
Pathophysiology and Treatment of Diabetic Cardiomyopathy and Heart Failure in Patients with Diabetes Mellitus

 ,
,  , , ,
, , ,
Abstract
:1. Complication of Diabetes and Heart Failure
2. History of Diabetic Cardiomyopathy
3. Pathophysiology and Mechanism of Heart Failure Associated with Diabetes
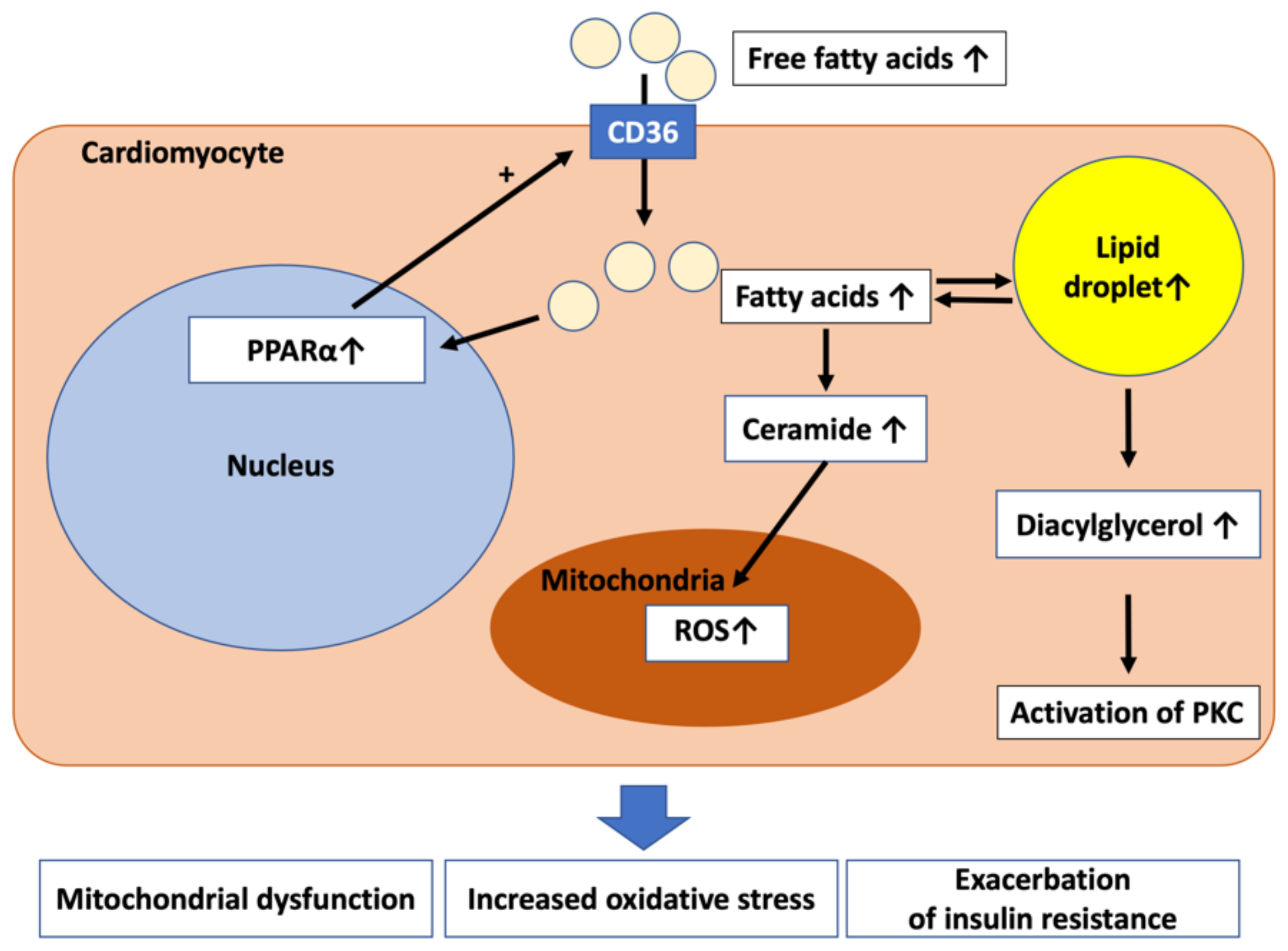
4. Lipotoxicity to the Myocardium
5. Increased Oxidative Stress in Myocardium
6. Mitochondrial Dysfunction
7. Inflammation
8. Abnormal Myocardial Calcium Handling
9. Autonomic Dysregulation in the Heart
10. Other Mechanisms
11. What Is an Effective Treatment?
11.1. Calorie Restriction
11.2. Sulfonylureas
11.3. Insulin
11.4. Thiazolidinediones
11.5. Dipeptidyl Peptidase-4 (DPP4) Inhibitors
11.6. Metformin
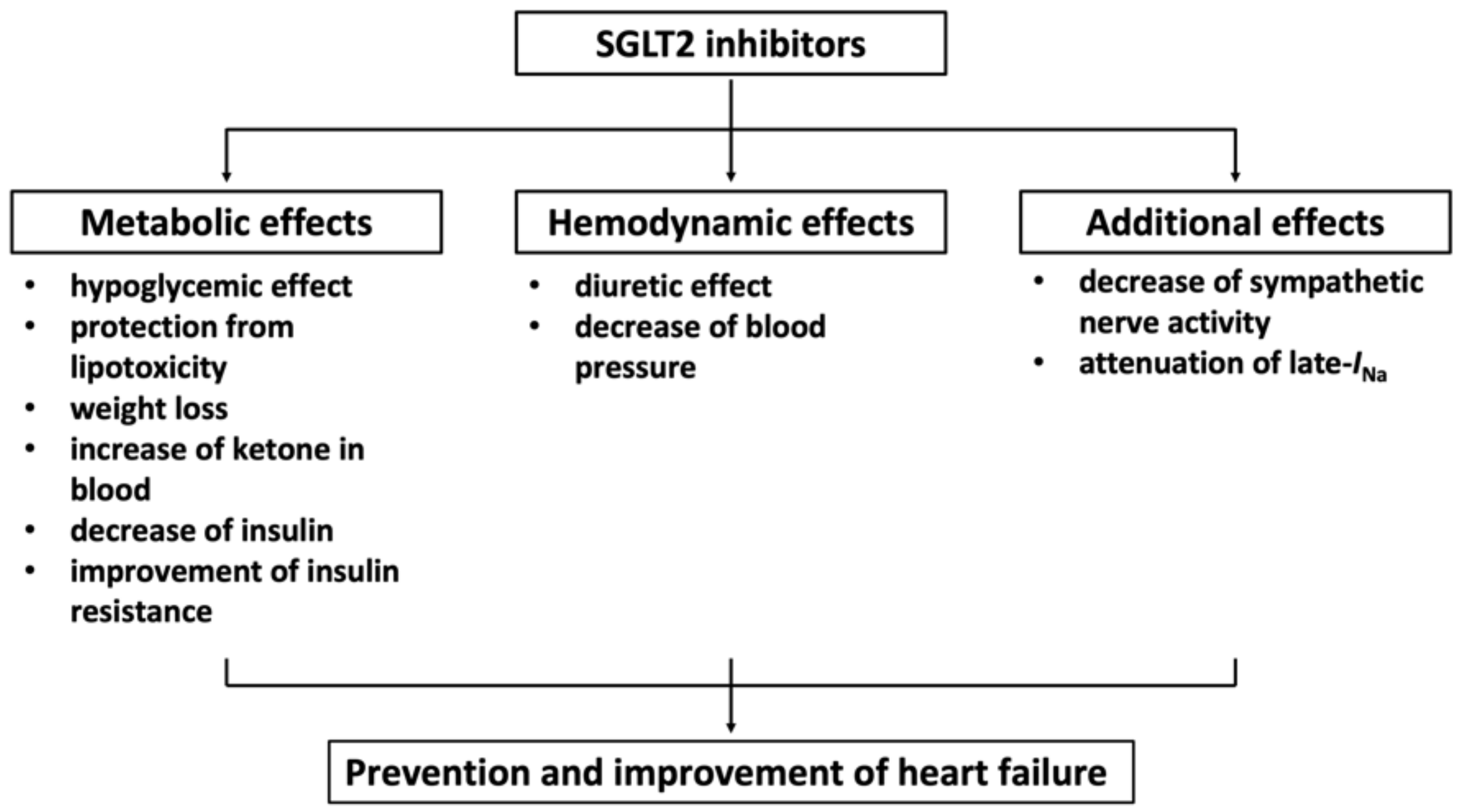
11.7. Sodium Glucose Cotransporter 2 (SGLT2) Inhibitor
11.8. Glucagon-like Peptide-1 (GLP-1) Receptor Agonists
12. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AMPK | AMP-activated protein kinase |
| BNP | brain natriuretic peptide |
| CD36 | cluster of differentiation 36 |
| DPP4 | dipeptidyl peptidase-4 |
| HFpEF | heart failure with preserved ejection fraction |
| HFrEF | heart failure with reduced ejection fraction |
| IL-1β | interleukin-1-beta |
| IL-18 | interleukin-18 |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| late-INa | late component of the cardiac sodium channel current |
| NF-kB | nuclear factor-kB |
| NLRP3 | nucleotide-binding oligomerization domain-like receptor family, pyrin domain-containing 3 |
| TXNIP | thioredoxin-interacting/-inhibiting protein (TXNIP) |
| PKC | protein kinase C |
| PPARα | peroxisome proliferator-activated receptor α |
| ROS | reactive oxygen species |
| SGLT2 | sodium glucose cotransporter 2 |
References
- Kannel, W.B.; Hjortland, M.; Castelli, W.P. Role of diabetes in congestive heart failure: The Framingham study. Am. J. Cardiol. 1974, 34, 29–34. [Google Scholar] [CrossRef]
- Cosentino, F.; Grant, P.J.; Aboyans, V.; Bailey, C.J.; Ceriello, A.; Delgado, V.; Federici, M.; Filippatos, G.; Grobbee, D.E.; Hansen, T.B.; et al. 2019 ESC Guidelines on diabetes, pre-diabetes, and cardiovascular diseases developed in collaboration with the EASD. Eur. Heart J. 2020, 41, 255–323. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.V.; Hill, J.A. Diabetic cardiomyopathy: Catabolism driving metabolism. Circulation 2015, 131, 771–773. [Google Scholar] [CrossRef]
- Lee, W.S.; Kim, J. Diabetic cardiomyopathy: Where we are and where we are going. Korean J. Intern. Med. 2017, 32, 404–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, G.; Hill, M.A.; Sowers, J.R. Diabetic Cardiomyopathy: An Update of Mechanisms Contributing to This Clinical Entity. Circ. Res. 2018, 122, 624–638. [Google Scholar] [CrossRef] [PubMed]
- Dillmann, W.H. Diabetic Cardiomyopathy. Circ. Res. 2019, 124, 1160–1162. [Google Scholar] [CrossRef] [PubMed]
- Bando, Y.K.; Murohara, T. Diabetes-related heart failure. Circ. J. 2014, 78, 576–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinaglia, T.; Oliveira, D.C.; Matos-Souza, J.R.; Sposito, A.C. Diabetic cardiomyopathy: Factual or factoid? Rev. Assoc. Med. Bras. 2019, 65, 61–69. [Google Scholar] [CrossRef]
- Yancy, C.W.; Jessup, M.; Bozkurt, B.; Butler, J.; Casey, D.E., Jr.; Drazner, M.H.; Fonarow, G.C.; Geraci, S.A.; Horwich, T.; Januzzi, J.L.; et al. 2013 ACCF/AHA guideline for the management of heart failure: A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J. Am. Coll. Cardiol. 2013, 62, e147–e239. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, M.; Sadoshima, J. Cardiomyopathy in obesity, insulin resistance and diabetes. J. Physiol. 2020, 598, 2977–2993. [Google Scholar] [CrossRef] [Green Version]
- Rubler, S.; Dlugash, J.; Yuceoglu, Y.Z.; Kumral, T.; Branwood, A.W.; Grishman, A. New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am. J. Cardiol. 1972, 30, 595–602. [Google Scholar] [CrossRef]
- Elliott, P.; Andersson, B.; Arbustini, E.; Bilinska, Z.; Cecchi, F.; Charron, P.; Dubourg, O.; Kuhl, U.; Maisch, B.; McKenna, W.J.; et al. Classification of the cardiomyopathies: A position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2008, 29, 270–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regan, T.J.; Lyons, M.M.; Ahmed, S.S.; Levinson, G.E.; Oldewurtel, H.A.; Ahmad, M.R.; Haider, B. Evidence for cardiomyopathy in familial diabetes mellitus. J. Clin. Investig. 1977, 60, 884–899. [Google Scholar] [CrossRef] [PubMed]
- Redfield, M.M.; Jacobsen, S.J.; Burnett, J.C., Jr.; Mahoney, D.W.; Bailey, K.R.; Rodeheffer, R.J. Burden of systolic and diastolic ventricular dysfunction in the community: Appreciating the scope of the heart failure epidemic. JAMA 2003, 289, 194–202. [Google Scholar] [CrossRef]
- Paulus, W.J.; Tschope, C. A novel paradigm for heart failure with preserved ejection fraction: Comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 2013, 62, 263–271. [Google Scholar] [CrossRef] [Green Version]
- Boudina, S.; Abel, E.D. Diabetic cardiomyopathy revisited. Circulation 2007, 115, 3213–3223. [Google Scholar] [CrossRef]
- van de Weijer, T.; Schrauwen-Hinderling, V.B.; Schrauwen, P. Lipotoxicity in type 2 diabetic cardiomyopathy. Cardiovasc. Res. 2011, 92, 10–18. [Google Scholar] [CrossRef] [Green Version]
- Tong, M.; Saito, T.; Zhai, P.; Oka, S.I.; Mizushima, W.; Nakamura, M.; Ikeda, S.; Shirakabe, A.; Sadoshima, J. Mitophagy is Essential for Maintaining Cardiac Function During High Fat Diet-Induced Diabetic Cardiomyopathy. Circ. Res. 2019, 124, 1360–1371. [Google Scholar] [CrossRef]
- Kenny, H.C.; Abel, E.D. Heart Failure in Type 2 Diabetes Mellitus. Circ. Res. 2019, 124, 121–141. [Google Scholar] [CrossRef]
- Russo, I.; Frangogiannis, N.G. Diabetes-associated cardiac fibrosis: Cellular effectors, molecular mechanisms and therapeutic opportunities. J. Mol. Cell. Cardiol. 2016, 90, 84–93. [Google Scholar] [CrossRef] [Green Version]
- Bodiga, V.L.; Eda, S.R.; Bodiga, S. Advanced glycation end products: Role in pathology of diabetic cardiomyopathy. Heart. Fail. Rev. 2014, 19, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Grubic Rotkvic, P.; Planinic, Z.; Liberati Prso, A.M.; Sikic, J.; Galic, E.; Rotkvic, L. The Mystery of Diabetic Cardiomyopathy: From Early Concepts and Underlying Mechanisms to Novel Therapeutic Possibilities. Int. J. Mol. Sci. 2021, 22, 5973. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, R.H.; Abel, E.D. Basic Mechanisms of Diabetic Heart Disease. Circ. Res. 2020, 126, 1501–1525. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, R.; Sugiyama, H.; Nakamura, K.; Tohgi, K.; Hongo, T.; Tsuchiya, M.; Momoki, N.; Nose, S.; Yutani, C.; Ikeda, Y.; et al. Electron Microscopy Revealed Massive Lipid Droplets in Cardiomyocytes in a Patient with Cardiogenic Shock Following a Fulminant Type 1 Diabetes Mellitus. Int. Heart J. 2021, 62, 197–200. [Google Scholar] [CrossRef]
- Shimabukuro, M. Cardiac adiposity and global cardiometabolic risk: New concept and clinical implication. Circ. J. 2009, 73, 27–34. [Google Scholar] [CrossRef] [Green Version]
- Mahabadi, A.A.; Berg, M.H.; Lehmann, N.; Kalsch, H.; Bauer, M.; Kara, K.; Dragano, N.; Moebus, S.; Jockel, K.H.; Erbel, R.; et al. Association of epicardial fat with cardiovascular risk factors and incident myocardial infarction in the general population: The Heinz Nixdorf Recall Study. J. Am. Coll. Cardiol. 2013, 61, 1388–1395. [Google Scholar] [CrossRef] [Green Version]
- Chokshi, A.; Drosatos, K.; Cheema, F.H.; Ji, R.; Khawaja, T.; Yu, S.; Kato, T.; Khan, R.; Takayama, H.; Knoll, R.; et al. Ventricular assist device implantation corrects myocardial lipotoxicity, reverses insulin resistance, and normalizes cardiac metabolism in patients with advanced heart failure. Circulation 2012, 125, 2844–2853. [Google Scholar] [CrossRef] [Green Version]
- Park, T.S.; Hu, Y.; Noh, H.L.; Drosatos, K.; Okajima, K.; Buchanan, J.; Tuinei, J.; Homma, S.; Jiang, X.C.; Abel, E.D.; et al. Ceramide is a cardiotoxin in lipotoxic cardiomyopathy. J. Lipid. Res. 2008, 49, 2101–2112. [Google Scholar] [CrossRef] [Green Version]
- Basu, R.; Oudit, G.Y.; Wang, X.; Zhang, L.; Ussher, J.R.; Lopaschuk, G.D.; Kassiri, Z. Type 1 diabetic cardiomyopathy in the Akita (Ins2WT/C96Y) mouse model is characterized by lipotoxicity and diastolic dysfunction with preserved systolic function. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H2096–H2108. [Google Scholar] [CrossRef] [Green Version]
- Barger, P.M.; Brandt, J.M.; Leone, T.C.; Weinheimer, C.J.; Kelly, D.P. Deactivation of peroxisome proliferator-activated receptor-alpha during cardiac hypertrophic growth. J. Clin. Investig. 2000, 105, 1723–1730. [Google Scholar] [CrossRef] [Green Version]
- Tuleta, I.; Frangogiannis, N.G. Diabetic fibrosis. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166044. [Google Scholar] [CrossRef] [PubMed]
- Faria, A.; Persaud, S.J. Cardiac oxidative stress in diabetes: Mechanisms and therapeutic potential. Pharmacol. Ther. 2017, 172, 50–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Z.; Wang, P.; Dong, C.; Zhang, J.; Wang, X.; Pei, H. Oxidative Stress Signaling Mediated Pathogenesis of Diabetic Cardiomyopathy. Oxid. Med. Cell. Longev. 2022, 2022, 5913374. [Google Scholar] [CrossRef] [PubMed]
- Gong, W.; Zhang, S.; Chen, Y.; Shen, J.; Zheng, Y.; Liu, X.; Zhu, M.; Meng, G. Protective role of hydrogen sulfide against diabetic cardiomyopathy via alleviating necroptosis. Free Radic. Biol. Med. 2022, 181, 29–42. [Google Scholar] [CrossRef]
- Zheng, H.; Zhu, H.; Liu, X.; Huang, X.; Huang, A.; Huang, Y. Mitophagy in Diabetic Cardiomyopathy: Roles and Mechanisms. Front. Cell Dev. Biol. 2021, 9, 750382. [Google Scholar] [CrossRef]
- Volpe CM, O.; Villar-Delfino, P.H.; Dos Anjos PM, F.; Nogueira-Machado, J.A. Cellular death, reactive oxygen species (ROS) and diabetic complications. Cell Death Dis. 2018, 9, 119. [Google Scholar] [CrossRef]
- Luo, B.; Li, B.; Wang, W.; Liu, X.; Xia, Y.; Zhang, C.; Zhang, M.; Zhang, Y.; An, F. NLRP3 gene silencing ameliorates diabetic cardiomyopathy in a type 2 diabetes rat model. PLoS ONE 2014, 9, e104771. [Google Scholar] [CrossRef]
- Pal, P.B.; Sonowal, H.; Shukla, K.; Srivastava, S.K.; Ramana, K.V. Aldose Reductase Mediates NLRP3 Inflammasome-Initiated Innate Immune Response in Hyperglycemia-Induced Thp1 Monocytes and Male Mice. Endocrinology 2017, 158, 3661–3675. [Google Scholar] [CrossRef]
- Zeng, C.; Wang, R.; Tan, H. Role of Pyroptosis in Cardiovascular Diseases and its Therapeutic Implications. Int. J. Biol. Sci. 2019, 15, 1345–1357. [Google Scholar] [CrossRef] [Green Version]
- Belke, D.D.; Swanson, E.A.; Dillmann, W.H. Decreased sarcoplasmic reticulum activity and contractility in diabetic db/db mouse heart. Diabetes 2004, 53, 3201–3208. [Google Scholar] [CrossRef] [Green Version]
- Van den Bergh, A.; Vanderper, A.; Vangheluwe, P.; Desjardins, F.; Nevelsteen, I.; Verreth, W.; Wuytack, F.; Holvoet, P.; Flameng, W.; Balligand, J.L.; et al. Dyslipidaemia in type II diabetic mice does not aggravate contractile impairment but increases ventricular stiffness. Cardiovasc. Res. 2008, 77, 371–379. [Google Scholar] [CrossRef] [Green Version]
- Agashe, S.; Petak, S. Cardiac Autonomic Neuropathy in Diabetes Mellitus. Methodist Debakey Cardiovasc. J. 2018, 14, 251–256. [Google Scholar] [CrossRef]
- Ramsey, M.W.; Goodfellow, J.; Jones, C.J.; Luddington, L.A.; Lewis, M.J.; Henderson, A.H. Endothelial control of arterial distensibility is impaired in chronic heart failure. Circulation 1995, 92, 3212–3219. [Google Scholar] [CrossRef] [PubMed]
- Katz, S.D.; Hryniewicz, K.; Hriljac, I.; Balidemaj, K.; Dimayuga, C.; Hudaihed, A.; Yasskiy, A. Vascular endothelial dysfunction and mortality risk in patients with chronic heart failure. Circulation 2005, 111, 310–314. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, K.; Miyoshi, T.; Yunoki, K.; Ito, H. Postprandial hyperlipidemia as a potential residual risk factor. J. Cardiol. 2016, 67, 335–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quagliaro, L.; Piconi, L.; Assaloni, R.; Martinelli, L.; Motz, E.; Ceriello, A. Intermittent high glucose enhances apoptosis related to oxidative stress in human umbilical vein endothelial cells: The role of protein kinase C and NAD(P)H-oxidase activation. Diabetes 2003, 52, 2795–2804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taegtmeyer, H.; Beauloye, C.; Harmancey, R.; Hue, L. Comment on Nolan et al. Insulin Resistance as a Physiological Defense Against Metabolic Stress: Implications for the Management of Subsets of Type 2 Diabetes. Diabetes 2015, 64, e37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groop, P.H.; Forsblom, C.; Thomas, M.C. Mechanisms of disease: Pathway-selective insulin resistance and microvascular complications of diabetes. Nat. Clin. Pract. Endocrinol. Metab. 2005, 1, 100–110. [Google Scholar] [CrossRef]
- Jin, W.L.; Azuma, K.; Mita, T.; Goto, H.; Kanazawa, A.; Shimizu, T.; Ikeda, F.; Fujitani, Y.; Hirose, T.; Kawamori, R.; et al. Repetitive hypoglycaemia increases serum adrenaline and induces monocyte adhesion to the endothelium in rat thoracic aorta. Diabetologia 2011, 54, 1921–1929. [Google Scholar] [CrossRef] [Green Version]
- Razavi Nematollahi, L.; Kitabchi, A.E.; Stentz, F.B.; Wan, J.Y.; Larijani, B.A.; Tehrani, M.M.; Gozashti, M.H.; Omidfar, K.; Taheri, E. Proinflammatory cytokines in response to insulin-induced hypoglycemic stress in healthy subjects. Metabolism 2009, 58, 443–448. [Google Scholar] [CrossRef]
- Yoshida, M.; Nakamura, K.; Miyoshi, T.; Yoshida, M.; Kondo, M.; Akazawa, K.; Kimura, T.; Ohtsuka, H.; Ohno, Y.; Miura, D.; et al. Combination therapy with pemafibrate (K-877) and pitavastatin improves vascular endothelial dysfunction in dahl/salt-sensitive rats fed a high-salt and high-fat diet. Cardiovasc. Diabetol. 2020, 19, 149. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Bohm, M.; Burri, H.; Butler, J.; Celutkiene, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, D.; Bozkurt, B.; Ramasubbu, K.; Deswal, A. Relationship of hemoglobin A1C and mortality in heart failure patients with diabetes. J. Am. Coll. Cardiol. 2009, 54, 422–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castagno, D.; Baird-Gunning, J.; Jhund, P.S.; Biondi-Zoccai, G.; MacDonald, M.R.; Petrie, M.C.; Gaita, F.; McMurray, J.J. Intensive glycemic control has no impact on the risk of heart failure in type 2 diabetic patients: Evidence from a 37,229 patient meta-analysis. Am. Heart J. 2011, 162, 938–948.e2. [Google Scholar] [CrossRef]
- Boussageon, R.; Bejan-Angoulvant, T.; Saadatian-Elahi, M.; Lafont, S.; Bergeonneau, C.; Kassai, B.; Erpeldinger, S.; Wright, J.M.; Gueyffier, F.; Cornu, C. Effect of intensive glucose lowering treatment on all cause mortality, cardiovascular death, and microvascular events in type 2 diabetes: Meta-analysis of randomised controlled trials. BMJ 2011, 343, d4169. [Google Scholar] [CrossRef] [Green Version]
- Redman, L.M.; Smith, S.R.; Burton, J.H.; Martin, C.K.; Il’yasova, D.; Ravussin, E. Metabolic Slowing and Reduced Oxidative Damage with Sustained Caloric Restriction Support the Rate of Living and Oxidative Damage Theories of Aging. Cell Metab. 2018, 27, 805–815.e4. [Google Scholar] [CrossRef] [Green Version]
- Tzoulaki, I.; Molokhia, M.; Curcin, V.; Little, M.P.; Millett, C.J.; Ng, A.; Hughes, R.I.; Khunti, K.; Wilkins, M.R.; Majeed, A.; et al. Risk of cardiovascular disease and all cause mortality among patients with type 2 diabetes prescribed oral antidiabetes drugs: Retrospective cohort study using UK general practice research database. BMJ 2009, 339, b4731. [Google Scholar] [CrossRef] [Green Version]
- Lago, R.M.; Singh, P.P.; Nesto, R.W. Congestive heart failure and cardiovascular death in patients with prediabetes and type 2 diabetes given thiazolidinediones: A meta-analysis of randomised clinical trials. Lancet 2007, 370, 1129–1136. [Google Scholar] [CrossRef]
- Oe, H.; Nakamura, K.; Kihara, H.; Shimada, K.; Fukuda, S.; Takagi, T.; Miyoshi, T.; Hirata, K.; Yoshikawa, J.; Ito, H.; et al. Comparison of effects of sitagliptin and voglibose on left ventricular diastolic dysfunction in patients with type 2 diabetes: Results of the 3D trial. Cardiovasc. Diabetol. 2015, 14, 83. [Google Scholar] [CrossRef] [Green Version]
- White, W.B.; Cannon, C.P.; Heller, S.R.; Nissen, S.E.; Bergenstal, R.M.; Bakris, G.L.; Perez, A.T.; Fleck, P.R.; Mehta, C.R.; Kupfer, S.; et al. Alogliptin after acute coronary syndrome in patients with type 2 diabetes. N. Engl. J. Med. 2013, 369, 1327–1335. [Google Scholar] [CrossRef] [Green Version]
- Green, J.B.; Bethel, M.A.; Armstrong, P.W.; Buse, J.B.; Engel, S.S.; Garg, J.; Josse, R.; Kaufman, K.D.; Koglin, J.; Korn, S.; et al. Effect of Sitagliptin on Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 232–242. [Google Scholar] [CrossRef] [Green Version]
- Rosenstock, J.; Perkovic, V.; Johansen, O.E.; Cooper, M.E.; Kahn, S.E.; Marx, N.; Alexander, J.H.; Pencina, M.; Toto, R.D.; Wanner, C.; et al. Effect of Linagliptin vs Placebo on Major Cardiovascular Events in Adults With Type 2 Diabetes and High Cardiovascular and Renal Risk: The CARMELINA Randomized Clinical Trial. JAMA 2019, 321, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Scirica, B.M.; Bhatt, D.L.; Braunwald, E.; Steg, P.G.; Davidson, J.; Hirshberg, B.; Ohman, P.; Frederich, R.; Wiviott, S.D.; Hoffman, E.B.; et al. Investigators, Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N. Engl. J. Med. 2013, 369, 1317–1326. [Google Scholar] [CrossRef] [Green Version]
- Packer, M. Do DPP-4 Inhibitors Cause Heart Failure Events by Promoting Adrenergically Mediated Cardiotoxicity? Clues From Laboratory Models and Clinical Trials. Circ. Res. 2018, 122, 928–932. [Google Scholar] [CrossRef]
- Eurich, D.T.; Weir, D.L.; Majumdar, S.R.; Tsuyuki, R.T.; Johnson, J.A.; Tjosvold, L.; Vanderloo, S.E.; McAlister, F.A. Comparative safety and effectiveness of metformin in patients with diabetes mellitus and heart failure: Systematic review of observational studies involving 34,000 patients. Circ. Heart Fail. 2013, 6, 395–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersson, C.; Olesen, J.B.; Hansen, P.R.; Weeke, P.; Norgaard, M.L.; Jorgensen, C.H.; Lange, T.; Abildstrom, S.Z.; Schramm, T.K.; Vaag, A.; et al. Metformin treatment is associated with a low risk of mortality in diabetic patients with heart failure: A retrospective nationwide cohort study. Diabetologia 2010, 53, 2546–2553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gundewar, S.; Calvert, J.W.; Jha, S.; Toedt-Pingel, I.; Ji, S.Y.; Nunez, D.; Ramachandran, A.; Anaya-Cisneros, M.; Tian, R.; Lefer, D.J. Activation of AMP-activated protein kinase by metformin improves left ventricular function and survival in heart failure. Circ. Res. 2009, 104, 403–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, H.; Asanuma, H.; Fujita, M.; Takahama, H.; Wakeno, M.; Ito, S.; Ogai, A.; Asakura, M.; Kim, J.; Minamino, T.; et al. Metformin prevents progression of heart failure in dogs: Role of AMP-activated protein kinase. Circulation 2009, 119, 2568–2577. [Google Scholar] [CrossRef] [Green Version]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef] [Green Version]
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; de Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Law, G.; Desai, M.; Matthews, D.R.; et al. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 644–657. [Google Scholar] [CrossRef]
- Tsutsui, H.; Isobe, M.; Ito, H.; Ito, H.; Okumura, K.; Ono, M.; Kitakaze, M.; Kinugawa, K.; Kihara, Y.; Goto, Y.; et al. JCS 2017/JHFS 2017 Guideline on Diagnosis and Treatment of Acute and Chronic Heart Failure- Digest Version. Circ. J. 2019, 83, 2084–2184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiviott, S.D.; Raz, I.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; Silverman, M.G.; Zelniker, T.A.; Kuder, J.F.; Murphy, S.A.; et al. Dapagliflozin and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2019, 380, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Martens, P.; Mathieu, C.; Verbrugge, F.H. Promise of SGLT2 Inhibitors in Heart Failure: Diabetes and Beyond. Curr. Treat. Options Cardiovasc. Med. 2017, 19, 23. [Google Scholar] [CrossRef] [PubMed]
- Sano, M. A new class of drugs for heart failure: SGLT2 inhibitors reduce sympathetic overactivity. J. Cardiol. 2018, 71, 471–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szekeres, Z.; Toth, K.; Szabados, E. The Effects of SGLT2 Inhibitors on Lipid Metabolism. Metabolites 2021, 11, 87. [Google Scholar] [CrossRef]
- Takata, T.; Isomoto, H. Pleiotropic Effects of Sodium-Glucose Cotransporter-2 Inhibitors: Renoprotective Mechanisms beyond Glycemic Control. Int. J. Mol. Sci. 2021, 22, 4374. [Google Scholar] [CrossRef]
- Kimura, T.; Nakamura, K.; Miyoshi, T.; Yoshida, M.; Akazawa, K.; Saito, Y.; Akagi, S.; Ohno, Y.; Kondo, M.; Miura, D.; et al. Inhibitory Effects of Tofogliflozin on Cardiac Hypertrophy in Dahl Salt-Sensitive and Salt-Resistant Rats Fed a High-Fat Diet. Int. Heart J. 2019, 60, 728–735. [Google Scholar] [CrossRef] [Green Version]
- Rahman, A.; Fujisawa, Y.; Nakano, D.; Hitomi, H.; Nishiyama, A. Effect of a selective SGLT2 inhibitor, luseogliflozin, on circadian rhythm of sympathetic nervous function and locomotor activities in metabolic syndrome rats. Clin. Exp. Pharmacol. Physiol. 2017, 44, 522–525. [Google Scholar] [CrossRef]
- Matthews, V.B.; Elliot, R.H.; Rudnicka, C.; Hricova, J.; Herat, L.; Schlaich, M.P. Role of the sympathetic nervous system in regulation of the sodium glucose cotransporter 2. J. Hypertens. 2017, 35, 2059–2068. [Google Scholar] [CrossRef]
- Sano, M.; Chen, S.; Imazeki, H.; Ochiai, H.; Seino, Y. Changes in heart rate in patients with type 2 diabetes mellitus after treatment with luseogliflozin: Subanalysis of placebo-controlled, double-blind clinical trials. J. Diabetes Investig. 2018, 9, 638–641. [Google Scholar] [CrossRef]
- Philippaert, K.; Kalyaanamoorthy, S.; Fatehi, M.; Long, W.; Soni, S.; Byrne, N.J.; Barr, A.; Singh, J.; Wong, J.; Palechuk, T.; et al. Cardiac Late Sodium Channel Current Is a Molecular Target for the Sodium/Glucose Cotransporter 2 Inhibitor Empagliflozin. Circulation 2021, 143, 2188–2204. [Google Scholar] [CrossRef] [PubMed]
- Makielski, J.C. Late sodium current: A mechanism for angina, heart failure, and arrhythmia. Trends Cardiovasc. Med. 2016, 26, 115–122. [Google Scholar] [CrossRef] [Green Version]
- Nie, J.; Duan, Q.; He, M.; Li, X.; Wang, B.; Zhou, C.; Wu, L.; Wen, Z.; Chen, C.; Wang, D.W.; et al. Ranolazine prevents pressure overload-induced cardiac hypertrophy and heart failure by restoring aberrant Na(+) and Ca(2+) handling. J. Cell. Physiol. 2019, 234, 11587–11601. [Google Scholar] [CrossRef] [PubMed]
- Lindegger, N.; Hagen, B.M.; Marks, A.R.; Lederer, W.J.; Kass, R.S. Diastolic transient inward current in long QT syndrome type 3 is caused by Ca2+ overload and inhibited by ranolazine. J. Mol. Cell. Cardiol. 2009, 47, 326–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Kober, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Belohlavek, J.; et al. Investigators, Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Pocock, S.J.; Carson, P.; Januzzi, J.; Verma, S.; Tsutsui, H.; Brueckmann, M.; et al. Cardiovascular and Renal Outcomes with Empagliflozin in Heart Failure. N. Engl. J. Med. 2020, 383, 1413–1424. [Google Scholar] [CrossRef] [PubMed]
- Ejiri, K.; Miyoshi, T.; Kihara, H.; Hata, Y.; Nagano, T.; Takaishi, A.; Toda, H.; Nanba, S.; Nakamura, Y.; Akagi, S.; et al. Effect of Luseogliflozin on Heart Failure With Preserved Ejection Fraction in Patients With Diabetes Mellitus. J. Am. Heart Assoc. 2020, 9, e015103. [Google Scholar] [CrossRef]
- Bhatt, D.L.; Szarek, M.; Steg, P.G.; Cannon, C.P.; Leiter, L.A.; McGuire, D.K.; Lewis, J.B.; Riddle, M.C.; Voors, A.A.; Metra, M.; et al. Sotagliflozin in Patients with Diabetes and Recent Worsening Heart Failure. N. Engl. J. Med. 2021, 384, 117–128. [Google Scholar] [CrossRef]
- Anker, S.D.; Butler, J.; Filippatos, G.; Ferreira, J.P.; Bocchi, E.; Bohm, M.; Brunner-La Rocca, H.P.; Choi, D.J.; Chopra, V.; Chuquiure-Valenzuela, E.; et al. Empagliflozin in Heart Failure with a Preserved Ejection Fraction. N. Engl. J. Med. 2021, 385, 1451–1461. [Google Scholar] [CrossRef]
- Volpe, M.; Patrono, C. The EMPEROR-Preserved study: End of the search for the “Phoenix” or beginning of a new season for trials in heart failure with preserved ejection fraction. Eur. Heart J. 2021, 42, 4621–4623. [Google Scholar] [CrossRef]
- Yanagawa, T. Is an Increase in Serum Magnesium One of the Causes of Cardiovascular Events Reduction in the EMPA-REG OUTCOME Study? J. Clin. Med. Res. 2017, 9, 449–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Gobbo, L.C.; Song, Y.; Poirier, P.; Dewailly, E.; Elin, R.J.; Egeland, G.M. Low serum magnesium concentrations are associated with a high prevalence of premature ventricular complexes in obese adults with type 2 diabetes. Cardiovasc. Diabetol. 2012, 11, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, K.M.; Lau, Y.M.; Dhandhania, V.; Cai, Z.J.; Lee, Y.K.; Lai, W.H.; Tse, H.F.; Siu, C.W. Empagliflozin Ammeliorates High Glucose Induced-Cardiac Dysfuntion in Human iPSC-Derived Cardiomyocytes. Sci. Rep. 2018, 8, 14872. [Google Scholar] [CrossRef] [PubMed]
- Palmer, S.C.; Tendal, B.; Mustafa, R.A.; Vandvik, P.O.; Li, S.; Hao, Q.; Tunnicliffe, D.; Ruospo, M.; Natale, P.; Saglimbene, V.; et al. Sodium-glucose cotransporter protein-2 (SGLT-2) inhibitors and glucagon-like peptide-1 (GLP-1) receptor agonists for type 2 diabetes: Systematic review and network meta-analysis of randomised controlled trials. BMJ 2021, 372, m4573. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
|
|
|
|
|
|
|
|
|
|
|
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakamura, K.; Miyoshi, T.; Yoshida, M.; Akagi, S.; Saito, Y.; Ejiri, K.; Matsuo, N.; Ichikawa, K.; Iwasaki, K.; Naito, T.; et al. Pathophysiology and Treatment of Diabetic Cardiomyopathy and Heart Failure in Patients with Diabetes Mellitus. Int. J. Mol. Sci. 2022, 23, 3587. https://doi.org/10.3390/ijms23073587
Nakamura K, Miyoshi T, Yoshida M, Akagi S, Saito Y, Ejiri K, Matsuo N, Ichikawa K, Iwasaki K, Naito T, et al. Pathophysiology and Treatment of Diabetic Cardiomyopathy and Heart Failure in Patients with Diabetes Mellitus. International Journal of Molecular Sciences. 2022; 23(7):3587. https://doi.org/10.3390/ijms23073587
Chicago/Turabian StyleNakamura, Kazufumi, Toru Miyoshi, Masashi Yoshida, Satoshi Akagi, Yukihiro Saito, Kentaro Ejiri, Naoaki Matsuo, Keishi Ichikawa, Keiichiro Iwasaki, Takanori Naito, and et al. 2022. "Pathophysiology and Treatment of Diabetic Cardiomyopathy and Heart Failure in Patients with Diabetes Mellitus" International Journal of Molecular Sciences 23, no. 7: 3587. https://doi.org/10.3390/ijms23073587
APA StyleNakamura, K., Miyoshi, T., Yoshida, M., Akagi, S., Saito, Y., Ejiri, K., Matsuo, N., Ichikawa, K., Iwasaki, K., Naito, T., Namba, Y., Yoshida, M., Sugiyama, H., & Ito, H. (2022). Pathophysiology and Treatment of Diabetic Cardiomyopathy and Heart Failure in Patients with Diabetes Mellitus. International Journal of Molecular Sciences, 23(7), 3587. https://doi.org/10.3390/ijms23073587