
Fecal Microbiota Transplantation Derived from Alzheimer’s Disease Mice Worsens Brain Trauma Outcomes in Wild-Type Controls

 , , and
, , and 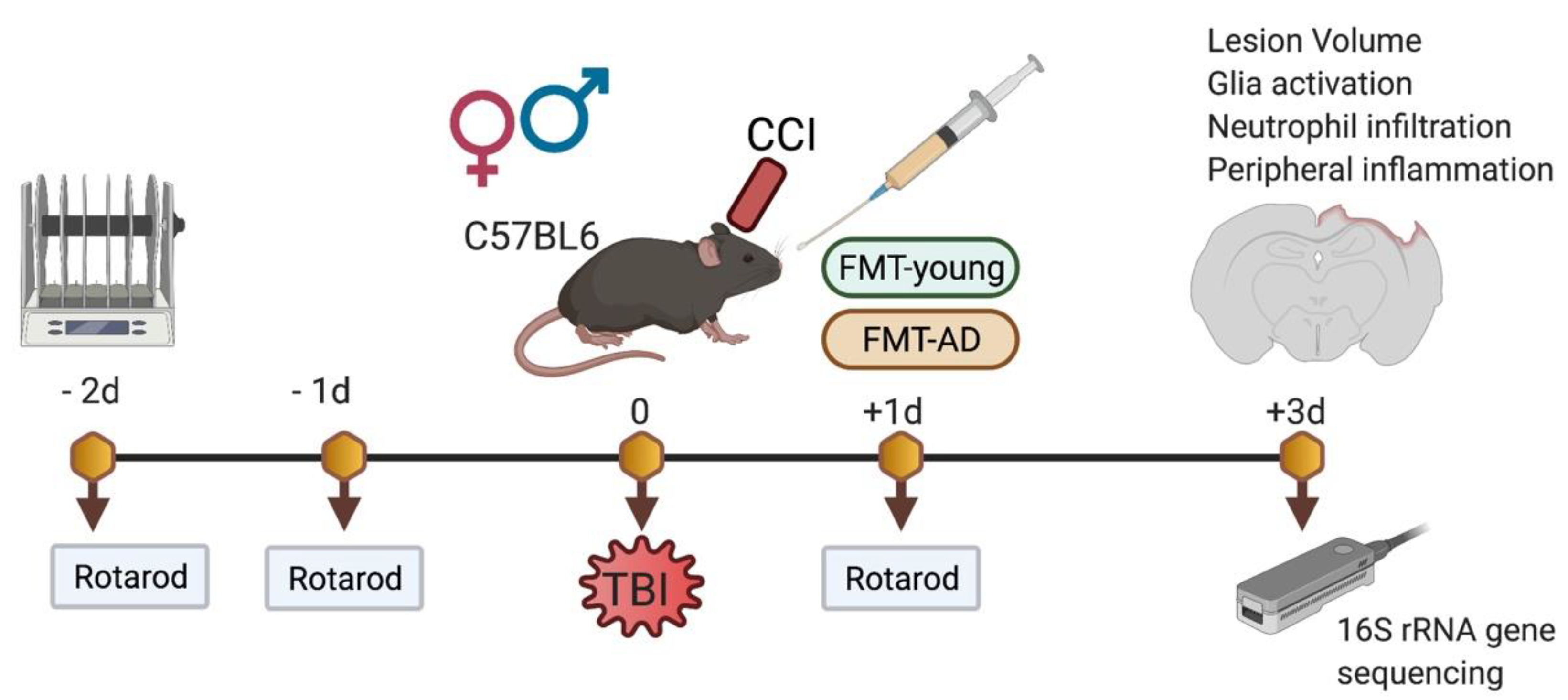{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
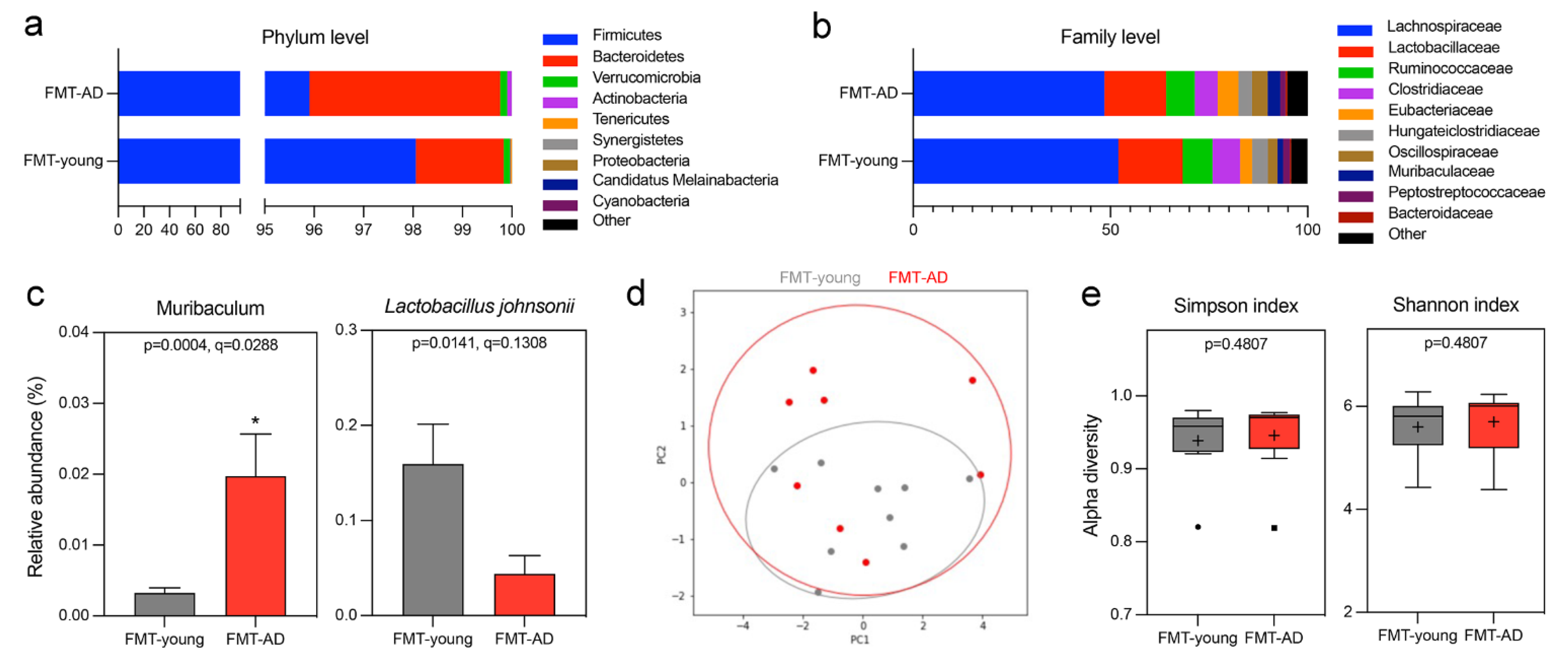
2.1. FMT-AD Induced Gut Microbiome Changes in C57BL/6 Recipient Mice Following TBI
2.2. Microbiota from AD Mice Aggravated the Lesion Size after TBI
2.3. Microbiota from AD Mice Impaired Motor Ability after TBI
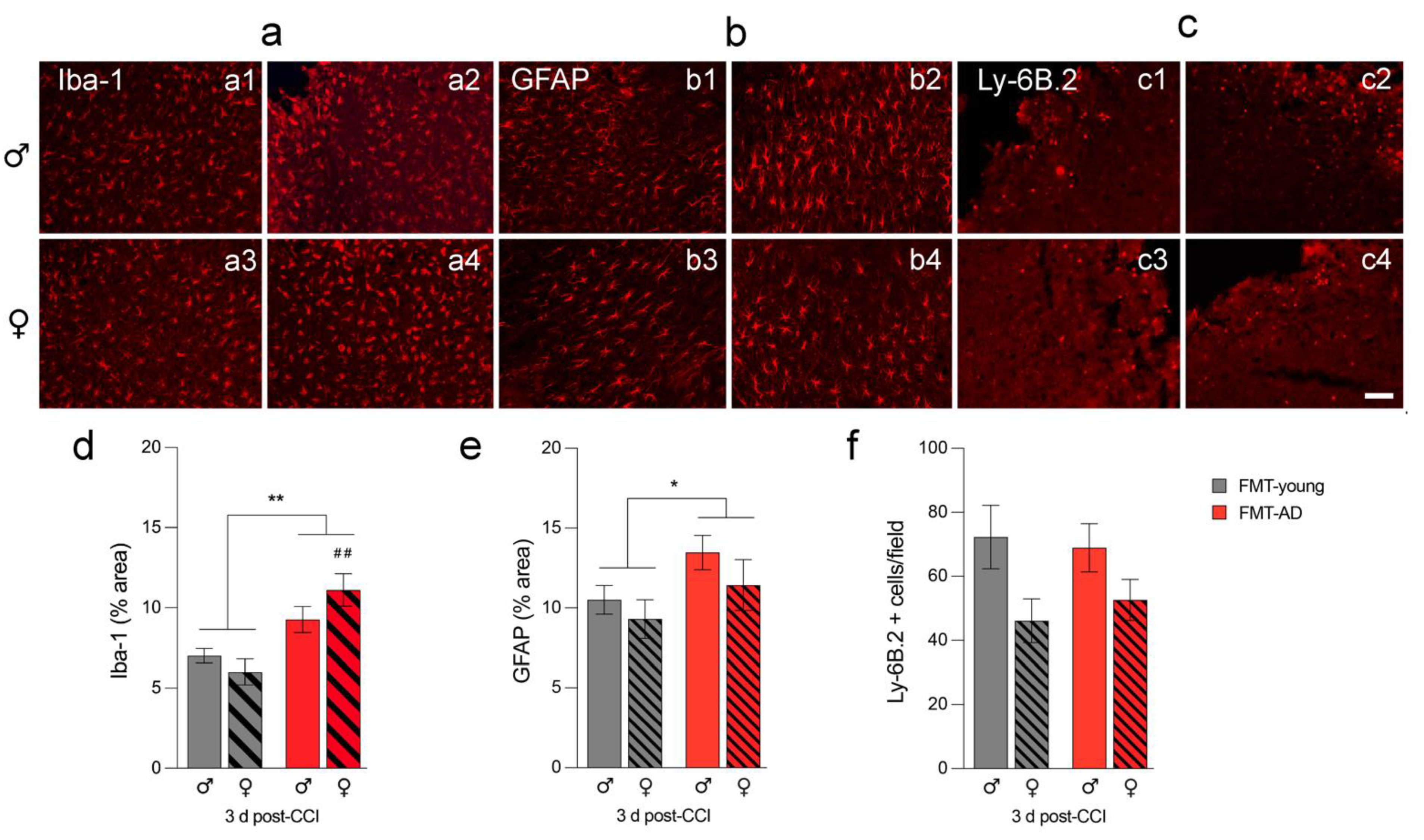
2.4. FMT-AD Led to Increased Neuroinflammation
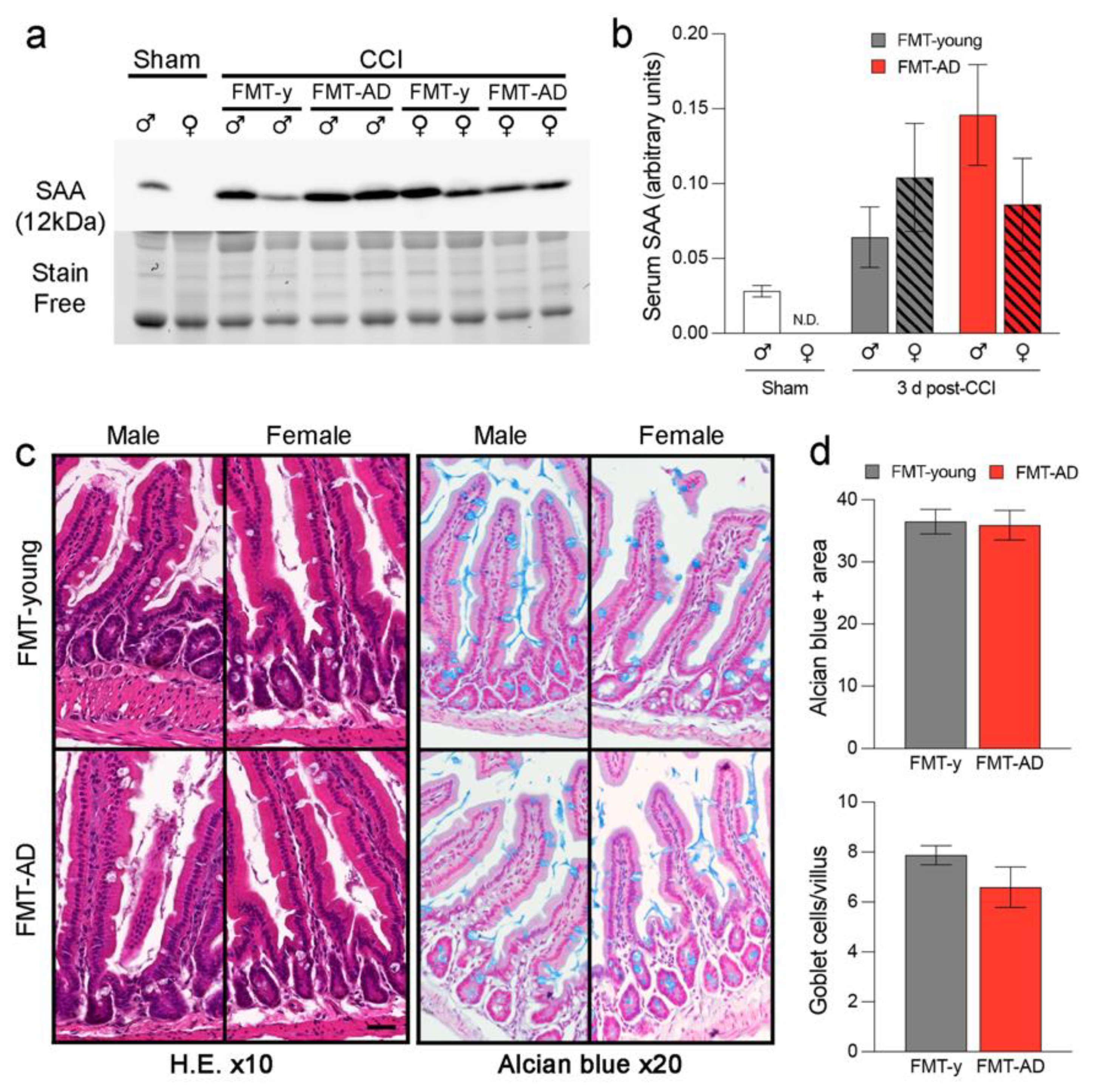
2.5. FMT-AD Did Not Increase Serum Amyloid A (SAA) Levels Nor Did It Induce Gut Alterations
3. Discussion
4. Materials and Methods
4.1. Mice and Traumatic Brain Injury Model
4.2. Fecal Microbiota Transplantation (FMT), Fecal Sample Collection, and DNA Extraction
4.3. Long Read 16S rRNA Gene Sequencing
4.4. Long-Read 16S Bioinformatic Analysis
4.5. Rotarod
4.6. Cresyl Violet Staining and Lesion Measurements
4.7. Immunofluorescence Analysis
4.8. Western Blot Analysis
4.9. Gut Histological Analysis
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–890. [Google Scholar] [CrossRef]
- De Strooper, B.; Karran, E. The Cellular Phase of Alzheimer’s Disease. Cell 2016, 164, 603–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buccellato, F.R.; D’Anca, M.; Fenoglio, C.; Scarpini, E.; Galimberti, D. Role of Oxidative Damage in Alzheimer’s Disease and Neurodegeneration: From Pathogenic Mechanisms to Biomarker Discovery. Antioxidants 2021, 10, 1353. [Google Scholar] [CrossRef] [PubMed]
- Jo, D.S.; Park, N.Y.; Cho, D.H. Peroxisome quality control and dysregulated lipid metabolism in neurodegenerative diseases. Exp. Mol. Med. 2020, 52, 1486–1495. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 30. [Google Scholar] [CrossRef]
- Webers, A.; Heneka, M.T.; Gleeson, P.A. The role of innate immune responses and neuroinflammation in amyloid accumulation and progression of Alzheimer’s disease. Immunol. Cell Biol. 2020, 98, 28–41. [Google Scholar] [CrossRef]
- Atri, A. The Alzheimer’s Disease Clinical Spectrum: Diagnosis and Management. Med. Clin. N. Am. 2019, 103, 263–293. [Google Scholar] [CrossRef]
- Villa, C.; Lavitrano, M.; Salvatore, E.; Combi, R. Molecular and Imaging Biomarkers in Alzheimer’s Disease: A Focus on Recent Insights. J. Pers. Med. 2020, 10, 61. [Google Scholar] [CrossRef]
- Torok, N.; Tanaka, M.; Vecsei, L. Searching for Peripheral Biomarkers in Neurodegenerative Diseases: The Tryptophan-Kynurenine Metabolic Pathway. Int. J. Mol. Sci. 2020, 21, 9338. [Google Scholar] [CrossRef]
- Vaz, M.; Silvestre, S. Alzheimer’s disease: Recent treatment strategies. Eur. J. Pharm. 2020, 887, 173554. [Google Scholar] [CrossRef]
- Fleminger, S.; Oliver, D.L.; Lovestone, S.; Rabe-Hesketh, S.; Giora, A. Head injury as a risk factor for Alzheimer’s disease: The evidence 10 years on; a partial replication. J. Neurol. Neurosurg. Psychiatry 2003, 74, 857–862. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.K.; Hou, S.W.; Lee, C.C.; Hsu, C.Y.; Huang, Y.S.; Su, Y.C. Increased risk of dementia in patients with mild traumatic brain injury: A nationwide cohort study. PLoS ONE 2013, 8, e62422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, D.E.; Kaup, A.; Kirby, K.A.; Byers, A.L.; Diaz-Arrastia, R.; Yaffe, K. Traumatic brain injury and risk of dementia in older veterans. Neurology 2014, 83, 312–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardner, R.C.; Burke, J.F.; Nettiksimmons, J.; Kaup, A.; Barnes, D.E.; Yaffe, K. Dementia risk after traumatic brain injury vs nonbrain trauma: The role of age and severity. JAMA Neurol. 2014, 71, 1490–1497. [Google Scholar] [CrossRef]
- Suhanov, A.V.; Pilipenko, P.I.; Korczyn, A.D.; Hofman, A.; Voevoda, M.I.; Shishkin, S.V.; Simonova, G.I.; Nikitin, Y.P.; Feigin, V.L. Risk factors for Alzheimer’s disease in Russia: A case-control study. Eur. J. Neurol. 2006, 13, 990–995. [Google Scholar] [CrossRef]
- McConeghy, K.W.; Hatton, J.; Hughes, L.; Cook, A.M. A review of neuroprotection pharmacology and therapies in patients with acute traumatic brain injury. CNS Drugs 2012, 26, 613–636. [Google Scholar] [CrossRef] [PubMed]
- Gruenbaum, S.E.; Zlotnik, A.; Gruenbaum, B.F.; Hersey, D.; Bilotta, F. Pharmacologic Neuroprotection for Functional Outcomes After Traumatic Brain Injury: A Systematic Review of the Clinical Literature. CNS Drugs 2016, 30, 791–806. [Google Scholar] [CrossRef]
- Simon, D.W.; McGeachy, M.J.; Bayir, H.; Clark, R.S.; Loane, D.J.; Kochanek, P.M. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat. Rev. Neurol. 2017, 13, 171–191. [Google Scholar] [CrossRef] [Green Version]
- Kokiko-Cochran, O.; Ransohoff, L.; Veenstra, M.; Lee, S.; Saber, M.; Sikora, M.; Teknipp, R.; Xu, G.; Bemiller, S.; Wilson, G.; et al. Altered Neuroinflammation and Behavior after Traumatic Brain Injury in a Mouse Model of Alzheimer’s Disease. J. Neurotrauma 2016, 33, 625–640. [Google Scholar] [CrossRef] [Green Version]
- Washington, P.M.; Morffy, N.; Parsadanian, M.; Zapple, D.N.; Burns, M.P. Experimental traumatic brain injury induces rapid aggregation and oligomerization of amyloid-beta in an Alzheimer’s disease mouse model. J. Neurotrauma 2014, 31, 125–134. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.T.; Hsiao, I.T.; Hsieh, C.J.; Chiang, Y.H.; Yen, T.C.; Chiu, W.T.; Lin, K.J.; Hu, C.J. Accumulation of amyloid in cognitive impairment after mild traumatic brain injury. J. Neurol. Sci. 2015, 349, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Roberts, G.W.; Gentleman, S.M.; Lynch, A.; Graham, D.I. beta A4 amyloid protein deposition in brain after head trauma. Lancet 1991, 338, 1422–1423. [Google Scholar] [CrossRef]
- Roberts, G.W.; Gentleman, S.M.; Lynch, A.; Murray, L.; Landon, M.; Graham, D.I. Beta amyloid protein deposition in the brain after severe head injury: Implications for the pathogenesis of Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 1994, 57, 419–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, M.; Snyder, C.; Corcoran, C.; Norton, M.C.; Lyketsos, C.G.; Tschanz, J.T. The association of traumatic brain injury with rate of progression of cognitive and functional impairment in a population-based cohort of Alzheimer’s disease: The Cache County Dementia Progression Study. Int. Psychogeriatr. 2014, 26, 1593–1601. [Google Scholar] [CrossRef] [Green Version]
- LoBue, C.; Wadsworth, H.; Wilmoth, K.; Clem, M.; Hart, J., Jr.; Womack, K.B.; Didehbani, N.; Lacritz, L.H.; Rossetti, H.C.; Cullum, C.M. Traumatic brain injury history is associated with earlier age of onset of Alzheimer disease. Clin. Neuropsychol. 2017, 31, 85–98. [Google Scholar] [CrossRef] [Green Version]
- Schofield, P.W.; Tang, M.; Marder, K.; Bell, K.; Dooneief, G.; Chun, M.; Sano, M.; Stern, Y.; Mayeux, R. Alzheimer’s disease after remote head injury: An incidence study. J. Neurol Neurosurg. Psychiatry 1997, 62, 119–124. [Google Scholar] [CrossRef] [Green Version]
- Hinson, H.E.; Rowell, S.; Schreiber, M. Clinical evidence of inflammation driving secondary brain injury: A systematic review. J. Trauma Acute Care Surg. 2015, 78, 184–191. [Google Scholar] [CrossRef] [Green Version]
- Shishido, H.; Ueno, M.; Sato, K.; Matsumura, M.; Toyota, Y.; Kirino, Y.; Tamiya, T.; Kawai, N.; Kishimoto, Y. Traumatic Brain Injury by Weight-Drop Method Causes Transient Amyloid-beta Deposition and Acute Cognitive Deficits in Mice. Behav. Neurol. 2019, 2019, 3248519. [Google Scholar] [CrossRef] [Green Version]
- Faul, M.; Coronado, V. Epidemiology of traumatic brain injury. Handb. Clin. Neurol. 2015, 127, 3–13. [Google Scholar] [CrossRef]
- Cheng, W.H.; Stukas, S.; Martens, K.M.; Namjoshi, D.R.; Button, E.B.; Wilkinson, A.; Bashir, A.; Robert, J.; Cripton, P.A.; Wellington, C.L. Age at injury and genotype modify acute inflammatory and neurofilament-light responses to mild CHIMERA traumatic brain injury in wild-type and APP/PS1 mice. Exp. Neurol. 2018, 301, 26–38. [Google Scholar] [CrossRef]
- Wang, Y.; Kasper, L.H. The role of microbiome in central nervous system disorders. Brain Behav. Immun. 2014, 38, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cryan, J.F.; Dinan, T.G. Mind-altering microorganisms: The impact of the gut microbiota on brain and behaviour. Nat. Rev. Neurosci. 2012, 13, 701–712. [Google Scholar] [CrossRef]
- Barko, P.C.; McMichael, M.A.; Swanson, K.S.; Williams, D.A. The Gastrointestinal Microbiome: A Review. J. Vet. Intern. Med. 2018, 32, 9–25. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Real, J.M.; Serino, M.; Blasco, G.; Puig, J.; Daunis-i-Estadella, J.; Ricart, W.; Burcelin, R.; Fernandez-Aranda, F.; Portero-Otin, M. Gut Microbiota Interacts With Brain Microstructure and Function. J. Clin. Endocrinol. Metab. 2015, 100, 4505–4513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bravo, J.A.; Forsythe, P.; Chew, M.V.; Escaravage, E.; Savignac, H.M.; Dinan, T.G.; Bienenstock, J.; Cryan, J.F. Ingestion of Lactobacillus strain regulates emotional behavior and central GABA receptor expression in a mouse via the vagus nerve. Proc. Natl. Acad. Sci. USA 2011, 108, 16050–16055. [Google Scholar] [CrossRef] [Green Version]
- Abuaish, S.; Al-Otaibi, N.M.; Abujamel, T.S.; Alzahrani, S.A.; Alotaibi, S.M.; AlShawakir, Y.A.; Aabed, K.; El-Ansary, A. Fecal Transplant and Bifidobacterium Treatments Modulate Gut Clostridium Bacteria and Rescue Social Impairment and Hippocampal BDNF Expression in a Rodent Model of Autism. Brain Sci. 2021, 11, 1038. [Google Scholar] [CrossRef]
- Lee, G.A.; Lin, Y.K.; Lai, J.H.; Lo, Y.C.; Yang, Y.S.H.; Ye, S.Y.; Lee, C.J.; Wang, C.C.; Chiang, Y.H.; Tseng, S.H. Maternal Immune Activation Causes Social Behavior Deficits and Hypomyelination in Male Rat Offspring with an Autism-Like Microbiota Profile. Brain Sci. 2021, 11, 1085. [Google Scholar] [CrossRef]
- Sun, P.; Su, L.; Zhu, H.; Li, X.; Guo, Y.; Du, X.; Zhang, L.; Qin, C. Gut Microbiota Regulation and Their Implication in the Development of Neurodegenerative Disease. Microorganisms 2021, 9, 2281. [Google Scholar] [CrossRef]
- Gonzalez-Sanmiguel, J.; Schuh, C.; Munoz-Montesino, C.; Contreras-Kallens, P.; Aguayo, L.G.; Aguayo, S. Complex Interaction between Resident Microbiota and Misfolded Proteins: Role in Neuroinflammation and Neurodegeneration. Cells 2020, 9, 2476. [Google Scholar] [CrossRef]
- Montagne, A.; Barnes, S.R.; Sweeney, M.D.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W.; Jacobs, R.E.; Liu, C.Y.; Amezcua, L.; et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron 2015, 85, 296–302. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Wang, Z.; Peng, J.; Gerner, S.T.; Yin, S.; Jiang, Y. Gut microbiota-brain interaction: An emerging immunotherapy for traumatic brain injury. Exp. Neurol. 2021, 337, 113585. [Google Scholar] [CrossRef] [PubMed]
- Murray, E.R.; Kemp, M.; Nguyen, T.T. The Microbiota-Gut-Brain Axis in Alzheimer’s Disease: A Review of Taxonomic Alterations and Potential Avenues for Interventions. Arch. Clin. Neuropsychol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Rieder, R.; Wisniewski, P.J.; Alderman, B.L.; Campbell, S.C. Microbes and mental health: A review. Brain Behav. Immun. 2017, 66, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Vogt, N.M.; Kerby, R.L.; Dill-McFarland, K.A.; Harding, S.J.; Merluzzi, A.P.; Johnson, S.C.; Carlsson, C.M.; Asthana, S.; Zetterberg, H.; Blennow, K.; et al. Gut microbiome alterations in Alzheimer’s disease. Sci. Rep. 2017, 7, 13537. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Miller, R.G.; Gascon, R.; Champion, S.; Katz, J.; Lancero, M.; Narvaez, A.; Honrada, R.; Ruvalcaba, D.; McGrath, M.S. Circulating endotoxin and systemic immune activation in sporadic amyotrophic lateral sclerosis (sALS). J. Neuroimmunol. 2009, 206, 121–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kesika, P.; Suganthy, N.; Sivamaruthi, B.S.; Chaiyasut, C. Role of gut-brain axis, gut microbial composition, and probiotic intervention in Alzheimer’s disease. Life Sci. 2021, 264, 118627. [Google Scholar] [CrossRef]
- Leblhuber, F.; Geisler, S.; Steiner, K.; Fuchs, D.; Schutz, B. Elevated fecal calprotectin in patients with Alzheimer’s dementia indicates leaky gut. J. Neural. Transm. 2015, 122, 1319–1322. [Google Scholar] [CrossRef]
- Friedland, R.P. Mechanisms of molecular mimicry involving the microbiota in neurodegeneration. J. Alzheimers Dis. 2015, 45, 349–362. [Google Scholar] [CrossRef] [Green Version]
- Megur, A.; Baltriukiene, D.; Bukelskiene, V.; Burokas, A. The Microbiota-Gut-Brain Axis and Alzheimer’s Disease: Neuroinflammation Is to Blame? Nutrients 2020, 13, 37. [Google Scholar] [CrossRef]
- Villapol, S.; Balarezo, M.G.; Affram, K.; Saavedra, J.M.; Symes, A.J. Neurorestoration after traumatic brain injury through angiotensin II receptor blockage. Brain 2015, 138, 3299–3315. [Google Scholar] [CrossRef] [Green Version]
- Zinger, A.; Soriano, S.; Baudo, G.; De Rosa, E.; Taraballi, F.; Villapol, S. Biomimetic Nanoparticles as a Theranostic Tool for Traumatic Brain Injury. Adv. Funct. Mater. 2021, 31, 2100722. [Google Scholar] [CrossRef] [PubMed]
- Villapol, S.; Byrnes, K.R.; Symes, A.J. Temporal dynamics of cerebral blood flow, cortical damage, apoptosis, astrocyte-vasculature interaction and astrogliosis in the pericontusional region after traumatic brain injury. Front. Neurol. 2014, 5, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soriano, S.; Moffet, B.; Wicker, E.; Villapol, S. Serum Amyloid A is Expressed in the Brain After Traumatic Brain Injury in a Sex-Dependent Manner. Cell Mol. Neurobiol. 2020, 40, 1199–1211. [Google Scholar] [CrossRef] [PubMed]
- Villapol, S.; Kryndushkin, D.; Balarezo, M.G.; Campbell, A.M.; Saavedra, J.M.; Shewmaker, F.P.; Symes, A.J. Hepatic expression of serum amyloid A1 is induced by traumatic brain injury and modulated by telmisartan. Am. J. Pathol. 2015, 185, 2641–2652. [Google Scholar] [CrossRef] [Green Version]
- Treangen, T.J.; Wagner, J.; Burns, M.P.; Villapol, S. Traumatic Brain Injury in Mice Induces Acute Bacterial Dysbiosis Within the Fecal Microbiome. Front. Immunol. 2018, 9, 2757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kigerl, K.A.; Hall, J.C.; Wang, L.; Mo, X.; Yu, Z.; Popovich, P.G. Gut dysbiosis impairs recovery after spinal cord injury. J. Exp. Med. 2016, 213, 2603–2620. [Google Scholar] [CrossRef]
- Houlden, A.; Goldrick, M.; Brough, D.; Vizi, E.S.; Lenart, N.; Martinecz, B.; Roberts, I.S.; Denes, A. Brain injury induces specific changes in the caecal microbiota of mice via altered autonomic activity and mucoprotein production. Brain Behav. Immun. 2016, 57, 10–20. [Google Scholar] [CrossRef]
- Liu, S.; Gao, J.; Zhu, M.; Liu, K.; Zhang, H.L. Gut Microbiota and Dysbiosis in Alzheimer’s Disease: Implications for Pathogenesis and Treatment. Mol. Neurobiol. 2020, 57, 5026–5043. [Google Scholar] [CrossRef]
- Lucarini, E.; Di Pilato, V.; Parisio, C.; Micheli, L.; Toti, A.; Pacini, A.; Bartolucci, G.; Baldi, S.; Niccolai, E.; Amedei, A.; et al. Visceral sensitivity modulation by faecal microbiota transplantation: The active role of gut bacteria in pain persistence. Pain 2021, 163, 861–877. [Google Scholar] [CrossRef]
- Sun, J.; Xu, J.; Ling, Y.; Wang, F.; Gong, T.; Yang, C.; Ye, S.; Ye, K.; Wei, D.; Song, Z.; et al. Fecal microbiota transplantation alleviated Alzheimer’s disease-like pathogenesis in APP/PS1 transgenic mice. Transl. Psychiatry 2019, 9, 189. [Google Scholar] [CrossRef] [Green Version]
- Du, D.; Tang, W.; Zhou, C.; Sun, X.; Wei, Z.; Zhong, J.; Huang, Z. Fecal Microbiota Transplantation Is a Promising Method to Restore Gut Microbiota Dysbiosis and Relieve Neurological Deficits after Traumatic Brain Injury. Oxid. Med. Cell. Longev. 2021, 2021, 5816837. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.M.; DeFazio, J.R.; Hyoju, S.K.; Sangani, K.; Keskey, R.; Krezalek, M.A.; Khodarev, N.N.; Sangwan, N.; Christley, S.; Harris, K.G.; et al. Fecal microbiota transplant rescues mice from human pathogen mediated sepsis by restoring systemic immunity. Nat. Commun. 2020, 11, 2354. [Google Scholar] [CrossRef]
- Benakis, C.; Brea, D.; Caballero, S.; Faraco, G.; Moore, J.; Murphy, M.; Sita, G.; Racchumi, G.; Ling, L.; Pamer, E.G.; et al. Commensal microbiota affects ischemic stroke outcome by regulating intestinal gammadelta T cells. Nat. Med. 2016, 22, 516–523. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yu, C.; Li, R.; Liu, K.; Jin, G.; Ge, R.; Tang, F.; Cui, S. High-altitude Tibetan fermented milk ameliorated cognitive dysfunction by modified gut microbiota in Alzheimer’s disease transgenic mice. Food Funct. 2020, 11, 5308–5319. [Google Scholar] [CrossRef] [PubMed]
- Shamsipour, S.; Sharifi, G.; Taghian, F. Impact of interval training with probiotic (L. plantarum/Bifidobacterium bifidum) on passive avoidance test, ChAT and BDNF in the hippocampus of rats with Alzheimer’s disease. Neurosci. Lett. 2021, 756, 135949. [Google Scholar] [CrossRef]
- Wang, H.; Sun, Y.; Xin, J.; Zhang, T.; Sun, N.; Ni, X.; Zeng, D.; Bai, Y. Lactobacillus johnsonii BS15 Prevents Psychological Stress-Induced Memory Dysfunction in Mice by Modulating the Gut-Brain Axis. Front. Microbiol. 2020, 11, 1941. [Google Scholar] [CrossRef]
- Yang, X.; Yu, D.; Xue, L.; Li, H.; Du, J. Probiotics modulate the microbiota-gut-brain axis and improve memory deficits in aged SAMP8 mice. Acta Pharm. Sin. B 2020, 10, 475–487. [Google Scholar] [CrossRef]
- McNamara, M.P.; Singleton, J.M.; Cadney, M.D.; Ruegger, P.M.; Borneman, J.; Garland, T. Early-life effects of juvenile Western diet and exercise on adult gut microbiome composition in mice. J. Exp. Biol. 2021, 224, jeb239699. [Google Scholar] [CrossRef]
- Dobranowski, P.A.; Tang, C.; Sauve, J.P.; Menzies, S.C.; Sly, L.M. Compositional changes to the ileal microbiome precede the onset of spontaneous ileitis in SHIP deficient mice. Gut Microbes 2019, 10, 578–598. [Google Scholar] [CrossRef] [Green Version]
- Cattaneo, A.; Cattane, N.; Galluzzi, S.; Provasi, S.; Lopizzo, N.; Festari, C.; Ferrari, C.; Guerra, U.P.; Paghera, B.; Muscio, C.; et al. Association of brain amyloidosis with pro-inflammatory gut bacterial taxa and peripheral inflammation markers in cognitively impaired elderly. Neurobiol. Aging 2017, 49, 60–68. [Google Scholar] [CrossRef] [Green Version]
- Soriano, S.; Curry, K.; Sadrameli, S.S.; Wang, Q.; Nute, M.; Reeves, E.; Kabir, R.; Wiese, J.; Criswell, A.; Schodrof, S.; et al. Alterations to the gut microbiome after sport-related concussion in a collegiate football players cohort: A pilot study. Brain Behav. Immun. Health 2022, 21, 100438. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Zhou, G.; Shao, B.; Zhou, H.; Xu, C.; Yan, F.; Wang, L.; Chen, G.; Li, J.; Fu, X. Gut Microbiota Dysbiosis Induced by Intracerebral Hemorrhage Aggravates Neuroinflammation in Mice. Front. Microbiol. 2021, 12, 647304. [Google Scholar] [CrossRef] [PubMed]
- Erny, D.; Hrabe de Angelis, A.L.; Jaitin, D.; Wieghofer, P.; Staszewski, O.; David, E.; Keren-Shaul, H.; Mahlakoiv, T.; Jakobshagen, K.; Buch, T.; et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat. Neurosci. 2015, 18, 965–977. [Google Scholar] [CrossRef] [PubMed]
- Tooley, K.L. Effects of the Human Gut Microbiota on Cognitive Performance, Brain Structure and Function: A Narrative Review. Nutrients 2020, 12, 3009. [Google Scholar] [CrossRef]
- Battaglia, S. Neurobiological advances of learned fear in humans. Adv. Clin. Exp. Med. 2022, 31, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, S.; Harrison, B.J.; Fullana, M.A. Does the human ventromedial prefrontal cortex support fear learning, fear extinction or both? A commentary on subregional contributions. Mol. Psychiatry 2021. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, S.; Garofalo, S.; di Pellegrino, G. Context-dependent extinction of threat memories: Influences of healthy aging. Sci. Rep. 2018, 8, 12592. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Jeon, S.H.; Ju, I.G.; Gee, M.S.; Do, J.; Oh, M.S.; Lee, J.K. Transplantation of gut microbiota derived from Alzheimer’s disease mouse model impairs memory function and neurogenesis in C57BL/6 mice. Brain Behav. Immun. 2021, 98, 357–365. [Google Scholar] [CrossRef]
- Curry, K.D.; Wang, Q.; Nute, M.G.; Tyshaieva, A.; Reeves, E.; Soriano, S.; Graeber, E.; Finzer, P.; Mendling, W.; Wu, Q.; et al. Emu: Species-Level Microbial Community Profiling for Full-Length Nanopore 16S Reads. bioRxiv 2021. [Google Scholar] [CrossRef]
- Villapol, S.; Loane, D.J.; Burns, M.P. Sexual dimorphism in the inflammatory response to traumatic brain injury. Glia 2017, 65, 1423–1438. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soriano, S.; Curry, K.; Wang, Q.; Chow, E.; Treangen, T.J.; Villapol, S. Fecal Microbiota Transplantation Derived from Alzheimer’s Disease Mice Worsens Brain Trauma Outcomes in Wild-Type Controls. Int. J. Mol. Sci. 2022, 23, 4476. https://doi.org/10.3390/ijms23094476
Soriano S, Curry K, Wang Q, Chow E, Treangen TJ, Villapol S. Fecal Microbiota Transplantation Derived from Alzheimer’s Disease Mice Worsens Brain Trauma Outcomes in Wild-Type Controls. International Journal of Molecular Sciences. 2022; 23(9):4476. https://doi.org/10.3390/ijms23094476
Chicago/Turabian StyleSoriano, Sirena, Kristen Curry, Qi Wang, Elsbeth Chow, Todd J. Treangen, and Sonia Villapol. 2022. "Fecal Microbiota Transplantation Derived from Alzheimer’s Disease Mice Worsens Brain Trauma Outcomes in Wild-Type Controls" International Journal of Molecular Sciences 23, no. 9: 4476. https://doi.org/10.3390/ijms23094476
APA StyleSoriano, S., Curry, K., Wang, Q., Chow, E., Treangen, T. J., & Villapol, S. (2022). Fecal Microbiota Transplantation Derived from Alzheimer’s Disease Mice Worsens Brain Trauma Outcomes in Wild-Type Controls. International Journal of Molecular Sciences, 23(9), 4476. https://doi.org/10.3390/ijms23094476