
Resolving the Mechanism of Acoustic Plasmon Instability in Graphene Doped by Alkali Metals

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
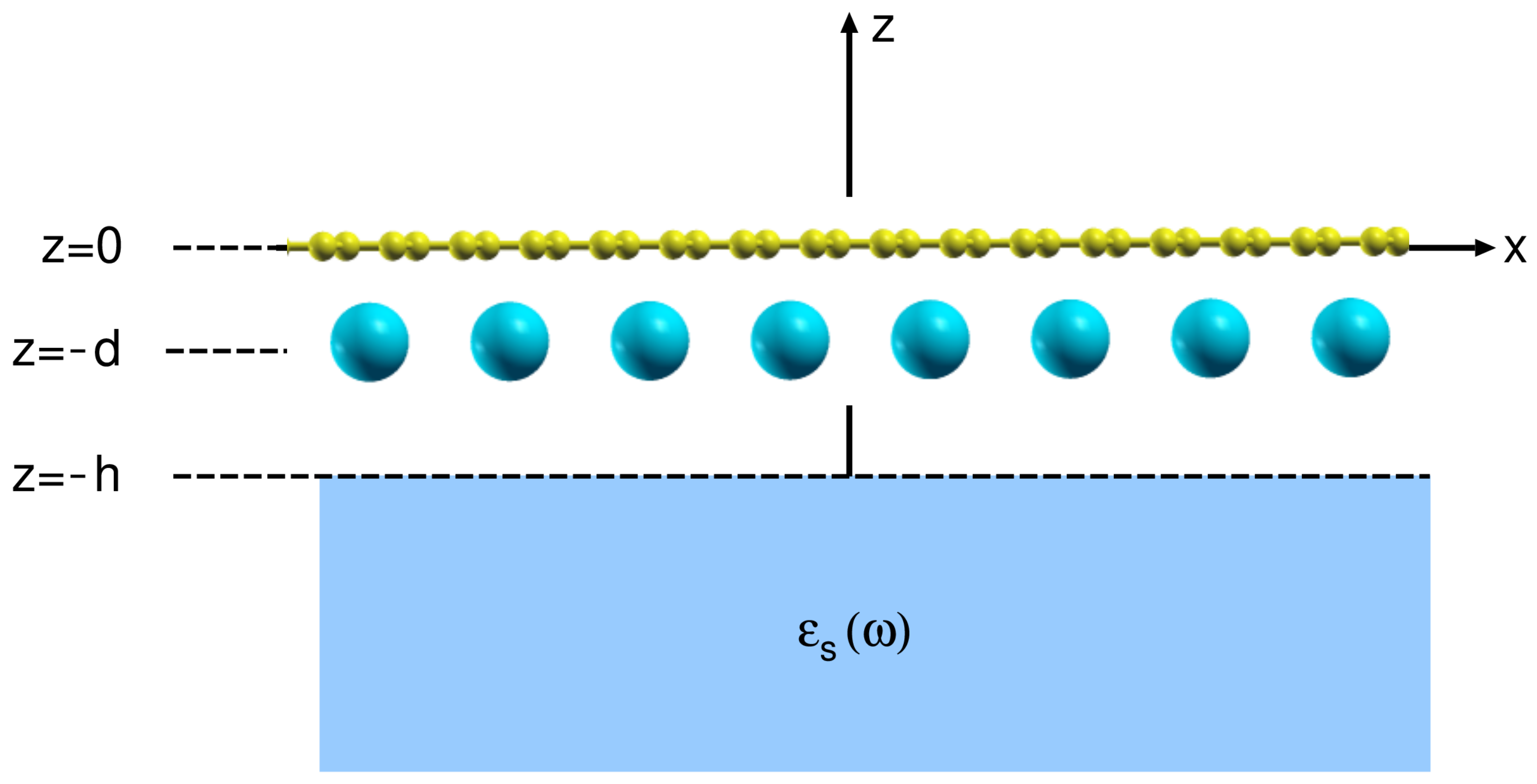
2. Theoretical Formulation
2.1. Calculation of the Surface Electronic Excitations Spectra
2.2. Calculation of the 2D Dynamical Polarizability Function
2.3. Calculation of the Substrate Dielectric Function
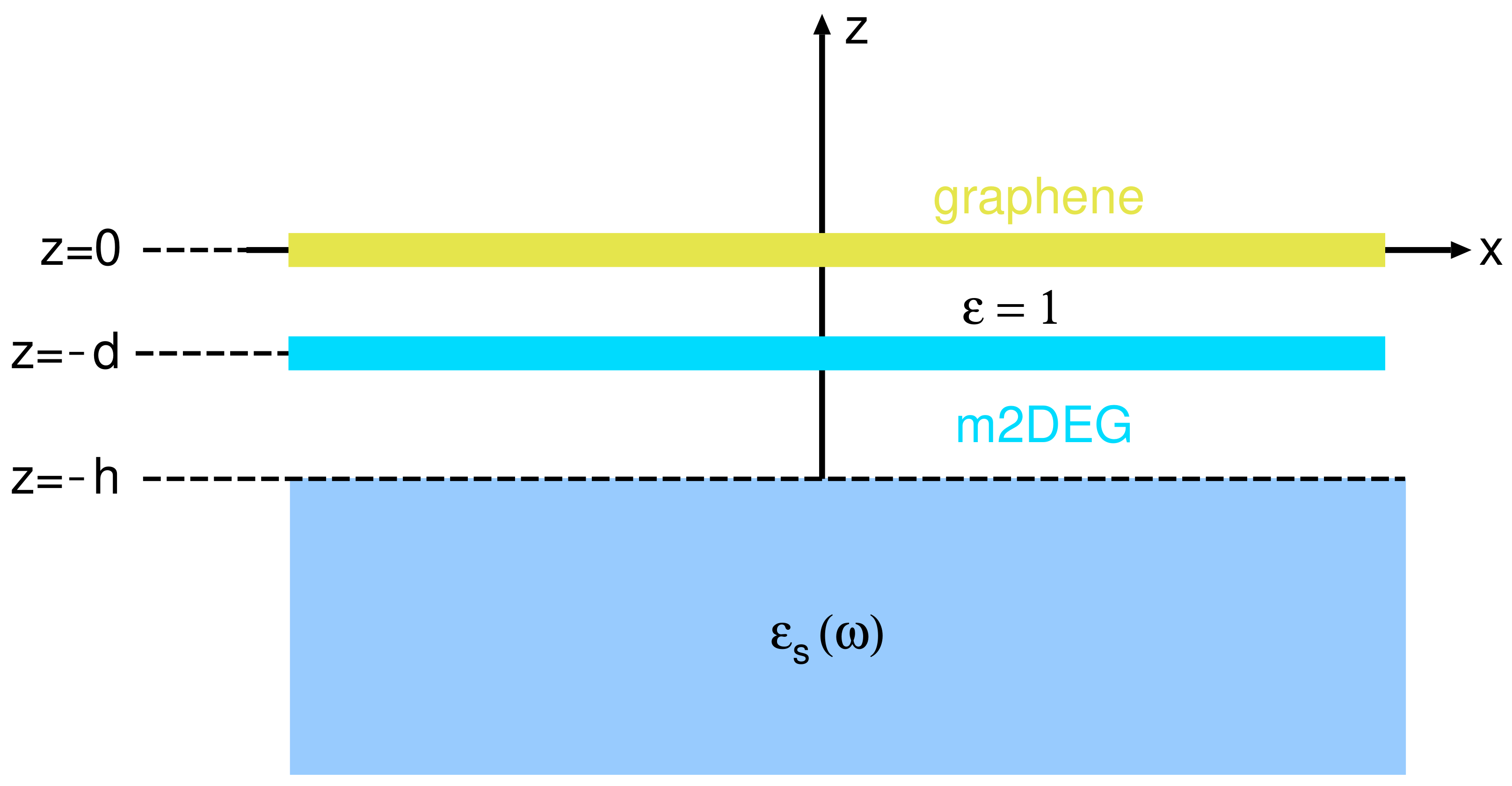
2.4. Reduced Model
2.5. Computational Details
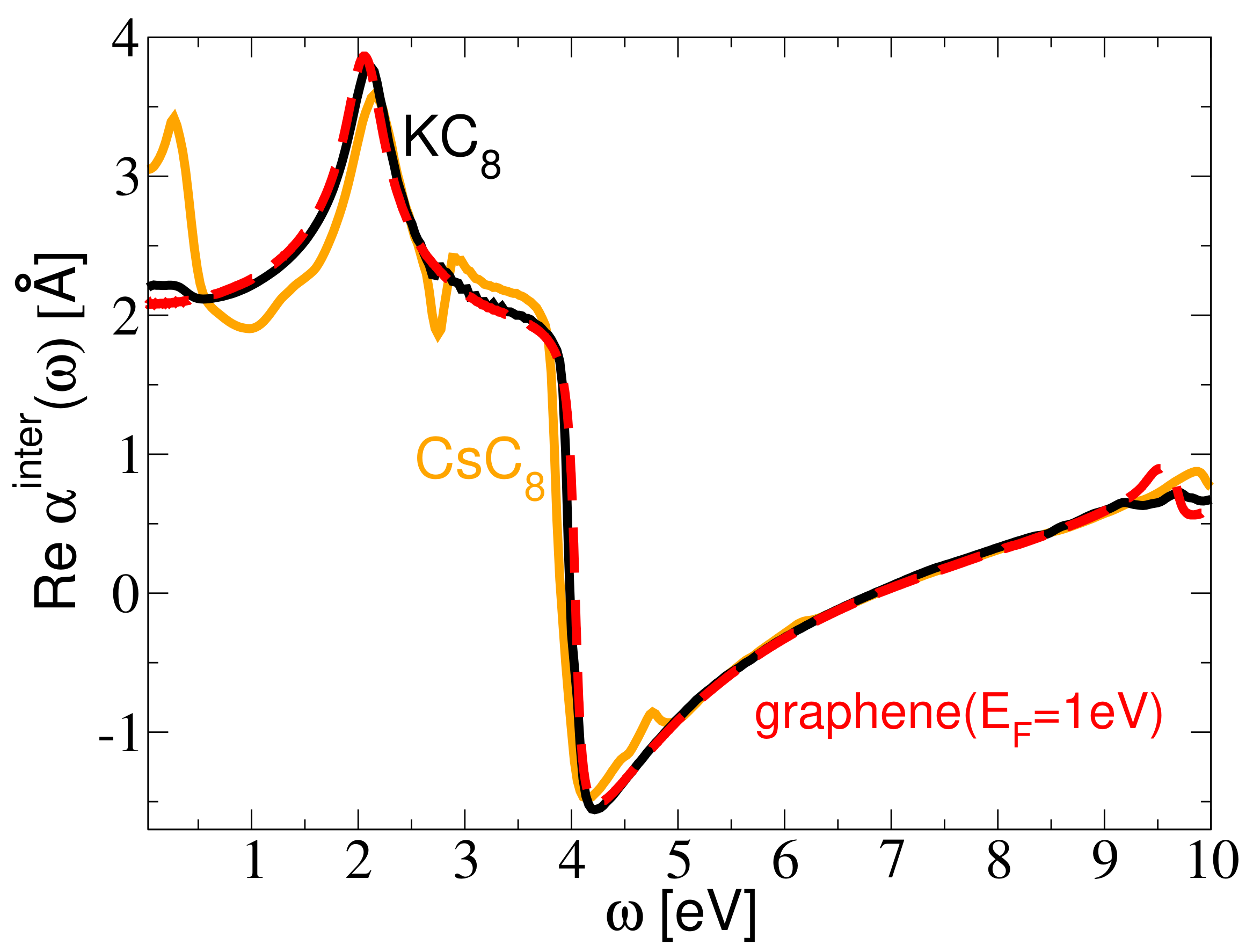
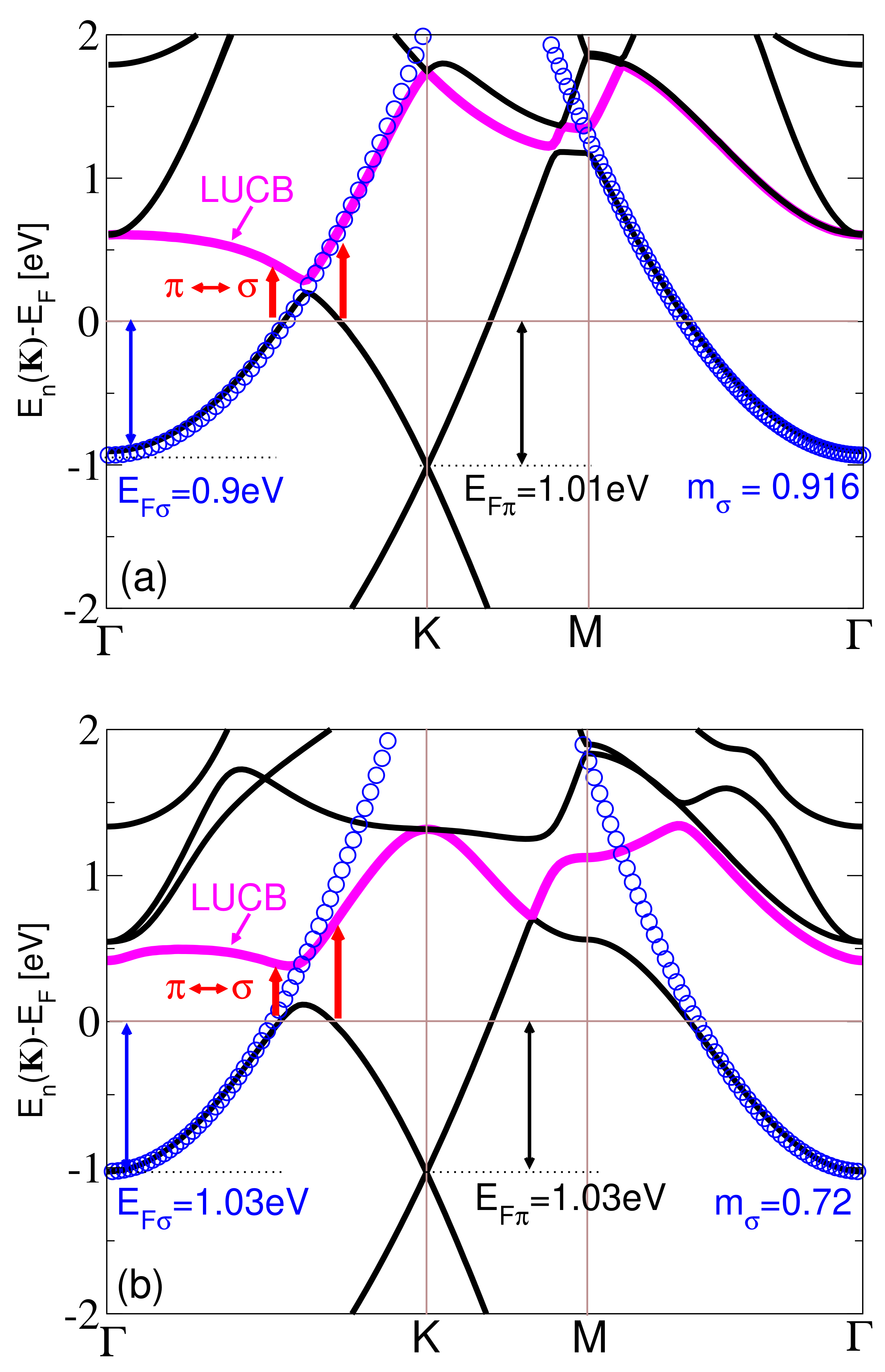
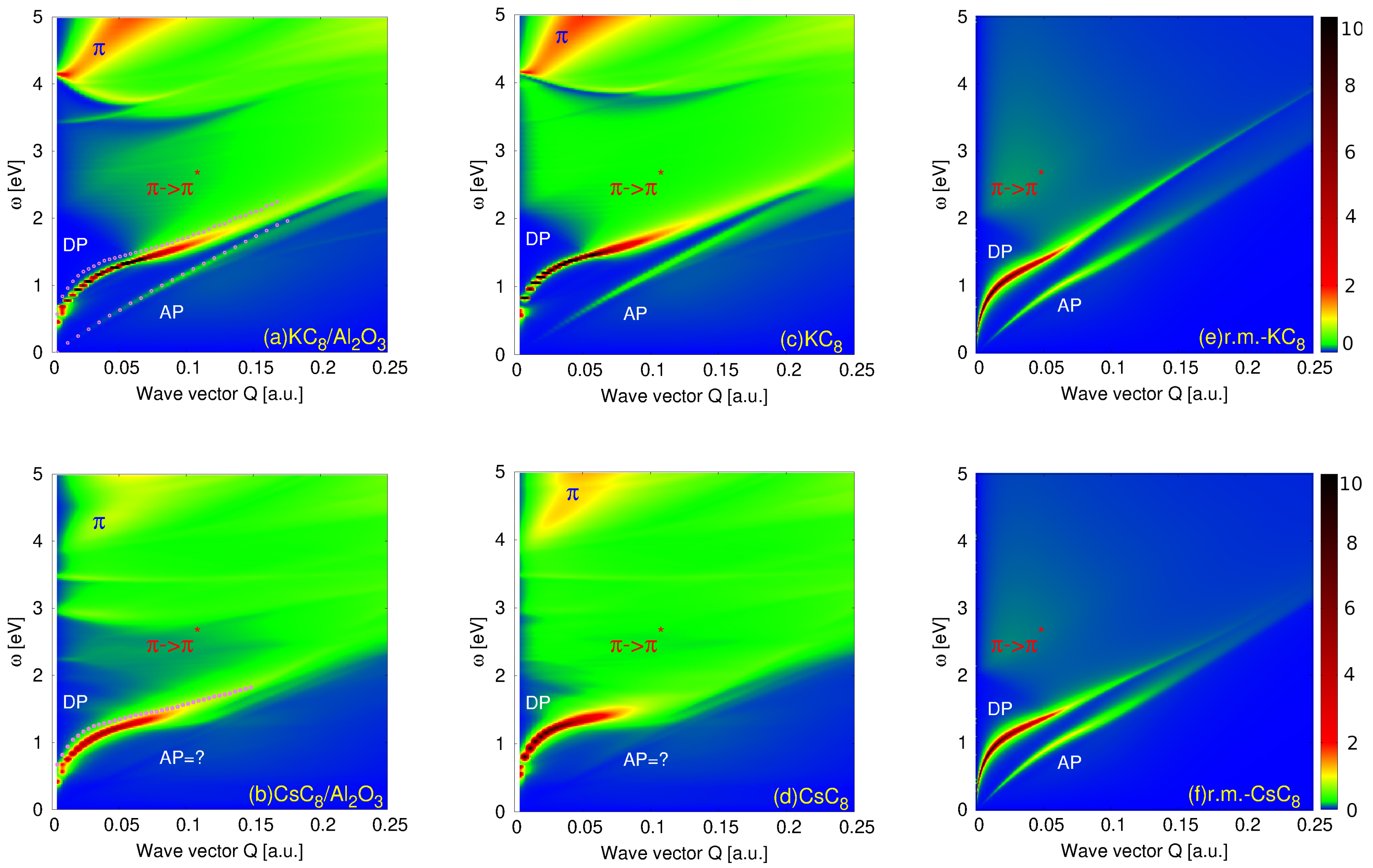
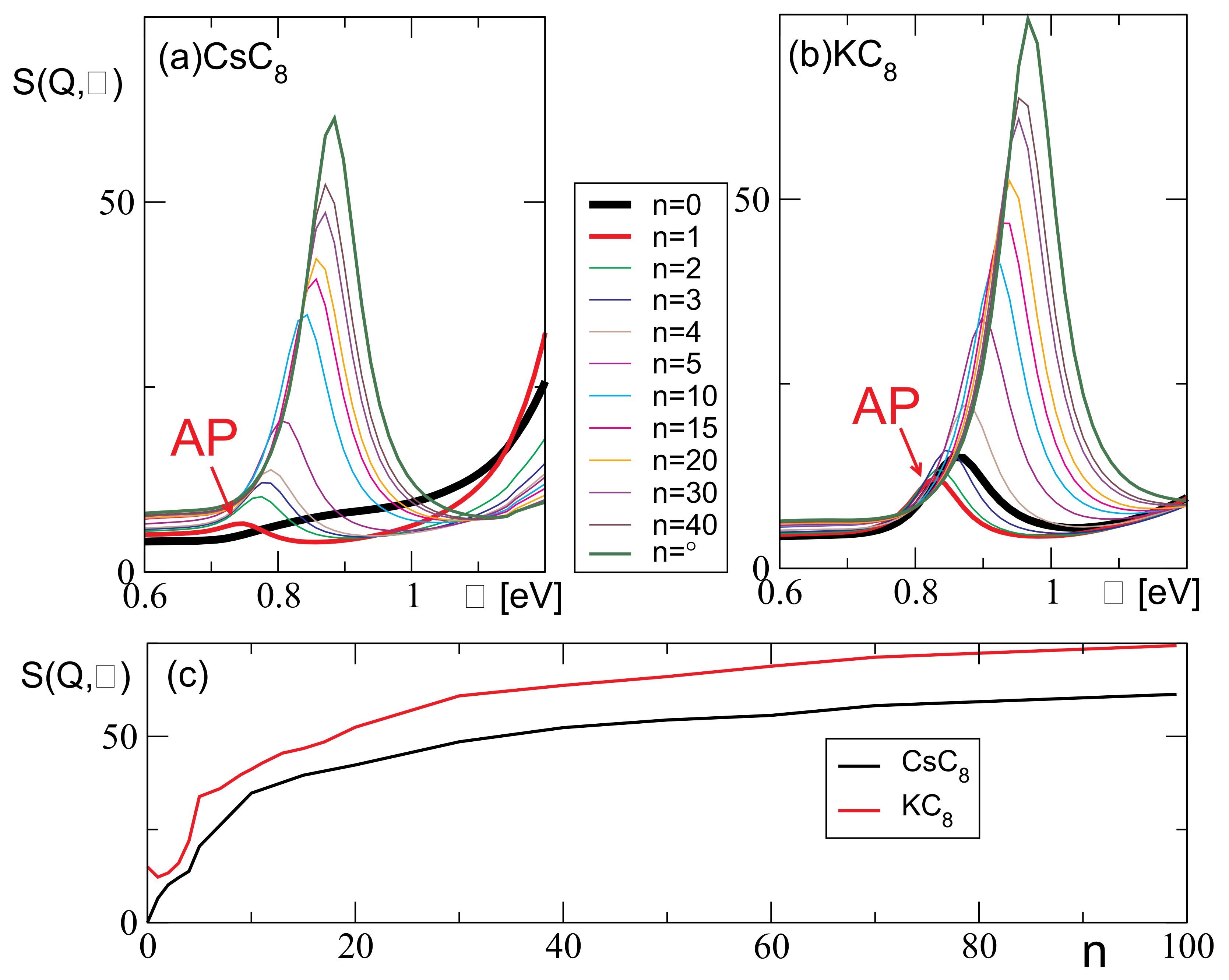
3. Results and Discussion
Resolving the Mechanism of the AP Instability
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, J.; Lin, Z.; Miao, G.; Zhong, W.; Xue, S.; Li, Y.; Tao, Z.; Wang, W.; Guo, J.; Zhu, X. Geometric Effect of High-Resolution Electron Energy Loss Spectroscopy on the Identification of Plasmons: An Example of Graphene. Surf. Sci. 2022, 721, 122067. [Google Scholar] [CrossRef]
- Sergey, G.M.; Lee, I.; Lee, S.; Ha, H.; Heiden, J.T.; Yoo, D.; Kim, T.; Low, T.; Lee, Y.H.; Oh, S.; et al. Real-space imaging of acoustic plasmons in large-area graphene grown by chemical vapor deposition. Nat. Commun. 2021, 12, 938. [Google Scholar]
- Fateev, D.V.; Polischuk, O.V.; Mashinsky, K.V.; Moiseenko, I.M.; Morozov, M.Y.; Popov, V.V. Terahertz Lasing with Weak Plasmon Modes in Periodic Graphene Structures. Phys. Rev. Appl. 2021, 15, 034043. [Google Scholar] [CrossRef]
- Kim, P.D.T.; Nguyen, M.V. Exchange-correlation effects and layer-thickness affect plasmon modes in gapped graphene-GaAs double-layer systems. Eur. Phys. J. B 2021, 94, 14. [Google Scholar]
- Chiu, C.; Chung, Y.; Yang, C.; Liu, C.; Lin, C. Coulomb decay rates in monolayer doped graphene. RSC Adv. 2020, 10, 2337–2346. [Google Scholar] [CrossRef] [Green Version]
- Wen, C.; Luo, J.; Xu, W.; Zhu, Z.; Qin, S.; Zhang, J. Enhanced Molecular Infrared Spectroscopy Employing Bilayer Graphene Acoustic Plasmon Resonator. Biosensors 2021, 11, 431. [Google Scholar] [CrossRef]
- Mohammadi, Y. Tunable plasmon modes in doped AA-stacked bilayer graphene. Superlattices Microstruct. 2021, 156, 106955. [Google Scholar] [CrossRef]
- Men, N.V.; Khanh, N.Q.; Phuong, D.T.K. Plasmon modes in double-layer gapped graphene. Phys. E Low-Dimens. Syst. Nanostruct. 2020, 118, 113859. [Google Scholar] [CrossRef]
- Men, N.V.; Phuong, D.T.K. Plasmon modes in double-layer gapped graphene at zero temperature. Phys. Lett. A 2020, 384, 126221. [Google Scholar] [CrossRef]
- Nguyen, V.-M. Temperature and inhomogeneity combination effects on collective excitations in three-layer graphene structures. Phys. E Low-Dimens. Syst. Nanostruct. 2022, 140, 115201. [Google Scholar] [CrossRef]
- Dong-Thi, K.P.; Nguyen, V.M. Plasmonic Excitations in 4-MLG Structures: Background Dielectric Inhomogeneity Effects. J. Low Temp. Phys. 2022, 206, 51. [Google Scholar] [CrossRef]
- Kim, P.D.T.; Van, M.N. Plasmon modes in N-layer graphene structures at zero temperature. J. Low Temp. Phys. 2020, 201, 311. [Google Scholar]
- Nguyen, V.M. Plasmon modes in N-layer gapped graphene. Phys. B Condens. Matter 2020, 578, 411876. [Google Scholar]
- Li, H.; Zhang, Y.; Xiao, H.; Qin, M.; Xia, S.; Wang, L. Investigation of acoustic plasmons in vertically stacked metal/dielectric/graphene heterostructures for multiband coherent perfect absorption. Opt. Express 2020, 28, 37577. [Google Scholar] [CrossRef]
- Kumar, A.; Reddy, A.L.M.; Mukherjee, A.; Dubey, M.; Zhan, X.; Singh, N.; Ci, L.; Billups, W.E.; Nagurny, J.; Mital, G.; et al. Direct Synthesis of Lithium-Intercalated Graphene for Electrochemical Energy Storage Application. ACS Nano 2011, 5, 4345. [Google Scholar] [CrossRef]
- Yang, S.L.; Sobota, J.A.; Howard, C.A.; Pickard, C.J.; Hashimoto, M.; Lu, D.H.; Mo, S.K.; Kirchmann, P.S.; Shen, Z.X. Superconducting graphene sheets in CaC6 enabled by phonon-mediated interband interactions. Nat. Commun. 2014, 5, 3493. [Google Scholar] [CrossRef]
- Caffrey, N.M.; Johansson, L.I.; Xia, C.; Armiento, R.; Abrikosov, I.A.; Jacobi, C. Structural and electronic properties of Li-intercalated graphene on SiC(0001). Phys. Rev. B 2016, 93, 195421. [Google Scholar] [CrossRef] [Green Version]
- Ichinokura, S.; Sugawara, K.; Takayama, A.; Takahashi, T.; Hasegawa, S. Superconducting Calcium-Intercalated Bilayer Graphene. ACS Nano 2016, 10, 2761. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Feng, X.; Zhang, W.; Ou, Y.; Chen, L.; He, K.; Wang, L.; Guo, L.; Liu, G.; Xue, Q.; et al. Superconducting Calcium-Intercalated Bilayer Graphene. Appl. Phys. Lett. 2013, 103, 062601. [Google Scholar] [CrossRef] [Green Version]
- Pervan, P.; Lazić, P.; Petrović, M.; Rakić, I.Š.; Pletikosić, I.; Kralj, M.; Milun, M.; Valla, T. Li adsorption versus graphene intercalation on Ir(111): From quenching to restoration of the Ir surface state. Phys. Rev. B 2015, 92, 245415. [Google Scholar] [CrossRef] [Green Version]
- Petrović, M.; Rakić, I.Š.; Runte, S.; Busse, C.; Sadowski, J.T.; Lazić, P.; Pletikosić, I.; Pan, Z.-H.; Milun, M.; Pervan, P.; et al. The mechanism of caesium intercalation of graphene. Nat. Commun. 2013, 4, 2772. [Google Scholar] [CrossRef] [Green Version]
- Marušić, L.; Despoja, V. Prediction of measurable two-dimensional plasmons in Li-intercalated graphene LiC2. Phys. Rev. B 2017, 95, 201408(R). [Google Scholar] [CrossRef]
- Despoja, V.; Marušić, L. UV-active plasmons in alkali and alkaline-earth intercalated graphene. Phys. Rev. B 2018, 97, 205426. [Google Scholar] [CrossRef] [Green Version]
- Despoja, V.; Novko, D.; Lončarić, I.; Golenić, N.; Marušić, L.; Silkin, V.M. Strong acoustic plasmons in chemically doped graphene induced by a nearby metal surface. Phys. Rev. B 2019, 100, 195401. [Google Scholar] [CrossRef] [Green Version]
- Despoja, V.; Jakovac, J.; Golenić, N.; Marušić, L. Bias-controlled plasmon switching in lithium-doped graphene on dielectric model Al2O3 substrate. NPJ 2D Mater. Appl. 2020, 4, 19. [Google Scholar] [CrossRef]
- Jablan, M.; Buljan, H.; Soljačić, M. Plasmonics in graphene at infrared frequencies. Phys. Rev. B 2009, 80, 245435. [Google Scholar] [CrossRef] [Green Version]
- Bonaccorso, F.; Sun, Z.; Hasan, T.; Ferrari, A.C. Graphene photonics and optoelectronics. Nat. Photonics 2010, 4, 611–622. [Google Scholar] [CrossRef] [Green Version]
- Vakil, A.; Engheta, N. Transformation Optics Using Graphene. Science 2011, 332, 1291–1294. [Google Scholar] [CrossRef] [Green Version]
- Hua-Qiang, W.; Chang-Yang, L.; Hong-Ming, L.; He, Q. Graphene applications in electronic and optoelectronic devices and circuits. Chin. Phys. B 2013, 22, 098106. [Google Scholar]
- Pospischil, A.; Furchi, M.M.; Mueller, T. Solar-energy conversion and light emission in an atomic monolayer p–n diode. Nat. Nanotechnol. 2014, 9, 257–261. [Google Scholar] [CrossRef]
- Ross, J.S.; Klement, P.; Jones, A.M.; Ghimire, N.J.; Yan, J.; Mandrus, D.G.; Taniguchi, T.; Watanabe, K.; Kitamura, K.; Yao, W.; et al. Electrically tunable excitonic light-emitting diodes based on monolayer WSe2 p-n junctions. Nat. Nanotechnol. 2014, 9, 268–272. [Google Scholar] [CrossRef]
- Koppens, F.H.L.; Mueller, T.; Avouris, P.; Ferrari, A.C.; Vitiello, M.S.; Polini, M. Photodetectors based on graphene, other two-dimensional materials and hybrid systems. Nat. Nanotechnol. 2014, 9, 780–793. [Google Scholar] [CrossRef]
- Jo, S.; Ubrig, N.; Berger, H.; Kuzmenko, A.B.; Morpurgo, A.F. Mono- and Bilayer WS2 Light-Emitting Transistors. Nano Lett. 2014, 14, 2019–2025. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Sanchez, O.; Llado, E.A.; Koman, V.; Morral, A.F.i.; Radenovic, A.; Kis, A. NEXT Light Generation and Harvesting in a van der Waals Heterostructure. ACS Nano 2014, 8, 3042. [Google Scholar] [CrossRef]
- Lee, C.-H.; Lee, G.-H.; Zande, A.M.V.; Chen, W.; Li, Y.; Han, M.; Cui, X.; Arefe, G.; Nuckolls, C.; Heinz, T.F.; et al. Atomically thin p-n junctions with van der Waals heterointerfaces. Nat. Nanotechnol. 2014, 9, 676–681. [Google Scholar] [CrossRef] [Green Version]
- Baugher, B.W.H.; Churchill, H.O.H.; Yang, Y.; Jarillo-Herrero, P. Optoelectronic Devices Based on Electrically Tunable P–n Diodes in a Monolayer Dichalcogenide. Nat. Nanotechnol. 2014, 9, 262–267. [Google Scholar] [CrossRef] [Green Version]
- Low, T.; Avouris, P. Graphene Plasmonics for Terahertz to Mid-Infrared Applications. ACS Nano 2014, 8, 1086–1101. [Google Scholar] [CrossRef] [Green Version]
- Britnell, L.; Ribeiro, R.M.; Eckmann, A.; Jalil, R.; Belle, B.D.; Mishchenko, A.; Kim, Y.J.; Gorbachev, R.V.; Georgiou, T.; Morozov, S.V.; et al. Absorption enhancement of thin layer black phosphorous in the mid-infrared with an all-dielectric metasurface. Science 2013, 340, 1311. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.; Du, D.; Lin, Y. Graphene and graphene-like 2D materials for optical biosensing and bioimaging: A review. 2D Mater. 2015, 2, 032004. [Google Scholar] [CrossRef]
- Zhu, A.Y.; Cubukcu, E. Graphene nanophotonic sensors. 2D Mater. 2015, 2, 032005. [Google Scholar] [CrossRef]
- Pitarke, J.M.; Nazarov, V.U.; Silkin, V.M.; Chulkov, E.V.; Zaremba, E.; Echenique, P.M. Theory of acoustic surface plasmons. Phys. Rev. B 2004, 70, 205403. [Google Scholar] [CrossRef] [Green Version]
- Diaconescu, B.; Pohl, K.; Vattuone, L.; Savio, L.; Hofmann, P.; Silkin, V.M.; Pitarke, J.M.; Chulkov, E.V.; Echenique, P.M.; Farías, D.; et al. Low-energy acoustic plasmons at metal surfaces. Nature 2007, 448, 57–59. [Google Scholar] [CrossRef]
- Silkin, V.M.; Pitarke, J.M.; Chulkov, E.V.; Diaconescu, B.; Pohl, K.; Vattuone, L.; Savio, L.; Hofmann, P.; Farías, D.; Rocca, M.; et al. Band structure effects on the Be(0001) acoustic surface plasmon energy dispersion. Phys. Stat. Sol. 2008, 205, 1307–1311. [Google Scholar] [CrossRef] [Green Version]
- Despoja, V.; Djordjević, T.; Karbunar, L.; Radović, I.; Mišković, Z.L. Ab initio study of the electron energy loss function in a graphene-sapphire-graphene composite system. Phys. Rev. B 2017, 96, 075433. [Google Scholar] [CrossRef] [Green Version]
- Falkovsky, L.A.; Pershoguba, S.S. Optical far-infrared properties of a graphene monolayer and multilayer. Phys. Rev. B 2007, 76, 153410. [Google Scholar] [CrossRef] [Green Version]
- Despoja, V.; Mowbray, D.J.; Vlahović, D.; Marušić, L. TDDFT study of time-dependent and static screening in graphene. Phys. Rev. B 2012, 86, 195429. [Google Scholar] [CrossRef] [Green Version]
- Despoja, V.; Dekanić, K.; Šunjić, M.; Marušić, L. Ab initio study of energy loss and wake potential in the vicinity of a graphene monolayer. Phys. Rev. B 2012, 86, 165419. [Google Scholar] [CrossRef] [Green Version]
- Despoja, V.; Novko, D.; Dekanić, K.; Šunjić, M.; Marušić, L. Two-dimensional and π plasmon spectra in pristine and doped graphene Phys. Rev. B 2013, 87, 075447. [Google Scholar] [CrossRef] [Green Version]
- Novko, D.; Despoja, V.; Šunjić, M. Changing character of electronic transitions in graphene: From single-particle excitations to plasmons. Phys. Rev. B 2015, 91, 195407. [Google Scholar] [CrossRef] [Green Version]
- Despoja, V.; Rukelj, Z.; Marušić, L. Ab initio study of electronic excitations and the dielectric function in molybdenum disulfide monolayer. Phys. Rev. B 2016, 94, 165446. [Google Scholar] [CrossRef] [Green Version]
- Novko, D.; Šunjić, M.; Despoja, V. Optical absorption and conductivity in quasi-two-dimensional crystals from first principles: Application to graphene. Phys. Rev. B 2016, 93, 125413. [Google Scholar] [CrossRef] [Green Version]
- Kalinic, A.; Radovic, I.; Karbunar, L.; Despoja, V.; Miskovic, Z.L. Wake effect in interactions of ions with graphene-sapphire-graphene composite system. Phys. E Low-Dimens. Syst. Nanostruct. 2021, 126, 114447. [Google Scholar] [CrossRef]
- Gjerding, M.N.; Cavalcante, L.S.R.; Chaves, A.; Thygesen, K.S. Enhancing and Controlling Plasmons in Janus MoSSe–Graphene Based van der Waals Heterostructures. J. Phys. Chem. C 2021, 124, 11609. [Google Scholar] [CrossRef]
- Wunsch, B.; Stauber, T.; Sols, F.; Guinea, F. Dynamical polarization of graphene at finite doping. New J. Phys. 2006, 8, 318. [Google Scholar] [CrossRef]
- Hwang, E.H.; Sarma, S.D. Dielectric function, screening, and plasmons in two-dimensional graphene. Phys. Rev. B 2007, 75, 205418. [Google Scholar] [CrossRef] [Green Version]
- Nersisyan, H.B.; Das, A.K. Interaction of fast charged projectiles with two-dimensional electron gas: Interaction and collisional-damping effects. Phys. Rev. E 2009, 80, 016402. [Google Scholar] [CrossRef]
- Falkovsky, L.A. Optical properties of graphene. J. Phys. Conf. Ser. 2008, 129, 012004. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. Quantum espresso: A Modul. Open-Source Softw. Proj. Quantum Simulations Materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef]
- Troullier, N.; Martins, J.L. Efficient pseudopotentials for plane-wave calculations. Phys. Rev. B 1991, 43, 1993. [Google Scholar] [CrossRef]
- Hamann, D.R. Optimized norm-conserving Vanderbilt pseudopotentials. Phys. Rev. B 2013, 88, 085117. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [Green Version]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188. [Google Scholar] [CrossRef]
- Perdew, J.P.; Zunger, A. Self-interaction correction to density-functional approximations for many-electron systems. Phys. Rev. B 1981, 23, 5048. [Google Scholar] [CrossRef] [Green Version]
- Saito, R.; Dresselhaus, G.; Dresselhaus, M.S. Physical Properties of Carbon Nanotubes; Imperial College Press: London, UK, 1998. [Google Scholar]
- Arakawa, E.T.; Williams, M.W. Optical properties of aluminum oxide in the vacuum ultraviolet. J. Phys. Chem. Solids 1968, 29, 735–744. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marušić, L.; Kalinić, A.; Radović, I.; Jakovac, J.; Mišković, Z.L.; Despoja, V. Resolving the Mechanism of Acoustic Plasmon Instability in Graphene Doped by Alkali Metals. Int. J. Mol. Sci. 2022, 23, 4770. https://doi.org/10.3390/ijms23094770
Marušić L, Kalinić A, Radović I, Jakovac J, Mišković ZL, Despoja V. Resolving the Mechanism of Acoustic Plasmon Instability in Graphene Doped by Alkali Metals. International Journal of Molecular Sciences. 2022; 23(9):4770. https://doi.org/10.3390/ijms23094770
Chicago/Turabian StyleMarušić, Leonardo, Ana Kalinić, Ivan Radović, Josip Jakovac, Zoran L. Mišković, and Vito Despoja. 2022. "Resolving the Mechanism of Acoustic Plasmon Instability in Graphene Doped by Alkali Metals" International Journal of Molecular Sciences 23, no. 9: 4770. https://doi.org/10.3390/ijms23094770
APA StyleMarušić, L., Kalinić, A., Radović, I., Jakovac, J., Mišković, Z. L., & Despoja, V. (2022). Resolving the Mechanism of Acoustic Plasmon Instability in Graphene Doped by Alkali Metals. International Journal of Molecular Sciences, 23(9), 4770. https://doi.org/10.3390/ijms23094770