
The Impact of Cytokines on Neutrophils’ Phagocytosis and NET Formation during Sepsis—A Review


Abstract
:1. Sepsis
2. The Role of Neutrophils during Sepsis
3. Cytokines—Overview
3.1. Receptors for Cytokines
3.2. Sepsis and Cytokines
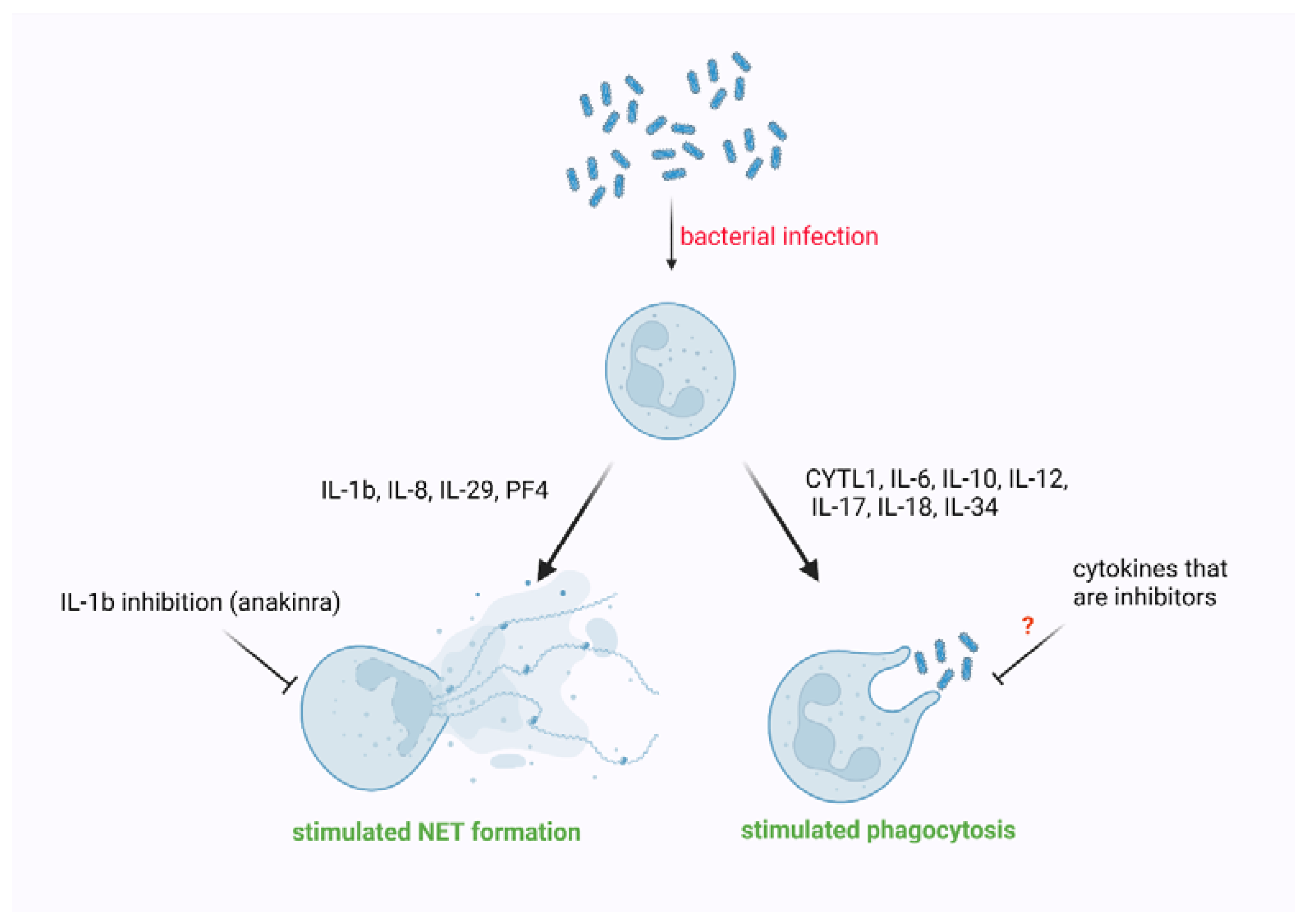
4. The Impact of Cytokines on Phagocytosis Performed by Neutrophils
5. The Impact of Cytokines on NET Formation
6. Conclusions
7. Study Selection
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
References
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.-D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef] [PubMed]
- American College of Chest Physicians and Society of Critical Care Medicine Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine Consensus Conference: Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Crit. Care Med. 1992, 20, 864–874. [Google Scholar] [CrossRef]
- Hotchkiss, R.S.; Monneret, G.; Payen, D. Sepsis-induced immunosuppression: From cellular dysfunctions to immunotherapy. Nat. Rev. Immunol. 2013, 13, 862–874. [Google Scholar] [CrossRef] [PubMed]
- Evans, L.; Rhodes, A.; Alhazzani, W.; Antonelli, M.; Coopersmith, C.M.; French, C.; Machado, F.R.; Mcintyre, L.; Ostermann, M.; Prescott, H.C.; et al. Surviving sepsis campaign: International guidelines for management of sepsis and septic shock 2021. Intensiv. Care Med. 2021, 47, 1181–1247. [Google Scholar] [CrossRef]
- Downing, N.L.; Rolnick, J.; Poole, S.F.; Hall, E.; Wessels, A.J.; Heidenreich, P.; Shieh, L. Electronic health record-based clinical decision support alert for severe sepsis: A randomised evaluation. BMJ Qual. Saf. 2019, 28, 762–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhee, C.; Dantes, R.; Epstein, L.; Murphy, D.J.; Seymour, C.W.; Iwashyna, T.J.; Kadri, S.S.; Angus, D.C.; Danner, R.L.; Fiore, A.E.; et al. Incidence and Trends of Sepsis in US Hospitals Using Clinical vs Claims Data, 2009–2014. JAMA 2017, 318, 1241–1249. [Google Scholar] [CrossRef]
- Fleischmann-Struzek, C.; Mellhammar, L.; Rose, N.; Cassini, A.; Rudd, K.E.; Schlattmann, P.; Allegranzi, B.; Reinhart, K. Incidence and mortality of hospital- and ICU-treated sepsis: Results from an updated and expanded systematic review and meta-analysis. Intensiv. Care Med. 2020, 46, 1552–1562. [Google Scholar] [CrossRef] [PubMed]
- Iskander, K.N.; Osuchowski, M.F.; Stearns-Kurosawa, D.; Kurosawa, S.; Stepien, D.; Valentine, C.; Remick, D. Sepsis: Multiple Abnormalities, Heterogeneous Responses, and Evolving Understanding. Physiol. Rev. 2013, 93, 1247–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, M.P.; Stenstrom, R.; Paquette, K.; Stabler, S.N.; Akhter, M.; Davidson, A.C.; Gavric, M.; Lawandi, A.; Jinah, R.; Saeed, Z.; et al. Blood Culture Results Before and After Antimicrobial Administration in Patients with Severe Manifestations of Sepsis: A Diagnostic Study. Ann. Intern. Med. 2019, 171, 547–554. [Google Scholar] [CrossRef]
- Vincent, J.-L.; Rello, J.; Marshall, J.K.; Silva, E.; Anzueto, A.; Martin, C.D.; Moreno, R.; Lipman, J.; Gomersall, C.; Sakr, Y.; et al. International Study of the Prevalence and Outcomes of Infection in Intensive Care Units. JAMA 2009, 302, 2323–2329. [Google Scholar] [CrossRef] [Green Version]
- Vincent, J.-L.; Sakr, Y.; Sprung, C.L.; Ranieri, V.M.; Reinhart, K.; Gerlach, H.; Moreno, R.; Carlet, J.; Le Gall, J.-R.; Payen, D. Sepsis in European intensive care units: Results of the SOAP study*. Crit. Care Med. 2006, 34, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Boyd, J.H.; Russell, J.A.; Fjell, C. The Meta-Genome of Sepsis: Host Genetics, Pathogens and the Acute Immune Response. J. Innate Immun. 2014, 6, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.F.; Malik, A.B. NF-kappa B activation as a pathological mechanism of septic shock and inflammation. Am. J. Physiol. Lung. Cell Mol. Physiol. 2006, 290, L622–L645. [Google Scholar] [CrossRef] [PubMed]
- Hotchkiss, R.S.; Karl, I.E. The Pathophysiology and Treatment of Sepsis. N. Engl. J. Med. 2003, 348, 138–150. [Google Scholar] [CrossRef] [Green Version]
- Simmons, J.; Pittet, J.-F. The coagulopathy of acute sepsis. Curr. Opin. Anaesthesiol. 2015, 28, 227–236. [Google Scholar] [CrossRef] [Green Version]
- Denstaedt, S.; Singer, B.; Standiford, T.J. Sepsis and Nosocomial Infection: Patient Characteristics, Mechanisms, and Modulation. Front. Immunol. 2018, 9, 2446. [Google Scholar] [CrossRef] [Green Version]
- Bezemer, R.; Bartels, S.A.; Bakker, J.; Ince, C. Clinical review: Clinical imaging of the sublingual microcirculation in the critically ill-Where do we stand? Crit. Care 2012, 16, 224. [Google Scholar] [CrossRef] [Green Version]
- Vincent, J.L.; Zhang, H.; Szabo, C.; Preiser, J.C. Effects of nitric oxide in septic shock. Am. J. Respir. Crit. Care Med. 2000, 161, 1781–1785. [Google Scholar] [CrossRef]
- Cecconi, M.; Moldawer, L.L.; Opal, S.M.; Reinhart, K.; Turnbull, I.R.; Vincent, J.L. Sepsis and septic shock. Lancet 2018, 392, 75–87. [Google Scholar] [CrossRef]
- Fink, M. Cytopathic hypoxia in sepsis. Acta Anaesthesiol. Scand. Suppl. 1997, 110, 87–95. [Google Scholar] [CrossRef]
- Singer, M. The role of mitochondrial dysfunction in sepsis-induced multi-organ failure. Virulence 2014, 5, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef] [PubMed]
- Borregaard, N. Neutrophils, from Marrow to Microbes. Immunity 2010, 33, 657–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papayannopoulos, V.; Zychlinsky, A. NETs: A new strategy for using old weapons. Trends Immunol. 2009, 30, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Sônego, F.; Castanheira, F.V.E.S.; Ferreira, R.G.; Kanashiro, A.; Leite, C.A.V.G.; Nascimento, D.C.; Colón, D.F.; Borges, V.D.F.; Alves-Filho, J.C.; Cunha, F.Q. Paradoxical Roles of the Neutrophil in Sepsis: Protective and Deleterious. Front. Immunol. 2016, 7, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luster, A.D. Chemokines—Chemotactic Cytokines That Mediate Inflammation. N. Engl. J. Med. 1998, 338, 436–445. [Google Scholar] [CrossRef]
- Alves-Filho, J.C.; de Freitas, A.; Spiller, F.; Souto, F.O.; Cunha, F.Q. The role of neutrophils in severe sepsis. Shock 2008, 30, 3–9. [Google Scholar] [CrossRef]
- Chishti, A.D.; Shenton, B.K.; Kirby, J.; Baudouin, S.V. Neutrophil chemotaxis and receptor expression in clinical septic shock. Intensiv. Care Med. 2004, 30, 605–611. [Google Scholar] [CrossRef]
- Arraes, S.M.A.; Freitas, M.S.; da Silva, S.V.; Neto, H.A.D.P.; Alves-Filho, J.C.; Martins, M.A.; Basile-Filho, A.; Tavares-Murta, B.M.; Barja-Fidalgo, C.; Cunha, F.Q. Impaired neutrophil chemotaxis in sepsis associates with GRK expression and inhibition of actin assembly and tyrosine phosphorylation. Blood 2006, 108, 2906–2913. [Google Scholar] [CrossRef] [Green Version]
- Alves-Filho, J.C.; De Freitas, A.; Russo, M.; Cunha, F.Q. Toll-like receptor 4 signaling leads to neutrophil migration impairment in polymicrobial sepsis*. Crit. Care Med. 2006, 34, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Rios-Santos, F.; Alves-Filho, J.C.; Souto, F.O.; Spiller, F.; Freitas, A.; Lotufo, C.M.C.; Soares, M.B.P.; Dos Santos, R.R.; Teixeira, M.M.; Cunha, F.D.Q. Down-regulation of CXCR2 on Neutrophils in Severe Sepsis is Mediated by Inducible Nitric Oxide Synthase-derived Nitric Oxide. Am. J. Respir. Crit. Care Med. 2007, 175, 490–497. [Google Scholar] [CrossRef] [PubMed]
- Cunha, F.Q.; Assreuy, J.; Moss, D.W.; Rees, D.; Leal, L.M.; Moncada, S.; Carrier, M.; O’Donnell, C.A.; Liew, F.Y. Differential induction of nitric oxide synthase in various organs of the mouse during endotoxaemia: Role of TNF-alpha and IL-1-beta. Immunology 1994, 81, 211–215. [Google Scholar] [PubMed]
- Mestriner, F.L.A.C.; Spiller, F.; Laure, H.J.; Souto, F.O.; Tavares-Murta, B.M.; Rosa, J.C.; Basile-Filho, A.; Ferreira, S.H.; Greene, L.J.; Cunha, F.Q. Acute-phase protein α-1-acid glycoprotein mediates neutrophil migration failure in sepsis by a nitric oxide-dependent mechanism. Proc. Natl. Acad. Sci. USA 2007, 104, 19595–19600. [Google Scholar] [CrossRef] [Green Version]
- Torres-Dueñas, D.; Celes, M.R.N.; Freitas, A.; Alves-Filho, J.C.; Spiller, F.; Dal-Secco, D.; Dalto, V.F.; Rossi, M.A.; Ferreira, S.H.; Cunha, F.Q. Peroxynitrite mediates the failure of neutrophil migration in severe polymicrobial sepsis in mice. J. Cereb. Blood Flow Metab. 2007, 152, 341–352. [Google Scholar] [CrossRef]
- Wu, Z.; Sawamura, T.; Kurdowska, A.K.; Ji, H.-L.; Idell, S.; Fu, J. LOX-1 Deletion Improves Neutrophil Responses, Enhances Bacterial Clearance, and Reduces Lung Injury in a Murine Polymicrobial Sepsis Model. Infect. Immun. 2011, 79, 2865–2870. [Google Scholar] [CrossRef] [Green Version]
- Freitas, A.; Alves-Filho, J.C.; Victoni, T.; Secher, T.; Lemos, H.P.; Sônego, F.; Cunha, F.Q.; Ryffel, B. IL-17 Receptor Signaling Is Required to Control Polymicrobial Sepsis. J. Immunol. 2009, 182, 7846–7854. [Google Scholar] [CrossRef] [Green Version]
- Hoesel, L.M.; Neff, T.A.; Neff, S.B.; Younger, J.; Olle, E.W.; Gao, H.; Pianko, M.; Bernacki, K.D.; Sarma, J.V.; Ward, P.A. Harmful and protective roles of neutrophils in sepsis. Shock 2005, 24, 40–47. [Google Scholar] [CrossRef]
- Brown, K.; Brain, S.; Pearson, J.; Edgeworth, J.; Lewis, S.; Treacher, D. Neutrophils in development of multiple organ failure in sepsis. Lancet 2006, 368, 157–169. [Google Scholar] [CrossRef]
- Zhang, S.; Rahman, M.; Qi, Z.; Thorlacius, H. Simvastatin antagonizes CD40L secretion, CXC chemokine formation, and pulmonary infiltration of neutrophils in abdominal sepsis. J. Leukoc. Biol. 2011, 89, 735–742. [Google Scholar] [CrossRef]
- Speyer, C.L.; Gao, H.; Rancilio, N.; Neff, T.A.; Huffnagle, G.B.; Sarma, J.V.; Ward, P.A. Novel Chemokine Responsiveness and Mobilization of Neutrophils during Sepsis. Am. J. Pathol. 2004, 165, 2187–2196. [Google Scholar] [CrossRef] [Green Version]
- Johnston, B.; Burns, A.R.; Suematsu, M.; Issekutz, T.B.; Woodman, R.C.; Kubes, P. Chronic inflammation upregulates chemokine receptors and induces neutrophil migration to monocyte chemoattractant protein-1. J. Clin. Investig. 1999, 103, 1269–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souto, F.O.; Alves-Filho, J.C.; Turato, W.M.; Auxiliadora-Martins, M.; Basile-Filho, A.; Cunha, F.Q. Essential Role of CCR2 in Neutrophil Tissue Infiltration and Multiple Organ Dysfunction in Sepsis. Am. J. Respir. Crit. Care Med. 2011, 183, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Martinod, K.; Fuchs, T.A.; Zitomersky, N.L.; Wong, S.L.; Demers, M.; Gallant, M.; Wang, Y.; Wagner, D.D. PAD4-deficiency does not affect bacteremia in polymicrobial sepsis and ameliorates endotoxemic shock. Blood 2015, 125, 1948–1956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, K.; Koike, Y.; Shimura, T.; Okigami, M.; Ide, S.; Toiyama, Y.; Okugawa, Y.; Inoue, Y.; Araki, T.; Uchida, K.; et al. In Vivo Characterization of Neutrophil Extracellular Traps in Various Organs of a Murine Sepsis Model. PLoS ONE 2014, 9, e111888. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.R.; Ma, A.C.; Tavener, S.A.; McDonald, B.; Goodarzi, Z.; Kelly, M.M.; Patel, K.D.; Chakrabarti, S.; McAvoy, E.; Sinclair, G.D.; et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 2007, 13, 463–469. [Google Scholar] [CrossRef]
- Gao, X.; Hao, S.; Yan, H.; Ding, W.; Li, K.; Li, J. Neutrophil extracellular traps contribute to the intestine damage in endotoxemic rats. J. Surg. Res. 2015, 195, 211–218. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, X.; Pelayo, R.; Monestier, M.; Ammollo, C.T.; Semeraro, F.; Taylor, F.B.; Esmon, N.L.; Lupu, F.; Esmon, C.T. Extracellular histones are major mediators of death in sepsis. Nat. Med. 2009, 15, 1318–1321. [Google Scholar] [CrossRef] [Green Version]
- Astiz, M.E.; DeGent, G.E.; Lin, R.Y.; Rackow, E.C. Microvascular function and rheologic changes in hyperdynamic sepsis. Crit. Care Med. 1995, 23, 265–271. [Google Scholar] [CrossRef]
- Rees, D.D.; Monkhouse, J.E.; Cambridge, D.; Moncada, S. Nitric oxide and the haemodynamic profile of endotoxin shock in the conscious mouse. J. Cereb. Blood Flow Metab. 1998, 124, 540–546. [Google Scholar] [CrossRef]
- Kooy, N.W.; Lewis, S.J.; Royall, J.A.; Yao, Z.Y.; Kelly, D.R.; Beckman, J.S. Extensive tyrosine nitration in human myocardial inflammation: Evidence for the presence of peroxynitrite. Crit. Care Med. 1997, 25, 812–819. [Google Scholar] [CrossRef] [PubMed]
- Loppnow, H. Cytokines: Classification, receptors, mechanisms of action. Internist 2001, 42, 13–14. [Google Scholar] [CrossRef] [PubMed]
- Schett, G.; Elewaut, D.; McInnes, I.; Dayer, J.-M.; Neurath, M.F. How Cytokine Networks Fuel Inflammation: Toward a cytokine-based disease taxonomy. Nat. Med. 2013, 19, 822–824. [Google Scholar] [CrossRef]
- Kouttab, N.M.; Mehta, S.; Morgan, J.; Tannir, N.; Sahasrabuddhe, C.; Maizel, A.L. Lymphokines and monokines as regulators of human lymphoproliferation. Clin. Chem. 1984, 30, 1539–1545. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.E.; Ahn, S.H.; Marks, R.M.; Monsanto, S.P.; Fazleabas, A.T.; Koti, M.; Tayade, C. IL-17A Modulates Peritoneal Macrophage Recruitment and M2 Polarization in Endometriosis. Front. Immunol. 2020, 11, 108. [Google Scholar] [CrossRef] [PubMed]
- Rahe, M.C.; Murtaugh, M.P. Interleukin-21 Drives Proliferation and Differentiation of Porcine Memory B Cells into Antibody Secreting Cells. PLoS ONE 2017, 12, e0171171. [Google Scholar] [CrossRef] [PubMed]
- Kiaei, M.; Kipiani, K.; Calingasan, N.Y.; Wille, E.; Chen, J.; Heissig, B.; Rafii, S.; Lorenzl, S.; Beal, M.F. Matrix metalloproteinase-9 regulates TNF-α and FasL expression in neuronal, glial cells and its absence extends life in a transgenic mouse model of amyotrophic lateral sclerosis. Exp. Neurol. 2007, 205, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.-X.; Leonard, W.J. Fine-Tuning Cytokine Signals. Annu. Rev. Immunol. 2019, 37, 295–324. [Google Scholar] [CrossRef]
- Spangler, J.B.; Moraga, I.; Mendoza, J.L.; Garcia, K.C. Insights into Cytokine–Receptor Interactions from Cytokine Engineering. Annu. Rev. Immunol. 2015, 33, 139–167. [Google Scholar] [CrossRef] [Green Version]
- Oppenheim, J.J. Cytokines: Past, present, and future. Int. J. Hematol. 2001, 74, 3–8. [Google Scholar] [CrossRef]
- Kasper, B.; Brandt, E.; Ernst, M.; Petersen, F. Neutrophil adhesion to endothelial cells induced by platelet factor 4 requires sequential activation of Ras, Syk, and JNK MAP kinases. Blood 2006, 107, 1768–1775. [Google Scholar] [CrossRef] [PubMed]
- Gotthardt, D.; Trifinopoulos, J.; Sexl, V.; Putz, E.M. JAK/STAT Cytokine Signaling at the Crossroad of NK Cell Development and Maturation. Front. Immunol. 2019, 10, 2590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrington, J.S.; Choi, A.M.; Nakahira, K. Mitochondrial DNA in Sepsis. Curr. Opin. Crit. Care 2017, 23, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Recovery Collaborative Group; Horby, P.; Lim, W.S.; Emberson, J.R.; Mafham, M.; Bell, J.L.; Linsell, L.; Staplin, N.; Brightling, C.; Ustianowski, A.; et al. Dexamethasone in Hospitalized Patients with COVID-19. N. Engl. J. Med. 2021, 384, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Gordon, A.C.; Mouncey, P.R.; Al-Beidh, F.; Rowan, K.M.; Nichol, A.D.; Arabi, Y.M.; Annane, D.; Beane, A.; van Bentum-Puijk, W.; Berry, L.R.; et al. Interleukin-6 Receptor Antagonists in Critically Ill Patients with COVID-19. N. Engl. J. Med. 2021, 384, 1491–1502. [Google Scholar]
- Wang, Y.; Zhu, K.; Dai, R.; Li, R.; Li, M.; Lv, X.; Yu, Q. Specific Interleukin-1 Inhibitors, Specific Interleukin-6 Inhibitors, and GM-CSF Blockades for COVID-19 (at the Edge of Sepsis): A Systematic Review. Front. Pharmacol. 2021, 12, 804250. [Google Scholar] [CrossRef]
- Marconi, V.C.; Ramanan, A.V.; de Bono, S.; Kartman, C.E.; Krishnan, V.; Liao, R.; Piruzeli, M.L.B.; Goldman, J.D.; Alatorre-Alexander, J.; Pellegrini, R.D.C.; et al. Efficacy and safety of baricitinib for the treatment of hospitalised adults with COVID-19 (COV-BARRIER): A randomised, double-blind, parallel-group, placebo-controlled phase 3 trial. Lancet Respir. Med. 2021, 9, 1407–1418. [Google Scholar] [CrossRef]
- Guimarães, P.O.; Quirk, D.; Furtado, R.H.; Maia, L.N.; Saraiva, J.F.; Antunes, M.O.; Filho, R.K.; Junior, V.M.; Soeiro, A.M.; Tognon, A.P.; et al. Tofacitinib in Patients Hospitalized with COVID-19 Pneumonia. N. Engl. J. Med. 2021, 385, 406–415. [Google Scholar] [CrossRef]
- Chaudhry, H.; Zhou, J.; Zhong, Y.; Ali, M.M.; McGuire, F.; Nagarkatti, P.S.; Nagarkatti, M. Role of cytokines as a double-edged sword in sepsis. In Vivo 2013, 27, 669–684. [Google Scholar]
- Cai, J.; Lin, Z. Correlation of blood high mobility group box-1 protein with mortality of patients with sepsis: A meta-analysis. Hearth Lung 2021, 50, 885–892. [Google Scholar] [CrossRef]
- Xu, X.-E.; Liu, L.; Wang, Y.-C.; Wang, C.-T.; Zheng, Q.; Liu, Q.-X.; Li, Z.-F.; Bai, X.-J.; Liu, X.-H. Caspase-1 inhibitor exerts brain-protective effects against sepsis-associated encephalopathy and cognitive impairments in a mouse model of sepsis. Brain Behav. Immun. 2019, 80, 859–870. [Google Scholar] [CrossRef] [PubMed]
- Sandquist, M.; Wong, H.R. Biomarkers of sepsis and their potential value in diagnosis, prognosis and treatment. Expert Rev. Clin. Immunol. 2014, 10, 1349–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashem, H.E.; Ibrahim, Z.H.; Ahmed, W.O. Diagnostic, Prognostic, Predictive, and Monitoring Role of Neutrophil CD11b and Monocyte CD14 in Neonatal Sepsis. Dis. Markers 2021, 2021, 4537760. [Google Scholar] [CrossRef] [PubMed]
- Tverring, J.; The FINNAKI Study Group; Vaara, S.T.; Fisher, J.; Poukkanen, M.; Pettilä, V.; Linder, A. Heparin-binding protein (HBP) improves prediction of sepsis-related acute kidney injury. Ann. Intensiv. Care 2017, 7, 105. [Google Scholar] [CrossRef] [Green Version]
- Jones, T.K.; Feng, R.; Kerchberger, V.; Reilly, J.P.; Anderson, B.J.; Shashaty, M.G.S.; Wang, F.; Dunn, T.G.; Riley, T.R.; Abbott, J.; et al. Plasma sRAGE Acts as a Genetically Regulated Causal Intermediate in Sepsis-associated Acute Respiratory Distress Syndrome. Am. J. Respir. Crit. Care Med. 2020, 201, 47–56. [Google Scholar] [CrossRef]
- Kumar, V. Toll-like receptors in sepsis-associated cytokine storm and their endogenous negative regulators as future immunomodulatory targets. Int. Immunopharmacol. 2020, 89, 107087. [Google Scholar] [CrossRef]
- Gouel-Chéron, A.; Allaouchiche, B.; Floccard, B.; Rimmelé, T.; Monneret, G. Early daily mHLA-DR monitoring predicts forthcoming sepsis in severe trauma patients. Intensive Care Med. 2015, 41, 2229–2230. [Google Scholar] [CrossRef]
- Tan, M.; Lu, Y.; Jiang, H.; Zhang, L. The diagnostic accuracy of procalcitonin and C-reactive protein for sepsis: A systematic review and meta-analysis. J. Cell. Biochem. 2019, 120, 5852–5859. [Google Scholar] [CrossRef]
- Keshari, R.S.; Silasi, R.; Popescu, N.I.; Regmi, G.; Chaaban, H.; Lambris, J.D.; Lupu, C.; Mollnes, T.E.; Lupu, F. CD14 inhibition improves survival and attenuates thrombo-inflammation and cardiopulmonary dysfunction in a baboon model of Escherichia coli sepsis. J. Thromb. Haemost. 2020, 19, 429–443. [Google Scholar] [CrossRef]
- Mewes, C.; Büttner, B.; Hinz, J.; Alpert, A.; Popov, A.-F.; Ghadimi, M.; Beissbarth, T.; Tzvetkov, M.; Jensen, O.; Runzheimer, J.; et al. CTLA-4 Genetic Variants Predict Survival in Patients with Sepsis. J. Clin. Med. 2019, 8, 70. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.; Li, X.; Ding, H.; Wang, K.; Liu, X.; Wang, Q.; Li, Y.; Zhou, M.; Chen, S.; Zhong, W.; et al. PD-1 in Tregs predicts the survival in sepsis patients using sepsis-3 criteria: A prospective, two-stage study. Int. Immunopharmacol. 2020, 89, 107175. [Google Scholar] [CrossRef] [PubMed]
- Németh, K.; Leelahavanichkul, A.; Yuen, P.S.; Mayer, B.; Parmelee, A.; Robey, P.G.; Leelahavanichkul, K.; Koller, B.H.; Brown, J.M. Bone marrow stromal cells attenuate sepsis via prostaglandin E(2)-dependent reprogramming of host macrophages to increase their interleukin-10 production. Nat. Med. 2009, 15, 42–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, J.G.; Simpson, L.J.; Ferreira, A.-M.; Rustagi, A.; Roque, J.; Asuni, A.; Ranganath, T.; Grant, P.M.; Subramanian, A.; Rosenberg-Hasson, Y.; et al. Cytokine profile in plasma of severe COVID-19 does not differ from ARDS and sepsis. JCI Insight 2020, 5, e140289. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Liu, Y.; Hou, H.; Yao, Y.; Meng, H. MiR-150 predicts survival in patients with sepsis and inhibits LPS-induced inflammatory factors and apoptosis by targeting NF-κB1 in human umbilical vein endothelial cells. Biochem. Biophys. Res. Commun. 2018, 500, 828–837. [Google Scholar] [CrossRef]
- Liu, Z.; Meng, Z.; Li, Y.; Zhao, J.; Wu, S.; Gou, S.; Wu, H. Prognostic accuracy of the serum lactate level, the SOFA score and the qSOFA score for mortality among adults with Sepsis. Scand. J. Trauma Resusc. Emerg. Med. 2019, 27, 51. [Google Scholar] [CrossRef]
- De Geus, H.R.; Betjes, M.G.; Schaick, R.V.; Groeneveld, J.A. Plasma NGAL similarly predicts acute kidney injury in sepsis and nonsepsis. Biomark. Med. 2013, 7, 415–421. [Google Scholar] [CrossRef]
- Jiang, N.; Huang, R.; Zhang, J.; Xu, D.; Li, T.; Sun, Z.; Su, L.; Peng, Z. TIMP2 mediates endoplasmic reticulum stress contributing to sepsis-induced acute kidney injury. FASEB J. 2022, 36, e22228. [Google Scholar] [CrossRef]
- Yang, C.; Xia, W.; Liu, X.; Lin, J.; Wu, A. Role of TXNIP/NLRP3 in sepsis-induced myocardial dysfunction. Int. J. Mol. Med. 2019, 44, 417–426. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.-F.; Qin, J.; Guo, X.-H. MiR-181-5p protects mice from sepsis via repressing HMGB1 in an experimental model. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 9712–9720. [Google Scholar]
- Schuetz, P.; Yano, K.; Sorasaki, M.; Ngo, L.; Hilaire, M.S.; Lucas, J.M.; Aird, W.; Shapiro, N.I. Influence of diabetes on endothelial cell response during sepsis. Diabetologia 2011, 54, 996–1003. [Google Scholar] [CrossRef] [Green Version]
- Besnier, E.; Brakenhielm, E.; Richard, V.; Tamion, F. Does anti-VEGF bevacizumab improve survival in experimental sepsis? Crit. Care 2017, 21, 163. [Google Scholar] [CrossRef] [Green Version]
- Iba, T.; Levi, M.; Levy, J.H. Sepsis-Induced Coagulopathy and Disseminated Intravascular Coagulation. Semin. Thromb. Hemost. 2020, 46, 89–95. [Google Scholar] [CrossRef] [PubMed]
- El Beshlawy, A.; Alaraby, I.; Hussein, H.A.; Abou-Elew, H.H.; Kader, M.S.E.M.A. Study of protein C, protein S, and antithrombin III in newborns with sepsis. Pediatr. Crit. Care Med. 2010, 11, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Semeraro, F.; Ammollo, C.T.; Caironi, P.; Masson, S.; Latini, R.; Panigada, M.; Semeraro, N.; Gattinoni, L.; Colucci, M. Low D-dimer levels in sepsis: Good or bad? Thromb. Res. 2019, 174, 13–15. [Google Scholar] [CrossRef] [PubMed]
- Carp, H. Mitochondrial N-formylmethionyl proteins as chemoattractants for neutrophils. J. Exp. Med. 1982, 155, 264–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, Y.; Murphy, P.M.; Wang, J.M. Formyl-peptide receptors revisited. Trends Immunol. 2002, 23, 541–548. [Google Scholar] [CrossRef]
- West, A.P.; Shadel, G.S. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat. Rev. Immunol. 2017, 17, 363–375. [Google Scholar] [CrossRef]
- Martinez-García, J.J.; Banaclocha, H.M.; Angosto, D.; De Torre-Minguela, C.; Baroja-Mazo, A.; Alarcón-Vila, C.; Martinez-Alarcon, L.; Amores-Iniesta, J.; Martín-Sánchez, F.; Ercole, G.A.; et al. P2X7 receptor induces mitochondrial failure in monocytes and compromises NLRP3 inflammasome activation during sepsis. Nat. Commun. 2019, 10, 2711. [Google Scholar] [CrossRef] [Green Version]
- Bermúdez-Mejía, C.; Torres-Cordón, M.F.; Becerra-Bayona, S.; Páez, C.M.; Vargas, C.I.; Cárdenas, M.E.; Serrano, S.E.; Baquero, I.; Martínez-Vega, R.; Schulz, R.; et al. Prognostic Value of MMP-9-1562 C/T Gene Polymorphism in Patients with Sepsis. Biomark. Insights 2019, 14, 1177271919847951. [Google Scholar] [CrossRef] [Green Version]
- Razak, A.; Hussain, A. Lactoferrin Supplementation to Prevent Late-Onset Sepsis in Preterm Infants: A Meta-Analysis. Am. J. Perinatol. 2021, 38, 283–290. [Google Scholar] [CrossRef]
- Benjamim, C.F.; Hogaboam, C.M.; Kunkel, S.L. The chronic consequences of severe sepsis. J. Leukoc. Biol. 2004, 75, 408–412. [Google Scholar] [CrossRef] [Green Version]
- Oberholzer, A.; Oberholzer, C.; Moldawer, L.L. Interleukin-10: A complex role in the pathogenesis of sepsis syndromes and its potential as an anti-inflammatory drug. Crit. Care Med. 2002, 30, S58–S63. [Google Scholar] [CrossRef] [PubMed]
- Steinhauser, M.L.; Kunkel, S.L.; Hogaboam, C.M. New Frontiers in Cytokine Involvement during Experimental Sepsis. ILAR J. 1999, 40, 142–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Küster, H.; Weiss, M.; Willeitner, A.E.; Detlefsen, S.; Jeremias, I.; Zbojan, J.; Geiger, R.; Lipowsky, G.; Simbruner, G. Interleukin-1 receptor antagonist and interleukin-6 for early diagnosis of neonatal sepsis 2 days before clinical manifestation. Lancet 1998, 352, 1271–1277. [Google Scholar] [CrossRef]
- Redl, H.; Schlag, G.; Ceska, M.; Davies, J.; Buurman, W.A. Interleukin-8 Release in Baboon Septicemia is Partially Dependent on Tumor Necrosis Factor. J. Infect. Dis. 1993, 167, 1464–1466. [Google Scholar] [CrossRef] [PubMed]
- Marty, C.; Misset, B.; Tamion, F.; Fitting, C.; Carlet, J.; Cavaillon, J.-M. Circulating interleukin-8 concentrations in patients with multiple organ failure of septic and nonseptic origin. Crit. Care Med. 1994, 22, 673–679. [Google Scholar] [CrossRef]
- Angus, D.C.; Linde-Zwirble, W.T.; Lidicker, J.; Clermont, G.; Carcillo, J.; Pinsky, M.R. Epidemiology of severe sepsis in the United States: Analysis of incidence, outcome, and associated costs of care. Crit. Care Med. 2001, 29, 1303–1310. [Google Scholar] [CrossRef]
- Friedman, G.; Jankowski, S.; Marchant, A.; Goldman, M.; Kahn, R.J.; Vincent, J.L. Blood interleukin 10 levels parallel the severity of septic shock. J. Crit. Care 1997, 12, 183–187. [Google Scholar] [CrossRef]
- Giannoudis, P.V.; Smith, R.M.; Perry, S.L.; Windsor, A.J.; Dickson, R.A.; Bellamy, M.C. Immediate IL-10 expression following major orthopaedic trauma: Relationship to anti-inflammatory response and subsequent development of sepsis. Intensiv. Care Med. 2000, 26, 1076–1081. [Google Scholar] [CrossRef]
- Gérard, C.; Bruyns, C.; Marchant, A.; Abramowicz, D.; Vandenabeele, P.; Delvaux, A.; Fiers, W.; Goldman, M.; Velu, T. Interleukin 10 reduces the release of tumor necrosis factor and prevents lethality in experimental endotoxemia. J. Exp. Med. 1993, 177, 547–550. [Google Scholar] [CrossRef]
- Kalechman, Y.; Gafter, U.; Gal, R.; Rushkin, G.; Yan, D.; Albeck, M.; Sredni, B. Anti-IL-10 Therapeutic Strategy Using the Immunomodulator AS101 in Protecting Mice from Sepsis-Induced Death: Dependence on Timing of Immunomodulating Intervention. J. Immunol. 2002, 169, 384–392. [Google Scholar] [CrossRef] [Green Version]
- Mazer, M.; Unsinger, J.; Drewry, A.; Walton, A.; Osborne, D.; Blood, T.; Hotchkiss, R.; Remy, K.E. IL-10 Has Differential Effects on the Innate and Adaptive Immune Systems of Septic Patients. J. Immunol. 2019, 203, 2088–2099. [Google Scholar] [CrossRef] [PubMed]
- Xue, H.; Li, S.; Zhao, X.; Guo, F.; Jiang, L.; Wang, Y.; Zhu, F. CYTL1 Promotes the Activation of Neutrophils in a Sepsis Model. Inflammation 2020, 43, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Onogawa, T.; Saito-Taki, T.; Yamamoto, H.; Wada, T. IL6 trans-signaling promotes functional recovery of hypofunctional phagocytes through STAT3 activation during peritonitis. Agents Actions 2013, 62, 797–810. [Google Scholar] [CrossRef] [PubMed]
- Mullen, P.G.; Windsor, A.C.; Walsh, C.J.; Fowler, A.A.; Sugerman, H.J. Tumor Necrosis Factor-α and Interleukin-6 Selectively Regulate Neutrophil Function in Vitro. J. Surg. Res. 1995, 58, 124–130. [Google Scholar] [CrossRef]
- Mittal, R.; Gonzalez-Gomez, I.; Panigrahy, A.; Goth, K.; Bonnet, R.; Prasadarao, N.V. IL-10 administration reduces PGE-2 levels and promotes CR3-mediated clearance of Escherichia coli K1 by phagocytes in meningitis. J. Exp. Med. 2010, 207, 1307–1319. [Google Scholar] [CrossRef] [Green Version]
- Moreno, S.E.; Alves-Filho, J.C.; Alfaya, T.M.; Da Silva, J.S.; Ferreira, S.H.; Liew, F.Y. IL-12, but Not IL-18, Is Critical to Neutrophil Activation and Resistance to Polymicrobial Sepsis Induced by Cecal Ligation and Puncture. J. Immunol. 2006, 177, 3218–3224. [Google Scholar] [CrossRef]
- Van De Veerdonk, F.; Kullberg, B.; Verschueren, I.; Hendriks, T.; Van Der Meer, J.; Joosten, L.; Netea, M. Differential effects of IL-17 pathway in disseminated candidiasis and zymosan-induced multiple organ failure. Shock 2010, 34, 407–411. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Luo, H.; Yan, X.; Song, Z.; Gao, X.; Xia, Y.; Zhang, L.; Yin, Y.; Cao, J. Interleukin-34 Ameliorates Survival and Bacterial Clearance in Polymicrobial Sepsis. Crit. Care Med. 2018, 46, e584–e590. [Google Scholar] [CrossRef]
- Gaber, T.; Hahne, M.; Strehl, C.; Hoff, P.; Dörffel, Y.; Feist, E.; Burmester, G.-R.; Buttgereit, F. Disentangling the effects of tocilizumab on neutrophil survival and function. Immunol. Res. 2015, 64, 665–676. [Google Scholar] [CrossRef]
- Mejia, L.A.F.; Cabrera-Rivera, G.L.; Ferat-Osorio, E.; Mancilla-Herrera, I.; Rosas, R.T.; Boscó-Garate, I.B.; Macías, C.I.R.L.; Isibasi, A.; Cérbulo-Vazquez, A.; Arriaga-Pizano, L.A. Function is Dissociated from Activation-Related Immunophenotype on Phagocytes From Patients With SIRS/Sepsis Syndrome. Shock 2019, 52, e68–e75. [Google Scholar] [CrossRef]
- Chen, Y.; Gong, F.-Y.; Li, Z.-J.; Gong, Z.; Zhou, Z.; Ma, S.-Y.; Gao, X.-M. A study on the risk of fungal infection with tofacitinib (CP-690550), a novel oral agent for rheumatoid arthritis. Sci. Rep. 2017, 7, 6779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gierlikowska, B.; Stachura, A.; Gierlikowski, W.; Demkow, U. Phagocytosis, Degranulation and Extracellular Traps Release by Neutrophils-The Current Knowledge, Pharmacological Modulation and Future Prospects. Front Pharmacol. 2021, 12, 666732. [Google Scholar] [CrossRef] [PubMed]
- Wadehn, H.; Raluy, L.P.; Kolman, J.; Duecker, C.; Trochimiuk, M.; Appl, B.; Boettcher, M.; Reinshagen, K.; Trah, J. Time- and dose-dependent inhibition of neutrophil extracellular trap formation by blocking of the interleukin-1 receptor. Central Eur. J. Immunol. 2021, 46, 419–426. [Google Scholar] [CrossRef]
- Yaqinuddin, A.; Kashir, J. Novel therapeutic targets for SARS-CoV-2-induced acute lung injury: Targeting a potential IL-1β/neutrophil extracellular traps feedback loop. Med. Hypotheses 2020, 143, 109906. [Google Scholar] [CrossRef] [PubMed]
- Abrams, S.T.; Morton, B.; Alhamdi, Y.; Alsabani, M.; Lane, S.; Welters, I.; Wang, G.; Toh, C.-H. A Novel Assay for Neutrophil Extracellular Trap Formation Independently Predicts Disseminated Intravascular Coagulation and Mortality in Critically Ill Patients. Am. J. Respir. Crit. Care Med. 2019, 200, 869–880. [Google Scholar] [CrossRef]
- Alsabani, M.; Abrams, S.T.; Cheng, Z.; Morton, B.; Lane, S.; Alosaimi, S.; Yu, W.; Wang, G.; Toh, C.H. Reduction of NETosis by targeting CXCR1/2 reduces thrombosis, lung injury, and mortality in experimental human and murine sepsis. Br. J. Anaesth. 2022, 128, 283–293. [Google Scholar] [CrossRef]
- Chrysanthopoulou, A.; Kambas, K.; Stakos, D.; Mitroulis, I.; Mitsios, A.; Vidali, V.; Angelidou, I.; Bochenek, M.; Arelaki, S.; Arampatzioglou, A.; et al. Interferon lambda1/IL-29 and inorganic polyphosphate are novel regulators of neutrophil-driven thromboinflammation. J. Pathol. 2017, 243, 111–122. [Google Scholar] [CrossRef]
- Rossaint, J.; Herter, J.M.; Van Aken, H.; Napirei, M.; Döring, Y.; Weber, C.; Soehnlein, O.; Zarbock, A. Synchronized integrin engagement and chemokine activation is crucial in neutrophil extracellular trap–mediated sterile inflammation. Blood J. Am. Soc. Hematol. 2014, 123, 2573–2584. [Google Scholar] [CrossRef]
- Gollomp, K.; Sarkar, A.; Harikumar, S.; Seeholzer, S.H.; Arepally, G.M.; Hudock, K.M.; Rauova, L.; Kowalska, M.A.; Poncz, M. Fc-modified HIT-like monoclonal antibody as a novel treatment for sepsis. Blood 2020, 135, 743–754. [Google Scholar] [CrossRef]
- Barnes, B.J.; Adrover, J.M.; Baxter-Stoltzfus, A.; Borczuk, A.; Cools-Lartigue, J.; Crawford, J.M.; Daßler-Plenker, J.; Guerci, P.; Huynh, C.; Knight, J.S.; et al. Targeting potential drivers of COVID-19: Neutrophil extracellular traps. J. Exp. Med. 2020, 217, e20200652. [Google Scholar] [CrossRef]
- Tomar, B.; Anders, H.-J.; Desai, J.; Mulay, S.R. Neutrophils and Neutrophil Extracellular Traps Drive Necroinflammation in COVID-19. Cells 2020, 9, 1383. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Ji, Q.; Zhu, Y.; Liu, D.; Fu, S.; Wang, X.; Tai, N. Peripheral 5-hydroxytryptophan aggravates lung injury in septic mice by inducing the formation of neutrophils extracellular trap. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue 2021, 33, 1423–1427. [Google Scholar] [PubMed]
- Kaufman, T.; Magosevich, D.; Moreno, M.C.; Guzman, M.A.; D’Atri, L.P.; Carestia, A.; Fandiño, M.E.; Fondevila, C.; Schattner, M. Nucleosomes and neutrophil extracellular traps in septic and burn patients. Clin. Immunol. 2017, 183, 254–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carestia, A.; Kaufman, T.; Rivadeneyra, L.; Landoni, V.I.; Pozner, R.G.; Negrotto, S.; D’Atri, L.P.; Gomez, R.; Schattner, M. Mediators and molecular pathways involved in the regulation of neutrophil extracellular trap formation mediated by activated platelets. J. Leukoc. Biol. 2016, 99, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Arase, H. Regulation of immune responses by neutrophils. Ann. N. Y. Acad. Sci. 2014, 1319, 66–81. [Google Scholar] [CrossRef]
- Donnelly, S.; MacGregor, I.; Zamani, A.; Gordon, M.W.; Robertson, C.E.; Steedman, D.J.; Little, K.; Haslett, C. Plasma elastase levels and the development of the adult respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 1995, 151, 1428–1433. [Google Scholar] [CrossRef]
- Dibbert, B.; Weber, M.; Nikolaizik, W.H.; Vogt, P.; Schöni, M.H.; Blaser, K.; Simon, H.-U. Cytokine-mediated Bax deficiency and consequent delayed neutrophil apoptosis: A general mechanism to accumulate effector cells in inflammation. Proc. Natl. Acad. Sci. USA 1999, 96, 13330–13335. [Google Scholar] [CrossRef] [Green Version]
- Taneja, R.; Parodo, J.; Jia, S.H.; Kapus, A.; Rotstein, O.D.; Marshall, J.C. Delayed neutrophil apoptosis in sepsis is associated with maintenance of mitochondrial transmembrane potential and reduced caspase-9 activity. Crit. Care Med. 2004, 32, 1460–1469. [Google Scholar] [CrossRef]
- Liu, F.-C.; Chuang, Y.-H.; Tsai, Y.-F.; Yu, H.-P. Role of Neutrophil Extracellular Traps Following Injury. Shock 2014, 41, 491–498. [Google Scholar] [CrossRef]
- Folco, E.J.; Mawson, T.L.; Vromman, A.; Bernardes-Souza, B.; Franck, G.; Persson, O.; Nakamura, M.; Newton, G.; Luscinskas, F.W.; Libby, P. Neutrophil Extracellular Traps Induce Endothelial Cell Activation and Tissue Factor Production Through Interleukin-1α and Cathepsin G. Arter. Thromb. Vasc. Biol. 2018, 38, 1901–1912. [Google Scholar] [CrossRef]
- Kanamaru, R.; Ohzawa, H.; Miyato, H.; Matsumoto, S.; Haruta, H.; Kurashina, K.; Saito, S.; Hosoya, Y.; Yamaguchi, H.; Yamashita, H.; et al. Low density neutrophils (LDN) in postoperative abdominal cavity assist the peritoneal recurrence through the production of neutrophil extracellular traps (NETs). Sci. Rep. 2018, 8, 632. [Google Scholar] [CrossRef] [PubMed]
- Furumoto, Y.; Smith, C.K.; Blanco, L.; Zhao, W.; Brooks, S.R.; Thacker, S.G.; Zarzour, A.; Sciume, G.; Tsai, W.L.; Trier, A.; et al. Tofacitinib Ameliorates Murine Lupus and Its Associated Vascular Dysfunction. Arthritis Rheumatol. 2017, 69, 148–160. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Type of Mediators | Examples | Ref. |
|---|---|---|
| Pro-inflammatory cytokines | IL-1β, IL-6, IL-8, TNF-α, MCP-1, IL-1RA, TNF-R1/2, HMGB-1 | [69,70,71] |
| Mediators of neutrophil activation | Proteins in neutrophil granules, CD64, CD11b, sCD14, TREM-1, HBP, sRAGE, TLR, suPAR, mHLA-DR | [72,73,74,75,76,77] |
| Acute phase proteins | CRP, procalcitonin, LBP, PTX3 | [78] |
| Complement system | C3b, C5a | [79] |
| Mediators of immunosuppression in sepsis | mHLA-DR, CTLA-4, PD-1, IL-10, IL-1Ra | [77,80,81,82,83] |
| Mediators of organ injury | Lactate, NGAL, TIMP2, troponins, microRNAs (miR-181, miR-150) | [84,85,86,87,88,89] |
| Mediators of endothelium injury | ICAM-1, VCAM-1, E-selectin, VEGF | [90,91] |
| Coagulation activation | Antithrombin, thrombomodulin, C-protein, S-protein, D-dimers | [92,93,94] |
| Mediators of mtDNA injury | N-formylmethionine containing mitochondrial proteins (CYOX-S-1, CYOX-S-2), proteins not containing N-formylrnethionine (CYOX-S-IV, CYOX-S-V, CYOX-S-VI, Complex II), formyl-peptide receptor (FPR) and its variant FPRL1 (FPR-like 1), unmethylated CpG, P2X7 receptor | [95,96,97,98] |
| Other | MMP-9, lactoferrin | [99,100] |
| Cytokine | Organism | Setting | Target | Effect | Ref. |
|---|---|---|---|---|---|
| CYTL1 | in vitro Human, in vivo Mice | Escherichia coli infection | phosphorylation of protein kinase B (Akt) | enhanced phagocytosis | [113] |
| IL-6 | Mice | Staphylococcus aureus infection | phosphorylation of STAT3 | augmented the uptake of bacteria and phagosome acidification | [114] |
| IL-6 | Human | Staphylococcus aureus infection | n.i. | enhanced phagocytosis and stimulated ROS generation | [115] |
| IL-10 | Mice | Escherichia coli induced meningitis | increased expression of CR3 | enhanced phagocytosis | [116] |
| IL-12 | Mice | polymicrobial sepsis induced by cecal ligation and puncture (CLP) | induction of IFNγ | enhanced phagocytosis | [117] |
| IL-17 | Mice | Candida albicans induced sepsis, and zymosan-induced multiple organ failure | n.i. | enhanced phagocytosis | [118] |
| IL-18 | Mice | polymicrobial sepsis induced by cecal ligation and puncture (CLP) | n.i. | did not affect phagocytosis | [117] |
| IL-34 | in vitro Human, in vivo Mice | sepsis | n.i. | enhanced phagocytosis | [119] |
| Cytokine | Organism | Setting | Target | Effect | Ref. |
|---|---|---|---|---|---|
| IL-1β | Human | (LPS)-induced and phorbol-12-myristate 13-acetate (PMA)-induced formation | n.i. | Increased NET formation | [124] |
| IL-1β | Human | SARS-CoV-2-induced acute lung injury, sepsis | NLRP3 | Increased NET formation | [125] |
| IL-8 | Human | Intensive care units (ICU), sepsis | Ras/Raf/MAPK | Increased NET formation | [126] |
| IL-8 | Human | E. coli-induced sepsis | CXCR1/2 | Increased NET formation | [127] |
| IL-8 | Mice | Experimental sepsis (caecal ligation and puncture or intraperitoneal injection of E. coli) | CXCR1/2 | Increased NET formation | [127] |
| IL-8 | Human | n.i. | n.i. | Increased NET formation | [25] |
| IL-29 | Mice | ferric chloride-induced thrombosis | mTOR | Activation of NETosis | [128] |
| PF4 | Human | acute lung injury (ALI) | n.i. | Increased NET formation | [129] |
| PF4 | Mice | Sepsis | n.i. | Increased NET formation | [130] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gierlikowska, B.; Stachura, A.; Gierlikowski, W.; Demkow, U. The Impact of Cytokines on Neutrophils’ Phagocytosis and NET Formation during Sepsis—A Review. Int. J. Mol. Sci. 2022, 23, 5076. https://doi.org/10.3390/ijms23095076
Gierlikowska B, Stachura A, Gierlikowski W, Demkow U. The Impact of Cytokines on Neutrophils’ Phagocytosis and NET Formation during Sepsis—A Review. International Journal of Molecular Sciences. 2022; 23(9):5076. https://doi.org/10.3390/ijms23095076
Chicago/Turabian StyleGierlikowska, Barbara, Albert Stachura, Wojciech Gierlikowski, and Urszula Demkow. 2022. "The Impact of Cytokines on Neutrophils’ Phagocytosis and NET Formation during Sepsis—A Review" International Journal of Molecular Sciences 23, no. 9: 5076. https://doi.org/10.3390/ijms23095076
APA StyleGierlikowska, B., Stachura, A., Gierlikowski, W., & Demkow, U. (2022). The Impact of Cytokines on Neutrophils’ Phagocytosis and NET Formation during Sepsis—A Review. International Journal of Molecular Sciences, 23(9), 5076. https://doi.org/10.3390/ijms23095076