] [PubMed]
- Larosche, I.; Letteron, P.; Fromenty, B.; Vadrot, N.; Abbey-Toby, A.; Feldmann, G.; Pessayre, D.; Mansouri, A. Tamoxifen inhibits topoisomerases, depletes mitochondrial DNA, and triggers steatosis in mouse liver. J. Pharmacol. Exp. Ther. 2007, 321, 526–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardoso, C.M.; Custodio, J.B.; Almeida, L.M.; Moreno, A.J. Mechanisms of the deleterious effects of tamoxifen on mitochondrial respiration rate and phosphorylation efficiency. Toxicol. Appl. Pharmacol. 2001, 176, 145–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribeiro, M.P.; Santos, A.E.; Custodio, J.B. Mitochondria: The gateway for tamoxifen-induced liver injury. Toxicology 2014, 323, 10–18. [Google Scholar] [CrossRef]
- Hesselbarth, N.; Pettinelli, C.; Gericke, M.; Berger, C.; Kunath, A.; Stumvoll, M.; Bluher, M.; Kloting, N. Tamoxifen affects glucose and lipid metabolism parameters, causes browning of subcutaneous adipose tissue and transient body composition changes in C57BL/6NTac mice. Biochem. Biophys. Res. Commun. 2015, 464, 724–729. [Google Scholar] [CrossRef]
- Reddy, J.K.; Hashimoto, T. Peroxisomal beta-oxidation and peroxisome proliferator-activated receptor alpha: An adaptive metabolic system. Annu. Rev. Nutr. 2001, 21, 193–230. [Google Scholar] [CrossRef] [PubMed]
- Ranea-Robles, P.; Violante, S.; Argmann, C.; Dodatko, T.; Bhattacharya, D.; Chen, H.; Yu, C.; Friedman, S.L.; Puchowicz, M.; Houten, S.M. Murine deficiency of peroxisomal L-bifunctional protein (EHHADH) causes medium-chain 3-hydroxydicarboxylic aciduria and perturbs hepatic cholesterol homeostasis. Cell Mol. Life Sci. 2021, 78, 5631–5646. [Google Scholar] [CrossRef] [PubMed]
- Hardwick, J.P. Cytochrome P450 omega hydroxylase (CYP4) function in fatty acid metabolism and metabolic diseases. Biochem. Pharmacol. 2008, 75, 2263–2275. [Google Scholar] [CrossRef]
- Le Quere, V.; Plee-Gautier, E.; Potin, P.; Madec, S.; Salaun, J.P. Human CYP4F3s are the main catalysts in the oxidation of fatty acid epoxides. J. Lipid Res. 2004, 45, 1446–1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Violante, S.; Achetib, N.; van Roermund, C.W.T.; Hagen, J.; Dodatko, T.; Vaz, F.M.; Waterham, H.R.; Chen, H.; Baes, M.; Yu, C.; et al. Peroxisomes can oxidize medium- and long-chain fatty acids through a pathway involving ABCD3 and HSD17B4. FASEB J. 2019, 33, 4355–4364. [Google Scholar] [CrossRef] [PubMed]
- Ranea-Robles, P.; Chen, H.; Stauffer, B.; Yu, C.; Bhattacharya, D.; Friedman, S.L.; Puchowicz, M.; Houten, S.M. The peroxisomal transporter ABCD3 plays a major role in hepatic dicarboxylic fatty acid metabolism and lipid homeostasis. J. Inherit. Metab. Dis. 2021, 44, 1419–1433. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Li, X.; Issop, L.; Culty, M.; Papadopoulos, V. ACBD2/ECI2-Mediated Peroxisome-Mitochondria Interactions in Leydig Cell Steroid Biosynthesis. Mol. Endocrinol. 2016, 30, 763–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozen, R.; Vockley, J.; Zhou, L.; Milos, R.; Willard, J.; Fu, K.; Vicanek, C.; Low-Nang, L.; Torban, E.; Fournier, B. Isolation and expression of a cDNA encoding the precursor for a novel member (ACADSB) of the acyl-CoA dehydrogenase gene family. Genomics 1994, 24, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Tafti, M.; Petit, B.; Chollet, D.; Neidhart, E.; de Bilbao, F.; Kiss, J.Z.; Wood, P.A.; Franken, P. Deficiency in short-chain fatty acid beta-oxidation affects theta oscillations during sleep. Nat. Genet. 2003, 34, 320–325. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Wicks, S.E.; Vandanmagsar, B.; Mendoza, T.M.; Bayless, D.S.; Salbaum, J.M.; Dearth, S.P.; Campagna, S.R.; Mynatt, R.L.; Noland, R.C. Extensive metabolic remodeling after limiting mitochondrial lipid burden is consistent with an improved metabolic health profile. J. Biol. Chem. 2019, 294, 12313–12327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumgartner, M.R.; Almashanu, S.; Suormala, T.; Obie, C.; Cole, R.N.; Packman, S.; Baumgartner, E.R.; Valle, D. The molecular basis of human 3-methylcrotonyl-CoA carboxylase deficiency. J. Clin. Investig. 2001, 107, 495–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, D.E.; Perez, L.; Ryan, R.O. 3-Methylglutaric acid in energy metabolism. Clin. Chim Acta 2020, 502, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Zandberg, L.; van Dyk, H.C.; van der Westhuizen, F.H.; van Dijk, A.A. A 3-methylcrotonyl-CoA carboxylase deficient human skin fibroblast transcriptome reveals underlying mitochondrial dysfunction and oxidative stress. Int. J. Biochem. Cell Biol. 2016, 78, 116–129. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, G.F.; Islam, K.; Pease, R.J. Mobilisation of triacylglycerol stores. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2000, 1483, 37–57. [Google Scholar] [CrossRef]
- Lian, J.; Wei, E.; Wang, S.P.; Quiroga, A.D.; Li, L.; Di Pardo, A.; van der Veen, J.; Sipione, S.; Mitchell, G.A.; Lehner, R. Liver specific inactivation of carboxylesterase 3/triacylglycerol hydrolase decreases blood lipids without causing severe steatosis in mice. Hepatology 2012, 56, 2154–2162. [Google Scholar] [CrossRef] [PubMed]
- Chalhoub, G.; Kolleritsch, S.; Maresch, L.K.; Taschler, U.; Pajed, L.; Tilp, A.; Eisner, H.; Rosina, P.; Kien, B.; Radner, F.P.W.; et al. Carboxylesterase 2 proteins are efficient diglyceride and monoglyceride lipases possibly implicated in metabolic disease. J. Lipid Res. 2021, 62, 100075. [Google Scholar] [CrossRef] [PubMed]
- Redzic, J.S.; Duff, M.R.; Blue, A.; Pitts, T.M.; Agarwal, P.; Eisenmesser, E.Z. Modulating Enzyme Function via Dynamic Allostery within Biliverdin Reductase B. Front. Mol. Biosci. 2021, 8, 691208. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.H.; O’Hara, L.; Atanassova, N.; Smith, S.E.; Curley, M.K.; Rebourcet, D.; Darbey, A.L.; Gannon, A.L.; Sharpe, R.M.; Smith, L.B. Low-dose tamoxifen treatment in juvenile males has long-term adverse effects on the reproductive system: Implications for inducible transgenics. Sci. Rep. 2017, 7, 8991. [Google Scholar] [CrossRef] [PubMed]
- Donocoff, R.S.; Teteloshvili, N.; Chung, H.; Shoulson, R.; Creusot, R.J. Optimization of tamoxifen-induced Cre activity and its effect on immune cell populations. Sci. Rep. 2020, 10, 15244. [Google Scholar] [CrossRef] [PubMed]
- Eaton, S.; Middleton, B.; Bartlett, K. Control of mitochondrial β-oxidation: Sensitivity of the trifunctional protein to [NAD+]/[NADH] and [acetyl-CoA]/[CoA]. Biochim. Biophys. Acta (BBA)-Protein Struct. Mol. Enzymol. 1998, 1429, 230–238. [Google Scholar] [CrossRef]
- De Medina, P.; Paillasse, M.R.; Segala, G.; Poirot, M.; Silvente-Poirot, S. Identification and pharmacological characterization of cholesterol-5,6epoxide hydrolase as a target for tamoxifen and AEBS ligands. Proc. Natl. Acad. Sci. USA 2010, 107, 13520–13525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mésange, F.; Sebbar, M.; Capdevielle, J.; Guillemot, J.C.; Ferrara, P.; Bayard, F.; Poirot, M.; Faye, J.C. Identification of two tamoxifen target proteins by photolabeling with 4-(2-morpholinoethoxy)benzophenone. Bioconjug. Chem. 2002, 13, 766–772. [Google Scholar] [CrossRef] [PubMed]
- Lian, J.; Nelson, R.; Lehner, R. Carboxylesterases in lipid metabolism: From mouse to human. Protein Cell 2018, 9, 178–195. [Google Scholar] [CrossRef] [PubMed]
- Touw, C.M.; Derks, T.G.; Bakker, B.M.; Groen, A.K.; Smit, G.P.; Reijngoud, D.J. From genome to phenome-Simple inborn errors of metabolism as complex traits. Biochim. Biophys. Acta 2014, 1842, 2021–2029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Yuan, J.; Yue, W.; Bi, Y.; Shen, X.; Gao, J.; Xu, X.; Lu, Z. GCN2 deficiency protects against high fat diet induced hepatic steatosis and insulin resistance in mice. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3257–3267. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Yu, Z.; Gao, J.; Luo, K.; Shen, X.; Cui, B.; Lu, Z. Inhibition of GCN2 alleviates hepatic steatosis and oxidative stress in obese mice: Involvement of NRF2 regulation. Redox Biol. 2022, 49, 102224. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xiong, Z.; Yan, W.; Gao, E.; Cheng, H.; Wu, G.; Liu, Y.; Zhang, L.; Li, C.; Wang, S.; et al. Branched chain amino acids exacerbate myocardial ischemia/reperfusion vulnerability via enhancing GCN2/ATF6/PPAR-alpha pathway-dependent fatty acid oxidation. Theranostics 2020, 10, 5623–5640. [Google Scholar] [CrossRef]
- Holecek, M. Why Are Branched-Chain Amino Acids Increased in Starvation and Diabetes? Nutrients 2020, 12, 3087. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Ou, Y.; Luo, R.; Wang, J.; Wang, D.; Guan, J.; Li, Y.; Xia, P.; Chen, P.R.; Liu, Y. SAR1B senses leucine levels to regulate mTORC1 signalling. Nature 2021, 596, 281–284. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.Y.; Zhang, X.N.; Xu, C.Z.; Zhou, D.H.; Chen, J.; Liu, Z.X.; Sun, Y.; Huang, W.; Qu, L.S. MCCC2 promotes HCC development by supporting leucine oncogenic function. Cancer Cell Int. 2021, 21, 22. [Google Scholar] [CrossRef] [PubMed]
- Park, B.S.; Im, H.L.; Yoon, N.A.; Tu, T.H.; Park, J.W.; Kim, J.G.; Lee, B.J. Developmentally regulated GTP-binding protein-2 regulates adipocyte differentiation. Biochem. Biophys. Res. Commun. 2021, 578, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Nie, T.; Li, K.; Wu, W.; Long, Q.; Feng, T.; Mao, L.; Gao, Y.; Liu, Q.; Gao, X.; et al. Hepatic CPT1A Facilitates Liver–Adipose Cross Talk via Induction of FGF21 in Mice. Diabetes 2022, 71, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Villarroya, F.; Peyrou, M.; Giralt, M. Transcriptional regulation of the uncoupling protein-1 gene. Biochimie 2017, 134, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Gromada, J.; Franklin, I.; Wollheim, C.B. Alpha-cells of the endocrine pancreas: 35 years of research but the enigma remains. Endocr. Rev. 2007, 28, 84–116. [Google Scholar] [CrossRef] [PubMed]
- Teunissen, S.F.; Jager, N.G.; Rosing, H.; Schinkel, A.H.; Schellens, J.H.; Beijnen, J.H. Development and validation of a quantitative assay for the determination of tamoxifen and its five main phase I metabolites in human serum using liquid chromatography coupled with tandem mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2011, 879, 1677–1685. [Google Scholar] [CrossRef] [PubMed]
- Tanji, K.; Miki, Y.; Maruyama, A.; Mimura, J.; Matsumiya, T.; Mori, F.; Imaizumi, T.; Itoh, K.; Wakabayashi, K. Trehalose intake induces chaperone molecules along with autophagy in a mouse model of Lewy body disease. Biochem. Biophys. Res. Commun. 2015, 465, 746–752. [Google Scholar] [CrossRef] [PubMed]
- Narita, H.; Tanji, K.; Miki, Y.; Mori, F.; Wakabayashi, K. Trehalose intake and exercise upregulate a glucose transporter, GLUT8, in the brain. Biochem. Biophys. Res. Commun. 2019, 514, 672–677. [Google Scholar] [CrossRef] [PubMed]
- Churukian, C.J. Lillie’s Oil Red O Method for Neutral Lipids. J. Histotechnol. 2013, 22, 309–311. [Google Scholar] [CrossRef]
- Mehlem, A.; Hagberg, C.E.; Muhl, L.; Eriksson, U.; Falkevall, A. Imaging of neutral lipids by oil red O for analyzing the metabolic status in health and disease. Nat. Protoc. 2013, 8, 1149–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seligman, A.M.; Karnovsky, M.J.; Wasserkrug, H.L.; Hanker, J.S. Nondroplet ultrastructural demonstration of cytochrome oxidase activity with a polymerizing osmiophilic reagent, diaminobenzidine (DAB). J. Cell Biol. 1968, 38, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Rak, M.; Benit, P.; Chretien, D.; Bouchereau, J.; Schiff, M.; El-Khoury, R.; Tzagoloff, A.; Rustin, P. Mitochondrial cytochrome c oxidase deficiency. Clin. Sci. 2016, 130, 393–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.H.; Liu, Y.; Chaitankar, V.; Pirooznia, M.; Xu, H. Electron transport chain biogenesis activated by a JNK-insulin-Myc relay primes mitochondrial inheritance in Drosophila. Elife 2019, 8, e49309. [Google Scholar] [CrossRef] [PubMed]
- Shinkai, Y.; Sumi, D.; Fukami, I.; Ishii, T.; Kumagai, Y. Sulforaphane, an activator of Nrf2, suppresses cellular accumulation of arsenic and its cytotoxicity in primary mouse hepatocytes. FEBS Lett. 2006, 580, 1771–1774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Fan, C.; Liang, A.; Fan, X.; Wang, R.; Li, P.; Qi, K. Effects of SCFA on the DNA methylation pattern of adiponectin and resistin in high-fat-diet-induced obese male mice. Br. J. Nutr. 2018, 120, 385–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirai, H.; Sato, E.; Sekimoto, A.; Uchida, T.; Oe, Y.; Ito, S.; Sato, H.; Takahashi, N. The effect of aldosterone on adiposity—The role of glucose absorption in the small intestine. Biochem. Biophys. Res. Commun. 2020, 531, 628–635. [Google Scholar] [CrossRef] [PubMed]
- Okuda, S.; Watanabe, Y.; Moriya, Y.; Kawano, S.; Yamamoto, T.; Matsumoto, M.; Takami, T.; Kobayashi, D.; Araki, N.; Yoshizawa, A.C.; et al. jPOSTrepo: An international standard data repository for proteomes. Nucleic Acids Res. 2017, 45, D1107–D1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.; Kasai, S.; Tatara, Y.; Yamazaki, H.; Mimura, J.; Mizuno, S.; Sugiyama, F.; Takahashi, S.; Sato, T.; Ozaki, T.; et al. Inducible Systemic Gcn1 Deletion in Mice Leads to Transient Body Weight Loss upon Tamoxifen Treatment Associated with Decrease of Fat and Liver Glycogen Storage. Int. J. Mol. Sci. 2022, 23, 3201. https://doi.org/10.3390/ijms23063201
Liu J, Kasai S, Tatara Y, Yamazaki H, Mimura J, Mizuno S, Sugiyama F, Takahashi S, Sato T, Ozaki T, et al. Inducible Systemic Gcn1 Deletion in Mice Leads to Transient Body Weight Loss upon Tamoxifen Treatment Associated with Decrease of Fat and Liver Glycogen Storage. International Journal of Molecular Sciences. 2022; 23(6):3201. https://doi.org/10.3390/ijms23063201
Chicago/Turabian StyleLiu, Jun, Shuya Kasai, Yota Tatara, Hiromi Yamazaki, Junsei Mimura, Seiya Mizuno, Fumihiro Sugiyama, Satoru Takahashi, Tsubasa Sato, Taku Ozaki, and et al. 2022. "Inducible Systemic Gcn1 Deletion in Mice Leads to Transient Body Weight Loss upon Tamoxifen Treatment Associated with Decrease of Fat and Liver Glycogen Storage" International Journal of Molecular Sciences 23, no. 6: 3201. https://doi.org/10.3390/ijms23063201
APA StyleLiu, J., Kasai, S., Tatara, Y., Yamazaki, H., Mimura, J., Mizuno, S., Sugiyama, F., Takahashi, S., Sato, T., Ozaki, T., Tanji, K., Wakabayashi, K., Maeda, H., Mizukami, H., Shinkai, Y., Kumagai, Y., Tomita, H., & Itoh, K. (2022). Inducible Systemic Gcn1 Deletion in Mice Leads to Transient Body Weight Loss upon Tamoxifen Treatment Associated with Decrease of Fat and Liver Glycogen Storage. International Journal of Molecular Sciences, 23(6), 3201. https://doi.org/10.3390/ijms23063201