
Pharmacological Effects of Panduratin A on Renal Cyst Development in In Vitro and In Vivo Models of Polycystic Kidney Disease

 , and
, and 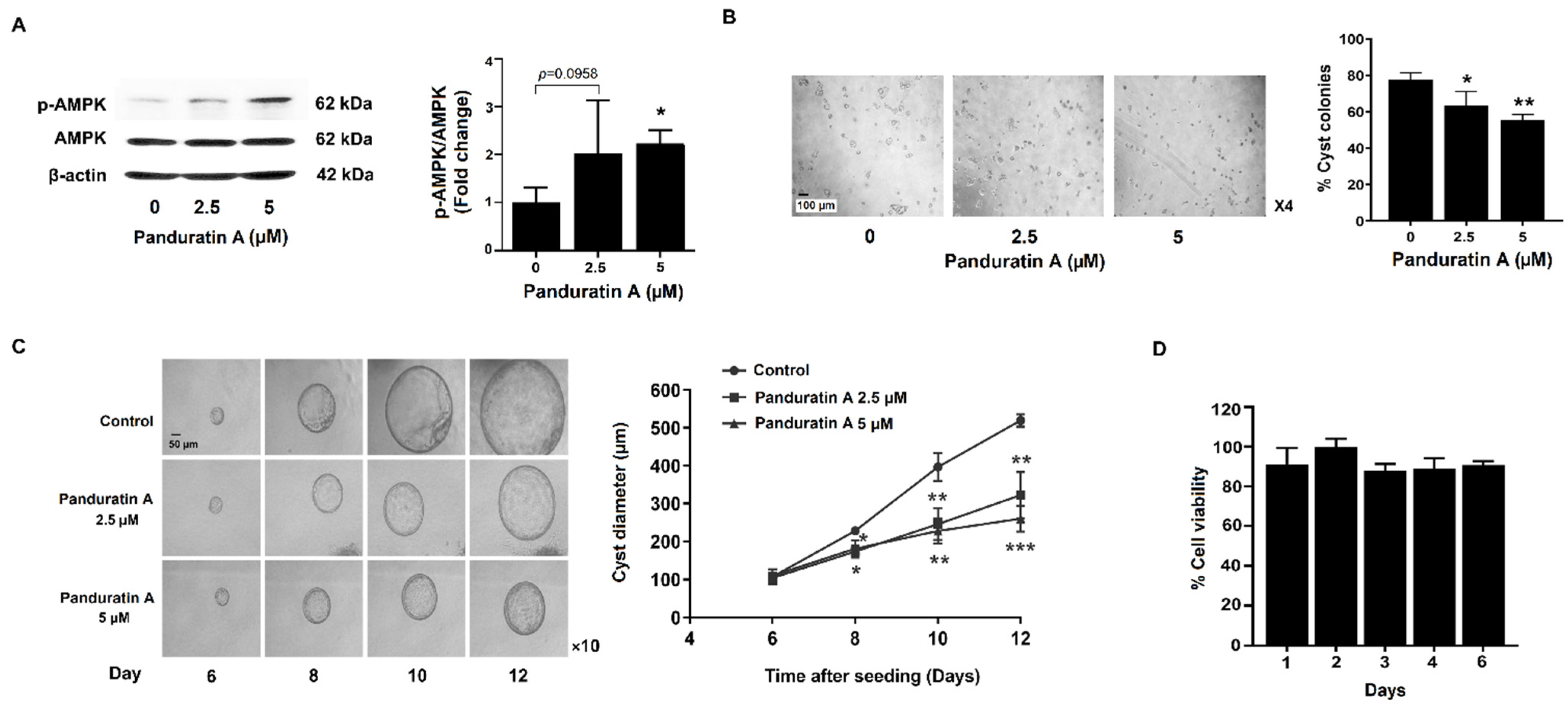{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Panduratin A Treatment Slows in Vitro Cystogenesis
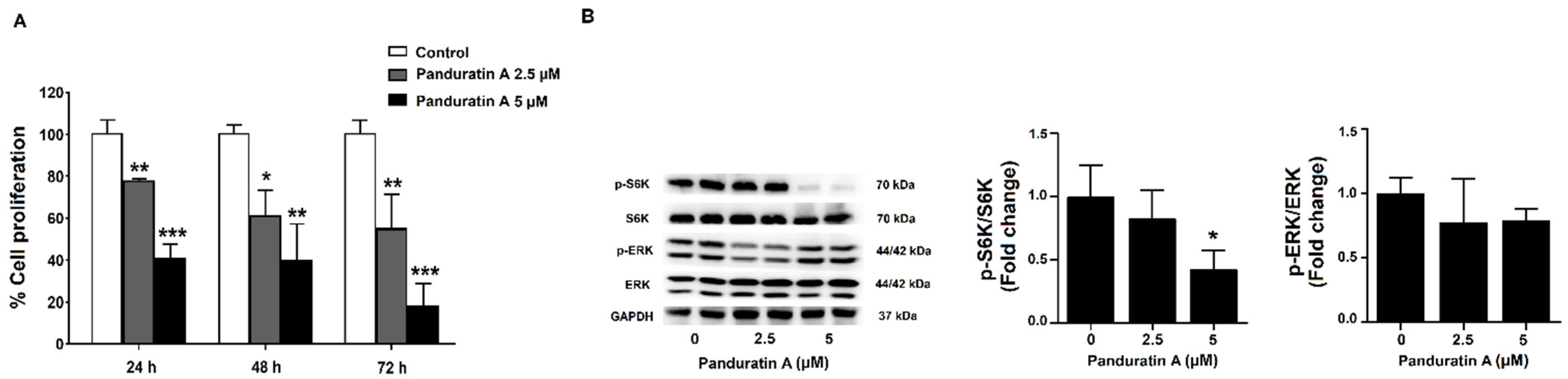
2.2. Panduratin A Treatment Reduces Cell Proliferation via Inhibition of mTOR Signaling in MDCK Cells
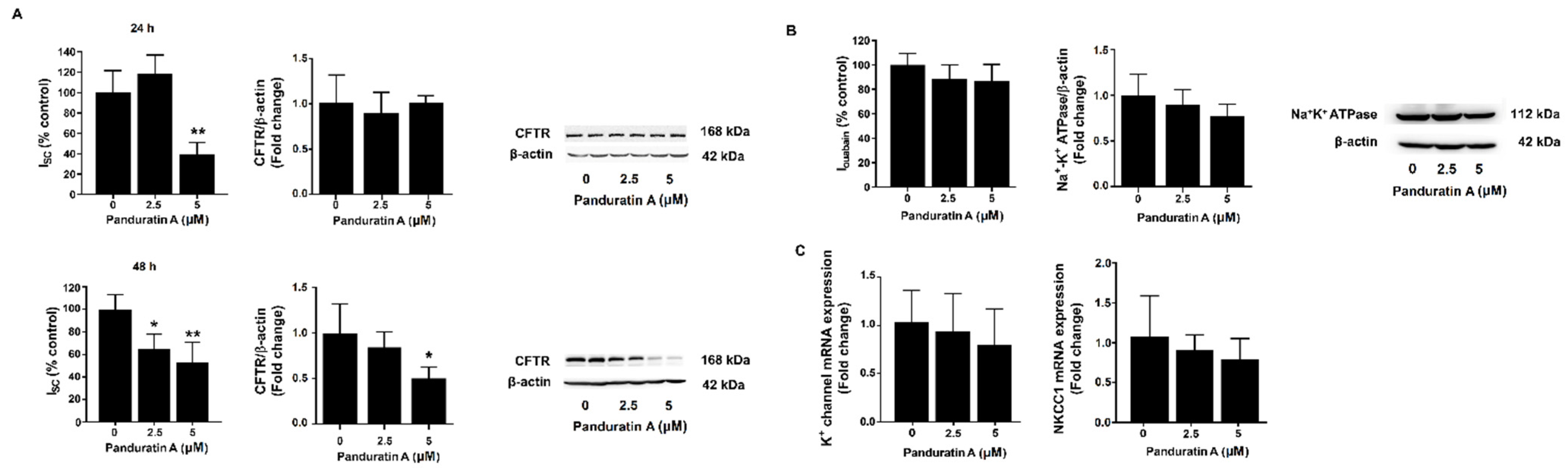
2.3. Panduratin A Inhibits CFTR-Mediated Chloride Secretion in MDCK Cells
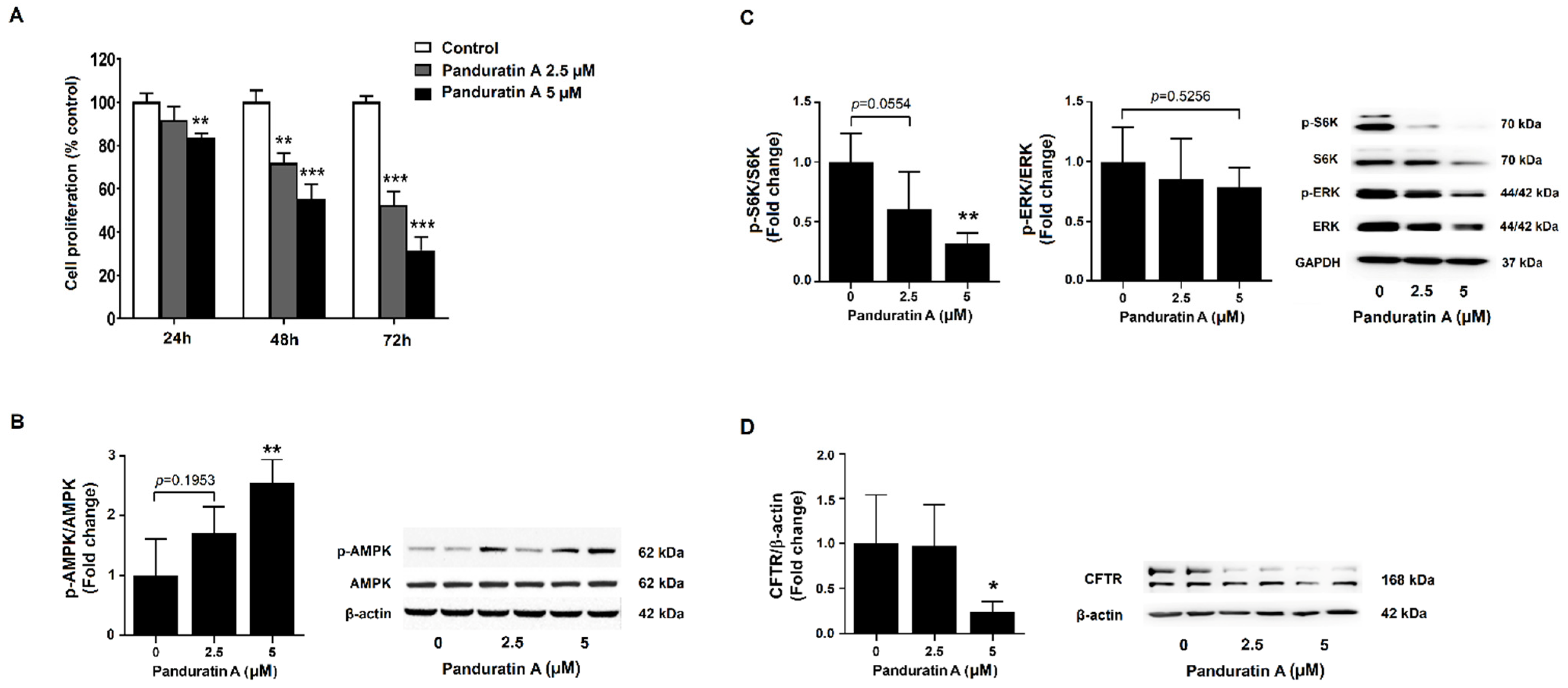
2.4. Panduratin A Treatment Reduces the Proliferation of Human ADPKD Cells
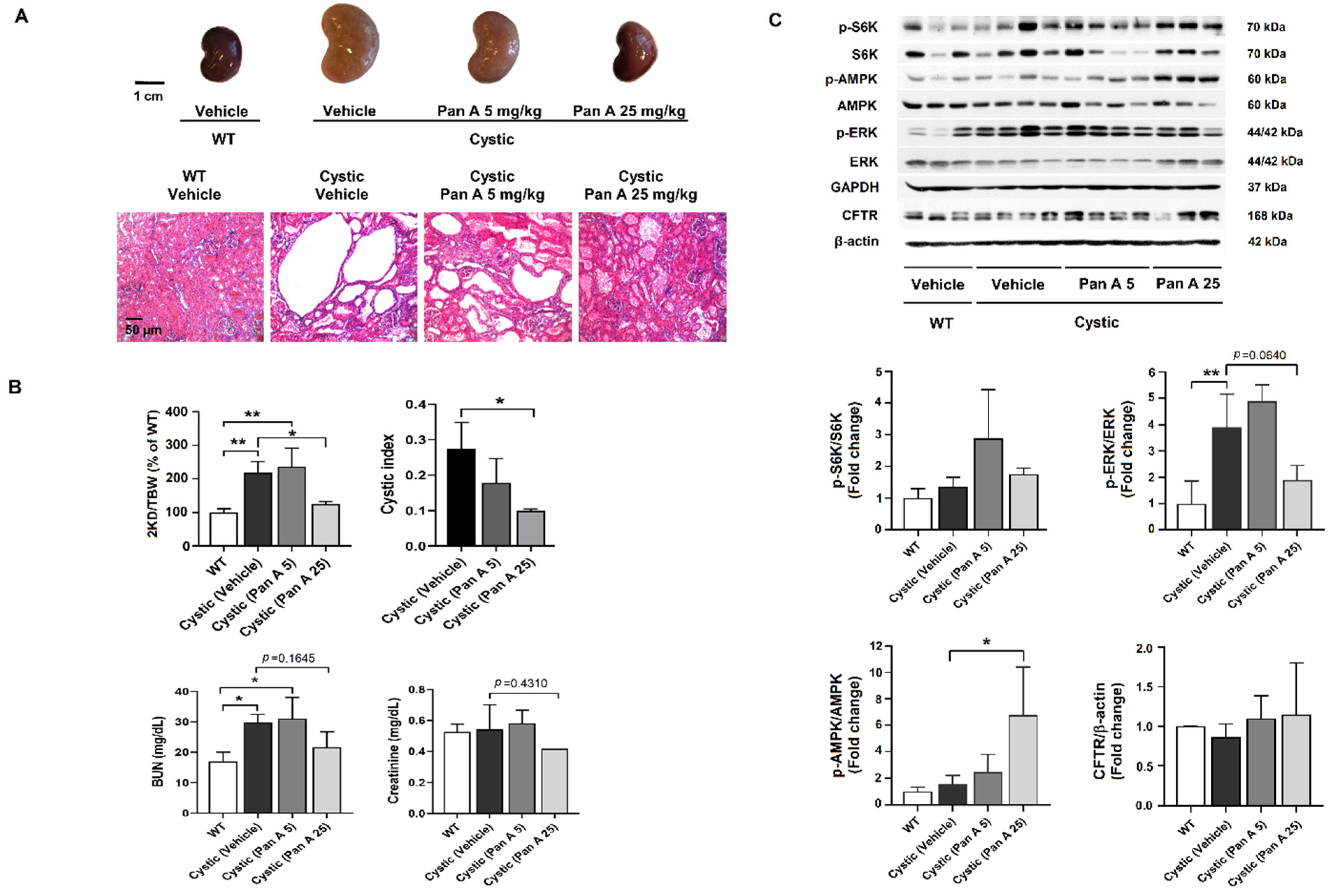
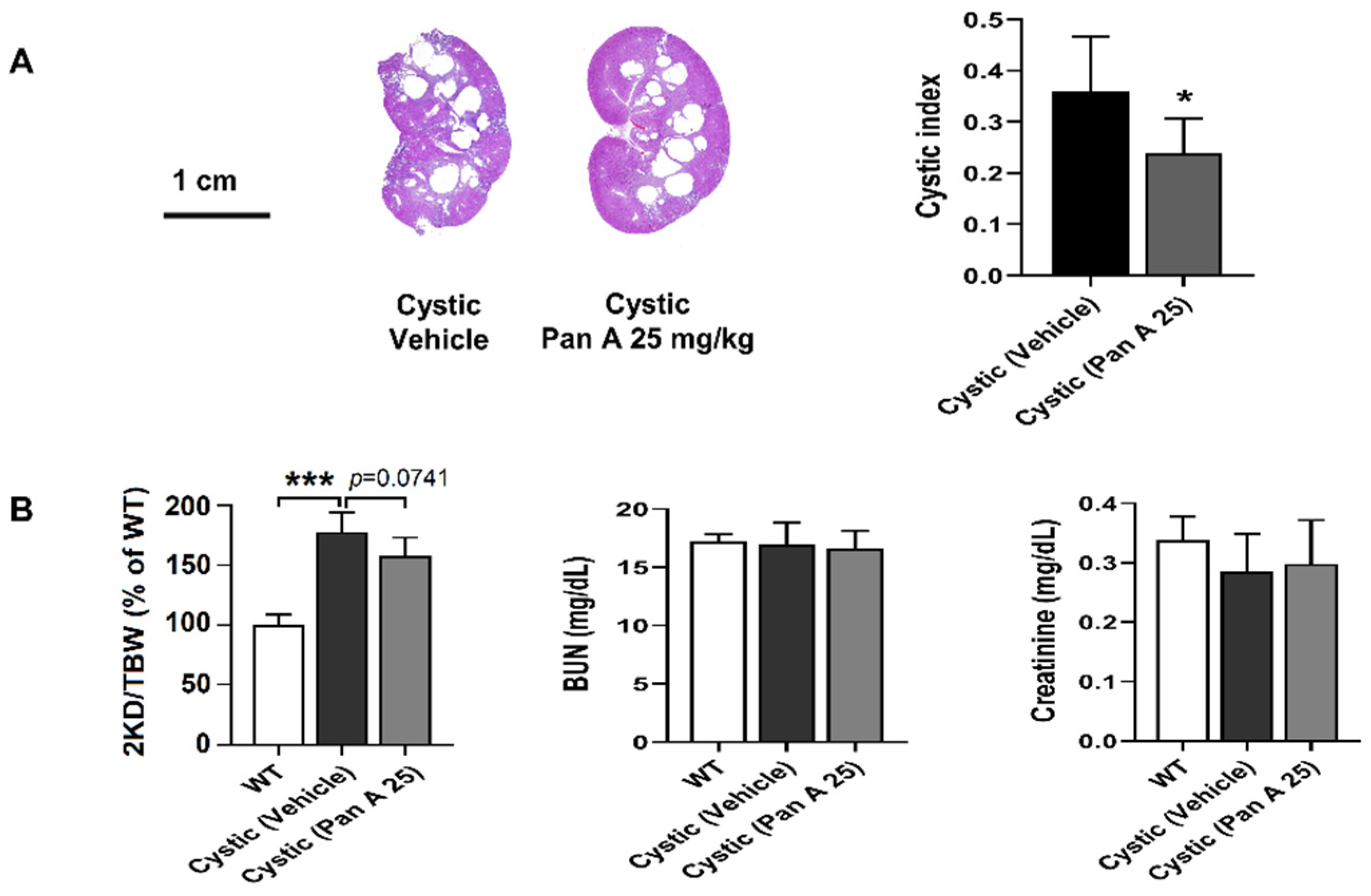
2.5. Effect of Panduratin A on Renal Cyst Progression in PKD Rat Models
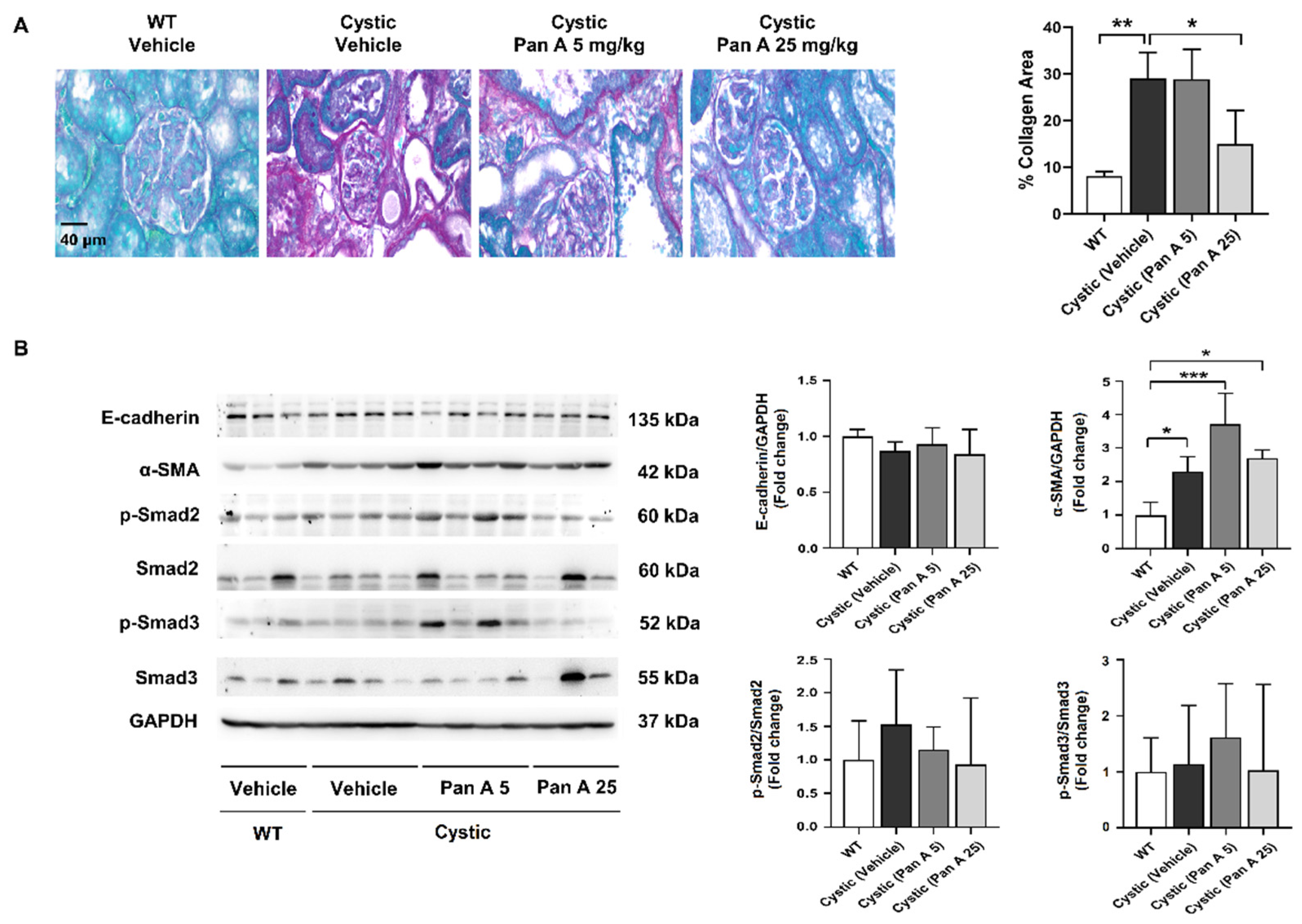
2.6. Effect of Panduratin A on Interstitial Fibrosis in Kidneys PKD Rats
3. Discussion
4. Materials and Methods
4.1. Preparation of Panduratin A
4.2. Chemicals
4.3. Cell Lines
4.4. Animals
4.5. MDCK Cell-Derived Cyst Formation and Growth
4.6. Cell Viability Assay
4.7. Cell Proliferation Assay
4.8. Real-Time PCR
4.9. Western Blot Analysis
4.10. Ussing Chamber Experiments
4.11. Histological Staining
4.12. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chapin, H.C.; Caplan, M.J. The cell biology of polycystic kidney disease. J. Cell Biol. 2010, 191, 701–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, P.C.; Torres, V.E. Polycystic kidney disease. Annu. Rev. Med. 2009, 60, 321–337. [Google Scholar] [CrossRef] [PubMed]
- Torres, V.E. Cyclic AMP, at the hub of the cystic cycle. Kidney Int. 2004, 66, 1283–1285. [Google Scholar] [CrossRef] [Green Version]
- Belibi, F.A.; Reif, G.; Wallace, D.P.; Yamaguchi, T.; Olsen, L.; Li, H.; Helmkamp, G.M., Jr.; Grantham, J.J. Cyclic AMP promotes growth and secretion in human polycystic kidney epithelial cells. Kidney Int. 2004, 66, 964–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, T.; Pelling, J.C.; Ramaswamy, N.T.; Eppler, J.W.; Wallace, D.P.; Nagao, S.; Rome, L.A.; Sullivan, L.P.; Grantham, J.J. cAMP stimulates the in vitro proliferation of renal cyst epithelial cells by activating the extracellular signal-regulated kinase pathway. Kidney Int. 2000, 57, 1460–1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvet, J.P. Strategies to Inhibit Cyst Formation in ADPKD. Clin. J. Am. Soc. Nephrol. 2008, 3, 1205–1211. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Findlay, I.A.; Sheppard, D.N. The relationship between cell proliferation, Cl− secretion, and renal cyst growth: A study using CFTR inhibitors. Kidney Int. 2004, 66, 1926–1938. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.; Sonawane, N.D.; Zhao, D.; Somlo, S.; Verkman, A.S. Small-Molecule CFTR Inhibitors Slow Cyst Growth in Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2008, 19, 1300–1310. [Google Scholar] [CrossRef] [Green Version]
- Raksaseri, P.; Chatsudthipong, V.; Muanprasat, C.; Soodvilai, S. Activation of liver X receptors reduces CFTR-mediated Cl− transport in kidney collecting duct cells. Am. J. Physiol. Ren. Physiol. 2013, 305, F583–F591. [Google Scholar] [CrossRef] [Green Version]
- Yoshihara, D.; Kurahashi, H.; Morita, M.; Kugita, M.; Hiki, Y.; Aukema, H.M.; Yamaguchi, T.; Calvet, J.P.; Wallace, D.P.; Nagao, S. PPAR-γ agonist ameliorates kidney and liver disease in an orthologous rat model of human autosomal recessive polycystic kidney disease. Am. J. Physiol. Ren. Physiol. 2011, 300, F465–F474. [Google Scholar] [CrossRef] [Green Version]
- Blazer-Yost, B.L.; Haydon, J.; Eggleston-Gulyas, T.; Chen, J.-H.; Wang, X.; Gattone, V.; Torres, V.E. Pioglitazone Attenuates Cystic Burden in the PCK Rodent Model of Polycystic Kidney Disease. PPAR Res. 2010, 2010, 274376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, B.; Liu, Y.; Mei, C.; Fu, L.; Xiong, X.; Zhang, Y.; Shen, X.; Hua, Z. Rosiglitazone attenuates development of polycystic kidney disease and prolongs survival in Han:SPRD rats. Clin. Sci. 2010, 119, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Bastos, A.P.; Onuchic, L.F. Molecular and cellular pathogenesis of autosomal dominant polycystic kidney disease. Braz. J. Med. Biol. Res. 2011, 44, 606–617. [Google Scholar] [CrossRef] [Green Version]
- Zafar, I.; Belibi, F.A.; He, Z.; Edelstein, C.L. Long-term rapamycin therapy in the Han:SPRD rat model of polycystic kidney disease (PKD). Nephrol. Dial. Transplant. 2009, 24, 2349–2353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Li, H.; Gao, X.; Yang, M.; Yuan, L.; Fu, L.; Wang, X.; Mei, C. Concomitant use of rapamycin and rosiglitazone delays the progression of polycystic kidney disease in Han:SPRD rats: A study of the mechanism of action. Am. J. Physiol. Ren. Physiol. 2018, 314, F844–F854. [Google Scholar] [CrossRef]
- Nantavishit, J.; Chatsudthipong, V.; Soodvilai, S. Lansoprazole reduces renal cyst in polycystic kidney disease via inhibition of cell proliferation and fluid secretion. Biochem. Pharmacol. 2018, 154, 175–182. [Google Scholar] [CrossRef]
- Tao, Y.; Kim, J.; Schrier, R.W.; Edelstein, C.L. Rapamycin Markedly Slows Disease Progression in a Rat Model of Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2004, 16, 46–51. [Google Scholar] [CrossRef] [Green Version]
- Shillingford, J.M.; Piontek, K.B.; Germino, G.; Weimbs, T. Rapamycin Ameliorates PKD Resulting from Conditional Inactivation of Pkd1. J. Am. Soc. Nephrol. 2010, 21, 489–497. [Google Scholar] [CrossRef] [Green Version]
- Chang, M.-Y.; Ong, A.C.M. New treatments for autosomal dominant polycystic kidney disease. Br. J. Clin. Pharmacol. 2013, 76, 524–535. [Google Scholar] [CrossRef] [Green Version]
- Takiar, V.; Nishio, S.; Seo-Mayer, P.; King, J.D.; Li, H.; Zhang, L.; Karihaloo, A.; Hallows, K.R.; Somlo, S.; Caplan, M.J. Activating AMP-activated protein kinase (AMPK) slows renal cystogenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 2462–2467. [Google Scholar] [CrossRef] [Green Version]
- Chiaravalli, M.; Rowe, I.; Mannella, V.; Quilici, G.; Canu, T.; Bianchi, V.; Gurgone, A.; Antunes, S.; D’Adamo, P.; Esposito, A.; et al. 2-Deoxy-d-Glucose Ameliorates PKD Progression. J. Am. Soc. Nephrol. 2015, 27, 1958–1969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Lee, M.S.; Jo, K.; Lee, K.E.; Hwang, J.K. Therapeutic potential of panduratin A, LKB1-dependent AMP-activated protein kinase stimulator, with activation of PPARα/δ for the treatment of obesity. J. Diabetes Obes. Metab. 2011, 13, 584–593. [Google Scholar] [CrossRef]
- Ghosh, D.; Parida, P. Multipotential Therapeutic Bioactive Compound: Panduratin A. Everyman’s Sci. 2020, LV 1&2, 24–29. [Google Scholar]
- Eng-Chong, T.; Yean-Kee, L.; Chin-Fei, C.; Choon-Han, H.; Sher-Ming, W.; Li-Ping, C.T.; Gen-Teck, F.; Khalid, N.; Rahman, N.A.; Karsani, S.A.; et al. Boesenbergia rotunda: From Ethnomedicine to Drug Discovery. Evid. Based Complement. Altern. Med. 2012, 2012, 473637. [Google Scholar] [CrossRef] [Green Version]
- Loghman-Adham, M.; Nauli, S.M.; Soto, C.E.; Kariuki, B.; Zhou, J. Immortalized epithelial cells from human autosomal dominant polycystic kidney cysts. Am. J. Physiol. Ren. Physiol. 2003, 285, F397–F412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thongnuanjan, P.; Soodvilai, S.; Fongsupa, S.; Chabang, N.; Vivithanaporn, P.; Tuchinda, P.; Soodvilai, S. Protective Effect of Panduratin A on Cisplatin-Induced Apoptosis of Human Renal Proximal Tubular Cells and Acute Kidney Injury in Mice. Biol. Pharm. Bull. 2021, 44, 830–837. [Google Scholar] [CrossRef]
- Lager, D.J.; Qian, Q.; Bengal, R.J.; Ishibashi, M.; Torres, V.E. The pck rat: A new model that resembles human autosomal dominant polycystic kidney and liver disease. Kidney Int. 2001, 59, 126–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norman, J. Fibrosis and progression of Autosomal Dominant Polycystic Kidney Disease (ADPKD). Rev. Biochim. Biophys. Acta 2011, 1812, 1327–1336. [Google Scholar] [CrossRef] [Green Version]
- Song, C.J.; Zimmerman, K.A.; Henke, S.J.; Yoder, B.K. Inflammation and Fibrosis in Polycystic Kidney Disease. J. Kidney Dev. Dis. 2017, 60, 323–344. [Google Scholar] [CrossRef]
- Chuang, H.-C.; Chou, C.-C.; Kulp, S.K.; Chen, C.-S. AMPK as a potential anticancer target-friend or foe? J. Curr. Pharm. Des. 2014, 20, 2607–2618. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.K.; Park, K.-G. Metabolic roles of AMPK and metformin in cancer cells. Mol. Cells 2013, 36, 279–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riwanto, M.; Kapoor, S.; Rodriguez, D.; Edenhofer, I.; Segerer, S.; Wüthrich, R.P. Inhibition of aerobic glycolysis attenuates disease progression in polycystic kidney disease. PLoS ONE 2016, 11, e0146654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seliger, S.L.; Abebe, K.Z.; Hallows, K.R.; Miskulin, D.C.; Perrone, R.D.; Watnick, T.; Bae, K.T. A Randomized Clinical Trial of Metformin to Treat Autosomal Dominant Polycystic Kidney Disease. Am. J. Nephrol. 2018, 47, 352–360. [Google Scholar] [CrossRef]
- Song, X.; Tsakiridis, E.; Steinberg, G.R.; Pei, Y. Targeting AMP-activated protein kinase (AMPK) for treatment of autosomal dominant polycystic kidney disease. Cell. Signal. 2020, 73, 109704. [Google Scholar] [CrossRef] [PubMed]
- Rowe, I.; Chiaravalli, M.; Mannella, V.; Ulisse, V.; Quilici, G.; Pema, M.; Song, X.W.; Xu, H.; Mari, S.; Qian, F.; et al. Defective glucose metabolism in polycystic kidney disease identifies a new therapeutic strategy. Nat. Med. 2013, 19, 488–493. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.-Y.; Ong, A.C. Mechanism-Based Therapeutics for Autosomal Dominant Polycystic Kidney Disease: Recent Progress and Future Prospects. Nephron Clin. Pract. 2012, 120, c25–c35. [Google Scholar] [CrossRef]
- Fan, L.X.; Zhou, X.; Sweeney, W.E.; Wallace, D.P.; Avner, E.D.; Grantham, J.J.; Li, X. Smac-Mimetic–Induced Epithelial Cell Death Reduces the Growth of Renal Cysts. J. Am. Soc. Nephrol. 2013, 24, 2010–2022. [Google Scholar] [CrossRef] [Green Version]
- Ikuma, M.; Welsh, M.J. Regulation of CFTR Cl − channel gating by ATP binding and hydrolysis. Proc. Natl. Acad. Sci. USA 2000, 97, 8675–8680. [Google Scholar] [CrossRef] [Green Version]
- Hwang, T.-C.; Kirk, K.L. The CFTR ion channel: Gating, regulation, and anion permeation. J. Cold Spring Harb. Perspect. Med. 2013, 3, a009498. [Google Scholar] [CrossRef] [Green Version]
- Bertrand, C.A.; Frizzell, R.A. The role of regulated CFTR trafficking in epithelial secretion. Am. J. Physiol. Ren. Physiol. 2003, 285, C1–C18. [Google Scholar] [CrossRef] [Green Version]
- Nagao, S.; Kugita, M.; Yoshihara, D.; Yamaguchi, T. Animal Models for Human Polycystic Kidney Disease. Exp. Anim. 2012, 61, 477–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schäfer, K.; Gretz, N.; Bader, M.; Oberbäumer, I.; Eckardt, K.-U.; Kriz, W.; Bachmann, S. Characterization of the Han:SPRD rat model for hereditary polycystic kidney disease. Kidney Int. 1994, 46, 134–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grantham, J.J.; Mulamalla, S.; Swenson-Fields, K.I. Why kidneys fail in autosomal dominant polycystic kidney disease. Nat. Rev. Nephrol. 2011, 7, 556–566. [Google Scholar] [CrossRef] [PubMed]
- Shimomura, Y.; Brock, W.J.; Ito, Y.; Morishita, K. Age-Related Alterations in Blood Biochemical Characterization of Hepatorenal Function in the PCK Rat: A Model of Polycystic Kidney Disease. Int. J. Toxicol. 2015, 34, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Salama, S.M.; Ibrahim, I.A.A.; Shahzad, N.; Al-Ghamdi, S.; Ayoub, N.; AlRashdi, A.S.; Abdulla, M.A.; Salehen, N.; Bilgen, M. Hepatoprotectivity of Panduratin A against liver damage: In vivo demonstration with a rat model of cirrhosis induced by thioacetamide. APMIS 2018, 126, 710–721. [Google Scholar] [CrossRef]
- Choi, S.; Kim, C.; Son, H.; Hwang, J.-K.; Kang, W. Estimation of an Appropriate Human Dose of Boesenbergia pandurata Extracts Based on Allometric Scaling Data of Panduratin A in Mice, Rats, and Dogs. J. Med. Food 2020, 23, 453–458. [Google Scholar] [CrossRef]
- Tuchinda, P.; Reutrakul, V.; Claeson, P.; Pongprayoon, U.; Sematong, T.; Santisuk, T.; Taylor, W.C. Anti-inflammatory cyclohexenyl chalcone derivatives in Boesenbergia pandurata. Phytochemistry 2002, 59, 169–173. [Google Scholar] [CrossRef]
- Owens, D.R. Spontaneous, Surgically and Chemically Induced Models of Disease. In The Laboratory Rat; Elsevier: Amsterdam, The Netherlands, 2006; pp. 711–732. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tonum, K.; Srimai, N.; Chabang, N.; Fongsupa, S.; Tuchinda, P.; Torres, J.A.; Weimbs, T.; Soodvilai, S. Pharmacological Effects of Panduratin A on Renal Cyst Development in In Vitro and In Vivo Models of Polycystic Kidney Disease. Int. J. Mol. Sci. 2022, 23, 4328. https://doi.org/10.3390/ijms23084328
Tonum K, Srimai N, Chabang N, Fongsupa S, Tuchinda P, Torres JA, Weimbs T, Soodvilai S. Pharmacological Effects of Panduratin A on Renal Cyst Development in In Vitro and In Vivo Models of Polycystic Kidney Disease. International Journal of Molecular Sciences. 2022; 23(8):4328. https://doi.org/10.3390/ijms23084328
Chicago/Turabian StyleTonum, Kanlayanee, Nipitpon Srimai, Napason Chabang, Somsak Fongsupa, Patoomratana Tuchinda, Jacob A. Torres, Thomas Weimbs, and Sunhapas Soodvilai. 2022. "Pharmacological Effects of Panduratin A on Renal Cyst Development in In Vitro and In Vivo Models of Polycystic Kidney Disease" International Journal of Molecular Sciences 23, no. 8: 4328. https://doi.org/10.3390/ijms23084328
APA StyleTonum, K., Srimai, N., Chabang, N., Fongsupa, S., Tuchinda, P., Torres, J. A., Weimbs, T., & Soodvilai, S. (2022). Pharmacological Effects of Panduratin A on Renal Cyst Development in In Vitro and In Vivo Models of Polycystic Kidney Disease. International Journal of Molecular Sciences, 23(8), 4328. https://doi.org/10.3390/ijms23084328