
Effects of B-Cell Lymphoma on the Immune System and Immune Recovery after Treatment: The Paradigm of Targeted Therapy

 and
and
Abstract
:1. Introduction
2. Effects of B-Cell Lymphoma/Lymphoproliferative Diseases on Immune Functions
2.1. Chronic Lymphocytic Leukemia
2.2. Indolent Lymphoma
2.3. Aggressive B-Cell Lymphoma
2.3.1. Diffuse Large B-Cell Lymphoma
2.3.2. Mantle Cell Lymphoma
3. Effects of Targeted Therapy on Immune Functions
3.1. Agents Targeting Hematological Cells
3.1.1. Anti-CD20 Direct Agents: Rituximab and Obinutuzumab
3.1.2. Other Antibodies
3.2. Bruton’s Tyrosine Kinase Inhibitors: Ibrutinib, Acalabrutinib, Zanubrutinib
3.3. PI3K Inhibitors
3.4. Bcl-2 Homology 3 (BH3) Mimetics: Antiapoptotic Protein B-Cell Lymphoma 2 (BCL-2) Inhibitors
4. Agents Targeting Immune Effector Cells and Enhance or Direct Antineoplastic Effect
4.1. Immune Checkpoint Inhibitors
4.2. Lenalidomide
5. Adoptive T/NK Cell Therapy
CAR-T Cell Therapy
6. Discussion
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Küppers, R. Mechanisms of B-cell lymphoma pathogenesis. Nat. Rev. Cancer 2005, 5, 251–262. [Google Scholar] [CrossRef]
- Perincheri, S. Tumor Microenvironment of Lymphomas and Plasma Cell Neoplasms: Broad Overview and Impact on Evaluation for Immune Based Therapies. Front. Oncol. 2021, 8, 719140. [Google Scholar] [CrossRef]
- National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®): B Cell Lymphomas; Version 4; NCCN: Plymouth Meeting, PA, USA, 2019. [Google Scholar]
- Wang, L.; Qin, W.; Huo, Y.-J.; Li, X.; Shi, Q.; Rasko, J.E.J.; Janin, A.; Zhao, W.-L. Advances in targeted therapy for malignant lymphoma. Signal Transduct. Target. Ther. 2020, 5, 1–46. [Google Scholar] [CrossRef] [Green Version]
- Chiorazzi, N.; Rai, K.R.; Ferrarini, M. Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2005, 352, 804–815. [Google Scholar] [CrossRef] [Green Version]
- Balducci, L.; Dolan, D. Chronic Lymphocytic Leukemia in the Elderly: Epidemiology and Proposed Patient-Related Approach. Cancer Control 2015, 22, 3–6. [Google Scholar] [CrossRef] [Green Version]
- Forconi, F.; Moss, P. Perturbation of the normal immune system in patients with CLL. Blood 2015, 126, 573–581. [Google Scholar] [CrossRef] [Green Version]
- Dearden, C. Disease-specific complications of chronic lymphocytic leukemia. Hematology 2008, 2008, 450–456. [Google Scholar] [CrossRef] [Green Version]
- Stevens, W.B.C.; Netea, M.G.; Kater, A.P.; Van Der Velden, W.J.F.M. Trained immunity: Consequences for lymphoid malignancies. Haematologica 2016, 101, 1460–1468. [Google Scholar] [CrossRef] [Green Version]
- Füst, G.; Miszlay, Z.; Czink, E.; Varga, L.; Pálóczi, K.; Szegedi, G.; Hollán, S.R. C1 and C4 abnormalities in chronic lymphocytic leukaemia and their significance. Immunol. Lett. 1987, 14, 255–259. [Google Scholar] [CrossRef]
- Middleton, O.; Cosimo, E.; Dobbin, E.; McCaig, A.M.; Clarke, C.L.; Brant, A.M.; Leach, M.T.; Michie, A.M.; Wheadon, H. Complement deficiencies limit CD20 monoclonal antibody treatment efficacy in CLL. Leukemia 2015, 29, 107–114. [Google Scholar] [CrossRef] [Green Version]
- Kontoyiannis, D.P.; Georgiadou, S.P.; Wierda, W.G.; Wright, S.; Albert, N.D.; Ferrajoli, A.; Keating, M.; Lewis, R.E. Impaired bactericidal but not fungicidal activity of polymorphonuclear neutrophils in patients with chronic lymphocytic leukemia. Leuk. Lymphoma 2013, 54, 1730–1733. [Google Scholar] [CrossRef]
- Maffei, R.; Bulgarelli, J.; Fiorcari, S.; Bertoncelli, L.; Martinelli, S.; Guarnotta, C.; Castelli, I.; Deaglio, S.; Debbia, G.; De Biasi, S.; et al. The monocytic population in chronic lymphocytic leukemia shows altered composition and deregulation of genes involved in phagocytosis and inflammation. Haematologica 2013, 98, 1115–1123. [Google Scholar] [CrossRef]
- Audrito, V.; Serra, S.; Brusa, D.; Mazzola, F.; Arruga, F.; Vaisitti, T.; Coscia, M.; Maffei, R.; Rossi, D.; Wang, T.; et al. Extracellular nicotinamide phosphoribosyltransferase (NAMPT) promotes M2 macrophage polarization in chronic lymphocytic leukemia. Blood 2015, 125, 111–123. [Google Scholar] [CrossRef]
- Huergo-Zapico, L.; Acebes-Huerta, A.; Gonzalez-Rodriguez, A.P.; Contesti, J.; Gonzalez-García, E.; Payer, A.R.; Villa-Alvarez, M.; Fernández-Guizán, A.; López-Soto, A.; Gonzalez, S. Expansion of NK cells and reduction of NKG2D expression in chronic Lymphocytic leukemia. correlation with progressive disease. PLoS ONE 2014, 9, e108326. [Google Scholar] [CrossRef]
- Riches, J.C.; Davies, J.K.; McClanahan, F.; Fatah, R.; Iqbal, S.; Agrawal, S.; Ramsay, A.G.; Gribben, J.G. T cells from CLL patients exhibit features of T-cell exhaustion but retain capacity for cytokine production. Blood 2013, 121, 1612–1621. [Google Scholar] [CrossRef]
- Long, M.; Beckwith, K.; Do, P.; Mundy, B.L.; Gordon, A.; Lehman, A.M.; Maddocks, K.J.; Cheney, C.; Jones, J.A.; Flynn, J.M.; et al. Ibrutinib treatment improves T cell number and function in CLL patients. J. Clin. Investig. 2017, 127, 3052–3064. [Google Scholar] [CrossRef]
- Mahnke, Y.D.; Brodie, T.M.; Sallusto, F.; Roederer, M.; Lugli, E. The who’ s who of T-cell differentiation: Human memory T-cell subsets. Eur. J. Immunol. 2013, 43, 2797–2809. [Google Scholar] [CrossRef]
- Andersen, M.H.; Schrama, D.; Thor Straten, P.; Becker, J.C. Cytotoxic T cells. J. Investig. Dermatol. 2006, 126, 32–41. [Google Scholar] [CrossRef] [Green Version]
- Palma, M.; Gentilcore, G.; Heimersson, K.; Mozaffari, F.; Näsman-Glaser, B.; Young, E.; Rosenquist, R.; Hansson, L.; Österborg, A.; Mellstedt, H. T cells in chronic lymphocytic leukemia display dysregulated expression of immune checkpoints and activation markers. Haematologica 2017, 102, 562–572. [Google Scholar] [CrossRef] [Green Version]
- Taghiloo, S.; Allahmoradi, E.; Tehrani, M.; Hossein-Nataj, H.; Shekarriz, R.; Janbabaei, G.; Abediankenari, S.; Asgarian-Omran, H. Frequency and functional characterization of exhausted CD8+ T cells in chronic lymphocytic leukemia. Eur. J. Haematol. 2017, 98, 622–631. [Google Scholar] [CrossRef]
- Aref, S.; Azmy, E.; Hakim, H.; El Khodary, T.; ElMenshawy, N.; Ebrahim, L. Regulatory T cells in chronic lymphocytic leukemia. Comp. Clin. Pathol. 2015, 24, 649–652. [Google Scholar] [CrossRef]
- Jitschin, R.; Braun, M.; Büttner, M.; Dettmer-Wilde, K.; Bricks, J.; Berger, J.; Eckart, M.J.; Krause, S.W.; Oefner, P.J.; Le Blanc, K.; et al. CLL-cells induce IDOhi CD14+HLA-DRlo myeloid-derived suppressor cells that inhibit T-cell responses and promote TRegs. Blood 2014, 124, 750–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solman, I.G.; Blum, L.K.; Hoh, H.Y.; Kipps, T.J.; Burger, J.A.; Barrientos, J.C.; O’Brien, S.; Mulligan, S.P.; Kay, N.E.; Hillmen, P.; et al. Ibrutinib restores immune cell numbers and function in first-line and relapsed/refractory chronic lymphocytic leukemia. Leuk. Res. 2020, 97, 106432. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef]
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef] [Green Version]
- Morrison, V.A. Infectious complications of chronic lymphocytic leukaemia: Pathogenesis, spectrum of infection, preventive approaches. Best Pract. Res. Clin. Haematol. 2010, 23, 145–153. [Google Scholar] [CrossRef]
- Grywalska, E.; Zaborek, M.; Łyczba, J.; Hrynkiewicz, R.; Bębnowska, D.; Becht, R.; Sosnowska-Pasiarska, B.; Smok-Kalwat, J.; Pasiarski, M.; Góźdź, S.; et al. Chronic Lymphocytic Leukemia-Induced Humoral Immunosuppression: A Systematic Review. Cells 2020, 9, 2398. [Google Scholar] [CrossRef]
- Rozman, C.; Montserrat, E.; Viñolas, N. Serum immunoglobulins in B-chronic lymphocytic leukemia. Natural history and prognostic significance. Cancer 1988, 61, 279–283. [Google Scholar] [CrossRef]
- Sinisalo, M.; Aittoniemi, J.; Käyhty, H.; Vilpo, J. Vaccination against infections in chronic lymphocytic leukemia. Leuk. Lymphoma 2003, 44, 649–652. [Google Scholar] [CrossRef]
- Pasiarski, M.; Rolinski, J.; Grywalska, E.; Stelmach-Goldys, A.; Korona-Glowniak, I.; Gozdz, S.; Hus, I.; Malm, A. Antibody and plasmablast response to 13-valent pneumococcal conjugate vaccine in chronic lymphocytic leukemia patients—Preliminary report. PLoS ONE 2014, 9, e114966. [Google Scholar] [CrossRef]
- DiLillo, D.J.; Weinberg, J.B.; Yoshizaki, A.; Horikawa, M.; Bryant, J.M.; Iwata, Y.; Matsushita, T.; Matta, K.M.; Chen, Y.; Venturi, G.M.; et al. Chronic lymphocytic leukemia and regulatory B cells share IL-10 competence and immunosuppressive function. Leukemia 2013, 27, 170–182. [Google Scholar] [CrossRef] [Green Version]
- Garaud, S.; Morva, A.; Lemoine, S.; Hillion, S.; Bordron, A.; Pers, J.-O.; Berthou, C.; Mageed, R.A.; Renaudineau, Y.; Youinou, P. CD5 promotes IL-10 production in chronic lymphocytic leukemia B cells through STAT3 and NFAT2 activation. J. Immunol. 2011, 186, 4835–4844. [Google Scholar] [CrossRef] [Green Version]
- Scarfò, L.; Chatzikonstantinou, T.; Rigolin, G.M.; Quaresmini, G.; Motta, M.; Vitale, C.; Garcia-Marco, J.A.; Hernández-Rivas, J.; Mirás, F.; Baile, M.; et al. COVID-19 severity and mortality in patients with chronic lymphocytic leukemia: A joint study by ERIC, the European Research Initiative on CLL, and CLL Campus. Leukemia 2020, 34, 2354–2363. [Google Scholar] [CrossRef]
- Hanssen, J.L.J.; Stienstra, J.; Boers, S.A.; Pothast, C.R.; Zaaijer, H.L.; Tjon, J.M.; Heemskerk, M.H.M.; Feltkamp, M.C.W.; Arend, S.M. Convalescent Plasma in a Patient with Protracted COVID-19 and Secondary Hypogammaglobulinemia Due to Chronic Lymphocytic Leukemia: Buying Time to Develop Immunity? Infect. Dis. Rep. 2021, 13, 855–864. [Google Scholar] [CrossRef]
- Mato, A.R.; Roeker, L.; Lamanna, N.; Allan, J.N.; Leslie, L.; Pagel, J.M.; Patel, K.; Osterborg, A.; Wojenski, D.; Kamdar, M.; et al. Outcomes of COVID-19 in patients with CLL: A multicenter international experience. Blood 2020, 136, 1134–1143. [Google Scholar] [CrossRef]
- Ysebaert, L.; Gross, E.; Kuhlein, E.; Blanc, A.; Corre, J.; Fournie, J.-J.; Laurent, G.; Quillet-Mary, A. Immune recovery after fludarabine–cyclophosphamide–rituximab treatment in B-chronic lymphocytic leukemia: Implication for maintenance immunotherapy. Leukemia 2010, 24, 1310–1316. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Calle, N.; Hartley, S.; Ahearne, M.; Kasenda, B.; Beech, A.; Knight, H.; Balotis, C.; Kennedy, B.; Wagner, S.; Dyer, M.; et al. Kinetics of T-cell subset reconstitution following treatment with bendamustine and rituximab for low-grade lymphoproliferative disease: A population-based analysis. Br. J. Haematol. 2019, 184, 957–968. [Google Scholar] [CrossRef]
- Yang, Z.-Z.; Novak, A.J.; Ziesmer, S.C.; Witzig, T.E.; Ansell, S.M. Attenuation of CD8+ T-cell function by CD4+CD25+ regulatory T cells in B-cell non-Hodgkin’s lymphoma. Cancer Res. 2006, 66, 10145–10152. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.-Z.; Grote, D.M.; Ziesmer, S.C.; Xiu, B.; Yates, N.R.; Secreto, F.J.; Hodge, L.S.; Witzig, T.E.; Novak, A.J.; Ansell, S.M. Soluble and membrane-bound TGF-β-mediated regulation of intratumoral T cell differentiation and function in B-cell non-Hodgkin lymphoma. PLoS ONE 2013, 8, e59456. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.-Z.; Grote, D.M.; Ziesmer, S.C.; Niki, T.; Hirashima, M.; Novak, A.J.; Witzig, T.E.; Ansell, S.M. IL-12 upregulates TIM-3 expression and induces T cell exhaustion in patients with follicular B cell non-Hodgkin lymphoma. J. Clin. Investig. 2012, 122, 1271–1282. [Google Scholar] [CrossRef]
- Yang, Z.-Z.; Kim, H.J.; Villasboas, J.C.; Chen, Y.-P.; Price-Troska, T.; Jalali, S.; Wilson, M.; Novak, A.J.; Ansell, S.M. Expression of LAG-3 defines exhaustion of intratumoral PD-1+ T cells and correlates with poor outcome in follicular lymphoma. Oncotarget 2017, 8, 61425–61439. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.-Z.; Grote, D.M.; Xiu, B.; Ziesmer, S.C.; Price-Troska, T.L.; Hodge, L.S.; Yates, D.M.; Novak, A.J.; Ansell, S.M. TGF-β upregulates CD70 expression and induces exhaustion of effector memory T cells in B-cell non-Hodgkin’s lymphoma. Leukemia 2014, 28, 1872–1884. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Tang, X.; Kim, H.J.; Jalali, S.; Pritchett, J.C.; Villasboas, J.C.; Novak, A.J.; Yang, Z.-Z.; Ansell, S.M. Expression of KLRG1 and CD127 defines distinct CD8+ subsets that differentially impact patient outcome in follicular lymphoma. J. Immunother. Cancer 2021, 9, e002662. [Google Scholar] [CrossRef]
- Tobin, J.W.; Keane, C.; Gunawardana, J.; Mollee, P.; Birch, S.; Hoang, T.; Lee, J.; Li, L.; Huang, L.; Murigneux, V.; et al. Progression of Disease within 24 Months in Follicular Lymphoma Is Associated with Reduced Intratumoral Immune Infiltration. J. Clin. Oncol. 2019, 37, 3300–3309. [Google Scholar] [CrossRef]
- Milcent, B.; Josseaume, N.; Petitprez, F.; Riller, Q.; Amorim, S.; Loiseau, P.; Toubert, A.; Brice, P.; Thieblemont, C.; Teillaud, J.-L.; et al. Recovery of central memory and naive peripheral T cells in Follicular Lymphoma patients receiving rituximab-chemotherapy based regimen. Sci. Rep. 2019, 9, 13471. [Google Scholar] [CrossRef] [Green Version]
- Christopoulos, P.; Pfeifer, D.; Bartholomé, K.; Follo, M.; Timmer, J.; Fisch, P.; Veelken, H. Definition and characterization of the systemic T-cell dysregulation in untreated indolent B-cell lymphoma and very early CLL. Blood 2011, 117, 3836–3846. [Google Scholar] [CrossRef] [Green Version]
- Muñoz, R.G.; Izquierdo-Gil, A.; Muñoz, A.; Roldan-Galiacho, V.; Rabasa, P.; Panizo, C. Lymphocyte recovery is impaired in patients with chronic lymphocytic leukemia and indolent non-Hodgkin lymphomas treated with bendamustine plus rituximab. Ann. Hematol. 2014, 93, 1879–1887. [Google Scholar] [CrossRef]
- Liang, X.-J.; Fu, R.-Y.; Wang, H.-N.; Yang, J.; Yao, N.; Liu, X.-D.; Wang, L. An Immune-Related Prognostic Classifier Is Associated with Diffuse Large B Cell Lymphoma Microenvironment. J. Immunol. Res. 2021, 2021, 1–26. [Google Scholar] [CrossRef]
- Roufaiel, M.E.K.N.; Wells, J.W.; Steptoe, R.J. Impaired T-Cell Function in B-Cell Lymphoma: A Direct Consequence of Events at the Immunological Synapse? Front. Immunol. 2015, 6, 258. [Google Scholar] [CrossRef] [Green Version]
- Moccia, A.A.; Thieblemont, C. Curing diffuse large B-cell lymphomas in elderly patients. Eur. J. Intern. Med. 2018, 58, 14–21. [Google Scholar] [CrossRef]
- Mancuso, S.; Carlisi, M.; Santoro, M.; Napolitano, M.; Raso, S.; Siragusa, S. Immunosenescence and lymphomagenesis. Immun. Ageing 2018, 15, 22. [Google Scholar] [CrossRef]
- Aiello, A.; Farzaneh, F.; Candore, G.; Caruso, C.; Davinelli, S.; Gambino, C.M.; Ligotti, M.E.; Zareian, N.; Accardi, G. Immunosenescence and Its Hallmarks: How to Oppose Aging Strategically? A Review of Potential Options for Therapeutic Intervention. Front. Immunol. 2019, 10, 2247. [Google Scholar] [CrossRef] [Green Version]
- Eyre, T.A.; Wilson, W.; Kirkwood, A.A.; Wolf, J.; Hildyard, C.; Plaschkes, H.; Griffith, J.; Fields, P.; Gunawan, A.; Oliver, R.; et al. Infection-related morbidity and mortality among older patients with DLBCL treated with full- or attenuated-dose R-CHOP. Blood Adv. 2021, 5, 2229–2236. [Google Scholar] [CrossRef]
- Howlader, N.; Mariotto, A.B.; Besson, C.; Suneja, G.; Robien, K.; Younes, N.; Engels, E.A. Cancer-specific mortality, cure fraction, and noncancer causes of death among diffuse large B-cell lymphoma patients in the immunochemotherapy era. Cancer 2017, 123, 3326–3334. [Google Scholar] [CrossRef] [Green Version]
- Shree, T.; Li, Q.; Glaser, S.L.; Brunson, A.; Maecker, H.T.; Haile, R.W.; Levy, R.; Keegan, T.H.M. Impaired Immune Health in Survivors of Diffuse Large B-Cell Lymphoma. J. Clin. Oncol. 2020, 38, 1664–1675. [Google Scholar] [CrossRef]
- Harrington, B.; Wheeler, E.; Hornbuckle, K.; Shana’Ah, A.Y.; Youssef, Y.; Smith, L.; Ii, Q.H.; Klamer, B.; Zhang, X.; Long, M.; et al. Modulation of immune checkpoint molecule expression in mantle cell lymphoma. Leuk. Lymphoma 2019, 60, 2498–2507. [Google Scholar] [CrossRef]
- Wang, L.; Qian, J.; Lu, Y.; Li, H.; Bao, H.; He, D.; Liu, Z.; Zheng, Y.; He, J.; Li, Y.; et al. Immune evasion of mantle cell lymphoma: Expression of B7-H1 leads to inhibited T-cell response to and killing of tumor cells. Haematologica 2013, 98, 1458–1466. [Google Scholar] [CrossRef] [Green Version]
- Maschmeyer, G.; De Greef, J.; Mellinghoff, S.C.; Nosari, A.; Thiebaut-Bertrand, A.; Bergeron, A.; Franquet, T.; Blijlevens, N.M.A.; Maertens, J.A. European Conference on Infections in Leukemia (ECIL). Infections associated with immunotherapeutic and molecular targeted agents in hematology and oncology. A position paper by the European Conference on Infections in Leukemia (ECIL). Leukemia 2019, 33, 844–862. [Google Scholar] [CrossRef] [Green Version]
- Sacco, K.A.; Abraham, R.S. Consequences of B-cell-depleting therapy: Hypogammaglobulinemia and impaired B-cell reconstitution. Immunotherapy 2018, 10, 713–728. [Google Scholar] [CrossRef]
- Chaiwatanatorn, K.; Lee, N.; Grigg, A.; Filshie, R.; Firkin, F. Delayed-onset neutropenia associated with rituximab therapy. Br. J. Haematol. 2003, 121, 913–918. [Google Scholar] [CrossRef]
- Evens, A.M.; Jovanovic, B.D.; Su, Y.-C.; Raisch, D.W.; Ganger, D.; Belknap, S.M.; Dai, M.-S.; Chiu, B.-C.C.; Fintel, B.; Cheng, Y.; et al. Rituximab-associated hepatitis B virus (HBV) reactivation in lymphoproliferative diseases: Meta-analysis and examination of FDA safety reports. Ann. Oncol. 2011, 22, 1170–1180. [Google Scholar] [CrossRef] [PubMed]
- Coiffier, B.; Lepage, E.; Brière, J.; Herbrecht, R.; Tilly, H.; Bouabdallah, R.; Morel, P.; Van Den Neste, E.; Salles, G.; Gaulard, P.; et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N. Engl. J. Med. 2002, 346, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Kamel, S.; O’Connor, S.; Lee, N.; Filshie, R.; Nandurkar, H.; Tam, C.S. High incidence of Pneumocystis jiroveciipneumonia in patients receiving biweekly rituximab and cyclophosphamide, adriamycin, vincristine, and prednisone. Leuk. Lymphoma 2010, 51, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Katsuya, H.; Suzumiya, J.; Sasaki, H.; Ishitsuka, K.; Shibata, T.; Takamatsu, Y.; Tamura, K. Addition of rituximab to cyclophosphamide, doxorubicin, vincristine, and prednisolone therapy has a high risk of developing interstitial pneumonia in patients with non-Hodgkin lymphoma. Leuk. Lymphoma 2009, 50, 1818–1823. [Google Scholar] [CrossRef] [PubMed]
- Shelton, E.; Yong, M.; Cohney, S. Late onset Pneumocystis pneumonia in patients receiving rituximab for humoral renal transplant rejection. Nephrology 2009, 14, 696–699. [Google Scholar] [CrossRef]
- Jiang, X.; Mei, X.; Feng, D.; Wang, X. Prophylaxis and Treatment of Pneumocystis jiroveci Pneumonia in Lymphoma Patients Subjected to Rituximab-Contained Therapy: A Systemic Review and Meta-Analysis. PLoS ONE 2015, 10, e0122171. [Google Scholar] [CrossRef]
- Mansharamani, N.G.; Balachandran, D.; Vernovsky, I.; Garland, R.; Koziel, H. Peripheral Blood CD4 + T-Lymphocyte Counts During Pneumocystis carinii Pneumonia in Immunocompromised Patients without HIV Infection. Chest 2000, 118, 712–720. [Google Scholar] [CrossRef]
- Moor, M.B.; Suter-Riniker, F.; Horn, M.P.; Aeberli, D.; Amsler, J.; Möller, B.; Njue, L.M.; Medri, C.; Angelillo-Scherrer, A.; Borradori, L.; et al. Humoral and cellular responses to mRNA vaccines against SARS-CoV-2 in patients with a history of CD20 B-cell-depleting therapy (RituxiVac): An investigator-initiated, single-centre, open-label study. Lancet Rheumatol. 2021, 3, e789–e797. [Google Scholar] [CrossRef]
- Ghielmini, M.; Rufibach, K.; Salles, G.; Leoncini-Franscini, L.; Léger-Falandry, C.; Cogliatti, S.; Fey, M.; Martinelli, G.; Stahel, R.; Lohri, A.; et al. Single agent rituximab in patients with follicular or mantle cell lymphoma: Clinical and biological factors that are predictive of response and event-free survival as well as the effect of rituximab on the immune system: A study of the Swiss Group for Clinical Cancer Research (SAKK). Ann. Oncol. 2005, 16, 1675–1682. [Google Scholar] [CrossRef]
- Ghielmini, M.; Schmitz, S.-F.H.; Cogliatti, S.B.; Pichert, G.; Hummerjohann, J.; Waltzer, U.; Fey, M.F.; Betticher, D.C.; Martinelli, G.; Peccatori, F.; et al. Prolonged treatment with rituximab in patients with follicular lymphoma significantly increases event-free survival and response duration compared with the standard weekly × 4 schedule. Blood 2004, 103, 4416–4423. [Google Scholar] [CrossRef] [Green Version]
- Kurokawa, T.; Hase, M.; Tokuman, N.; Yoshida, T. Immune reconstitution of B-cell lymphoma patients receiving CHOP-based chemotherapy containing rituximab. Hematol. Oncol. 2011, 29, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Okamoto, M.; Inaguma, Y.; Okamoto, A.; Ando, M.; Ando, Y.; Tsuge, M.; Tomono, A.; Kakumae, Y.; Hayashi, T.; et al. Influence of R-CHOP Therapy on Immune System Restoration in Patients with B-Cell Lymphoma. Oncology 2016, 91, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Anolik, J.H.; Friedberg, J.W.; Zheng, B.; Barnard, J.; Owen, T.; Cushing, E.; Kelly, J.; Milner, E.C.; Fisher, R.I.; Sanz, I. B cell reconstitution after rituximab treatment of lymphoma recapitulates B cell ontogeny. Clin. Immunol. 2007, 122, 139–145. [Google Scholar] [CrossRef] [PubMed]
- García-Muñoz, R.; Aguinaga, L.; Feliu, J.; Anton-Remirez, J.; Jorge-Del-Val, L.; Casajús-Navasal, A.; Nebot-Villacampa, M.J.; Daroca-Fernandez, I.; Domínguez-Garrido, E.; Rabasa, P.; et al. Obinutuzumab induces depletion of NK cells in patients with chronic lymphocytic leukemia. Immunotherapy 2018, 10, 491–499. [Google Scholar] [CrossRef]
- Salles, G.; Duell, J.; Barca, E.G.; Tournilhac, O.; Jurczak, W.; Liberati, A.M.; Nagy, Z.; Obr, A.; Gaidano, G.; André, M.; et al. Tafasitamab plus lenalidomide in relapsed or refractory diffuse large B-cell lymphoma (L-MIND): A multicentre, prospective, single-arm, phase 2 study. Lancet Oncol. 2020, 21, 978–988. [Google Scholar] [CrossRef]
- Viardot, A.; Goebeler, M.-E.; Hess, G.; Neumann, S.; Pfreundschuh, M.; Adrian, N.; Zettl, F.; Libicher, M.; Sayehli, C.; Stieglmaier, J.; et al. Phase 2 study of the bispecific T-cell engager (BiTE) antibody blinatumomab in relapsed/refractory diffuse large B-cell lymphoma. Blood 2016, 127, 1410–1416. [Google Scholar] [CrossRef]
- Goebeler, M.-E.; Knop, S.; Viardot, A.; Kufer, P.; Topp, M.S.; Einsele, H.; Noppeney, R.; Hess, G.; Kallert, S.; Mackensen, A.; et al. Bispecific T-Cell Engager (BiTE) Antibody Construct Blinatumomab for the Treatment of Patients with Relapsed/Refractory Non-Hodgkin Lymphoma: Final Results from a Phase I Study. J. Clin. Oncol. 2016, 34, 1104–1111. [Google Scholar] [CrossRef]
- Nägele, V.; Kratzer, A.; Zugmaier, G.; Holland, C.; Hijazi, Y.; Topp, M.S.; Gökbuget, N.; Baeuerle, P.A.; Kufer, P.; Wolf, A.; et al. Changes in clinical laboratory parameters and pharmacodynamic markers in response to blinatumomab treatment of patients with relapsed/refractory ALL. Exp. Hematol. Oncol. 2017, 6, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Zugmaier, G.; Topp, M.S.; Alekar, S.V.; Viardot, A.; Horst, H.-A.; Neumann, S.; Stelljes, M.; Bargou, R.C.; Goebeler, M.; Wessiepe, D.; et al. Long-term follow-up of serum immunoglobulin levels in blinatumomab-treated patients with minimal residual disease-positive B-precursor acute lymphoblastic leukemia. Blood Cancer J. 2014, 4, e244. [Google Scholar] [CrossRef]
- Caimi, P.; Kahal, B.S.; Hamadani, M.; Carlo-Stella, C.; He, S.; Ungar, D.; Feingold, J.; Ardeshna, K.M.; Radford, J.; Solh, M.; et al. Safety and Efficacy of Adct-402 (Loncastuximab Tesirine), a Novel Antibody Drug Conjugate, in Relapsed/Refractory Follicular Lymphoma and Mantle Cell Lymphoma: Interim Results from the Phase 1 First-in-Human Study. Blood 2018, 132, 2874. [Google Scholar] [CrossRef]
- Zinzani, P.L.; Minotti, G. Anti-CD19 monoclonal antibodies for the treatment of relapsed or refractory B-cell malignancies: A narrative review with focus on diffuse large B-cell lymphoma. J. Cancer Res. Clin. Oncol. 2021, 148, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Advani, R.H.; Buggy, J.J.; Sharman, J.P.; Smith, S.M.; Boyd, T.E.; Grant, B.; Kolibaba, K.S.; Furman, R.R.; Rodriguez, S.; Chang, B.Y.; et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J. Clin. Oncol. 2013, 31, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.P.; Dammeijer, F.; Hendriks, R.W. Role of Bruton’s tyrosine kinase in B cells and malignancies. Mol. Cancer 2018, 17, 57. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.M.; Baran, A.M.; Meacham, P.J.; Feldman, M.M.; Valencia, H.E.; Newsom-Stewart, C.; Gupta, N.; Janelsins, M.C.; Barr, P.M.; Zent, C.S. Analysis of the risk of infection in patients with chronic lymphocytic leukemia in the era of novel therapies. Leuk. Lymphoma 2018, 59, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Varughese, T.; Taur, Y.; Cohen, N.; Palomba, M.L.; Seo, S.K.; Hohl, T.M.; Redelman-Sidi, G. Serious infections in patients receiving ibrutinib for treatment of lymphoid cancer. Clin. Infect. Dis. 2018, 67, 687–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ball, S.; Das, A.; Vutthikraivit, W.; Edwards, P.J.; Hardwicke, F.; Short, N.J.; Borthakur, G.; Maiti, A. Risk of Infection Associated with Ibrutinib in Patients with B-Cell Malignancies: A Systematic Review and Meta-analysis of Randomized Controlled Trials. Clin. Lymphoma Myeloma Leuk. 2020, 20, 87–97.e5. [Google Scholar] [CrossRef]
- Mauro, F.R.; Giannarelli, D.; Visentin, A.; Reda, G.; Sportoletti, P.; Frustaci, A.M.; Chiarenza, A.; Ciolli, S.; Vitale, C.; Laurenti, L.; et al. Prognostic Impact and Risk Factors of Infections in Patients with Chronic Lymphocytic Leukemia Treated with Ibrutinib. Cancers 2021, 13, 3240. [Google Scholar] [CrossRef]
- Reddy, Y.; Baig, M.; Kalva, N.; Puli, S.; Dhillon, S. Cytomegalovirus Proctitis in a Patient with Chronic Lymphocytic Leukemia on Ibrutinib Therapy: A Case Report. Cureus 2020, 12, e7837. [Google Scholar] [CrossRef] [Green Version]
- De la Asunción, C.S.; Terol, M.J.; Saus, A.; Olea, B.; Giménez, E.; Albert, E.; López-Jiménez, J.; Andreu, R.; García, D.; Fox, L.; et al. Cytomegalovirus-specific T-cell immunity and DNAemia in patients with chronic lymphocytic leukaemia undergoing treatment with ibrutinib. Br. J. Haematol. 2021, 195, 637–641. [Google Scholar] [CrossRef]
- Dubovsky, J.A.; Beckwith, K.A.; Natarajan, G.; Woyach, J.A.; Jaglowski, S.; Zhong, Y.; Hessler, J.D.; Liu, T.-M.; Chang, B.Y.; Larkin, K.M.; et al. Ibrutinib is an irreversible molecular inhibitor of ITK driving a Th1-selective pressure in T lymphocytes. Blood 2013, 122, 2539–2549. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Tian, X.; Lee, Y.S.; Gunti, S.; Lipsky, A.; Herman, S.E.M.; Salem, D.; Stetler-Stevenson, M.; Yuan, C.; Kardava, L.; et al. Partial reconstitution of humoral immunity and fewer infections in patients with chronic lymphocytic leukemia treated with ibrutinib. Blood 2015, 126, 2213–2219. [Google Scholar] [CrossRef] [PubMed]
- Yin, Q.; Sivina, M.; Robins, H.; Yusko, E.; Vignali, M.; O’Brien, S.; Keating, M.J.; Ferrajoli, A.; Estrov, Z.; Jain, N.; et al. Ibrutinib Therapy Increases T Cell Repertoire Diversity in Patients with Chronic Lymphocytic Leukemia. J. Immunol. 2017, 198, 1740–1747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coutré, S.E.; Furman, R.R.; Flinn, I.W.; Burger, J.A.; Blum, K.; Sharman, J.; Jones, J.; Wierda, W.; Zhao, W.; Heerema, N.A.; et al. Extended Treatment with Single-Agent Ibrutinib at the 420 mg Dose Leads to Durable Responses in Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma. Clin. Cancer Res. 2017, 23, 1149–1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burger, J.A.; Barr, P.M.; Robak, T.; Owen, C.; Ghia, P.; Tedeschi, A.; Bairey, O.; Hillmen, P.; Coutre, S.E.; Devereux, S.; et al. Long-term efficacy and safety of first-line ibrutinib treatment for patients with CLL/SLL: 5 years of follow-up from the phase 3 RESONATE-2 study. Leukemia 2020, 34, 787–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munir, T.; Brown, J.R.; O’Brien, S.; Barrientos, J.C.; Barr, P.M.; Reddy, N.M.; Coutre, S.; Tam, C.S.; Mulligan, S.P.; Jaeger, U.; et al. Final analysis from RESONATE: Up to six years of follow-up on ibrutinib in patients with previously treated chronic lymphocytic leukemia or small lymphocytic lymphoma. Am. J. Hematol. 2019, 94, 1353–1363. [Google Scholar] [CrossRef] [Green Version]
- Byrd, J.C.; Furman, R.R.; Coutre, S.E.; Burger, J.A.; Blum, K.A.; Coleman, M.; Wierda, W.G.; Jones, J.A.; Zhao, W.; Heerema, N.A.; et al. Three-year follow-up of treatment-naïve and previously treated patients with CLL and SLL receiving single-agent ibrutinib. Blood 2015, 125, 2497–2506. [Google Scholar] [CrossRef]
- Shanafelt, T.D.; Wang, X.V.; Kay, N.E.; Hanson, C.A.; O’Brien, S.; Barrientos, J.; Jelinek, D.F.; Braggio, E.; Leis, J.F.; Zhang, C.C.; et al. Ibrutinib–Rituximab or Chemoimmunotherapy for Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2019, 381, 432–443. [Google Scholar] [CrossRef]
- Gauthier, J.; Hirayama, A.V.; Purushe, J.; Hay, K.A.; Lymp, J.; Li, D.H.; Yeung, C.C.S.; Sheih, A.; Pender, B.S.; Hawkins, R.M.; et al. Feasibility and efficacy of CD19-targeted CAR T cells with concurrent ibrutinib for CLL after ibrutinib failure. Blood 2020, 135, 1650–1660. [Google Scholar] [CrossRef]
- Sagiv-Barfi, I.; Kohrt, H.E.K.; Czerwinski, D.K.; Ng, P.P.; Chang, B.Y.; Levy, R. Therapeutic antitumor immunity by checkpoint blockade is enhanced by ibrutinib, an inhibitor of both BTK and ITK. Proc. Natl. Acad. Sci. USA 2015, 112, E966–E972. [Google Scholar] [CrossRef] [Green Version]
- Moreno, C.; Muñoz, C.; Terol, M.J.; Hernández-Rivas, J.Á.; Villanueva, M. Restoration of the immune function as a complementary strategy to treat Chronic Lymphocytic Leukemia effectively. J. Exp. Clin. Cancer Res. 2021, 40, 321. [Google Scholar] [CrossRef]
- Palma, M.; Mulder, T.A.; Österborg, A. BTK Inhibitors in Chronic Lymphocytic Leukemia: Biological Activity and Immune Effects. Front. Immunol. 2021, 12, 686768. [Google Scholar] [CrossRef] [PubMed]
- Ng, Y.-S.; Wardemann, H.; Chelnis, J.; Cunningham-Rundles, C.; Meffre, E. Bruton’s tyrosine kinase is essential for human B cell tolerance. J. Exp. Med. 2004, 200, 927–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, J.E.; Handunnetti, S.M.; Ludford-Menting, M.; Sharpe, C.; Blombery, P.; Anderson, M.A.; Roberts, A.W.; Seymour, J.F.; Tam, C.S.; Ritchie, D.S.; et al. Immune recovery in patients with mantle cell lymphoma receiving long-term ibrutinib and venetoclax combination therapy. Blood Adv. 2020, 4, 4849–4859. [Google Scholar] [CrossRef]
- Rogers, K.A.; Ruppert, A.S.; Bingman, A.; Andritsos, L.A.; Awan, F.T.; Blum, K.A.; Flynn, J.M.; Jaglowski, S.M.; Lozanski, G.; Maddocks, K.J.; et al. Incidence and description of autoimmune cytopenias during treatment with ibrutinib for chronic lymphocytic leukemia. Leukemia 2016, 30, 346–350. [Google Scholar] [CrossRef] [PubMed]
- Vitale, C.; Ahn, I.E.; Sivina, M.; Ferrajoli, A.; Wierda, W.G.; Estrov, Z.; Konoplev, S.N.; Jain, N.; Brien, S.O.; Farooqui, M.; et al. Autoimmune cytopenias in patients with chronic lymphocytic leukemia treated with ibrutinib. Haematologica 2016, 101, e254–e258. [Google Scholar] [CrossRef] [Green Version]
- Zhu, S.; Gokhale, S.; Jung, J.; Spirollari, E.; Tsai, J.; Arceo, J.; Wu, B.W.; Victor, E.; Xie, P. Multifaceted Immunomodulatory Effects of the BTK Inhibitors Ibrutinib and Acalabrutinib on Different Immune Cell Subsets—Beyond B Lymphocytes. Front. Cell Dev. Biol. 2021, 9, 727531. [Google Scholar] [CrossRef]
- Byrd, J.C.; Wierda, W.G.; Schuh, A.; Devereux, S.; Chaves, J.M.; Brown, J.R.; Hillmen, P.; Martin, P.; Awan, F.T.; Stephens, D.M.; et al. Acalabrutinib monotherapy in patients with relapsed/refractory chronic lymphocytic leukemia: Updated phase 2 results. Blood 2020, 135, 1204–1213. [Google Scholar] [CrossRef]
- Zou, Y.; Zhu, H.; Li, X.; Xia, Y.; Miao, K.; Zhao, S.; Wu, Y.; Wang, L.; Xu, W.; Li, J. The impacts of zanubrutinib on immune cells in patients with chronic lymphocytic leukemia/small lymphocytic lymphoma. Hematol. Oncol. 2019, 37, 392–400. [Google Scholar] [CrossRef]
- Chellappa, S.; Kushekhar, K.; Munthe, L.A.; Tjønnfjord, G.E.; Aandahl, E.M.; Okkenhaug, K.; Taskén, K. The PI3K p110δ Isoform Inhibitor Idelalisib Preferentially Inhibits Human Regulatory T Cell Function. J. Immunol. 2019, 202, 1397–1405. [Google Scholar] [CrossRef]
- Gopal, A.K.; Kahl, B.S.; De Vos, S.; Wagner-Johnston, N.D.; Schuster, S.J.; Jurczak, W.; Flinn, I.; Flowers, C.R.; Martin, P.; Viardot, A.; et al. PI3Kδ Inhibition by Idelalisib in Patients with Relapsed Indolent Lymphoma. N. Engl. J. Med. 2014, 370, 1008–1018. [Google Scholar] [CrossRef] [Green Version]
- Teh, B.W.; Tam, C.S.; Handunnetti, S.; Worth, L.J.; Slavin, M.A. Infections in patients with chronic lymphocytic leukaemia: Mitigating risk in the era of targeted therapies. Blood Rev. 2018, 32, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Cuneo, A.; Barosi, G.; Danesi, R.; Fagiuoli, S.; Ghia, P.; Marzano, A.; Montillo, M.; Poletti, V.; Viale, P.; Zinzani, P.L. Management of adverse events associated with idelalisib treatment in chronic lymphocytic leukemia and follicular lymphoma: A multidisciplinary position paper. Hematol. Oncol. 2019, 37, 3–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dreyling, M.; Santoro, A.; Mollica, L.; Leppä, S.; Follows, G.A.; Lenz, G.; Kim, W.S.; Nagler, A.; Panayiotidis, P.; Demeter, J.; et al. Phosphatidylinositol 3-Kinase Inhibition by Copanlisib in Relapsed or Refractory Indolent Lymphoma. J. Clin. Oncol. 2017, 35, 3898–3905. [Google Scholar] [CrossRef] [PubMed]
- Flinn, I.W.; Miller, C.B.; Ardeshna, K.M.; Tetreault, S.; Assouline, S.E.; Mayer, J.; Merli, M.; Lunin, S.D.; Pettitt, A.R.; Nagy, Z.; et al. DYNAMO: A Phase II Study of Duvelisib (IPI-145) in Patients with Refractory Indolent Non-Hodgkin Lymphoma. J. Clin. Oncol. 2019, 37, 912–922. [Google Scholar] [CrossRef]
- Davids, M.S.; Roberts, A.; Seymour, J.F.; Pagel, J.M.; Kahl, B.S.; Wierda, W.G.; Puvvada, S.; Kipps, T.J.; Anderson, M.A.; Salem, A.H.; et al. Phase I First-in-Human Study of Venetoclax in Patients with Relapsed or Refractory Non-Hodgkin Lymphoma. J. Clin. Oncol. 2017, 35, 826–833. [Google Scholar] [CrossRef] [Green Version]
- Zinzani, P.L.; Flinn, I.W.; Yuen, S.L.S.; Topp, M.S.; Rusconi, C.; Fleury, I.; Le Dû, K.; Arthur, C.; Pro, B.; Gritti, G.; et al. Venetoclax-rituximab with or without bendamustine vs. bendamustine-rituximab in relapsed/refractory follicular lymphoma. Blood 2020, 136, 2628–2637. [Google Scholar]
- Leverson, J.D.; Phillips, D.C.; Mitten, M.J.; Boghaert, E.R.; Diaz, D.; Tahir, S.K.; Belmont, L.D.; Nimmer, P.; Xiao, Y.; Ma, X.M.; et al. Exploiting selective BCL-2 family inhibitors to dissect cell survival dependencies and define improved strategies for cancer therapy. Sci. Transl. Med. 2015, 7, 279ra40. [Google Scholar] [CrossRef]
- Davids, M.S.; Hallek, M.; Wierda, W.; Roberts, A.W.; Stilgenbauer, S.; Jones, J.A.; Gerecitano, J.F.; Kim, S.Y.; Potluri, J.; Busman, T.; et al. Comprehensive Safety Analysis of Venetoclax Monotherapy for Patients with Relapsed/Refractory Chronic Lymphocytic Leukemia. Clin. Cancer Res. 2018, 24, 4371–4379. [Google Scholar] [CrossRef] [Green Version]
- Coutre, S.; Choi, M.; Furman, R.R.; Eradat, H.; Heffner, L.; Jones, J.A.; Chyla, B.; Zhou, L.; Agarwal, S.; Waskiewicz, T.; et al. Venetoclax for patients with chronic lymphocytic leukemia who progressed during or after idelalisib therapy. Blood 2018, 131, 1704–1711. [Google Scholar] [CrossRef] [Green Version]
- Mathew, R.; Haribhai, D.; Kohlhapp, F.; Duggan, R.; Ellis, P.; Riehm, J.J.; Robinson, V.A.; Shi, Y.Y.; Bhathena, A.; Leverson, J.D.; et al. The BCL-2-Selective Inhibitor Venetoclax Spares Activated T-Cells during Anti-Tumor Immunity. Blood 2018, 132, 3704. [Google Scholar] [CrossRef]
- De Weerdt, I.; Hofland, T.; De Boer, R.; Dobber, J.A.; Dubois, J.; Van Nieuwenhuize, D.; Mobasher, M.; De Boer, F.; Hoogendoorn, M.; Velders, G.A.; et al. Distinct immune composition in lymph node and peripheral blood of CLL patients is reshaped during venetoclax treatment. Blood Adv. 2019, 3, 2642–2652. [Google Scholar] [CrossRef] [PubMed]
- Seymour, J.F.; Ma, S.; Brander, D.M.; Choi, M.Y.; Barrientos, J.; Davids, M.S.; Anderson, M.A.; Beaven, A.W.; Rosen, S.T.; Tam, C.S.; et al. Venetoclax plus rituximab in relapsed or refractory chronic lymphocytic leukaemia: A phase 1b study. Lancet Oncol. 2017, 18, 230–240. [Google Scholar] [CrossRef] [Green Version]
- Alatrash, G.; Daver, N.; Mittendorf, E.A. Targeting Immune Checkpoints in Hematologic Malignancies. Pharmacol. Rev. 2016, 68, 1014–1025. [Google Scholar] [CrossRef] [PubMed]
- Esfahani, K.; Roudaia, L.; Buhlaiga, N.; Del Rincon, S.V.; Papneja, N.; Miller, W.H., Jr. A review of cancer immunotherapy: From the past, to the present, to the future. Curr. Oncol. 2020, 27, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Berger, R.; Rotem-Yehudar, R.; Slama, G.; Landes, S.; Kneller, A.; Leiba, M.; Koren-Michowitz, M.; Shimoni, A.; Nagler, A. Phase I Safety and Pharmacokinetic Study of CT-011, a Humanized Antibody Interacting with PD-1, in Patients with Advanced Hematologic Malignancies. Clin. Cancer Res. 2008, 14, 3044–3051. [Google Scholar] [CrossRef] [Green Version]
- Lesokhin, A.M.; Ansell, S.M.; Armand, P.; Scott, E.C.; Halwani, A.; Gutierrez, M.; Millenson, M.M.; Cohen, A.D.; Schuster, S.J.; Lebovic, D.; et al. Nivolumab in Patients with Relapsed or Refractory Hematologic Malignancy: Preliminary Results of a Phase Ib Study. J. Clin. Oncol. 2016, 34, 2698–2704. [Google Scholar] [CrossRef] [Green Version]
- Armand, P.; Janssens, A.; Gritti, G.; Radford, J.; Timmerman, J.; Pinto, A.; Vilchez, S.M.; Johnson, P.; Cunningham, D.; Leonard, J.P.; et al. Efficacy and safety results from CheckMate 140, a phase 2 study of nivolumab for relapsed/refractory follicular lymphoma. Blood 2021, 137, 637–645. [Google Scholar] [CrossRef]
- Karam, J.-D.; Noel, N.; Voisin, A.-L.; Lanoy, E.; Michot, J.-M.; Lambotte, O. Infectious complications in patients treated with immune checkpoint inhibitors. Eur. J. Cancer 2020, 141, 137–142. [Google Scholar] [CrossRef]
- Deshpande, R.P.; Sharma, S.; Watabe, K. The Confounders of Cancer Immunotherapy: Roles of Lifestyle, Metabolic Disorders and Sociological Factors. Cancers 2020, 12, 2983. [Google Scholar] [CrossRef]
- Lee, B.-N.; Gao, H.; Cohen, E.N.; Badoux, X.; Wierda, W.G.; Estrov, Z.; Faderl, S.H.; Keating, M.J.; Ferrajoli, A.; Reuben, J.M.; et al. Treatment with lenalidomide modulates T-cell immunophenotype and cytokine production in patients with chronic lymphocytic leukemia. Cancer 2011, 117, 3999–4008. [Google Scholar] [CrossRef] [Green Version]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Brudno, J.N.; Kochenderfer, J.N. Toxicities of chimeric antigen receptor T cells: Recognition and management. Blood 2016, 127, 3321–3330. [Google Scholar] [CrossRef] [Green Version]
- Zahid, A.; Siegler, E.L.; Kenderian, S.S. CART Cell Toxicities: New Insight into Mechanisms and Management. Clin. Hematol. Int. 2020, 2, 149–155. [Google Scholar] [CrossRef]
- Stewart, A.G.; Henden, A.S. Infectious complications of CAR T-cell therapy: A clinical update. Ther. Adv. Infect. Dis. 2021, 8, 20499361211036773. [Google Scholar] [CrossRef]
- Bupha-Intr, O.; Haeusler, G.; Chee, L.; Thursky, K.; Slavin, M.; Teh, B. CAR-T cell therapy and infection: A review. Expert Rev. Anti-Infect. Ther. 2020, 19, 749–758. [Google Scholar] [CrossRef]
- Gudiol, C.; Lewis, R.E.; Strati, P.; Kontoyiannis, D.P. Chimeric antigen receptor T-cell therapy for the treatment of lymphoid malignancies: Is there an excess risk for infection? Lancet Haematol. 2021, 8, e216–e228. [Google Scholar] [CrossRef]
- Hill, J.A.; Li, D.A.; Hay, K.; Green, M.L.; Cherian, S.; Chen, X.; Riddell, S.R.; Maloney, D.G.; Boeckh, M.; Turtle, C.J. Infectious complications of CD19-targeted chimeric antigen receptor–modified T-cell immunotherapy. Blood 2018, 131, 121–130. [Google Scholar] [CrossRef]
- Haidar, G.; Garner, W.; Hill, J.A. Infections after anti-CD19 chimeric antigen receptor T-cell therapy for hematologic malignancies: Timeline, prevention, and uncertainties. Curr. Opin. Infect. Dis. 2020, 33, 449–457. [Google Scholar] [CrossRef]
- Fried, S.; Avigdor, A.; Bielorai, B.; Meir, A.; Besser, M.J.; Schachter, J.; Shimoni, A.; Nagler, A.; Toren, A.; Jacoby, E. Early and late hematologic toxicity following CD19 CAR-T cells. Bone Marrow Transplant. 2019, 54, 1643–1650. [Google Scholar] [CrossRef]
- Garner, W.; Samanta, P.; Haidar, G. Invasive Fungal Infections after Anti-CD19 Chimeric Antigen Receptor-Modified T-Cell Therapy: State of the Evidence and Future Directions. J. Fungi 2021, 7, 156. [Google Scholar] [CrossRef] [PubMed]
- Thakkar, A.; Cui, Z.; Peeke, S.Z.; Shah, N.; Pradhan, K.; Lombardo, A.; Khatun, F.; Mustafa, J.; De Castro, A.; Gillick, K.; et al. Patterns of leukocyte recovery predict infectious complications after CD19 CAR-T cell therapy in a real-world setting. Stem Cell Investig. 2021, 8, 18. [Google Scholar] [CrossRef]
- Wudhikarn, K.; Palomba, M.L.; Pennisi, M.; Garcia-Recio, M.; Flynn, J.R.; Devlin, S.M.; Afuye, A.; Silverberg, M.L.; Maloy, M.A.; Shah, G.L.; et al. Infection during the first year in patients treated with CD19 CAR T cells for diffuse large B cell lymphoma. Blood Cancer J. 2020, 10, 79. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.J.; Svoboda, J.; Chong, E.A.; Nasta, S.D.; Mato, A.R.; Anak, Ö.; Brogdon, J.L.; Pruteanu-Malinici, I.; Bhoj, V.; Landsburg, D.; et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N. Engl. J. Med. 2017, 377, 2545–2554. [Google Scholar] [CrossRef] [PubMed]
- Logue, J.M.; Zucchetti, E.; Bachmeier, C.A.; Krivenko, G.S.; Larson, V.; Ninh, D.; Grillo, G.; Cao, B.; Kim, J.; Chavez, J.C.; et al. Immune reconstitution and associated infections following axicabtagene ciloleucel in relapsed or refractory large B-cell lymphoma. Haematologica 2021, 106, 978–986. [Google Scholar] [CrossRef] [Green Version]
- Baird, J.H.; Epstein, D.J.; Tamaresis, J.S.; Ehlinger, Z.; Spiegel, J.Y.; Craig, J.; Claire, G.K.; Frank, M.J.; Muffly, L.; Shiraz, P.; et al. Immune reconstitution and infectious complications following axicabtagene ciloleucel therapy for large B-cell lymphoma. Blood Adv. 2021, 5, 143–155. [Google Scholar] [CrossRef]
- Pizzi, M.; Boi, M.; Bertoni, F.; Inghirami, G. Emerging therapies provide new opportunities to reshape the multifaceted interactions between the immune system and lymphoma cells. Leukemia 2016, 30, 1805–1815. [Google Scholar] [CrossRef]
- Menter, T.; Tzankov, A. Mechanisms of Immune Evasion and Immune Modulation by Lymphoma Cells. Front. Oncol. 2018, 8, 54. [Google Scholar] [CrossRef] [Green Version]
- Friman, V.; Winqvist, O.; Blimark, C.; Langerbeins, P.; Chapel, H.; Dhalla, F. Secondary immunodeficiency in lymphoproliferative malignancies. Hematol. Oncol. 2016, 34, 121–132. [Google Scholar] [CrossRef]
- Griggio, V.; Perutelli, F.; Salvetti, C.; Boccellato, E.; Boccadoro, M.; Vitale, C.; Coscia, M. Immune Dysfunctions and Immune-Based Therapeutic Interventions in Chronic Lymphocytic Leukemia. Front. Immunol. 2020, 11, 594556. [Google Scholar] [CrossRef]
- Crassini, K.R.; Best, O.G.; Mulligan, S.P. Immune failure, infection and survival in chronic lymphocytic leukemia. Haematologica 2018, 103, e329. [Google Scholar] [CrossRef] [PubMed]
- Elias, R.; Karantanos, T.; Sira, E.; Hartshorn, K.L. Immunotherapy comes of age: Immune aging & checkpoint inhibitors. J. Geriatr. Oncol. 2017, 8, 229–235. [Google Scholar] [CrossRef] [PubMed]


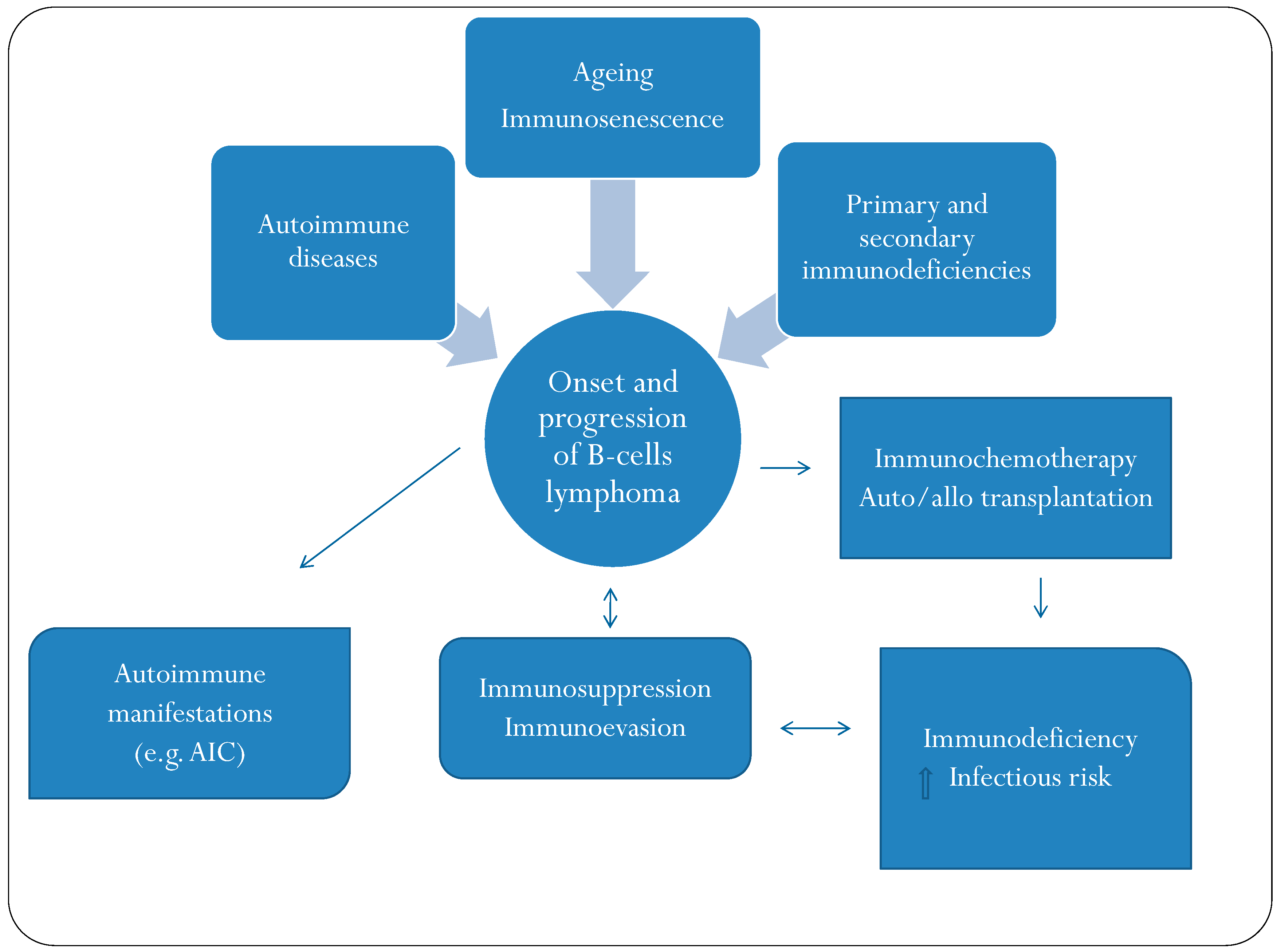{kind=link}
| Study | Disease | Design | Patients, n. | Functional Effect |
|---|---|---|---|---|
| Long et al. [17] | CLL | Prospective | 19 | CD4+ and CD8+ expansion; decreased Treg/CD4+ T ratio |
| RESONATE RESONATE-2, Solman et al. [24] | Naïve and relapsed/refractory CLL | Prospective | 55 50 | Normalization of B cells, regulatory T cells, effector/memory CD4+ and CD8+ T cells, NK and MDSC counts |
| Solano de la Asuncion et al. [90] | CLL | Multicenter observational | 23 | CMV-specific T-cell expansion |
| Sun et al. [92] | Naïve and relapsed/refractory CLL | Prospective | 84 | Increase in serum IgA |
| Yin et al. [93] | CLL | Prospective | 15 | Normalization of T-cell numbers and T-cell-related cytokine levels; increase in T-cell repertoire diversity |
| AIM trial Ibrutinib + Venetoclax Davis et al. [104] | MCL | Prospective | 24 | Normalization of CD4+ and CD8+ effector and central memory T cells and natural killer cells |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mancuso, S.; Mattana, M.; Carlisi, M.; Santoro, M.; Siragusa, S. Effects of B-Cell Lymphoma on the Immune System and Immune Recovery after Treatment: The Paradigm of Targeted Therapy. Int. J. Mol. Sci. 2022, 23, 3368. https://doi.org/10.3390/ijms23063368
Mancuso S, Mattana M, Carlisi M, Santoro M, Siragusa S. Effects of B-Cell Lymphoma on the Immune System and Immune Recovery after Treatment: The Paradigm of Targeted Therapy. International Journal of Molecular Sciences. 2022; 23(6):3368. https://doi.org/10.3390/ijms23063368
Chicago/Turabian StyleMancuso, Salvatrice, Marta Mattana, Melania Carlisi, Marco Santoro, and Sergio Siragusa. 2022. "Effects of B-Cell Lymphoma on the Immune System and Immune Recovery after Treatment: The Paradigm of Targeted Therapy" International Journal of Molecular Sciences 23, no. 6: 3368. https://doi.org/10.3390/ijms23063368
APA StyleMancuso, S., Mattana, M., Carlisi, M., Santoro, M., & Siragusa, S. (2022). Effects of B-Cell Lymphoma on the Immune System and Immune Recovery after Treatment: The Paradigm of Targeted Therapy. International Journal of Molecular Sciences, 23(6), 3368. https://doi.org/10.3390/ijms23063368