
Inverse Mixed-Solvent Molecular Dynamics for Visualization of the Residue Interaction Profile of Molecular Probes



Abstract
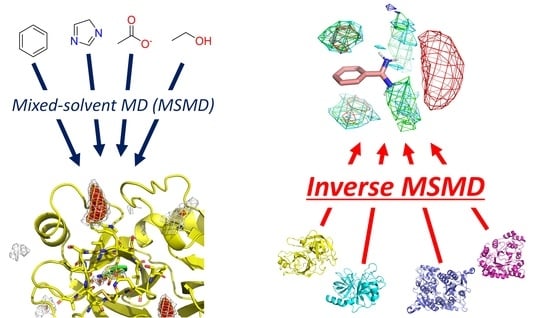
:
1. Introduction
2. Materials and Methods
2.1. Preparation of Proteins
2.2. Preparation of Probes
2.3. Mixed-Solvent Molecular Dynamics (MSMD)
2.3.1. Initial System Generation
2.3.2. Minimization, Heating, and Equilibration
2.3.3. Production Run
2.4. Inverse MSMD: Construction of Residue Interaction Profile
2.4.1. Determination of Preferable Protein Surfaces
2.4.2. Extraction of Residue Environments at Preferable Protein Surfaces
2.4.3. Description of Spatial Statistics for Each Type of Residue
2.5. Implementation
3. Results
3.1. Benzamidine: Evaluation of the Method
3.2. Catechol: Interaction Analysis of Hydroxy Groups
3.3. Benzene: Interaction Analysis of Phenyl Group Itself
4. Discussion
4.1. Comparison to Co-Crystallized Structures
4.1.1. Benzamidine
4.1.2. Catechol
4.2. Detection of Aromatic Residues’ Profile
4.3. Consideration of Binding Stability
- Strong binding affinity between a probe molecule and the protein surface, which allows a single probe molecule to bind stably to the surface.
- Frequent access of probe molecules to the protein surface, which makes multiple probe molecules bind to the protein surface alternatively.
4.4. Substituent Evaluation with Residue Interaction Profiles
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Berdigaliyev, N.; Aljofan, M. An overview of drug discovery and development. Future Sci. 2020, 12, 939–947. [Google Scholar] [CrossRef]
- Yoshino, R.; Yasuo, N.; Inaoka, D.K.; Hagiwara, Y.; Ohno, K.; Orita, M.; Inoue, M.; Shiba, T.; Harada, S.; Honma, T.; et al. Pharmacophore Modeling for Anti-Chagas Drug Design Using the Fragment Molecular Orbital Method. PLoS ONE 2015, 10, e0125829. [Google Scholar] [CrossRef]
- Yoshino, R.; Yasuo, N.; Hagiwara, Y.; Ishida, T.; Inaoka, D.K.; Amano, Y.; Tateishi, Y.; Ohno, K.; Namatame, I.; Niimi, T.; et al. In silico, in vitro, X-ray crystallography, and integrated strategies for discovering spermidine synthase inhibitors for Chagas disease. Sci. Rep. 2017, 7, 6666. [Google Scholar] [CrossRef] [Green Version]
- Chiba, S.; Ikeda, K.; Ishida, T.; Gromiha, M.M.; Taguchi, Y.-H.; Iwadate, M.; Umeyama, H.; Hsin, K.-Y.; Kitano, H.; Yamamoto, K.; et al. Identification of potential inhibitors based on compound proposal contest: Tyrosine-protein kinase Yes as a target. Sci. Rep. 2015, 5, 17209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiba, S.; Ishida, T.; Ikeda, K.; Mochizuki, M.; Teramoto, R.; Taguchi, Y.-H.; Iwadate, M.; Umeyama, H.; Ramakrishnan, C.; Thangakani, A.M.; et al. An iterative compound screening contest method for identifying target protein inhibitors using the tyrosine-protein kinase Yes. Sci. Rep. 2017, 7, 12038. [Google Scholar] [CrossRef]
- Cavasotto, C.N.; Di Filippo, J.I. Artificial intelligence in the early stages of drug discovery. Arch. Biochem. Biophys. 2021, 698, 108730. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; IJzerman, A.P.; van Westen, G.J.P. Computational Approaches for De Novo Drug Design: Past, Present, and Future. In Methods in Molecular Biology; Cartwright, H., Ed.; Humana: New York, NY, USA, 2021; Volume 2190, pp. 139–165. [Google Scholar] [CrossRef]
- Lin, X.; Li, X.; Lin, X. A Review on Applications of Computational Methods in Drug Screening and Design. Molecules 2020, 25, 1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimber, T.B.; Chen, Y.; Volkamer, A. Deep Learning in Virtual Screening: Recent Applications and Developments. Int. J. Mol. Sci. 2021, 22, 4435. [Google Scholar] [CrossRef] [PubMed]
- Maia, E.H.B.; Assis, L.C.; Oliveira, T.A.; Silva, A.M.; Taranto, A.G. Structure-Based Virtual Screening: From Classical to Artificial Intelligence. Front. Chem. 2020, 8, 343. [Google Scholar] [CrossRef]
- Barelier, S.; Eidam, O.; Fish, I.; Hollander, J.; Figaroa, F.; Nachane, R.; Irwin, J.J.; Shoichet, B.K.; Siegal, G. Increasing Chemical Space Coverage by Combining Empirical and Computational Fragment Screens. ACS Chem. Biol. 2014, 9, 1528–1535. [Google Scholar] [CrossRef] [Green Version]
- Tidten-Luksch, N.; Grimaldi, R.; Torrie, L.S.; Frearson, J.A.; Hunter, W.N.; Brenk, R. IspE Inhibitors Identified by a Combination of In Silico and In Vitro High-Throughput Screening. PLoS ONE 2012, 7, e35792. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Ranganathan, A.; IJzerman, A.P.; Siegal, G.; Carlsson, J. Complementarity between in Silico and Biophysical Screening Approaches in Fragment-Based Lead Discovery against the A2A Adenosine Receptor. J. Chem. Inf. Model. 2013, 53, 2701–2714. [Google Scholar] [CrossRef]
- Damm-Ganamet, K.L.; Bembenek, S.D.; Venable, J.W.; Castro, G.G.; Mangelschots, L.; Peeters, D.C.G.; Mcallister, H.M.; Edwards, J.P.; Disepio, D.; Mirzadegan, T. A Prospective Virtual Screening Study: Enriching Hit Rates and Designing Focus Libraries to Find Inhibitors of PI3Kδ and PI3Kγ. J. Med. Chem. 2016, 59, 4302–4313. [Google Scholar] [CrossRef] [PubMed]
- Fujitani, H.; Tanida, Y.; Matsuura, A. Massively parallel computation of absolute binding free energy with well-equilibrated states. Phys. Rev. E 2009, 79, 021914. [Google Scholar] [CrossRef] [PubMed]
- Abel, R.; Wang, L.; Harder, E.D.; Berne, B.J.; Friesner, F.A. Advancing Drug Discovery through Enhanced Free Energy Calculations. Acc. Chem. Res. 2017, 50, 1625–1632. [Google Scholar] [CrossRef]
- Kuhn, M.; Firth-Clark, S.; Tosco, P.; Mey, A.S.J.S.; Mackey, M.; Michel, J. Assessment of Binding Affinity via Alchemical Free-Energy Calculations. J. Chem. Inf. Model. 2020, 60, 3120–3130. [Google Scholar] [CrossRef]
- Ryde, U.; Söderhjelm, P. Ligand-Binding Affinity Estimates Supported by Quantum-Mechanical Methods. Chem. Rev. 2016, 116, 5520–5566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berman, H.; Henrick, K.; Nakamura, H.; Markley, J.L. The worldwide Protein Data Bank (wwPDB): Ensuring a single, uniform archive of PDB data. Nucleic Acids Res. 2007, 35, D301–D303. [Google Scholar] [CrossRef] [Green Version]
- Imai, Y.N.; Inoue, Y.; Yamamoto, Y. Propensities of Polar and Aromatic Amino Acids in Noncanonical Interactions: Nonbonded Contacts Analysis of Protein−Ligand Complexes in Crystal Structures. J. Med. Chem. 2007, 50, 1189–1196. [Google Scholar] [CrossRef]
- Wang, L.; Xie, Z.; Wipf, P.; Xie, X.-Q. Residue Preference Mapping of Ligand Fragments in the Protein Data Bank. J. Chem. Inf. Model. 2011, 51, 807–815. [Google Scholar] [CrossRef]
- Kasahara, K.; Shirota, M.; Kinoshita, K. Comprehensive Classification and Diversity Assessment of Atomic Contacts in Protein–Small Ligand Interactions. J. Chem. Inf. Model. 2013, 53, 241–248. [Google Scholar] [CrossRef]
- Zariquiey, F.S.; Souza, J.V.; Bronowska, A.K. Cosolvent Analysis Toolkit (CAT): A robust hotspot identification platform for cosolvent simulations of proteins to expand the druggable proteome. Sci. Rep. 2019, 9, 19118. [Google Scholar] [CrossRef] [Green Version]
- Ghanakota, P.; van Vlijmen, H.; Sherman, W.; Beuming, T. Large-Scale Validation of Mixed-Solvent Simulations to Assess Hotspots at Protein−Protein Interaction Interfaces. J. Chem. Inf. Model. 2018, 58, 784–793. [Google Scholar] [CrossRef]
- Schmidt, D.; Boehm, M.; McClendon, C.L.; Torella, R.; Gohlke, H. Cosolvent-Enhanced Sampling and Unbiased Identification of Cryptic Pockets Suitable for Structure-Based Drug Design. J. Chem. Theory Comput. 2019, 15, 3331–3343. [Google Scholar] [CrossRef]
- Kimura, S.R.; Hu, H.P.; Ruvinsky, A.M.; Sherman, W.; Favia, A.D. Deciphering Cryptic Binding Sites on Proteins by Mixed-Solvent Molecular Dynamics. J. Chem. Inf. Model. 2017, 57, 1388–1401. [Google Scholar] [CrossRef]
- Soga, S.; Shirai, H.; Kobori, M.; Hirayama, N. Identification of the Druggable Concavity in Homology Models Using the PLB Index. J. Chem. Inf. Model. 2007, 47, 2287–2292. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.F.; Honig, B.; Shaw, D.E.; Friesner, R.A. A Hierarchical Approach to All-Atom Protein Loop Prediction. Proteins Struct. Funct. Genet. 2004, 55, 2287–2292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsson, M.H.M.; Sondergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent treatment of internal and surface residues in empirical pKa predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the Amber biomolecular simulation package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Revision B.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Yanagisawa, K.; Moriwaki, Y.; Terada, T.; Shimizu, K. EXPRORER: Rational Cosolvent Set Construction Method for Cosolvent Molecular Dynamics Using Large-Scale Computation. J. Chem. Inf. Model. 2021, 61, 2744–2753. [Google Scholar] [CrossRef]
- Martínez, L.; Andrade, R.; Birgin, E.G.; Martínez, J.M. PACKMOL: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Hess, B. P-LINCS: A parallel linear constraint solver for molecular simulation. J. Chem. Theory Comput. 2008, 4, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [Green Version]
- Bussi, G.; Parrinello, M. Stochastic thermostats: Comparison of local and global schemes. Comput. Phys. Commun. 2008, 179, 26–29. [Google Scholar] [CrossRef] [Green Version]
- Bussi, G.; Zykova-Timan, T.; Parrinello, M. Isothermal-isobaric molecular dynamics using stochastic velocity rescaling. J. Chem. Phys. 2009, 130, 074101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berendsen, H.J.C.; Postma, J.P.M.; Van Gunsteren, W.F.; Dinola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to super-computers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Shirts, M.R.; Klein, C.; Swails, J.M.; Yin, J.; Gilson, M.K.; Mobley, D.L.; Case, D.A.; Zhong, E.D. Lessons learned from comparing molecular dynamics engines on the SAMPL5 dataset. J. Comput. Aided Mol. Des. 2017, 31, 147–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- MacKerell, A.D., Jr.; Jo, S.; Lakkaraju, S.K.; Lind, C.; Yu, W. Identification and characterization of fragment binding sites for allosteric ligand design using the site identification by ligand competitive saturation hotspots approach (SILCS-Hotspots). Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129519. [Google Scholar] [CrossRef]
- Cock, P.J.A.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef] [PubMed]
- Richard, D.T.; Malcolm, M.C.; Alastair, D.G.L. Rings in drugs. J. Med. Chem. 2014, 57, 5845–5859. [Google Scholar] [CrossRef]
- Harder, M.; Kuhn, B.; Diederich, F. Efficient Stacking on Protein Amide Fragments. ChemMedChem 2013, 8, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Ghanakota, P.; DasGupta, D.; Carlson, H.A. Free Energies and Entropies of Binding Sites Identified by MixMD Cosolvent Simulations. J. Chem. Inf. Model. 2019, 59, 2035–2045. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB Code | Chain ID | Protein Name |
|---|---|---|
| 1ZUA | X | Aldo-keto reductase family 1 member B10 |
| 1E0X | A | Endo-1,4-β-xylanase A precursor |
| 1BK9 | Phospholipase A2, acidic | |
| 1TU6 | A | Cathepsin K precursor |
| 1W4P | A | Ribonuclease pancreatic precursor |
| 1JZF | A | Azurin precursor |
| 1YMS | A | β-lactamase CTX-M-9a |
| 2WEA | Penicillopepsin | |
| 1HEE | A | Carboxypeptidase A1 precursor |
| 1WBI | A | Avidin-related protein 2 precursor |
| 1CXV | A | Collagenase 3 precursor |
| 1H4G | A | Glycoside hydrolase |
| 1TT1 | A | Glutamate receptor, ionotropic kainate 2 precursor |
| 2CYB | A | Tyrosyl-tRNA synthetase |
| 1H60 | A | Pentaerythritol tetranitrate reductase |
| Type | Residues |
|---|---|
| Acidic | Asp, Glu |
| Basic | Arg, His, Lys |
| Hydrophilic | Asn, Cys, Gln, Ser, Thr |
| Hydrophobic | Ala, Ile, Leu, Met, Pro, Val |
| Aromatic | Phe, Trp, Tyr |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yanagisawa, K.; Yoshino, R.; Kudo, G.; Hirokawa, T. Inverse Mixed-Solvent Molecular Dynamics for Visualization of the Residue Interaction Profile of Molecular Probes. Int. J. Mol. Sci. 2022, 23, 4749. https://doi.org/10.3390/ijms23094749
Yanagisawa K, Yoshino R, Kudo G, Hirokawa T. Inverse Mixed-Solvent Molecular Dynamics for Visualization of the Residue Interaction Profile of Molecular Probes. International Journal of Molecular Sciences. 2022; 23(9):4749. https://doi.org/10.3390/ijms23094749
Chicago/Turabian StyleYanagisawa, Keisuke, Ryunosuke Yoshino, Genki Kudo, and Takatsugu Hirokawa. 2022. "Inverse Mixed-Solvent Molecular Dynamics for Visualization of the Residue Interaction Profile of Molecular Probes" International Journal of Molecular Sciences 23, no. 9: 4749. https://doi.org/10.3390/ijms23094749
APA StyleYanagisawa, K., Yoshino, R., Kudo, G., & Hirokawa, T. (2022). Inverse Mixed-Solvent Molecular Dynamics for Visualization of the Residue Interaction Profile of Molecular Probes. International Journal of Molecular Sciences, 23(9), 4749. https://doi.org/10.3390/ijms23094749