
CRISPR/Cas9-Mediated Allele-Specific Disruption of a Dominant COL6A1 Pathogenic Variant Improves Collagen VI Network in Patient Fibroblasts

 , , , , , and
, , , , , and
Abstract
:
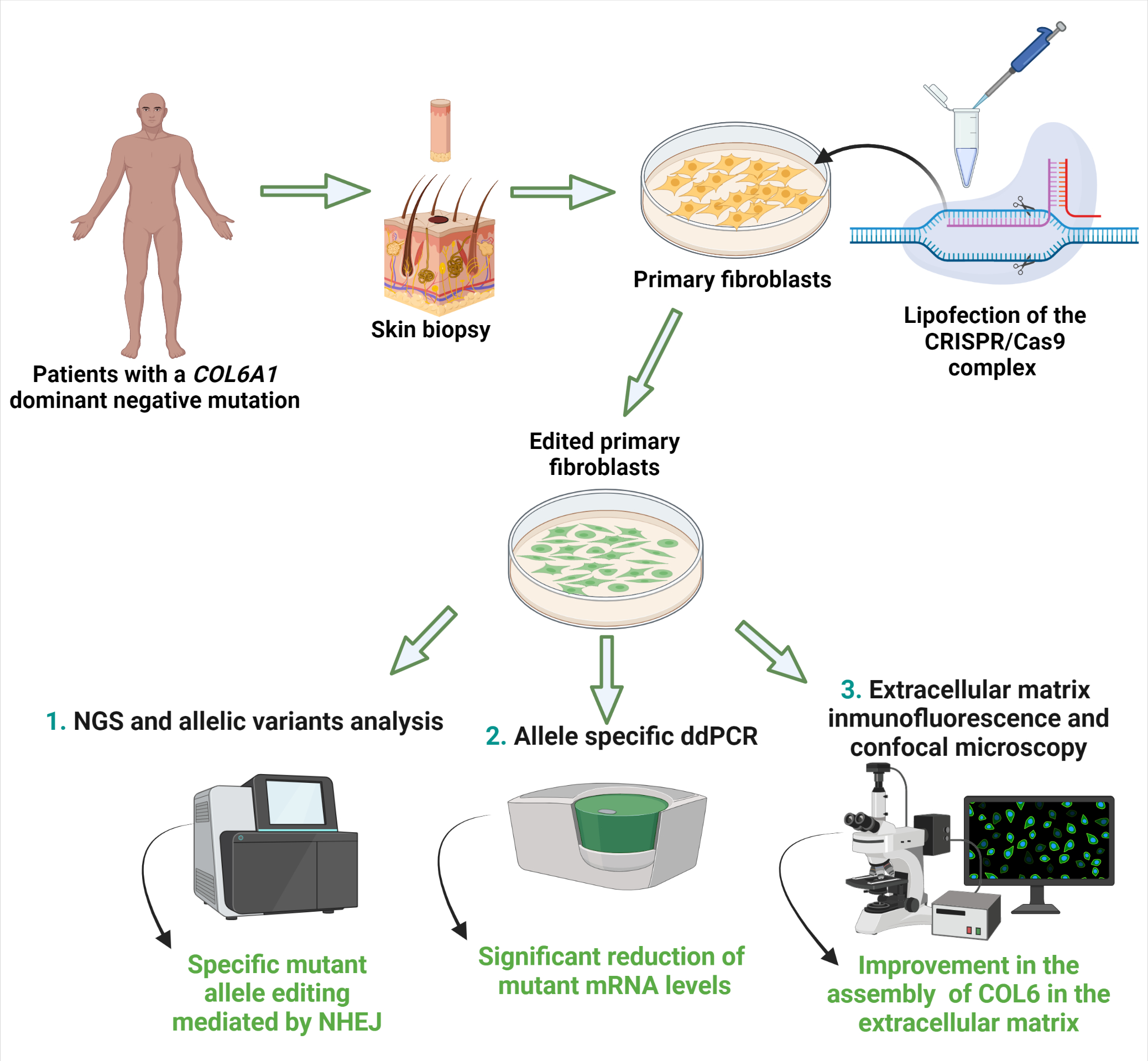{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Design and Analysis of crRNAs Targeting Exon 10 of COL6A1
2.2. CRISPR/Cas9 Transfection and Next Generation Sequencing of Fibroblasts
2.3. NHEJ-Mediated COL6A1 Editing of Patient Fibroblasts Reduces the RNA Levels of the Mutant Allele
2.4. Silencing of the Mutant Allele Partially Restores Collagen VI Intensity and Microfibrillar Structure in the Extracellular Matrix
3. Discussion
4. Materials and Methods
4.1. Human Fibroblasts
4.2. crRNAs and ssDNA Donor Template Design
4.3. Cell transfection
4.4. Fluorescence-Activated Cell Sorting
4.5. Genomic DNA Extraction
4.6. RNA Extraction and Retrotranscription
4.7. Characterization of Gene-Edited Allelic Variants
4.8. Digital Droplet PCR
4.9. Extracellular Matrix Immunostaining
4.10. Confocal and Super-Resolution Imaging
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Graziano, A.; Bianco, F.; D’Amico, A.; Moroni, I.; Messina, S.; Bruno, C.; Pegoraro, E.; Mora, M.; Astrea, G.; Magri, F.; et al. Prevalence of congenital muscular dystrophy in Italy: A population study. Neurology 2015, 84, 904–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimenez-Mallebrera, C.; Brown, S.C.; Sewry, C.A.; Muntoni, F. Congenital muscular dystrophy: Molecular and cellular aspects. Cell. Mol. Life Sci. 2005, 62, 809–823. [Google Scholar] [CrossRef] [PubMed]
- Mohassel, P.; Foley, A.R.; Bönnemann, C.G. Extracellular matrix-driven congenital muscular dystrophies. Matrix Biol. 2018, 71–72, 188–204. [Google Scholar] [CrossRef] [PubMed]
- Natera-de Benito, D.; Foley, A.R.; Domínguez-González, C.; Ortez, C.; Jain, M.; Mebrahtu, A.; Donkervoort, S.; Hu, Y.; Fink, M.; Yun, P.; et al. Association of Initial Maximal Motor Ability With Long-term Functional Outcome in Patients With COL6-Related Dystrophies. Neurology 2021, 96, e1413–e1424. [Google Scholar] [CrossRef]
- Batra, A.; Lott, D.J.; Willcocks, R.; Forbes, S.C.; Triplett, W.; Dastgir, J.; Yun, P.; Reghan Foley, A.; Bönnemann, C.G.; Vandenborne, K.; et al. Lower Extremity Muscle Involvement in the Intermediate and Bethlem Myopathy Forms of COL6-Related Dystrophy and Duchenne Muscular Dystrophy: A Cross-Sectional Study. J. Neuromuscul. Dis. 2020, 7, 407–417. [Google Scholar] [CrossRef]
- Bönnemann, C.G. The collagen VI-related myopathies: Muscle meets its matrix. Nat. Rev. Neurol. 2011, 7, 379–390. [Google Scholar] [CrossRef] [Green Version]
- Adam, M.P.; Ardinger, H.H.; Pagon, R.A.; Wallace, S.E.; Bean, L.J.H.; Mirzaa, G.; Amemiya, A. GeneReviews; University of Washington: Seattle, WA, USA.
- Camacho Vanegas, O.; Bertini, E.; Zhang, R.Z.; Petrini, S.; Minosse, C.; Sabatelli, P.; Giusti, B.; Chu, M.L.; Pepe, G. Ullrich scleroatonic muscular dystrophy is caused by recessive mutations in collagen type VI. Proc. Natl. Acad. Sci. USA 2001, 98, 7516–7521. [Google Scholar] [CrossRef] [Green Version]
- Prockop, D.J. Mutations that alter the primary structure of type I collagen. The perils of a system for generating large structures by the principle of nucleated growth. J. Biol. Chem. 1990, 265, 15349–15352. [Google Scholar] [CrossRef]
- Lamandé, S.R.; Bateman, J.F. Collagen VI disorders: Insights on form and function in the extracellular matrix and beyond. Matrix Biol. 2018, 71–72, 348–367. [Google Scholar] [CrossRef]
- Engvall, E.; Hessle, H.; Klier, G. Molecular assembly, secretion, and matrix deposition of type VI collagen. J. Cell Biol. 1986, 102, 703–710. [Google Scholar] [CrossRef] [Green Version]
- Jimenez-Mallebrera, C.; Maioli, M.A.; Kim, J.; Brown, S.C.; Feng, L.; Lampe, A.K.; Bushby, K.; Hicks, D.; Flanigan, K.M.; Bonnemann, C.; et al. A comparative analysis of collagen VI production in muscle, skin and fibroblasts from 14 Ullrich congenital muscular dystrophy patients with dominant and recessive COL6A mutations. Neuromuscul. Disord. 2006, 16, 571–582. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, R.J.; Foley, A.R.; Dastgir, J.; Asman, S.; Dunn, D.M.; Zou, Y.; Hu, Y.; Donkervoort, S.; Flanigan, K.M.; Swoboda, K.J.; et al. Position of glycine substitutions in the triple helix of COL6A1, COL6A2, and COL6A3 is correlated with severity and mode of inheritance in collagen VI myopathies. Hum. Mutat. 2013, 34, 1558–1567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolduc, V.; Foley, A.R.; Solomon-Degefa, H.; Sarathy, A.; Donkervoort, S.; Hu, Y.; Chen, G.S.; Sizov, K.; Nalls, M.; Zhou, H.; et al. A recurrent COL6A1 pseudoexon insertion causes muscular dystrophy and is effectively targeted by splice-correction therapies. JCI Insight 2019, 4, e124403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foley, A.R.; Hu, Y.; Zou, Y.; Yang, M.; Medne, L.; Leach, M.; Conlin, L.K.; Spinner, N.; Shaikh, T.H.; Falk, M.; et al. Large genomic deletions: A novel cause of Ullrich congenital muscular dystrophy. Ann. Neurol. 2011, 69, 206–211. [Google Scholar] [CrossRef] [Green Version]
- Higuchi, I.; Shiraishi, T.; Hashiguchi, T.; Suehara, M.; Niiyama, T.; Nakagawa, M.; Arimura, K.; Maruyama, I.; Osame, M. Frameshift mutation in the collagen VI gene causes Ullrich’s disease. Ann. Neurol. 2001, 50, 261–265. [Google Scholar] [CrossRef]
- Bolduc, V.; Zou, Y.; Ko, D.; Bönnemann, C.G. siRNA-mediated Allele-specific Silencing of a COL6A3 Mutation in a Cellular Model of Dominant Ullrich Muscular Dystrophy. Mol. Ther. Nucleic Acids 2014, 3, e147. [Google Scholar] [CrossRef]
- Noguchi, S.; Ogawa, M.; Kawahara, G.; Malicdan, M.C.; Nishino, I. Allele-specific Gene Silencing of Mutant mRNA Restores Cellular Function in Ullrich Congenital Muscular Dystrophy Fibroblasts. Mol. Ther. Nucleic Acids 2014, 3, e171. [Google Scholar] [CrossRef]
- Marrosu, E.; Ala, P.; Muntoni, F.; Zhou, H. Gapmer Antisense Oligonucleotides Suppress the Mutant Allele of COL6A3 and Restore Functional Protein in Ullrich Muscular Dystrophy. Mol. Ther. Nucleic Acids 2017, 8, 416–427. [Google Scholar] [CrossRef] [Green Version]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [Green Version]
- Jacobi, A.M.; Rettig, G.R.; Turk, R.; Collingwood, M.A.; Zeiner, S.A.; Quadros, R.M.; Harms, D.W.; Bonthuis, P.J.; Gregg, C.; Ohtsuka, M.; et al. Simplified CRISPR tools for efficient genome editing and streamlined protocols for their delivery into mammalian cells and mouse zygotes. Methods 2017, 121–122, 16–28. [Google Scholar] [CrossRef] [PubMed]
- Mojica, F.J.M.; Montoliu, L. On the Origin of CRISPR-Cas Technology: From Prokaryotes to Mammals. Trends Microbiol. 2016, 24, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Adli, M. The CRISPR tool kit for genome editing and beyond. Nat. Commun. 2018, 9, 1911. [Google Scholar] [CrossRef] [PubMed]
- Wojtal, D.; Kemaladewi, D.U.; Malam, Z.; Abdullah, S.; Wong, T.W.; Hyatt, E.; Baghestani, Z.; Pereira, S.; Stavropoulos, J.; Mouly, V.; et al. Spell Checking Nature: Versatility of CRISPR/Cas9 for Developing Treatments for Inherited Disorders. Am. J. Hum. Genet. 2016, 98, 90–101. [Google Scholar] [CrossRef] [Green Version]
- Breaking-Cas. Available online: http://www.bioinfogp.cnb.csic.es/tools/breakingcas/ (accessed on 4 April 2020).
- Fernández, A.; Morín, M.; Muñoz-Santos, D.; Josa, S.; Montero, A.; Rubio-Fernández, M.; Cantero, M.; Fernández, J.; Del Hierro, M.J.; Castrillo, M.; et al. Simple Protocol for Generating and Genotyping Genome-Edited Mice With CRISPR-Cas9 Reagents. Curr. Protoc. Mouse Biol. 2020, 10, e69. [Google Scholar] [CrossRef] [PubMed]
- Fananas-Baquero, S.; Quintana-Bustamante, O.; Morin, M.; Fernandez, S.; Fernandez, V.; Bueren, J.A.; Moreno-Pelayo, M.A.; Segovia, J.C. Highly Precise Single-Stranded Template Repair of a Pyruvate Kinase Deficiency-Causing Mutation in 732 Patient-Derived Lymphoblastic Cell Line. In Proceedings of the 23rd Annual Meeting of the American Society for Gene and Cell Therapy, Virtual Format, 12–15 May 2020; p. 363. [Google Scholar]
- Quintana-Bustamante, O.; Fananas-Baquero, S.; Morin, M.; Fernandez, S.; Fernandez, V.; Bueren, J.A.; Moreno-Pelayo, M.A.; Segovia, J.C. Highly precise gene editing correction of a Pyruvate Kinase deficiency-causing mutation in patient-derived lymphoblastic cells using single stranded oligodeoxinucleotides. In Proceedings of the ESGCT 27th Annual Congress, Barcelona, Spain, 15–19 October 2019; p. 729. [Google Scholar]
- Cervera, S.T.; Rodríguez-Martín, C.; Fernández-Tabanera, E.; Melero-Fernández de Mera, R.M.; Morin, M.; Fernández-Peñalver, S.; Iranzo-Martínez, M.; Amhih-Cardenas, J.; García-García, L.; González-González, L.; et al. Therapeutic Potential of EWSR1–FLI1 Inactivation by CRISPR/Cas9 in Ewing Sarcoma. Cancers 2021, 13, 3783. [Google Scholar] [CrossRef] [PubMed]
- Gualandi, F.; Manzati, E.; Sabatelli, P.; Passarelli, C.; Bovolenta, M.; Pellegrini, C.; Perrone, D.; Squarzoni, S.; Pegoraro, E.; Bonaldo, P.; et al. Antisense-induced messenger depletion corrects a COL6A2 dominant mutation in Ullrich myopathy. Hum. Gene Ther. 2012, 23, 1313–1318. [Google Scholar] [CrossRef]
- Bazaga, A.; Mònica, R.; Badosa, C.; Jiménez-Mallebrera, C.; Porta, J.M. A Convolutional Neural Network for the automatic diagnosis of collagen VI-related muscular dystrophies. Appl. Soft Comput. 2019, 85, 105772. [Google Scholar] [CrossRef] [Green Version]
- Barlow, A.M.; Mostaço-Guidolin, L.B.; Osei, E.T.; Booth, S.; Hackett, T.L. Super resolution measurement of collagen fibers in biological samples: Validation of a commercial solution for multiphoton microscopy. PLoS ONE 2020, 15, e0229278. [Google Scholar] [CrossRef] [Green Version]
- Ridler, C. CRISPR therapy shows promise in Duchenne muscular dystrophy. Nat. Rev. Neurol. 2018, 14, 632–633. [Google Scholar] [CrossRef]
- Miyaoka, Y.; Berman, J.R.; Cooper, S.B.; Mayerl, S.J.; Chan, A.H.; Zhang, B.; Karlin-Neumann, G.A.; Conklin, B.R. Systematic quantification of HDR and NHEJ reveals effects of locus, nuclease, and cell type on genome-editing. Sci. Rep. 2016, 6, 23549. [Google Scholar] [CrossRef] [PubMed]
- Osborn, M.J.; Starker, C.G.; McElroy, A.N.; Webber, B.R.; Riddle, M.J.; Xia, L.; DeFeo, A.P.; Gabriel, R.; Schmidt, M.; von Kalle, C.; et al. TALEN-based gene correction for epidermolysis bullosa. Mol. Ther. 2013, 21, 1151–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izmiryan, A.; Danos, O.; Hovnanian, A. Meganuclease-Mediated COL7A1 Gene Correction for Recessive Dystrophic Epidermolysis Bullosa. J. Investig. Dermatol. 2016, 136, 872–875. [Google Scholar] [CrossRef] [PubMed]
- Hainzl, S.; Peking, P.; Kocher, T.; Murauer, E.M.; Larcher, F.; Del Rio, M.; Duarte, B.; Steiner, M.; Klausegger, A.; Bauer, J.W.; et al. COL7A1 Editing via CRISPR/Cas9 in Recessive Dystrophic Epidermolysis Bullosa. Mol. Ther. 2017, 25, 2573–2584. [Google Scholar] [CrossRef] [Green Version]
- Newby, G.A.; Liu, D.R. In vivo somatic cell base editing and prime editing. Mol. Ther. 2021, 29, 3107–3124. [Google Scholar] [CrossRef]
- Yin, H.; Song, C.Q.; Dorkin, J.R.; Zhu, L.J.; Li, Y.; Wu, Q.; Park, A.; Yang, J.; Suresh, S.; Bizhanova, A.; et al. Therapeutic genome editing by combined viral and non-viral delivery of CRISPR system components in vivo. Nat. Biotechnol. 2016, 34, 328–333. [Google Scholar] [CrossRef]
- Shao, Y.; Wang, L.; Guo, N.; Wang, S.; Yang, L.; Li, Y.; Wang, M.; Yin, S.; Han, H.; Zeng, L.; et al. Cas9-nickase-mediated genome editing corrects hereditary tyrosinemia in rats. J. Biol. Chem. 2018, 293, 6883–6892. [Google Scholar] [CrossRef] [Green Version]
- Guan, Y.; Ma, Y.; Li, Q.; Sun, Z.; Ma, L.; Wu, L.; Wang, L.; Zeng, L.; Shao, Y.; Chen, Y.; et al. CRISPR/Cas9-mediated somatic correction of a novel coagulator factor IX gene mutation ameliorates hemophilia in mouse. EMBO Mol. Med. 2016, 8, 477–488. [Google Scholar] [CrossRef]
- Yin, H.; Xue, W.; Chen, S.; Bogorad, R.L.; Benedetti, E.; Grompe, M.; Koteliansky, V.; Sharp, P.A.; Jacks, T.; Anderson, D.G. Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat. Biotechnol. 2014, 32, 551–553. [Google Scholar] [CrossRef] [Green Version]
- Martin, R.M.; Ikeda, K.; Cromer, M.K.; Uchida, N.; Nishimura, T.; Romano, R.; Tong, A.J.; Lemgart, V.T.; Camarena, J.; Pavel-Dinu, M.; et al. Highly Efficient and Marker-free Genome Editing of Human Pluripotent Stem Cells by CRISPR-Cas9 RNP and AAV6 Donor-Mediated Homologous Recombination. Cell Stem Cell 2019, 24, 821–828.e825. [Google Scholar] [CrossRef]
- Gomez-Ospina, N.; Scharenberg, S.G.; Mostrel, N.; Bak, R.O.; Mantri, S.; Quadros, R.M.; Gurumurthy, C.B.; Lee, C.; Bao, G.; Suarez, C.J.; et al. Human genome-edited hematopoietic stem cells phenotypically correct Mucopolysaccharidosis type I. Nat. Commun. 2019, 10, 4045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dever, D.P.; Bak, R.O.; Reinisch, A.; Camarena, J.; Washington, G.; Nicolas, C.E.; Pavel-Dinu, M.; Saxena, N.; Wilkens, A.B.; Mantri, S.; et al. CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells. Nature 2016, 539, 384–389. [Google Scholar] [CrossRef] [PubMed]
- Bonafont, J.; Mencía, A.; Chacón-Solano, E.; Srifa, W.; Vaidyanathan, S.; Romano, R.; Garcia, M.; Hervás-Salcedo, R.; Ugalde, L.; Duarte, B.; et al. Correction of recessive dystrophic epidermolysis bullosa by homology-directed repair-mediated genome editing. Mol. Ther. 2021, 29, 2008–2018. [Google Scholar] [CrossRef] [PubMed]
- Izmiryan, A.; Ganier, C.; Bovolenta, M.; Schmitt, A.; Mavilio, F.; Hovnanian, A. Ex Vivo COL7A1 Correction for Recessive Dystrophic Epidermolysis Bullosa Using CRISPR/Cas9 and Homology-Directed Repair. Mol. Ther. Nucleic Acids 2018, 12, 554–567. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.P.; Newby, G.A.; Liu, D.R. Precision genome editing using cytosine and adenine base editors in mammalian cells. Nat. Protoc. 2021, 16, 1089–1128. [Google Scholar] [CrossRef] [PubMed]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef]
- Jalil, S.; Keskinen, T.; Maldonado, R.; Sokka, J.; Trokovic, R.; Otonkoski, T.; Wartiovaara, K. Simultaneous high-efficiency base editing and reprogramming of patient fibroblasts. Stem Cell Rep. 2021, 16, 3064–3075. [Google Scholar] [CrossRef]
- Gaudelli, N.M.; Lam, D.K.; Rees, H.A.; Solá-Esteves, N.M.; Barrera, L.A.; Born, D.A.; Edwards, A.; Gehrke, J.M.; Lee, S.J.; Liquori, A.J.; et al. Directed evolution of adenine base editors with increased activity and therapeutic application. Nat. Biotechnol. 2020, 38, 892–900. [Google Scholar] [CrossRef]
- Iyer, V.; Shen, B.; Zhang, W.; Hodgkins, A.; Keane, T.; Huang, X.; Skarnes, W.C. Off-target mutations are rare in Cas9-modified mice. Nat. Methods 2015, 12, 479. [Google Scholar] [CrossRef]
- Seruggia, D.; Fernández, A.; Cantero, M.; Pelczar, P.; Montoliu, L. Functional validation of mouse tyrosinase non-coding regulatory DNA elements by CRISPR-Cas9-mediated mutagenesis. Nucleic Acids Res. 2015, 43, 4855–4867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paco, S.; Kalko, S.G.; Jou, C.; Rodríguez, M.A.; Corbera, J.; Muntoni, F.; Feng, L.; Rivas, E.; Torner, F.; Gualandi, F.; et al. Gene expression profiling identifies molecular pathways associated with collagen VI deficiency and provides novel therapeutic targets. PLoS ONE 2013, 8, e77430. [Google Scholar] [CrossRef] [PubMed]
- Paco, S.; Casserras, T.; Rodríguez, M.A.; Jou, C.; Puigdelloses, M.; Ortez, C.I.; Diaz-Manera, J.; Gallardo, E.; Colomer, J.; Nascimento, A.; et al. Transcriptome Analysis of Ullrich Congenital Muscular Dystrophy Fibroblasts Reveals a Disease Extracellular Matrix Signature and Key Molecular Regulators. PLoS ONE 2015, 10, e0145107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guadagnin, E.; Mohassel, P.; Johnson, K.R.; Yang, L.; Santi, M.; Uapinyoying, P.; Dastgir, J.; Hu, Y.; Dillmann, A.; Cookson, M.R.; et al. Transcriptome analysis of collagen VI-related muscular dystrophy muscle biopsies. Ann. Clin. Transl. Neurol. 2021, 8, 2184–2198. [Google Scholar] [CrossRef]
- Lee, K.; Conboy, M.; Park, H.M.; Jiang, F.; Kim, H.J.; Dewitt, M.A.; Mackley, V.A.; Chang, K.; Rao, A.; Skinner, C.; et al. Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA. Nat. Biomed. Eng. 2017, 1, 889–901. [Google Scholar] [CrossRef] [Green Version]
- Chemello, F.; Bassel-Duby, R.; Olson, E.N. Correction of muscular dystrophies by CRISPR gene editing. J. Clin. Investig. 2020, 130, 2766–2776. [Google Scholar] [CrossRef]
- Oliveros, J.C.; Franch, M.; Tabas-Madrid, D.; San-León, D.; Montoliu, L.; Cubas, P.; Pazos, F. Breaking-Cas-interactive design of guide RNAs for CRISPR-Cas experiments for ENSEMBL genomes. Nucleic Acids Res. 2016, 44, W267–W271. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
López-Márquez, A.; Morín, M.; Fernández-Peñalver, S.; Badosa, C.; Hernández-Delgado, A.; Natera-de Benito, D.; Ortez, C.; Nascimento, A.; Grinberg, D.; Balcells, S.; et al. CRISPR/Cas9-Mediated Allele-Specific Disruption of a Dominant COL6A1 Pathogenic Variant Improves Collagen VI Network in Patient Fibroblasts. Int. J. Mol. Sci. 2022, 23, 4410. https://doi.org/10.3390/ijms23084410
López-Márquez A, Morín M, Fernández-Peñalver S, Badosa C, Hernández-Delgado A, Natera-de Benito D, Ortez C, Nascimento A, Grinberg D, Balcells S, et al. CRISPR/Cas9-Mediated Allele-Specific Disruption of a Dominant COL6A1 Pathogenic Variant Improves Collagen VI Network in Patient Fibroblasts. International Journal of Molecular Sciences. 2022; 23(8):4410. https://doi.org/10.3390/ijms23084410
Chicago/Turabian StyleLópez-Márquez, Arístides, Matías Morín, Sergio Fernández-Peñalver, Carmen Badosa, Alejandro Hernández-Delgado, Daniel Natera-de Benito, Carlos Ortez, Andrés Nascimento, Daniel Grinberg, Susanna Balcells, and et al. 2022. "CRISPR/Cas9-Mediated Allele-Specific Disruption of a Dominant COL6A1 Pathogenic Variant Improves Collagen VI Network in Patient Fibroblasts" International Journal of Molecular Sciences 23, no. 8: 4410. https://doi.org/10.3390/ijms23084410
APA StyleLópez-Márquez, A., Morín, M., Fernández-Peñalver, S., Badosa, C., Hernández-Delgado, A., Natera-de Benito, D., Ortez, C., Nascimento, A., Grinberg, D., Balcells, S., Roldán, M., Moreno-Pelayo, M. Á., & Jiménez-Mallebrera, C. (2022). CRISPR/Cas9-Mediated Allele-Specific Disruption of a Dominant COL6A1 Pathogenic Variant Improves Collagen VI Network in Patient Fibroblasts. International Journal of Molecular Sciences, 23(8), 4410. https://doi.org/10.3390/ijms23084410