
Neutrophil Death in Myeloproliferative Neoplasms: Shedding More Light on Neutrophils as a Pathogenic Link to Chronic Inflammation



Abstract
:1. Introduction
2. Neutrophils
MPN Neutrophils
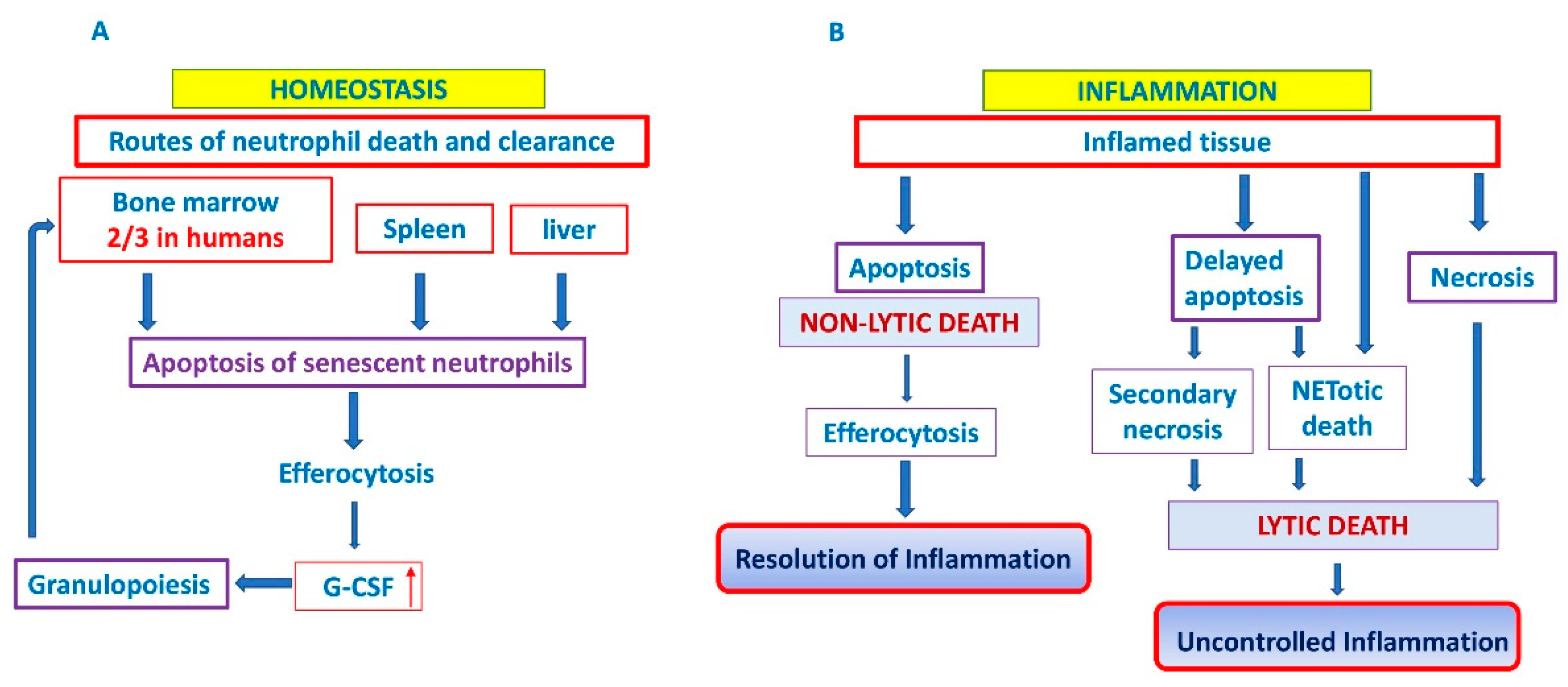
3. Neutrophil Cell Death
3.1. Apoptosis
3.2. Clearance of Apoptotic Neutrophils
4. The Interplay between Bone Marrow and Neutrophils
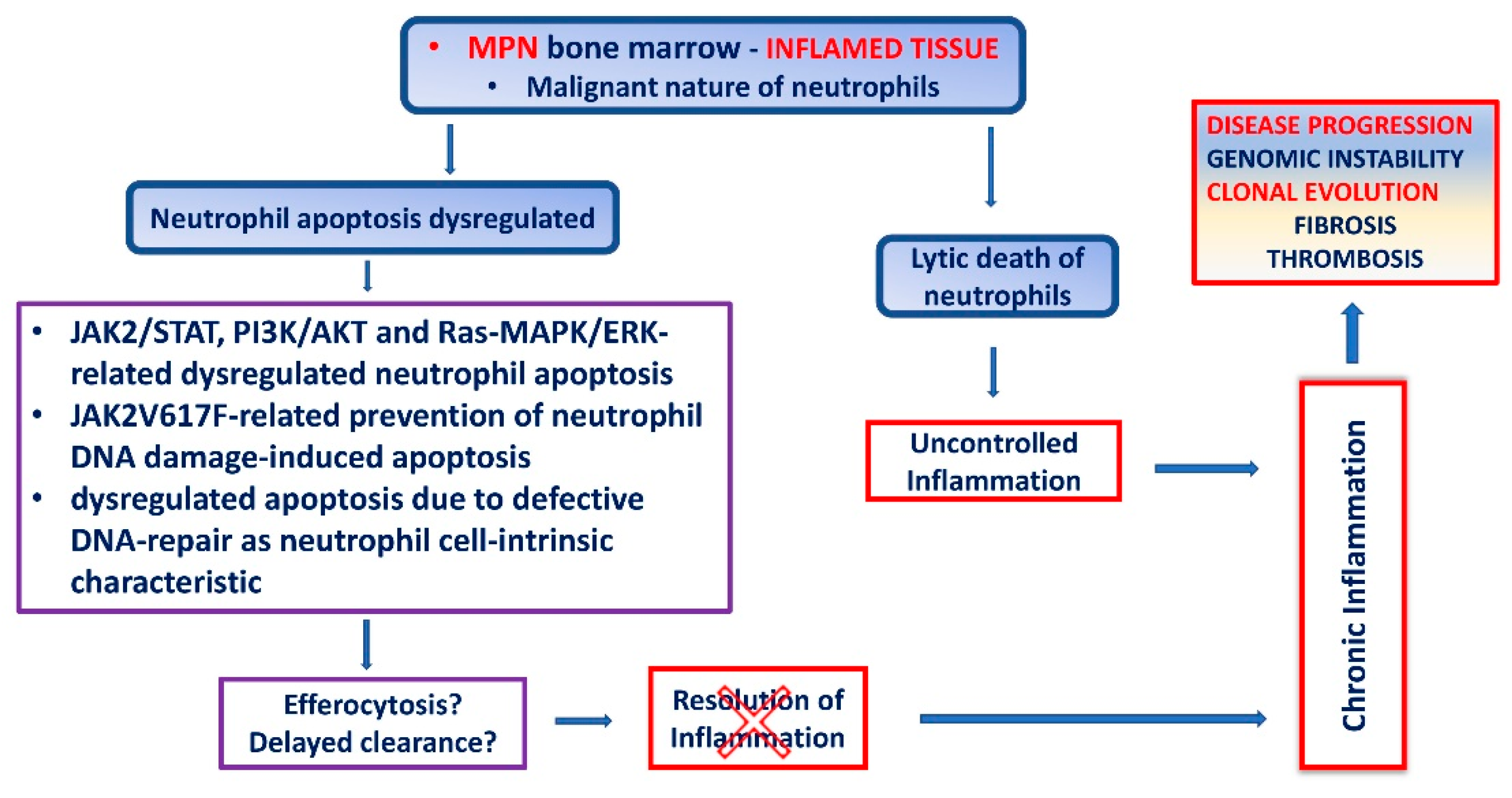
5. Apoptosis of MPN Neutrophils
6. Chronic Inflammation in MPN Due to Dysregulated Neutrophil Cell Death
7. Impaired Neutrophil Apoptosis in MPN as a Consequence of Defective DNA Repair Mechanisms
8. Targeting Neutrophil Apoptosis in MPN Therapy
9. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| AIM2 | absence in melanoma 2 |
| BCL-2 | B-cell lymphoma-2 |
| CALR | calreticulin |
| CDKs | cyclin-dependent kinases |
| cfDNA | cell-free DNA |
| COPD | chronic obstructive pulmonary disease |
| DAMPs | damage associated molecular patterns |
| DC | dendritic cells |
| DSB | double-strand breaks |
| dsDNA | double-stranded DNA |
| ET | essential thrombocythemia |
| HPC | hematopoietic progenitor cell |
| HSC | hematopoietic stem cell |
| JAK2 | Janus kinase 2 |
| LCN2 | lipocalin-2 |
| MAPK | mitogen activated protein kinase |
| MCL-1 | myeloid cell leukemia sequence 1 |
| MMP9 | matrix metalloproteinase 9 |
| MNDA | myeloid nuclear differentiation antigen |
| MPL | myeloproliferative leukemia virus, thrombopoietin receptor |
| MPN | myeloproliferative neoplasms |
| MPO | myeloperoxidase |
| NE | neutrophil elastase |
| NET | neutrophil extracellular trap |
| NGAL | neutrophil gelatinase-associated lipocalin |
| NLRP3 | NOD-like receptor pyrin domain containing 3 |
| NLRs | NOD-like receptors |
| PAMPs | pathogen associated molecular patterns |
| PCNA | proliferating cell nuclear antigen |
| PI3k | phosphatydilinositol-3-kinase |
| PMF | primary myelofibrosis |
| PRRs | pattern recognition receptors |
| PV | polycythemia vera |
| RNS | reactive nitrogen species |
| ROS | reactive oxygen species |
| SSB | single-strand breaks |
| STAT | signal transducers and activators of transcription |
References
- Cool, T.; Forsberg, E.C. Chasing Mavericks: The quest for defining developmental waves of hematopoiesis. Curr. Top. Dev. Biol. 2019, 132, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Adamson, J.W.; Fialkow, P.J.; Murphy, S.; Prchal, J.F.; Steinmann, L. Polycythemia Vera: Stem-Cell and Probable Clonal Origin of the Disease. N. Engl. J. Med. 1976, 295, 913–916. [Google Scholar] [CrossRef] [PubMed]
- Spivak, J.L. Narrative Review: Thrombocytosis, Polycythemia Vera, and JAK2 Mutations: The Phenotypic Mimicry of Chronic Myeloproliferation. Ann. Intern. Med. 2010, 152, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Saeidi, K. Myeloproliferative neoplasms: Current molecular biology and genetics. Crit. Rev. Oncol. 2016, 98, 375–389. [Google Scholar] [CrossRef] [PubMed]
- James, C.; Ugo, V.; Le Couédic, J.-P.; Staerk, J.; Delhommeau, F.; Lacout, C.; Garçon, L.; Raslova, H.; Berger, R.; Bennaceur-Griscelli, A.; et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005, 434, 1144–1148. [Google Scholar] [CrossRef]
- Kralovics, R.; Passamonti, F.; Buser, A.S.; Teo, S.-S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A Gain-of-Function Mutation of JAK2 in Myeloproliferative Disorders. N. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar] [CrossRef] [Green Version]
- Levine, R.L.; Wadleigh, M.; Cools, J.; Ebert, B.L.; Wernig, G.; Huntly, B.J.; Boggon, T.J.; Wlodarska, I.; Clark, J.J.; Moore, S.; et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005, 7, 387–397. [Google Scholar] [CrossRef] [Green Version]
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fourouclas, N.; Swanton, S.; Vassiliou, G.S.; Bench, A.J.; Boyd, E.M.; Curtin, N.; et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005, 365, 1054–1061. [Google Scholar] [CrossRef]
- Scott, L.; Tong, W.; Levine, R.L.; Scott, M.A.; Beer, P.A.; Stratton, M.R.; Futreal, P.A.; Erber, W.; McMullin, M.F.; Harrison, C.N.; et al. JAK2 Exon 12 Mutations in Polycythemia Vera and Idiopathic Erythrocytosis. N. Engl. J. Med. 2007, 356, 459–468. [Google Scholar] [CrossRef] [Green Version]
- Pardanani, A.D.; Levine, R.L.; Lasho, T.; Pikman, Y.; Mesa, R.A.; Wadleigh, M.; Steensma, D.P.; Elliott, M.A.; Wolanskyj, A.P.; Hogan, W.J.; et al. MPL515 mutations in myeloproliferative and other myeloid disorders: A study of 1182 patients. Blood 2006, 108, 3472–3476. [Google Scholar] [CrossRef] [Green Version]
- Pikman, Y.; Lee, B.H.; Mercher, T.; McDowell, E.; Ebert, B.L.; Gozo, M.; Cuker, A.; Wernig, G.; Moore, S.; Galinsky, I.; et al. MPLW515L Is a Novel Somatic Activating Mutation in Myelofibrosis with Myeloid Metaplasia. PLoS Med. 2006, 3, e270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klampfl, T.; Gisslinger, H.; Harutyunyan, A.S.; Nivarthi, H.; Rumi, E.; Milosevic, J.D.; Them, N.C.C.; Berg, T.; Gisslinger, B.; Pietra, D.; et al. Somatic Mutations of Calreticulin in Myeloproliferative Neoplasms. N. Engl. J. Med. 2013, 369, 2379–2390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nangalia, J.; Massie, C.E.; Baxter, E.J.; Nice, F.L.; Gundem, G.; Wedge, D.C.; Avezov, E.; Li, J.; Kollmann, K.; Kent, D.G.; et al. Somatic CALR Mutations in Myeloproliferative Neoplasms with Nonmutated JAK2. N. Engl. J. Med. 2013, 369, 2391–2405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delhommeau, F.; Jeziorowska, D.; Marzac, C.; Casadevall, N. Molecular aspects of myeloproliferative neoplasms. Int. J. Hematol. 2010, 91, 165–173. [Google Scholar] [CrossRef]
- Skoda, R.C.; Duek, A.; Grisouard, J. Pathogenesis of myeloproliferative neoplasms. Exp. Hematol. 2015, 43, 599–608. [Google Scholar] [CrossRef] [Green Version]
- Skov, V.; Larsen, T.S.; Thomassen, M.; Riley, C.H.; Jensen, M.K.; Bjerrum, O.W.; Kruse, T.A.; Hasselbalch, H. Molecular profiling of peripheral blood cells from patients with polycythemia vera and related neoplasms: Identification of deregulated genes of significance for inflammation and immune surveillance. Leuk. Res. 2012, 36, 1387–1392. [Google Scholar] [CrossRef]
- Hasselbalch, H.C. Perspectives on chronic inflammation in essential thrombocythemia, polycythemia vera, and myelofibrosis: Is chronic inflammation a trigger and driver of clonal evolution and development of accelerated atherosclerosis and second cancer? Blood 2012, 119, 3219–3225. [Google Scholar] [CrossRef] [Green Version]
- Bjørn, M.E.; Hasselbalch, H. The Role of Reactive Oxygen Species in Myelofibrosis and Related Neoplasms. Mediat. Inflamm. 2015, 2015, 648090. [Google Scholar] [CrossRef] [Green Version]
- Mambet, C.; Matei, L.; Necula, L.G.; Diaconu, C.C. A link between the driver mutations and dysregulated apoptosis in BCR-ABL1 negative myeloproliferative neoplasms. J. Immunoass. Immunochem. 2016, 37, 331–345. [Google Scholar] [CrossRef]
- Grimwade, L.F.; Happerfield, L.; Tristram, C.; McIntosh, G.; Rees, M.; Bench, A.J.; Boyd, E.M.; Hall, M.; Quinn, A.; Piggott, N.; et al. Phospho-STAT5 and phospho-Akt expression in chronic myeloproliferative neoplasms. Br. J. Haematol. 2009, 147, 495–506. [Google Scholar] [CrossRef]
- Marty, C.; Lacout, C.; Droin, N.; Le Couédic, J.-P.; Ribrag, V.; Solary, E.; Vainchenker, W.; Villeval, J.-L.; Plo, I. A role for reactive oxygen species in JAK2V617F myeloproliferative neoplasm progression. Leukemia 2013, 27, 2187–2195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rampal, R.; Al-Shahrour, F.; Abdel-Wahab, O.; Patel, J.P.; Brunel, J.-P.; Mermel, C.H.; Bass, A.J.; Pretz, J.; Ahn, J.; Hricik, T.; et al. Integrated genomic analysis illustrates the central role of JAK-STAT pathway activation in myeloproliferative neoplasm pathogenesis. Blood 2014, 123, e123–e133. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Hutchison, R.E.; Mohi, G. Critical requirement for Stat5 in a mouse model of polycythemia vera. Blood 2012, 119, 3539–3549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermouet, S.; Bigot-Corbel, E.; Gardie, B. Pathogenesis of Myeloproliferative Neoplasms: Role and Mechanisms of Chronic Inflammation. Mediat. Inflamm. 2015, 2015, 145293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleppe, M.; Koche, R.; Zou, L.; van Galen, P.; Hill, C.; Dong, L.; De Groote, S.; Papalexi, E.; Somasundara, A.V.H.; Cordner, K.; et al. Dual Targeting of Oncogenic Activation and Inflammatory Signaling Increases Therapeutic Efficacy in Myeloproliferative Neoplasms. Cancer Cell 2017, 33, 29–43.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasselbalch, H.C.; Bjørn, M.E. MPNs as Inflammatory Diseases: The Evidence, Consequences, and Perspectives. Mediat. Inflamm. 2015, 2015, e102476. [Google Scholar] [CrossRef] [Green Version]
- Petruk, C.; Mathias, J. The Myeloproliferative Neoplasm Landscape: A Patient’s Eye View. Adv. Ther. 2020, 37, 2050–2070. [Google Scholar] [CrossRef] [Green Version]
- Hasselbalch, H.C. Chronic inflammation as a promotor of mutagenesis in essential thrombocythemia, polycythemia vera and myelofibrosis. A human inflammation model for cancer development? Leuk. Res. 2013, 37, 214–220. [Google Scholar] [CrossRef]
- Hasselbalch, H.C. The platelet–cancer loop in myeloproliferative cancer. Is thrombocythemia an enhancer of cancer invasiveness and metastasis in essential thrombocythemia, polycythemia vera and myelofibrosis? Leuk. Res. 2014, 38, 1230–1236. [Google Scholar] [CrossRef]
- Fisher, D.A.C.; Fowles, J.S.; Zhou, A.; Oh, S.T. Inflammatory Pathophysiology as a Contributor to Myeloproliferative Neoplasms. Front. Immunol. 2021, 12, 683401. [Google Scholar] [CrossRef]
- Fleischman, A.G.; Aichberger, K.J.; Luty, S.B.; Bumm, T.G.; Petersen, C.L.; Doratotaj, S.; Vasudevan, K.B.; Latocha, D.H.; Yang, F.; Press, R.; et al. TNFα facilitates clonal expansion of JAK2V617F positive cells in myeloproliferative neoplasms. Blood 2011, 118, 6392–6398. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Xia, L.; Liu, Y.-C.; Hochman, T.; Bizzari, L.; Aruch, D.; Lew, J.; Weinberg, R.; Goldberg, J.D.; Hoffman, R. Lipocalin produced by myelofibrosis cells affects the fate of both hematopoietic and marrow microenvironmental cells. Blood 2015, 126, 972–982. [Google Scholar] [CrossRef] [Green Version]
- Fisher, D.A.; Malkova, O.; Engle, E.K.; Miner, C.; Fulbright, M.C.; Behbehani, G.K.; Collins, T.B.; Bandyopadhyay, S.; Zhou, A.; Nolan, G.P.; et al. Mass cytometry analysis reveals hyperactive NF Kappa B signaling in myelofibrosis and secondary acute myeloid leukemia. Leukemia 2016, 31, 1962–1974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Battista, V.; Bochicchio, M.T.; Giordano, G.; Napolitano, M.; Lucchesi, A. Genetics and Pathogenetic Role of Inflammasomes in Philadelphia Negative Chronic Myeloproliferative Neoplasms: A Narrative Review. Int. J. Mol. Sci. 2021, 22, 561. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Yan, S.; Liu, N.; He, N.; Zhang, A.; Meng, S.; Ji, C.; Ma, D.; Ye, J. Genetic polymorphisms and expression of NLRP3 inflammasome-related genes are associated with Philadelphia chromosome-negative myeloproliferative neoplasms. Hum. Immunol. 2020, 81, 606–613. [Google Scholar] [CrossRef] [PubMed]
- Longhitano, L.; Volti, G.L.; Giallongo, C.; Spampinato, M.; Barbagallo, I.; Di Rosa, M.; Romano, A.; Avola, R.; Tibullo, D.; Palumbo, G.A. The Role of Inflammation and Inflammasome in Myeloproliferative Disease. J. Clin. Med. 2020, 9, 2334. [Google Scholar] [CrossRef] [PubMed]
- Tourneur, L.; Witko-Sarsat, V. Inflammasome activation: Neutrophils go their own way. J. Leukoc. Biol. 2019, 105, 433–436. [Google Scholar] [CrossRef]
- Lange, C.; Hemmrich, G.; Klostermeier, U.C.; López-Quintero, J.A.; Miller, D.J.; Rahn, T.; Weiss, Y.; Bosch, T.C.; Rosenstiel, P. Defining the Origins of the NOD-Like Receptor System at the Base of Animal Evolution. Mol. Biol. Evol. 2010, 28, 1687–1702. [Google Scholar] [CrossRef] [Green Version]
- Sharma, B.R.; Karki, R.; Kanneganti, T. Role of AIM2 inflammasome in inflammatory diseases, cancer and infection. Eur. J. Immunol. 2019, 49, 1998–2011. [Google Scholar] [CrossRef] [Green Version]
- Liew, E.L.; Araki, M.; Hironaka, Y.; Mori, S.; Tan, T.Z.; Morishita, S.; Edahiro, Y.; Ohsaka, A.; Komatsu, N. Identification of AIM2 as a downstream target of JAK2V617F. Exp. Hematol. Oncol. 2015, 5, 2. [Google Scholar] [CrossRef] [Green Version]
- Arranz, L.; Sánchez-Aguilera, A.; Pérez, D.M.; Isern, J.; Langa, X.; Tzankov, A.; Lundberg, P.; Muntión, S.; Tzeng, Y.-S.; Lai, D.-M.; et al. Neuropathy of haematopoietic stem cell niche is essential for myeloproliferative neoplasms. Nature 2014, 512, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Găman, M.-A.; Cozma, M.-A.; Dobrică, E.-C.; Crețoiu, S.M.; Găman, A.M.; Diaconu, C.C. Liquid Biopsy and Potential Liquid Biopsy-Based Biomarkers in Philadelphia-Negative Classical Myeloproliferative Neoplasms: A Systematic Review. Life 2021, 11, 677. [Google Scholar] [CrossRef] [PubMed]
- Ceneli, O.; Haznedar, R.; Ongun, C.; Altan, N. Evaluation of Superoxide Dismutase Enzyme Activity of Polymorphonuclear Leucocytes, Erythrocytes and Thrombocytes in Patients with Chronic Myeloproliferative Disorders. J. Int. Med. Res. 2009, 37, 1365–1374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vener, C.; Novembrino, C.; Catena, F.B.; Fracchiolla, N.S.; Gianelli, U.; Savi, F.; Radaelli, F.; Fermo, E.; Cortelezzi, A.; Lonati, S. Oxidative stress is increased in primary and post−polycythemia vera myelofibrosis. Exp. Hematol. 2010, 38, 1058–1065. [Google Scholar] [CrossRef]
- Durmuş, A.; Mentese, A.; Yilmaz, M.; Sümer, A.; Akalin, I.; Topal, C.; Alver, A. The thrombotic events in polycythemia vera patients may be related to increased oxidative stress. Med. Princ. Pract. 2014, 23, 253–258. [Google Scholar] [CrossRef] [Green Version]
- Musolino, C.; Allegra, A.; Saija, A.; Alonci, A.; Russo, S.; Spatari, G.; Penna, G.; Gerace, D.; Cristani, M.; David, A.; et al. Changes in advanced oxidation protein products, advanced glycation end products, and s-nitrosylated proteins, in patients affected by polycythemia vera and essential thrombocythemia. Clin. Biochem. 2012, 45, 1439–1443. [Google Scholar] [CrossRef]
- Djikic, D.; Markovic, D.; Bogdanovic, A.; Mitrovic-Ajtic, O.; Suboticki, T.; Diklic, M.; Beleslin-Cokic, B.; Bjelica, S.; Kovacic, M.; Cokic, V.P. Oxidative and Nitrosative Stress in Myeloproliferative Neoplasms: The Impact on the AKT/MTOR Signaling Pathway. JBUON 2018, 23, 1481–1491. [Google Scholar]
- Yalcin, S.; Marinkovic, D.; Mungamuisk, S.K.; Zhang, X.; Tong, W.; Sellers, R.; Ghaffari, S. ROS-mediated amplification of AKT/mTOR signalling pathway leads to myeloproliferative syndrome in Foxo3−/− mice. EMBO J. 2010, 29, 4118–4131. [Google Scholar] [CrossRef] [Green Version]
- Hurtado-Nedelec, M.; Csillag-Grange, M.-J.; Boussetta, T.; Belambri, S.A.; Fay, M.; Cassinat, B.; Gougerot-Pocidalo, M.-A.; Dang, P.M.-C.; El-Benna, J. Increased reactive oxygen species production and p47phox phosphorylation in neutrophils from myeloproliferative disorders patients with JAK2 (V617F) mutation. Haematologica 2013, 98, 1517–1524. [Google Scholar] [CrossRef]
- Socoro-Yuste, N.; Mondet, J.; Plo, I.; Mossuz, P.; Čokić, V.P. Quantitative Proteome Heterogeneity in Myeloproliferative Neoplasm Subtypes and Association with JAK2 Mutation Status. Mol. Cancer Res. 2017, 15, 852–861. [Google Scholar] [CrossRef] [Green Version]
- Fialkow, L.; Wang, Y.; Downey, G.P. Reactive oxygen and nitrogen species as signaling molecules regulating neutrophil function. Free Radic. Biol. Med. 2007, 42, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Bittencourt, R.I.; Vassallo, J.; Chauffaille, M.D.L.L.F.; Xavier, S.G.; Pagnano, K.; Nascimento, A.C.K.; De Souza, C.A.; Chiattone, C.S. Philadelphia-negative chronic myeloproliferative neoplasms. Rev. Bras. Hematol. Hemoter. 2012, 34, 140–149. [Google Scholar] [CrossRef] [Green Version]
- Simon, H.-U. Neutrophil apoptosis pathways and their modifications in inflammation. Immunol. Rev. 2003, 193, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Korkmaz, B.; Horwitz, M.S.; Jenne, D.E.; Gauthier, F. Neutrophil Elastase, Proteinase 3, and Cathepsin G as Therapeutic Targets in Human Diseases. Pharmacol. Rev. 2010, 62, 726–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adrover, J.M.; Aroca-Crevillén, A.; Crainiciuc, G.; Ostos, F.; Rojas-Vega, Y.; Rubio-Ponce, A.; Cilloniz, C.; Bonzón-Kulichenko, E.; Calvo, E.; Rico, D.; et al. Programmed ‘disarming’ of the neutrophil proteome reduces the magnitude of inflammation. Nat. Immunol. 2020, 21, 135–144. [Google Scholar] [CrossRef] [PubMed]
- McKenna, E.; Mhaonaigh, A.U.; Wubben, R.; Dwivedi, A.; Hurley, T.; Kelly, L.A.; Stevenson, N.J.; Little, M.A.; Molloy, E.J. Neutrophils: Need for Standardized Nomenclature. Front. Immunol. 2021, 12, 602963. [Google Scholar] [CrossRef]
- Mcdonald, P.P. Transcriptional Regulation in Neutrophils: Teaching Old Cells New Tricks. Adv. Immunol. 2004, 82, 1–48. [Google Scholar] [CrossRef]
- Rosales, C. Neutrophil: A Cell with Many Roles in Inflammation or Several Cell Types? Front. Physiol. 2018, 9, 113. [Google Scholar] [CrossRef]
- Sallmyr, A.; Miller, A.; Gabdoulkhakova, A.; Safronova, V.; Henriksson, G.; Bredberg, A.; Sallmyr, A.M.A. Expression of DNA-dependent protein kinase in human granulocytes. Cell Res. 2004, 14, 331–340. [Google Scholar] [CrossRef]
- Salati, S.; Bianchi, E.; Zini, R.; Tenedini, E.; Quaglino, D.; Manfredini, R.; Ferrari, S. Eosinophils, but not neutrophils, exhibit an efficient DNA repair machinery and high nucleolar activity. Haematologica 2007, 92, 1311–1318. [Google Scholar] [CrossRef] [Green Version]
- Ponath, V.; Heylmann, D.; Haak, T.; Woods, K.; Becker, H.; Kaina, B. Compromised DNA Repair and Signalling in Human Granulocytes. J. Innate Immun. 2018, 11, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Stark, M.A.; Huo, Y.; Burcin, T.L.; Morris, M.A.; Olson, T.S.; Ley, K. Phagocytosis of Apoptotic Neutrophils Regulates Granulopoiesis via IL-23 and IL-17. Immunity 2005, 22, 285–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cossío, I.; Lucas, D.; Hidalgo, A. Neutrophils as regulators of the hematopoietic niche. Blood 2019, 133, 2140–2148. [Google Scholar] [CrossRef] [PubMed]
- Al Zaid Siddiquee, K.; Turkson, J. STAT3 as a target for inducing apoptosis in solid and hematological tumors. Cell Res. 2008, 18, 254–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.; Roberts, L.; Chen, Z.; Merta, P.J.; Glaser, K.B.; Shah, O.J. JAK2V617F Drives Mcl-1 Expression and Sensitizes Hematologic Cell Lines to Dual Inhibition of JAK2 and Bcl-xL. PLoS ONE 2015, 10, e0114363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Zhao, H.; Sun, A.; Lu, S.; Liu, B.; Tang, F.; Feng, Y.; Zhao, L.; Yang, R.; Han, Z.C. Early Down-Regulation of Bcl-XL Expression during Megakaryocytic Differentiation of Thrombopoietin-Induced CD34+ Bone Marrow Cells in Essential Thrombocythemia. Haematologica 2004, 89, 1199–1206. [Google Scholar] [CrossRef] [PubMed]
- Florena, A.M.; Tripodo, C.; Di Bernardo, A.; Iannitto, E.; Guarnotta, C.; Porcasi, R.; Ingrao, S.; Abbadessa, V.; Franco, V. Different immunophenotypical apoptotic profiles characterise megakaryocytes of essential thrombocythaemia and primary myelofibrosis. J. Clin. Pathol. 2009, 62, 331–338. [Google Scholar] [CrossRef]
- Koopmans, S.M.; Schouten, H.C.; Van Marion, A.M. Anti-Apoptotic Pathways in Bone Marrow and Megakaryocytes in Myeloproliferative Neoplasia. Pathobiology 2014, 81, 60–68. [Google Scholar] [CrossRef]
- Panopoulos, A.D.; Watowich, S.S. Granulocyte colony-stimulating factor: Molecular mechanisms of action during steady state and ‘emergency’ hematopoiesis. Cytokine 2008, 42, 277–288. [Google Scholar] [CrossRef] [Green Version]
- Greenlee-Wacker, M.C. Clearance of apoptotic neutrophils and resolution of inflammation. Immunol. Rev. 2016, 273, 357–370. [Google Scholar] [CrossRef] [Green Version]
- Dancey, J.T.; Deubelbeiss, K.A.; Harker, L.A.; Finch, C.A. Neutrophil kinetics in man. J. Clin. Investig. 1976, 58, 705–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bratton, D.L.; Henson, P.M. Neutrophil clearance: When the party is over, clean-up begins. Trends Immunol. 2011, 32, 350–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, B.B.; Allcock, R.J.; Mirzai, B.; Malherbe, J.A.; Choudry, F.A.; Frontini, M.; Chuah, H.; Liang, J.; Kavanagh, S.E.; Howman, R.; et al. Megakaryocytes in Myeloproliferative Neoplasms Have Unique Somatic Mutations. Am. J. Pathol. 2017, 187, 1512–1522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noetzli, L.J.; French, S.L.; Machlus, K. New Insights into the Differentiation of Megakaryocytes from Hematopoietic Progenitors. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1288–1300. [Google Scholar] [CrossRef] [PubMed]
- Falanga, A.; Marchetti, M.; Barbui, T.; Smith, C.W. Pathogenesis of Thrombosis in Essential Thrombocythemia and Polycythemia Vera: The Role of Neutrophils. Semin. Hematol. 2005, 42, 239–247. [Google Scholar] [CrossRef]
- Oku, S.; Takenaka, K.; Kuriyama, T.; Shide, K.; Kumano, T.; Kikushige, Y.; Urata, S.; Yamauchi, T.; Iwamoto, C.; Shimoda, H.K.; et al. JAK2 V617F uses distinct signalling pathways to induce cell proliferation and neutrophil activation. Br. J. Haematol. 2010, 150, 334–344. [Google Scholar] [CrossRef]
- Mesa, R.A.; Tefferi, A.; Lasho, T.S.; Loegering, D.; McClure, R.F.; Powell, H.L.; Dai, N.T.; Steensma, D.; Kaufmann, S. Janus kinase 2 (V617F) mutation status, signal transducer and activator of transcription-3 phosphorylation and impaired neutrophil apoptosis in myelofibrosis with myeloid metaplasia. Leukemia 2006, 20, 1800–1808. [Google Scholar] [CrossRef] [Green Version]
- Tognon, R.; Nunes, N.D.S.; De Castro, F.A. Desregulação da apoptose em neoplasias mieloproliferativas crônicas. Einstein 2013, 11, 540–544. [Google Scholar] [CrossRef] [Green Version]
- Kruger, P.; Saffarzadeh, M.; Weber, A.; Rieber, N.; Radsak, M.; Von Bernuth, H.; Benarafa, C.; Roos, D.; Skokowa, J.; Hartl, D. Neutrophils: Between Host Defence, Immune Modulation, and Tissue Injury. PLoS Pathog. 2015, 11, e1004651. [Google Scholar] [CrossRef] [Green Version]
- Anderson, R.; Tintinger, G.R.; Feldman, C. Inflammation and cancer: The role of the human neutrophil. S. Afr. J. Sci. 2014, 110, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Robertson, J.D.; Ward, J.R.; Avila-Olias, M.; Battaglia, G.; Renshaw, S.A. Targeting Neutrophilic Inflammation Using Polymersome-Mediated Cellular Delivery. J. Immunol. 2017, 198, 3596–3604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etecchio, C.; Micheletti, A.; Cassatella, M.A. Neutrophil-Derived Cytokines: Facts Beyond Expression. Front. Immunol. 2014, 5, 508. [Google Scholar] [CrossRef] [Green Version]
- Tecchio, C.; Cassatella, M.A. Neutrophil-derived chemokines on the road to immunity. Semin. Immunol. 2016, 28, 119–128. [Google Scholar] [CrossRef]
- Tamassia, N.; Bianchetto-Aguilera, F.; Arruda-Silva, F.; Gardiman, E.; Gasperini, S.; Calzetti, F.; Cassatella, M.A. Cytokine production by human neutrophils: Revisiting the “dark side of the moon”. Eur. J. Clin. Investig. 2018, 48, e12952. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.Y.; Xu, M.; Roboz, J.; Lu, M.; Mascarenhas, J.; Hoffman, R. The Effect of CXCL12 Processing on CD34+ Cell Migration in Myeloproliferative Neoplasms. Cancer Res. 2010, 70, 3402–3410. [Google Scholar] [CrossRef] [Green Version]
- Gigon, L.; Yousefi, S.; Karaulov, A.; Simon, H.-U. Mechanisms of toxicity mediated by neutrophil and eosinophil granule proteins. Allergol. Int. 2020, 70, 30–38. [Google Scholar] [CrossRef]
- Yousefi, S.; Mihalache, C.C.; Kozlowski, E.O.; Schmid, I.; Simon, H.-U. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ. 2009, 16, 1438–1444. [Google Scholar] [CrossRef]
- Yousefi, S.; Stojkov, D.; Germic, N.; Simon, D.; Wang, X.; Benarafa, C.; Simon, H. Untangling “NETosis” from NETs. Eur. J. Immunol. 2019, 49, 221–227. [Google Scholar] [CrossRef] [Green Version]
- Tsai, C.-Y.; Hsieh, S.-C.; Liu, C.-W.; Lu, C.-S.; Wu, C.-H.; Liao, H.-T.; Chen, M.-H.; Li, K.-J.; Shen, C.-Y.; Kuo, Y.-M.; et al. Cross-Talk among Polymorphonuclear Neutrophils, Immune, and Non-Immune Cells via Released Cytokines, Granule Proteins, Microvesicles, and Neutrophil Extracellular Trap Formation: A Novel Concept of Biology and Pathobiology for Neutrophils. Int. J. Mol. Sci. 2021, 22, 3119. [Google Scholar] [CrossRef]
- Bakele, M.; Joos, M.; Burdi, S.; Allgaier, N.; Pöschel, S.; Fehrenbacher, B.; Schaller, M.; Marcos, V.; Kümmerle-Deschner, J.; Rieber, N.; et al. Localization and Functionality of the Inflammasome in Neutrophils. J. Biol. Chem. 2014, 289, 5320–5329. [Google Scholar] [CrossRef] [Green Version]
- Ley, K.; Hoffman, H.M.; Kubes, P.; Cassatella, M.A.; Zychlinsky, A.; Hedrick, C.C.; Catz, S.D. Neutrophils: New insights and open questions. Sci. Immunol. 2018, 3, eaat4579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Summers, C.; Rankin, S.M.; Condliffe, A.M.; Singh, N.; Peters, A.M.; Chilvers, E.R. Neutrophil kinetics in health and disease. Trends Immunol. 2010, 31, 318–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lekovic, D.; Gotic, M.; Skoda, R.; Beleslin-Cokic, B.; Milic, N.; Mitrovic-Ajtic, O.; Nienhold, R.; Sefer, D.; Suboticki, T.; Buac, M.; et al. Bone marrow microvessel density and plasma angiogenic factors in myeloproliferative neoplasms: Clinicopathological and molecular correlations. Ann. Hematol. 2016, 96, 393–404. [Google Scholar] [CrossRef] [PubMed]
- Moliterno, A.R.; Williams, D.M.; Rogers, O.; Isaacs, M.A.; Spivak, J.L. Phenotypic variability within the JAK2 V617F-positive MPD: Roles of progenitor cell and neutrophil allele burdens. Exp. Hematol. 2008, 36, 1480–1486.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasselbalch, H.C. The role of cytokines in the initiation and progression of myelofibrosis. Cytokine Growth Factor Rev. 2013, 24, 133–145. [Google Scholar] [CrossRef]
- Cho, C.; Yoon, J.; Kim, D.; Kim, S.; Sung, H.J.; Lee, S.R. Association of peripheral blood neutrophil gelatinase-associated lipocalin levels with bone marrow neutrophil gelatinase-associated lipocalin levels and neutrophil count in hematologic malignancy. J. Clin. Lab. Anal. 2019, 33, e22920. [Google Scholar] [CrossRef]
- Cho, C.; Cha, J.; Chang, E.; Nam, M.; Park, S.; Sung, H.J.; Lee, S.R. Analysis of bone marrow supernatant neutrophil gelatinase-associated lipocalin and hematological parameters in hematological malignancy. J. Clin. Lab. Anal. 2020, 34, e23253. [Google Scholar] [CrossRef]
- Oh, S.T. Neutralize the neutrophils! Neutrophil β1/β2 integrin activation contributes to JAK2-V617F-driven thrombosis. J. Clin. Investig. 2018, 128, 4248–4250. [Google Scholar] [CrossRef]
- Oyarzún, C.P.M.; Heller, P.G. Platelets as Mediators of Thromboinflammation in Chronic Myeloproliferative Neoplasms. Front. Immunol. 2019, 10, 1373. [Google Scholar] [CrossRef] [Green Version]
- Ferrer-Marín, F.; Cuenca-Zamora, E.; Guijarro-Carrillo, P.; Teruel-Montoya, R. Emerging Role of Neutrophils in the Thrombosis of Chronic Myeloproliferative Neoplasms. Int. J. Mol. Sci. 2021, 22, 1143. [Google Scholar] [CrossRef]
- Schmitt, A.; Drouin, A.; Massé, J.-M.; Guichard, J.; Shagraoui, H.; Cramer, E.M. Polymorphonuclear Neutrophil and Megakaryocyte Mutual Involvement in Myelofibrosis Pathogenesis. Leuk. Lymphoma 2002, 43, 719–724. [Google Scholar] [CrossRef] [PubMed]
- Futosi, K.; Fodor, S.; Mócsai, A. Neutrophil cell surface receptors and their intracellular signal transduction pathways. Int. Immunopharmacol. 2013, 17, 638–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Figueroa, E.; Álvarez-Carrasco, P.; Ortega, E.; Maldonado-Bernal, C. Neutrophils: Many Ways to Die. Front. Immunol. 2021, 12, 631821. [Google Scholar] [CrossRef]
- Mayadas, T.N.; Cullere, X.; Lowell, C.A. The Multifaceted Functions of Neutrophils. Annu. Rev. Pathol. Mech. Dis. 2014, 9, 181–218. [Google Scholar] [CrossRef] [Green Version]
- Athens, J.W.; Haab, O.P.; Raab, S.O.; Mauer, A.M.; Ashenbrucker, H.; Cartwright, G.E.; Wintrobe, M.M. Leukokinetic Studies. IV. The Total Blood, Circulating and Marginal Granulocyte Pools and the Granulocyte Turnover Rate in Normal Subjects. J. Clin. Investig. 1961, 40, 989–995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCracken, J.M.; Allen, L.-A.H. Regulation of Human Neutrophil Apoptosis and Lifespan in Health and Disease. J. Cell Death 2014, 7, 15–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, A.D.; DeLeo, F.R. Neutrophil apoptosis and the resolution of infection. Immunol. Res. 2008, 43, 25–61. [Google Scholar] [CrossRef]
- Dąbrowska, D.; Jabłońska, E.; Iwaniuk, A.; Garley, M. Many Ways–One Destination: Different Types of Neutrophils Death. Int. Rev. Immunol. 2018, 38, 18–32. [Google Scholar] [CrossRef]
- Kajiume, T.; Kobayashi, M. Human granulocytes undergo cell death via autophagy. Cell Death Discov. 2018, 4, 111. [Google Scholar] [CrossRef]
- Lawrence, S.M.; Corriden, R.; Nizet, V. How Neutrophils Meet Their End. Trends Immunol. 2020, 41, 531–544. [Google Scholar] [CrossRef]
- Brostjan, C.; Oehler, R. The role of neutrophil death in chronic inflammation and cancer. Cell Death Discov. 2020, 6, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papaliagkas, V.; Anogianaki, A.; Anogianakis, G.; Ilonidis, G. The proteins and the mechanisms of apoptosis: A mini-review of the fundamentals. Hippokratia 2007, 11, 108–113. [Google Scholar] [PubMed]
- Vermeren, S.; Karmakar, U.; Rossi, A.G. Immune complex-induced neutrophil functions: A focus on cell death. Eur. J. Clin. Investig. 2018, 48, e12948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef]
- Moulding, D.; Akgul, C.; Derouet, M.; White, M.; Edwards, S.W. BCL-2 family expression in human neutrophils during delayed and accelerated apoptosis. J. Leukoc. Biol. 2001, 70, 783–792. [Google Scholar]
- Edwards, S.; Derouet, M.; Howse, M.; Moots, R. Regulation of neutrophil apoptosis by Mcl-1. Biochem. Soc. Trans. 2004, 32, 489–492. [Google Scholar] [CrossRef] [Green Version]
- Witko-Sarsat, V.; Pederzoli-Ribeil, M.; Hirsh, E.; Sozzani, S.; Cassatella, M.A. Regulating neutrophil apoptosis: New players enter the game. Trends Immunol. 2011, 32, 117–124. [Google Scholar] [CrossRef]
- Leitch, A.E.; Duffin, R.; Haslett, C.; Rossi, A.G. Relevance of granulocyte apoptosis to resolution of inflammation at the respiratory mucosa. Mucosal Immunol. 2008, 1, 350–363. [Google Scholar] [CrossRef]
- Porter, A.G.; Jänicke, R.U. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999, 6, 99–104. [Google Scholar] [CrossRef]
- Anderton, H.; Wicks, I.P.; Silke, J. Cell death in chronic inflammation: Breaking the cycle to treat rheumatic disease. Nat. Rev. Rheumatol. 2020, 16, 496–513. [Google Scholar] [CrossRef]
- Szczepura, K.R.; Ruparelia, P.; Solanki, C.K.; Balan, K.; Newbold, P.; Summers, C.; Chilvers, E.R.; Peters, A.M. Measuring whole-body neutrophil redistribution using a dedicated whole-body counter and ultra-low doses of 111Indium. Eur. J. Clin. Investig. 2010, 41, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Furze, R.C.; Rankin, S.M. Neutrophil mobilization and clearance in the bone marrow. Immunology 2008, 125, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Akashi, K.; Traver, D.; Miyamoto, T.; Weissman, I.L. A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature 2000, 404, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Dresch, C.; Flandrin, G.; Breton-Gorius, J. Phagocytosis of neutrophil polymorphonuclears by macrophages in human bone marrow: Importance in granulopoiesis. J. Clin. Pathol. 1980, 33, 1110–1113. [Google Scholar] [CrossRef] [Green Version]
- Saverymuttu, S.H.; Peters, A.M.; Keshavarzian, A.; Reavy, H.J.; Lavender, J.P. The kinetics of 111Indium distribution following injection of 111Indium labelled autologous granulocytes in man. Br. J. Haematol. 1985, 61, 675–685. [Google Scholar] [CrossRef]
- Martin, C.; Burdon, P.C.; Bridger, G.; Gutierrez-Ramos, J.-C.; Williams, T.J.; Rankin, S.M. Chemokines Acting via CXCR2 and CXCR4 Control the Release of Neutrophils from the Bone Marrow and Their Return following Senescence. Immunity 2003, 19, 583–593. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Hossain, M.; Thanabalasuriar, A.; Gunzer, M.; Meininger, C.; Kubes, P. Visualizing the function and fate of neutrophils in sterile injury and repair. Science 2017, 358, 111–116. [Google Scholar] [CrossRef] [Green Version]
- Arienti, S.; Barth, N.; Dorward, D.A.; Rossi, A.G.; Dransfield, I. Regulation of Apoptotic Cell Clearance During Resolution of Inflammation. Front. Pharmacol. 2019, 10, 891. [Google Scholar] [CrossRef] [Green Version]
- Fadok, V.A.; Bratton, D.L.; Konowal, A.; Freed, P.W.; Westcott, J.Y.; Henson, P.M. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J. Clin. Investig. 1998, 101, 890–898. [Google Scholar] [CrossRef] [Green Version]
- Ortega-Gomez, A.; Perretti, M.; Soehnlein, O. Resolution of inflammation: An integrated view. EMBO Mol. Med. 2013, 5, 661–674. [Google Scholar] [CrossRef]
- Le Bousse-Kerdilès, M.; Martyré, M. Involvement of the fibrogenic cytokines, TGF-β and bFGF, in the pathogenesis of idiopathic myelofibrosis. Pathol. Biol. 2001, 49, 153–157. [Google Scholar] [CrossRef]
- Luo, H.R.; Loison, F. Constitutive neutrophil apoptosis: Mechanisms and regulation. Am. J. Hematol. 2008, 83, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Grecian, R.; Whyte, M.K.B.; Walmsley, S. The role of neutrophils in cancer. Br. Med. Bull. 2018, 128, 5–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, R.D.; Hardisty, G.; Regan, K.H.; Smith, M.; Robb, C.T.; Duffin, R.; Mackellar, A.; Felton, J.M.; Paemka, L.; McCullagh, B.N.; et al. Delayed neutrophil apoptosis enhances NET formation in cystic fibrosis. Thorax 2017, 73, 134–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fadok, V.A.; Bratton, D.L.; Guthrie, L.; Henson, P.M. Differential Effects of Apoptotic Versus Lysed Cells on Macrophage Production of Cytokines: Role of Proteases. J. Immunol. 2001, 166, 6847–6854. [Google Scholar] [CrossRef]
- De Filippo, K.; Rankin, S.M. CXCR4, the master regulator of neutrophil trafficking in homeostasis and disease. Eur. J. Clin. Investig. 2018, 48, e12949. [Google Scholar] [CrossRef] [Green Version]
- De Filippo, K.; Rankin, S.M. The Secretive Life of Neutrophils Revealed by Intravital Microscopy. Front. Cell Dev. Biol. 2020, 8, 603230. [Google Scholar] [CrossRef]
- Liew, P.X.; Kubes, P. The Neutrophil’s Role During Health and Disease. Physiol. Rev. 2019, 99, 1223–1248. [Google Scholar] [CrossRef]
- Nagase, H.; Miyamasu, M.; Yamaguchi, M.; Imanishi, M.; Tsuno, N.H.; Matsushima, K.; Yamamoto, K.; Morita, Y.; Hirai, K. Cytokine-mediated regulation of CXCR4 expression in human neutrophils. J. Leukoc. Biol. 2002, 71, 711–717. [Google Scholar]
- Eash, K.J.; Greenbaum, A.; Gopalan, P.K.; Link, D.C. CXCR2 and CXCR4 antagonistically regulate neutrophil trafficking from murine bone marrow. J. Clin. Investig. 2010, 120, 2423–2431. [Google Scholar] [CrossRef] [Green Version]
- Hong, C.-W. Current Understanding in Neutrophil Differentiation and Heterogeneity. Immune Netw. 2017, 17, 298–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aroca-Crevillén, A.; Adrover, J.M.; Hidalgo, A. Circadian Features of Neutrophil Biology. Front. Immunol. 2020, 11, 576. [Google Scholar] [CrossRef] [PubMed]
- Casanova-Acebes, M.; Pitaval, C.; Weiss, L.A.; Nombela-Arrieta, C.; Chèvre, R.; Gonzalez, N.A.; Kunisaki, Y.; Zhang, D.; van Rooijen, N.; Silberstein, L.E.; et al. Rhythmic Modulation of the Hematopoietic Niche through Neutrophil Clearance. Cell 2013, 153, 1025–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bekkering, S. Another look at the life of a neutrophil. World J. Hematol. 2013, 2, 44. [Google Scholar] [CrossRef]
- Hidalgo, A.; Chilvers, E.R.; Summers, C.; Koenderman, L. The Neutrophil Life Cycle. Trends Immunol. 2019, 40, 584–597. [Google Scholar] [CrossRef] [PubMed]
- Brown, V.; Elborn, J.S.; Bradley, J.; Ennis, M. Dysregulated apoptosis and NFκB expression in COPD subjects. Respir. Res. 2009, 10, 24. [Google Scholar] [CrossRef]
- Bartneck, M.; Wang, J. Therapeutic Targeting of Neutrophil Granulocytes in Inflammatory Liver Disease. Front. Immunol. 2019, 10, 2257. [Google Scholar] [CrossRef]
- Arnold, M.; Kahwash, S.B. Phagocytized Neutrophil Fragments in the Bone Marrow: A Phenomenon Most Commonly Associated with Hodgkin Lymphoma. ISRN Hematol. 2014, 2014, 363854. [Google Scholar] [CrossRef] [Green Version]
- Gaiolla, R.D.; Domingues, M.A.C.; Niéro-Melo, L.; de Oliveira, D.E. Serum Levels of Interleukins 6, 10, and 13 Before and after Treatment of Classic Hodgkin Lymphoma. Arch. Pathol. Lab. Med. 2011, 135, 483–489. [Google Scholar] [CrossRef]
- Kowalska, M.; Tajer, J.; Chechlinska, M.; Fuksiewicz, M.; Kotowicz, B.; Syczewska, M.; Walewski, J.; Kaminska, J. Discriminant analysis involving serum cytokine levels and prediction of the response to therapy of patients with Hodgkin lymphoma. Tumor Biol. 2012, 33, 1733–1738. [Google Scholar] [CrossRef]
- Kawano, Y.; Fukui, C.; Shinohara, M.; Wakahashi, K.; Ishii, S.; Suzuki, T.; Sato, M.; Asada, N.; Kawano, H.; Minagawa, K.; et al. G-CSF-induced sympathetic tone provokes fever and primes antimobilizing functions of neutrophils via PGE2. Blood 2017, 129, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Casanova-Acebes, M.; Nicolás-Ávila, J.A.; Li, J.L.; García-Silva, S.; Balachander, A.; Rubio-Ponce, A.; Weiss, L.A.; Adrover, J.M.; Burrows, K.; A-González, N.; et al. Neutrophils instruct homeostatic and pathological states in naive tissues. J. Exp. Med. 2018, 215, 2778–2795. [Google Scholar] [CrossRef] [PubMed]
- Andersen, M.; Sajid, Z.; Pedersen, R.K.; Gudmand-Hoeyer, J.; Ellervik, C.; Skov, V.; Kjær, L.; Pallisgaard, N.; Kruse, T.A.; Thomassen, M.; et al. Mathematical modelling as a proof of concept for MPNs as a human inflammation model for cancer development. PLoS ONE 2017, 12, e0183620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luque, L.F.M.; Blackmon, A.L.; Ramanathan, G.; Fleischman, A.G. Key Role of Inflammation in Myeloproliferative Neoplasms: Instigator of Disease Initiation, Progression. and Symptoms. Curr. Hematol. Malig. Rep. 2019, 14, 145–153. [Google Scholar] [CrossRef]
- Diaconu, C.C.; Gurban, P.; Mambet, C.; Chivu-Economescu, M.; Necula, L.G.; Matei, L.; Dragu, D.; Nedeianu, S.; Neagu, A.I.; Tatic, A.; et al. Programmed Cell Death Deregulation in BCR-ABL1-Negative Myeloproliferative Neoplasms; IntechOpen: London, UK, 2020. [Google Scholar] [CrossRef] [Green Version]
- Zeuner, A.; Pedini, F.; Signore, M.; Ruscio, G.; Messina, C.; Tafuri, A.; Girelli, G.; Peschle, C.; De Maria, R. Increased death receptor resistance and FLIPshort expression in polycythemia vera erythroid precursor cells. Blood 2006, 107, 3495–3502. [Google Scholar] [CrossRef] [Green Version]
- Tognon, R.; Gasparotto, E.P.; Neves, R.P.; Nunes, N.S.; Ferreira, A.F.; Palma, P.V.; Kashima, S.; Covas, D.T.; Santana, M.; Souto, E.X.; et al. Deregulation of apoptosis-related genes is associated with PRV1 overexpression and JAK2 V617F allele burden in Essential Thrombocythemia and Myelofibrosis. J. Hematol. Oncol. 2012, 5, 2. [Google Scholar] [CrossRef] [Green Version]
- Gasparotto, E.P.L.; Tognon, R.; Ferreira, A.F.; Oliveira, G.L.V.; Palma, P.V.B.; Zanichelli, M.A.; Souto, E.X.; Velano, C.E.E.; Simões, B.P.; Carrara, R.D.C.V.; et al. Deregulated expression of A1, Bcl-2, Bcl-xL, and Mcl-1 antiapoptotic proteins and Bid, Bad, and Bax proapoptotic genes in polycythemia vera patients. Braz. J. Pharm. Sci. 2011, 47, 873–886. [Google Scholar] [CrossRef] [Green Version]
- Epling-Burnette, P.K.; Zhong, B.; Bai, F.; Jiang, K.; Bailey, R.D.; Garcia, R.; Jove, R.; Djeu, J.Y.; Loughran, T.P.; Wei, S. Cooperative Regulation of Mcl-1 by Janus Kinase/STAT and Phosphatidylinositol 3-Kinase Contribute to Granulocyte-Macrophage Colony-Stimulating Factor-Delayed Apoptosis in Human Neutrophils. J. Immunol. 2001, 166, 7486–7495. [Google Scholar] [CrossRef]
- Tognon, R.; Gasparotto, E.P.L.; Leroy, J.M.G.; Oliveira, G.L.V.; Neves, R.P.; Carrara, R.D.C.V.; Kashima, S.; Covas, D.T.; Santana, M.; Souto, E.X.; et al. Differential expression of apoptosis-related genes from death receptor pathway in chronic myeloproliferative diseases. J. Clin. Pathol. 2010, 64, 75–82. [Google Scholar] [CrossRef] [Green Version]
- Pellagatti, A.; Vetrie, D.; Langford, C.F.; Gama, S.; Eagleton, H.; Wainscoat, J.S.; Boultwood, J. Gene expression profiling in polycythemia vera using cDNA microarray technology. Cancer Res. 2003, 63, 3940–3944. [Google Scholar]
- Gallardo, M.; Barrio, S.; Fernandez, M.; Paradela, A.; Arenas, A.; Toldos, O.; Ayala, R.; Albizua, E.; Jimenez, A.; Redondo, S.; et al. Proteomic analysis reveals heat shock protein 70 has a key role in polycythemia Vera. Mol. Cancer 2013, 12, 142–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hui, W.; Ye, F.; Zhang, W.; Liu, C.; Cui, M.; Li, W.; Xu, J.; Zhang, D.Y. Aberrant expression of signaling proteins in essential thrombocythemia. Ann. Hematol. 2013, 92, 1229–1238. [Google Scholar] [CrossRef] [PubMed]
- Cokic, V.P.; Mossuz, P.; Han, J.; Socoro, N.; Beleslin-Cokic, B.B.; Mitrović, O.; Subotički, T.; Diklic, M.; Lekovic, D.; Gotić, M.; et al. Microarray and Proteomic Analyses of Myeloproliferative Neoplasms with a Highlight on the mTOR Signaling Pathway. PLoS ONE 2015, 10, e0135463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tognon, R.; Nunes, N.S.; Ambrosio, L.; Souto, E.X.; Perobelli, L.; Simoes, B.; Souza, M.C.L.; Chauffaille, M.D.L.; De Castro, F.A. Apoptosis- and cell cycle-related genes methylation profile in myeloproliferative neoplasms. Leuk. Lymphoma 2015, 57, 1201–1204. [Google Scholar] [CrossRef] [PubMed]
- Amulic, B.; Cazalet, C.; Hayes, G.L.; Metzler, K.D.; Zychlinsky, A. Neutrophil Function: From Mechanisms to Disease. Annu. Rev. Immunol. 2012, 30, 459–489. [Google Scholar] [CrossRef]
- Bian, Z.; Guo, Y.; Ha, B.; Zen, K.; Liu, Y. Regulation of the Inflammatory Response: Enhancing Neutrophil Infiltration under Chronic Inflammatory Conditions. J. Immunol. 2011, 188, 844–853. [Google Scholar] [CrossRef] [Green Version]
- Zhao, R.; Follows, G.A.; Beer, P.A.; Scott, L.M.; Huntly, B.J.; Green, A.R.; Alexander, D.R. Inhibition of the Bcl-xLDeamidation Pathway in Myeloproliferative Disorders. N. Engl. J. Med. 2008, 359, 2778–2789. [Google Scholar] [CrossRef] [Green Version]
- Scott, L.M.; Rebel, V. JAK2 and genomic instability in the myeloproliferative neoplasms: A case of the chicken or the egg? Am. J. Hematol. 2012, 87, 1028–1036. [Google Scholar] [CrossRef] [Green Version]
- Kagoya, Y.; Yoshimi, A.; Tsuruta-Kishino, T.; Arai, S.; Satoh, T.; Akira, S.; Kurokawa, M. JAK2V617F+ myeloproliferative neoplasm clones evoke paracrine DNA damage to adjacent normal cells through secretion of lipocalin-2. Blood 2014, 124, 2996–3006. [Google Scholar] [CrossRef] [Green Version]
- Fleischman, A.G. Inflammation as a Driver of Clonal Evolution in Myeloproliferative Neoplasm. Mediat. Inflamm. 2015, 2015, 606819. [Google Scholar] [CrossRef] [Green Version]
- Oyarzún, C.P.M.; Carestia, A.; Lev, P.R.; Glembotsky, A.C.; Ríos, M.A.C.; Moiraghi, B.; Molinas, F.C.; Marta, R.F.; Schattner, M.; Heller, P.G. Neutrophil extracellular trap formation and circulating nucleosomes in patients with chronic myeloproliferative neoplasms. Sci. Rep. 2016, 6, 38738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolach, O.; Sellar, R.S.; Martinod, K.; Cherpokova, D.; McConkey, M.; Chappell, R.J.; Silver, A.J.; Adams, D.; Castellano, C.A.; Schneider, R.K.; et al. Increased neutrophil extracellular trap formation promotes thrombosis in myeloproliferative neoplasms. Sci. Transl. Med. 2018, 10, 1–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guy, A.; Favre, S.; Labrouche-Colomer, S.; Deloison, L.; Gourdou-Latyszenok, V.; Renault, M.-A.; Riviere, E.; James, C. High circulating levels of MPO-DNA are associated with thrombosis in patients with MPN. Leukemia 2019, 33, 2544–2548. [Google Scholar] [CrossRef] [PubMed]
- Craver, B.M.; Ramanathan, G.; Hoang, S.; Chang, X.; Luque, L.F.M.; Brooks, S.; Lai, H.Y.; Fleischman, A.G. N-acetylcysteine inhibits thrombosis in a murine model of myeloproliferative neoplasm. Blood Adv. 2020, 4, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, A.; Jouault, H.; Guichard, J.; Wendling, F.; Drouin, A.; Cramer, E.M. Pathologic Interaction between Megakaryocytes and Polymorphonuclear Leukocytes in Myelofibrosis. Blood 2000, 96, 1342–1347. [Google Scholar] [CrossRef]
- Kjeldsen, L.; Johnsen, A.H.; Sengeløv, H.; Borregaard, N. Isolation and primary structure of NGAL, a novel protein associated with human neutrophil gelatinase. J. Biol. Chem. 1993, 268, 10425–10432. [Google Scholar] [CrossRef]
- Maréchal, A.; Zou, L. DNA Damage Sensing by the ATM and ATR Kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef]
- Heylmann, D.; Rödel, F.; Kindler, T.; Kaina, B. Radiation sensitivity of human and murine peripheral blood lymphocytes, stem and progenitor cells. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2014, 1846, 121–129. [Google Scholar] [CrossRef]
- Jeggo, P.A.; Löbrich, M. DNA double-strand breaks: Their cellular and clinical impact? Oncogene 2007, 26, 7717–7719. [Google Scholar] [CrossRef]
- Plo, I.; Nakatake, M.; Malivert, L.; de Villartay, J.-P.; Giraudier, S.; Villeval, J.-L.; Wiesmuller, L.; Vainchenker, W. JAK2 stimulates homologous recombination and genetic instability: Potential implication in the heterogeneity of myeloproliferative disorders. Blood 2008, 112, 1402–1412. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Oxley, D.; Smith, T.S.; Follows, G.A.; Green, A.; Alexander, D.R. DNA Damage–Induced Bcl-xL Deamidation Is Mediated by NHE-1 Antiport Regulated Intracellular pH. PLoS Biol. 2006, 5, e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.S.; Verstovsek, S.; Pemmaraju, N. Novel Therapies in Myeloproliferative Neoplasms: Beyond JAK Inhibitor Monotherapy. J. Immunother. Precis. Oncol. 2021, 4, 117–128. [Google Scholar] [CrossRef]
- Wei, A.H.; Roberts, A.W.; Spencer, A.; Rosenberg, A.; Siegel, D.; Walter, R.B.; Caenepeel, S.; Hughes, P.; McIver, Z.; Mezzi, K.; et al. Targeting MCL-1 in hematologic malignancies: Rationale and progress. Blood Rev. 2020, 44, 100672. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.G.; Sawatzky, D.A.; Walker, A.; Ward, C.; Sheldrake, T.A.; Riley, N.A.; Caldicott, A.; Martinez-Losa, M.; Walker, T.R.; Duffin, R.; et al. Cyclin-dependent kinase inhibitors enhance the resolution of inflammation by promoting inflammatory cell apoptosis. Nat. Med. 2006, 12, 1056–1064. [Google Scholar] [CrossRef]
- Leitch, A.E.; Riley, N.A.; Sheldrake, T.A.; Festa, M.; Fox, S.; Duffin, R.; Haslett, C.; Rossi, A.G. The cyclin-dependent kinase inhibitor R-roscovitine down-regulates Mcl-1 to override pro-inflammatory signalling and drive neutrophil apoptosis. Eur. J. Immunol. 2010, 40, 1127–1138. [Google Scholar] [CrossRef]
- Chu, D.; Dong, X.; Shi, X.; Zhang, C.Y.; Wang, Z. Neutrophil-Based Drug Delivery Systems. Adv. Mater. 2018, 30, e1706245. [Google Scholar] [CrossRef]
- Bisso, P.W.; Gaglione, S.; Guimarães, P.P.G.; Mitchell, M.J.; Langer, R. Nanomaterial Interactions with Human Neutrophils. ACS Biomater. Sci. Eng. 2018, 4, 4255–4265. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Human Bone Marrow | ||
|---|---|---|
| Homeostasis | MPN | |
| Granulopoiesis | 55% to 60% of the bone marrow is dedicated to neutrophil production [104] | Increased myeloproliferation, increased neutrophil number [3,28] |
| Neutrophil death and clearance | Up to two-thirds of circulating neutrophils [121] | Deregulated [77,78] |
| Neutrophil marginal pool | 25% of total blood pool [121] | Disbalanced? |
| Neutrophil reserve pool | 6 × 1011 cells [138]; contains ~20 times the number of neutrophils in circulation [71] | Disbalanced? |
| Pro-Apoptotic | MPN | Anti-Apoptotic | MPN |
|---|---|---|---|
| BID ↓, BID ↓ | ET [157] | C-FLIP ↑ | PV [160] |
| BIM ↓ | ET [157] | CORO1A ↑ | MPN [164] |
| BAD ↑ | ET, PMF [157] | FAIM ↑ | PMF [160] |
| DR5 ↓ | ET [160] | A1 ↑ | ET, PMF [157] |
| PYCARD ↑ | PMF [164] | BCL-2 ↑ | ET, PMF [157] |
| FAS ↑ | PV [160] | BCL-W ↑ | ET, PMF [157] |
| TRAIL ↑ | PV [160] | BCL-XL ↑ | ET, PMF [157] |
| BCL-XL ↑ | PMF [157] | ||
| S100 ↑ | MPN [164] | RAC2 ↑ | MPN [164] |
| TP53 methylation | ET, PMF [165] |
| Neutrophil Pro-Inflammatory Capacity | MPN | |
|---|---|---|
| Cytokine production | proinflammatory IL-1β, IL-6, IL-12, TNFα, MCP-1, lipocalin 2, oncostatin M and anti-inflammatory IL-1ra, TGF-β [57,82,84] | Increased [24,32,96,97] |
| Chemokine production | IL-8, GRO-α, MIP-1α and β, Mip-3 α/β, IP-10, MIG, I-TAC, Mip-3 [82,83] | Increased [24] |
| ROS and RNS production | Superoxide, H2O2, NO [51] | Increased [28,47,49] |
| NET components release | cfDNA, mitochondrial DNA, extracellular histones, granule proteins [87,88,89] | Increased [172,173,174,175] |
| NLRP3 and AIM2 inflammasome | process and release proinflammatory cytokines IL-1β and IL-18 [90] | Increased expression [35] |
| Neutrophil granules and secretory vesicles content | MPO, MMP9, proteinase 3, cathepsin G, neutrophil gelatinase-associated lipocalin-2, neutrophil elastase [54,55,56,177] | Increased [32,85,96,97,174] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marković, D.; Maslovarić, I.; Djikić, D.; Čokić, V.P. Neutrophil Death in Myeloproliferative Neoplasms: Shedding More Light on Neutrophils as a Pathogenic Link to Chronic Inflammation. Int. J. Mol. Sci. 2022, 23, 1490. https://doi.org/10.3390/ijms23031490
Marković D, Maslovarić I, Djikić D, Čokić VP. Neutrophil Death in Myeloproliferative Neoplasms: Shedding More Light on Neutrophils as a Pathogenic Link to Chronic Inflammation. International Journal of Molecular Sciences. 2022; 23(3):1490. https://doi.org/10.3390/ijms23031490
Chicago/Turabian StyleMarković, Dragana, Irina Maslovarić, Dragoslava Djikić, and Vladan P. Čokić. 2022. "Neutrophil Death in Myeloproliferative Neoplasms: Shedding More Light on Neutrophils as a Pathogenic Link to Chronic Inflammation" International Journal of Molecular Sciences 23, no. 3: 1490. https://doi.org/10.3390/ijms23031490
APA StyleMarković, D., Maslovarić, I., Djikić, D., & Čokić, V. P. (2022). Neutrophil Death in Myeloproliferative Neoplasms: Shedding More Light on Neutrophils as a Pathogenic Link to Chronic Inflammation. International Journal of Molecular Sciences, 23(3), 1490. https://doi.org/10.3390/ijms23031490