
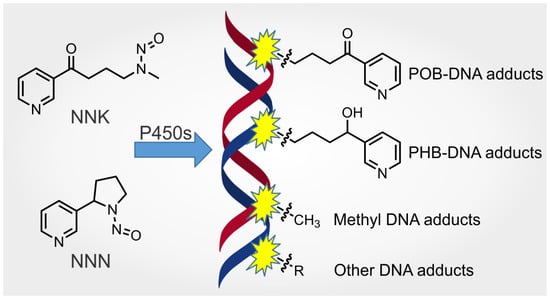
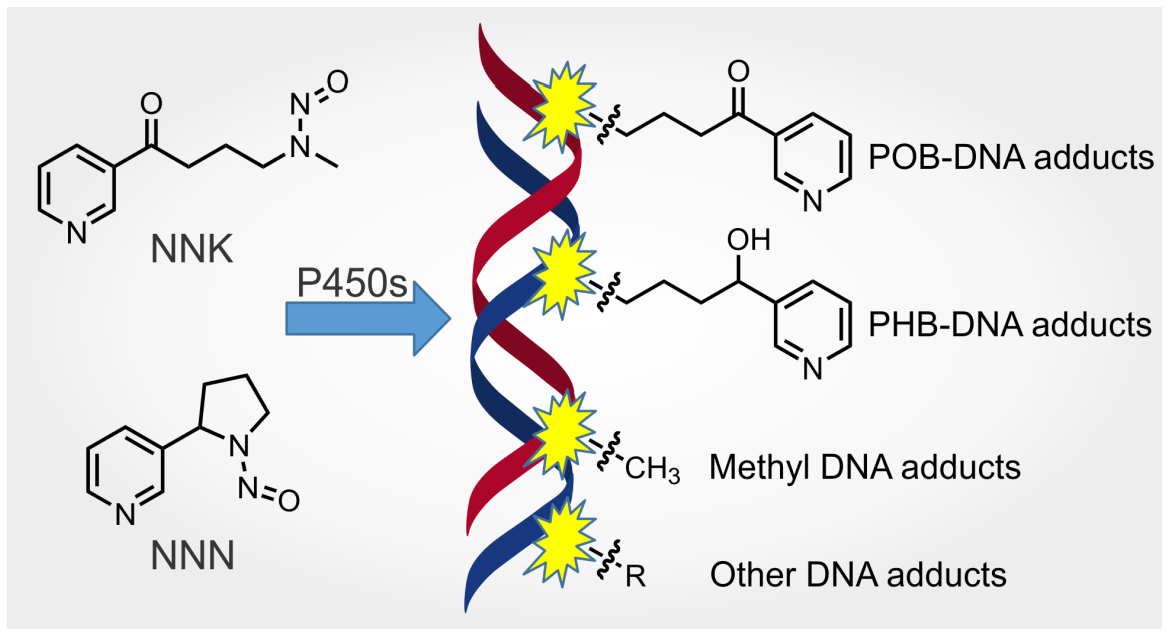
Metabolism and DNA Adduct Formation of Tobacco-Specific N-Nitrosamines


Abstract
:
1. Introduction
2. Human Exposure to Carcinogenic Tobacco-Specific N-Nitrosamines
3. Metabolism and DNA Adduct Formation of NNK and Its Metabolite, NNAL
3.1. Occurrence and Carcinogenicity
3.2. Metabolism
3.2.1. Metabolic Activation and Detoxification Pathways
3.2.2. Stereochemistry of NNAL and Its Glucuronides
3.2.3. Stereochemistry of Hydroxy Acid Formation
3.3. DNA Adducts Formed by NNK Metabolism
3.3.1. POB DNA Adducts
- (1)
- HPB-releasing DNA Adducts

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Animal Data | |||||||
|---|---|---|---|---|---|---|---|
| Animal Species | Administration Pathway | Exposure Amount | Exposure Duration | Target Tissue DNA | HPB-Releasing Adducts a | Ref. | |
| Female A/J mice | Single i.p. injection | 10 μmol/mouse [5-3H]NNK | 12–144 h | Lung | 8.4 pmol/μmol Gua @ 24 h time point | [79] | |
| Male F344 rats | Once daily i.p. injection | 15–5000 μg/kg/day of [5-3H]NNK or [C3H3]NNK | 4 days | Liver | 18–3400 fmol/mg DNA | [77] | |
| Lung | 58–2180 fmol/mg DNA | ||||||
| Single s.c. injection | 2 μmol/rat [5-3H]NNK | 24 h | Liver | 0.67 ± 0.1 pmol/mg DNA | [75] | ||
| Lung | Present | ||||||
| Single s.c. injection | 0.81 mg/kg of [5-3H]NNK | 24 h | Liver | 2.1 ± 0.1 pmol/μmol Gua | [76] | ||
| Lung | 1.6 pmol/μmol Gua | ||||||
| In the drinking water | 5 ppm NNK | 10 weeks | Lung | 9 ± 3 (pmol/mg DNA) | [24] | ||
| 30 weeks | 9 ± 3 | ||||||
| 50 weeks | 6 ± 1 | ||||||
| 70 weeks | 5 ± 2 | ||||||
| 5 ppm (S)-NNAL | 10 weeks | Lung | 5 ± 3 (pmol/mg DNA) | ||||
| 30 weeks | 10 ± 5 | ||||||
| 50 weeks | 11 ± 3 | ||||||
| 70 weeks | 5 ± 1 | ||||||
| 5 ppm (R)-NNAL | 10 weeks | Lung | 1 ± 0 (pmol/mg DNA) | ||||
| 30 weeks | 1 ± 1 | ||||||
| 50 weeks | 2 ± 1 | ||||||
| 70 weeks | 1 ± 1 | ||||||
| Female A/J mice | Single i.p. injection | 10 μmol/mouse [5-3H]NNN | 24 h | Liver | 22.6 (or 25.1) pmol/μmol Gua | [80] | |
| 11 μmol/mouse [5-3H]NNN | Lung | 5.6 pmol/μmol Gua | [79] | ||||
| Male F344 rats | Single s.c. injection | 0.35 μmol/rat [5-3H]NNN | 24 h | Liver | 0.08 ± 0.1 pmol/mg DNA | [75] | |
| Once daily i.p. injection | 0.407 μmol/rat [5-3H]NNN | 3 days | Liver | 0.54 pmol/mg DNA | [81] | ||
| Lung | 0.50 | ||||||
| Nasal mucosa | 0.02 | ||||||
| Esophagus | <0.005 | ||||||
| Kidney | <0.005 | ||||||
| Human Data | |||||||
| Sample source | Smoking status | Subject number | Tissues | HPB-releasing adducts | Note | Ref. | |
| Autopsy samples | Smokers verified by blood nicotine and cotinine, and medical record if necessary | 9 | Tracheobronchus | 16 ± 18 fmol/mg DNA | [80] | ||
| Peripheral lung tissues | 11 ± 16 | ||||||
| Nonsmokers | 8 | Tracheobronchus | 0.9 ± 1.7 | Only 1 subject had significant HPB levels. | |||
| Peripheral lung tissues | 0.9 ± 2.3 | ||||||
| TobPRAC Biorepository | Smokers (>10 cigs/day for at least 1 year) verified by urinary NNN and NNAL. | 30 | Mouthwash oral cells | 12.0 ± 35.1 pmol/mg DNA | [82] | ||
| Buccal cells | 45 ± 57 | ||||||
| Nonsmokers | 15 | Mouthwash oral cells | 0.23 ± 0.43 | Only 3 subjects had significant HPB levels. | |||
| Patients with HNSCC | Smokers (determined by lifetime tobacco use questionnaire) | 30 | Buccal cells | 8.19 ± 17.8 (median 1.51) pmol/mg DNA | [83] | ||
| Patients without HNSCC | 35 | 4.53 ± 14.36 (median 0.23) | |||||
| Patients with lung cancers | Self-reported smokers | 21 | Peripheral lung tissues | 404 ± 258 fmol/mg DNA | [84] | ||
| Self-reported nonsmokers | 11 | 59 ± 56 | |||||
| Tumor-free sudden death victims | Smokers (>15 ng cotinine/mL blood or >100 ng cotinine/mL urine) | 32 | Lung | 92 ± 148 fmol/mg DNA | Primarily road traffic accidents, suicide and sudden cardiac arrest. | [85] | |
| 29 | Mucosa of esophagus | 138 ± 208 | |||||
| 12 | Mucosa of cardia | 93.6 ± 91.9 | |||||
| Nonsmokers verified by blood or urinary cotinine | 56 | Lung | 61 ± 66 | ||||
| 53 | Mucosa of esophagus | 131 ± 130 | |||||
| 18 | Mucosa of cardia | 117 ± 110 | |||||
| Patients with or without upper gastrointestinal disorders | Self-reported smokers | 7 | Mucosal biopsies of the lower esophagus | 4.80 ± 3.57 pmol/mg DNA | One outlier of patient #55 with ulcerative gastritis was excluded due to its exceptionally high level of HPB-releasing adducts (36.98 pmol/mg DNA) | [86] | |
| Self-reported nonsmokers | 7 | 2.86 ± 2.44 | |||||
- (2)
- POB DNA Base Adducts
- (3)
- POB DNA Phosphate Adducts
3.3.2. PHB DNA Adducts
- (1)
- PHB DNA Base Adducts
- (2)
- PHB DNA Phosphate Adducts
3.3.3. Methyl DNA Adducts
- (1)
- Methyl DNA Base Adducts
- (2)
- Methyl DNA Phosphate Adducts
3.3.4. Formaldehyde-Derived DNA Adducts
3.3.5. Summary of DNA Adducts Formed by NNK Metabolism
3.4. Mutagenicity and Genotoxicity of POB and PHB DNA Adducts
3.5. NNK-Derived DNA Adduct Formation in Lung Carcinogenesis
3.6. Human DNA Adducts Related to NNK Metabolism
3.6.1. HPB-Releasing DNA Adducts in Human Tissues
3.6.2. Methyl Phosphate Adducts in Human Lung
4. Metabolism and DNA Adduct Formation of NNN
4.1. Occurrence and Carcinogenicity
4.2. Metabolism
4.2.1. NNN α-Hydroxylation
- (1)
- Overall Metabolic Profiles of NNN in Laboratory Animals
- (2)
- Preferential α-Hydroxylation of NNN in Target Tissues
- (3)
- Selectivity of α-Hydroxylation of NNN in Target Tissues
4.2.2. NNN β-Hydroxylation
4.2.3. NNN Detoxification Pathways
4.2.4. Other Possible Metabolic Pathways
4.2.5. Stereochemistry of NNN Metabolism
4.3. DNA Adducts Formed by NNN Metabolism
4.3.1. DNA Adducts Formed by NNN 2′-Hydroxylation
- (1)
- POB DNA Base Adducts Formed by NNN 2′-Hydroxylation
- (2)
- POB DNA Phosphate Adducts Formed by NNN 2′-Hydroxylation
4.3.2. DNA Adducts Formed by NNN 5′-Hydroxylation
- (1)
- DNA Base Adducts Formed by NNN 5′-Hydroxylation
- (2)
- DNA Phosphate Adducts Formed by NNN 5′-Hydroxylation
4.4. Mutagenicity and Genotoxicity of NNN-Specific DNA Adducts
5. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 5′-AcetoxyNNN | 5′-acetoxy-N′-nitrosonornicotine |
| AGT | O6-alkylguanine-DNA alkyltransferease |
| AKRs | aldo-keto reductases |
| AMMN | acetoxymethylmethylnitrosamine |
| dIno | 2′-deoxyinosine |
| dUrd | 2′-deoxyuridine |
| E. Coli | Escherichia coli |
| HPB | 4-hydroxy-1-(3-pyridyl)-1-butanone |
| 11β-HSD1 | 11β-hydroxysteroid dehydrogenase type 1 |
| IARC | International Agency for Research on Cancer |
| i.p. | intraperitoneal |
| i.v. | intravenous |
| N2-POBb-dGuo | N2-(4-(3-pyridyl)-4-oxobut-2-yl)-2′-deoxyguanosine |
| Py-Py-dI | 2-[2-(3-pyridyl)-N-pyrrolidinyl]-2′-deoxyinosine |
| Py-Py-dN | 6-[2-(3-pyridyl)-N-pyrrolidinyl]-2′-deoxynebularine |
| N2-Py-Py(OH)-dI | N2-[2-(3-pyridyl)-N-pyrrolidinyl-5-hydroxy]-2′-deoxyinosine |
| N6-Py-Py(OH)-dN | N6-[2-(3-Pyridyl)-N-pyrrolidinyl-5-hydroxy]-2′-deoxynebularine |
| N2-Py-THF-dGuo | N2-[5-(3-pyridyl)tetrahydrofuran-2-yl]-2′-deoxyguanosine |
| N6-HPB-dAdo | N6-[4-hydroxy-1-(pyridine-3-yl)butyl]-2′-deoxyadenosine |
| N6-OPB-dAdo | N6-[4-oxo-1-(pyridine-3-yl)butyl]-2′-deoxyadenosine |
| N6,N6-DHB-dAdo | N6,N6-(2,3-dihydroxybutan-1,4-diyl)-2′-deoxyadenosine |
| NAB | N′-nitrosoanabasine |
| NADP | nicotinamide adenosine dinucleotide phosphate |
| NAT | N′-nitrosoanatabine |
| NER | nucleotide excision repair |
| NHANES | National Health and Nutrition Examination Survey |
| NNAL | 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol |
| NNALOAc | 4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanol |
| NNAL-N-Gluc | 4-(methylnitrosamino)-1-(3-pyridyl-N-β-D-glycopyranuronosyl)-1-butanolonium inner salt |
| NNAL-O-Gluc | 4-(methylnitrosamino)-1-(3-pyridyl)-1-(O-β-D-glucopyranuronosyl)butane |
| NNC | nitrosamide N′-nitrosonorcotinine |
| NNK | 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone |
| NNKOAc | 4-((acetoxymethyl)nitrosamino)-1-(3-pyridyl)-1-butanone |
| NNN | N′-nitrosonornicotine |
| OPB | 4-oxo-4-(pyridin-3-yl)butanal |
| OPB diazonium ion | 4-oxo-1-(pyridine-3-yl)butyl diazonium ion |
| PATH study | Population Assessment of Tobacco and Health study |
| PHB | pyridylhydroxybutyl |
| POB | pyridyloxobutyl |
| pol κ | polymerase κ |
| ppm | parts per million |
| s.c. | subcutaneous |
| S. typhinurium | Salmonella typhinurium |
| TLS | translesion synthesis |
| TSNAs | tobacco-specific N-nitrosamines |
References
- Islami, F.; Goding Sauer, A.; Miller, K.D.; Siegel, R.L.; Fedewa, S.A.; Jacobs, E.J.; McCullough, M.L.; Patel, A.V.; Ma, J.; Soerjomataram, I.; et al. Proportion and number of cancer cases and deaths attributable to potentially modifiable risk factors in the United States. CA Cancer J. Clin. 2018, 68, 31–54. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hecht, S.S. Carcinogenic components of tobacco and tobacco smoke: A 2022 update. Food Chem. Toxicol. 2022. manuscript submitted. [Google Scholar]
- Lang, G.; Vuarnoz, A. Matrix-bound 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in tobacco: Quantification and evidence for an origin from lignin-incorporated alkaloids. J. Nat. Prod. 2015, 78, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.; Ji, H.; Fannin, F.F.; Bush, L.P. Contribution of Nicotine and Nornicotine toward the Production of N′-Nitrosonornicotine in Air-Cured Tobacco (Nicotiana tabacum). J. Nat. Prod. 2016, 79, 754–759. [Google Scholar] [CrossRef] [PubMed]
- Hecht, S.S.; Chen, C.B.; Ornaf, R.M.; Jacobs, E.; Adams, J.D.; Hoffmann, D. Reaction of nicotine and sodium nitrite: Formation of nitrosamines and fragmentation of the pyrrolidine ring. J. Org. Chem. 1978, 43, 72–76. [Google Scholar] [CrossRef]
- Hecht, S.S. Biochemistry, biology, and carcinogenicity of tobacco-specific N-nitrosamines. Chem. Res. Toxicol. 1998, 11, 559–603. [Google Scholar] [CrossRef]
- Hecht, S.S.; Stepanov, I.; Carmella, S.G. Exposure and metabolic activation biomarkers of carcinogenic tobacco-specific nitrosamines. Acc. Chem. Res. 2016, 49, 106–114. [Google Scholar] [CrossRef] [Green Version]
- Konstantinou, E.; Fotopoulou, F.; Drosos, A.; Dimakopoulou, N.; Zagoriti, Z.; Niarchos, A.; Makrynioti, D.; Kouretas, D.; Farsalinos, K.; Lagoumintzis, G.; et al. Tobacco-specific nitrosamines: A literature review. Food Chem. Toxicol. 2018, 118, 198–203. [Google Scholar] [CrossRef]
- Hecht, S.S.; Hatsukami, D.K. Smokeless tobacco and cigarette smoking: Chemical mechanisms and cancer prevention. Nat. Rev. Cancer 2022, 22, 143–155. [Google Scholar] [CrossRef]
- Shah, K.A.; Karnes, H.T. A review of the analysis of tobacco-specific nitrosamines in biological matrices. Crit. Rev. Toxicol. 2010, 40, 305–327. [Google Scholar] [CrossRef]
- Ma, B.; Stepanov, I.; Hecht, S.S. Recent studies on DNA adducts resulting from human exposure to tobacco smoke. Toxics 2019, 7, 16. [Google Scholar] [CrossRef] [Green Version]
- Gushgari, A.J.; Halden, R.U. Critical review of major sources of human exposure to N-nitrosamines. Chemosphere 2018, 210, 1124–1136. [Google Scholar] [CrossRef]
- Klus, H.; Kuhn, H. Determination of nornicotine nitrosamine in the smoke condensate of nornicotine-rich cigarettes. Fachl. Mitt. Oesterreichischen 1973, 14, 251–257. [Google Scholar]
- Hoffmann, D.; Hecht, S.S.; Ornaf, R.M.; Wynder, E.L. N′-nitrosonornicotine in tobacco. Science 1974, 186, 265–267. [Google Scholar] [CrossRef]
- Edwards, S.H.; Rossiter, L.M.; Taylor, K.M.; Holman, M.R.; Zhang, L.; Ding, Y.S.; Watson, C.H. Tobacco-specific nitrosamines in the tobacco and mainstream smoke of U.S. commercial cigarettes. Chem. Res. Toxicol. 2017, 30, 540–551. [Google Scholar] [CrossRef]
- Nasrin, S.; Chen, G.; Watson, C.J.W.; Lazarus, P. Comparison of tobacco-specific nitrosamine levels in smokeless tobacco products: High levels in products from Bangladesh. PLoS ONE 2020, 15, e0233111. [Google Scholar] [CrossRef]
- Lawler, T.S.; Stanfill, S.B.; Tran, H.T.; Lee, G.E.; Chen, P.X.; Kimbrell, J.B.; Lisko, J.G.; Fernandez, C.; Caudill, S.P.; de Castro, B.R.; et al. Chemical analysis of snus products from the United States and northern Europe. PLoS ONE 2020, 15, e0227837. [Google Scholar] [CrossRef]
- International Agency for Research on Cancer. Smokeless Tobacco and Some Tobacco-specific N-Nitrosamines. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; IARC: Lyon, France, 2007; Volume 89. [Google Scholar]
- Hecht, S.S.; Chen, C.B.; Hirota, N.; Ornaf, R.M.; Tso, T.C.; Hoffmann, D. Tobacco-specific nitrosamines: Formation from nicotine in vitro and during tobacco curing and carcinogenicity in strain A mice. J. Natl. Cancer Inst. 1978, 60, 819–824. [Google Scholar] [CrossRef]
- International Agency for Research on Cancer. Personal Habits and Indoor Combustions: N′-Nitrosonornicotine and 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; IARC: Lyon, France, 2012; Volume 100E, pp. 319–331. [Google Scholar]
- Hecht, S.S.; Rivenson, A.; Braley, J.; DiBello, J.; Adams, J.D.; Hoffmann, D. Induction of oral cavity tumors in F344 rats by tobacco-specific nitrosamines and snuff. Cancer Res. 1986, 46, 4162–4166. [Google Scholar]
- Kovi, R.C.; Johnson, C.S.; Balbo, S.; Hecht, S.S.; O’Sullivan, M.G. Metastasis to the F344 rat pancreas from lung cancer induced by 4-(methylnitrosamino)- 1-(3-pyridyl)-1-butanone and enantiomers of its metabolite 4-(methylnitrosamino)-1-(3-pyridyl)- 1-butanol, constituents of tobacco products. Toxicol. Pathol. 2018, 46, 184–192. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, D.; Rivenson, A.; Abbi, R.; Wynder, E.L. A study of tobacco carcinogenesis: Effect of the fat content of the diet on the carcinogenic activity of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in F344 rats. Cancer Res. 1993, 53, 2758–2761. [Google Scholar] [PubMed]
- Balbo, S.; Johnson, C.S.; Kovi, R.C.; James-Yi, S.A.; O’Sullivan, M.G.; Wang, M.; Le, C.T.; Khariwala, S.S.; Upadhyaya, P.; Hecht, S.S. Carcinogenicity and DNA adduct formation of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and enantiomers of its metabolite 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol in F-344 rats. Carcinogenesis 2014, 35, 2798–2806. [Google Scholar] [CrossRef] [Green Version]
- Breyer-Pfaff, U.; Martin, H.J.; Ernst, M.; Maser, E. Enantioselectivity of carbonyl reduction of 4-methylnitrosamino-1-(3-pyridyl)-1-butanone by tissue fractions from human and rat and by enzymes isolated from human liver. Drug Metab. Dispos. 2004, 32, 915–922. [Google Scholar] [PubMed]
- Hecht, S.S.; Trushin, N.; Reid-Quinn, C.A.; Burak, E.S.; Jones, A.B.; Southers, J.L.; Gombar, C.T.; Carmella, S.G.; Anderson, L.M.; Rice, J.M. Metabolism of the tobacco-specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in the patas monkey: Pharmacokinetics and characterization of glucuronide metabolites. Carcinogenesis 1993, 14, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Richter, E.; Engl, J.; Friesenegger, S.; Tricker, A.R. Biotransformation of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in lung tissue from mouse, rat, hamster, and man. Chem. Res. Toxicol. 2009, 22, 1008–1017. [Google Scholar] [CrossRef] [PubMed]
- Belinsky, S.A.; White, C.M.; Trushin, N.; Hecht, S.S. Cell specificity for the pulmonary metabolism of tobacco-specific nitrosamines in the Fischer rat. Carcinogenesis 1989, 10, 2269–2274. [Google Scholar] [CrossRef]
- Castonguay, A.; Lin, D.; Stoner, G.D.; Radok, P.; Furuya, K.; Hecht, S.S.; Schut, H.A.J.; Klaunig, J.E. A study of chemical carcinogenesis. 45. Comparative carcinogenicity in A/J mice and metabolism by cultured mouse peripheral lung of N′-nitrosonornicotine, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone, and their analogs. Cancer Res. 1983, 43, 1223–1229. [Google Scholar]
- Castonguay, A.; Stoner, G.D.; Schut, H.A.; Hecht, S.S. Metabolism of tobacco-specific N-nitrosamines by cultured human tissues. Proc. Natl. Acad. Sci. USA 1983, 80, 6694–6697. [Google Scholar] [CrossRef] [Green Version]
- Hecht, S.S.; Trushin, N. DNA and hemoglobin alkylation by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and its major metabolite 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol in F344 rats. Carcinogenesis 1988, 9, 1665–1668. [Google Scholar] [CrossRef]
- Upadhyaya, P.; Kenney, P.M.; Hochalter, J.B.; Wang, M.; Hecht, S.S. Tumorigenicity and metabolism of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol enantiomers and metabolites in the A/J mouse. Carcinogenesis 1999, 20, 1577–1582. [Google Scholar] [CrossRef] [Green Version]
- Adams, J.D.; LaVoie, E.J.; Hoffmann, D. On the pharmacokinetics of tobacco-specific N-nitrosamines in Fischer rats. Carcinogenesis 1985, 6, 509–511. [Google Scholar] [CrossRef]
- Hu, S.C.; Bryant, M.S.; Sepehr, E.; Kang, H.K.; Trbojevich, R.; Lagaud, G.; Mehta, D.; Ding, W.; Mittelstaedt, R.A.; Pearce, M.G.; et al. Toxicokinetic and genotoxicity study of NNK in male Sprague Dawley rats following nose-only inhalation exposure, intraperitoneal injection, and oral gavage. Toxicol. Sci. 2021, 182, 10–28. [Google Scholar] [CrossRef]
- Jalas, J.R.; Hecht, S.S.; Murphy, S.E. Cytochrome P450 enzymes as catalysts of metabolism of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone, a tobacco specific carcinogen. Chem. Res. Toxicol. 2005, 18, 95–110. [Google Scholar] [CrossRef]
- Murphy, S.E.; Spina, D.A.; Nunes, M.G.; Pullo, D.A. Glucuronidation of 4-((hydroxymethyl)nitrosamino)-1-(3-pyridyl)-1-butanone, a metabolically activated form of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone, by phenobarbital-treated rats. Chem. Res. Toxicol. 1995, 8, 772–779. [Google Scholar] [CrossRef]
- Peterson, L.A.; Urban, A.M.; Vu, C.C.; Cummings, M.E.; Brown, L.C.; Warmka, J.K.; Li, L.; Wattenberg, E.V.; Patel, Y.; Stram, D.O.; et al. Role of aldehydes in the toxic and mutagenic effects of nitrosamines. Chem. Res. Toxicol. 2013, 26, 1464–1473. [Google Scholar] [CrossRef] [Green Version]
- Carmella, S.G.; Borukhova, A.; Akerkar, S.A.; Hecht, S.S. Analysis of human urine for pyridine-N-oxide metabolites of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone, a tobacco-specific lung carcinogen. Cancer Epidemiol. Biomark. Prev. 1997, 6, 113–120. [Google Scholar]
- Carmella, S.G.; Akerkar, S.; Hecht, S.S. Metabolites of the tobacco-specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in smokers’ urine. Cancer Res. 1993, 53, 721–724. [Google Scholar]
- Carmella, S.G.; Han, S.; Villalta, P.W.; Hecht, S.S. Analysis of total 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol in smokers’ blood. Cancer Epidemiol. Biomark. Prev. 2005, 14, 2669–2672. [Google Scholar] [CrossRef] [Green Version]
- Church, T.R.; Anderson, K.E.; Caporaso, N.E.; Geisser, M.S.; Le, C.T.; Zhang, Y.; Benoit, A.R.; Carmella, S.G.; Hecht, S.S. A prospectively measured serum biomarker for a tobacco-specific carcinogen and lung cancer in smokers. Cancer Epidemiol. Biomark. Prev. 2009, 18, 260–266. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.M.; Koh, W.P.; Murphy, S.E.; Fan, Y.; Wang, R.; Carmella, S.G.; Han, S.; Wickham, K.; Gao, Y.T.; Yu, M.C.; et al. Urinary levels of tobacco-specific nitrosamine metabolites in relation to lung cancer development in two prospective cohorts of cigarette smokers. Cancer Res. 2009, 69, 2990–2995. [Google Scholar] [CrossRef] [Green Version]
- Milunsky, A.; Carmella, S.G.; Ye, M.; Hecht, S.S. A tobacco-specific carcinogen in the fetus. Prenat. Diagn. 2000, 20, 307–310. [Google Scholar] [CrossRef]
- Idris, A.M.; Nair, J.; Friesen, M.; Ohshima, H.; Brouet, I.; Faustman, E.M.; Bartsch, H. Carcinogenic tobacco-specific nitrosamines are present at unusually high levels in the saliva of oral snuff users in Sudan. Carcinogenesis 1992, 13, 1001–1005. [Google Scholar] [CrossRef] [PubMed]
- Cartanyà-Hueso, À.; Lidón-Moyano, C.; Fu, M.; Perez-Ortuño, R.; Ballbè, M.; Matilla-Santander, N.; Martín-Sánchez, J.C.; Pascual, J.A.; Fernández, E.; Martínez-Sánchez, J.M. Comparison of TSNAs concentration in saliva according to type of tobacco smoked. Environ. Res. 2019, 172, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Ortuño, R.; Martínez-Sánchez, J.M.; Fu, M.; Ballbè, M.; Quirós, N.; Fernández, E.; Pascual, J.A. Assessment of tobacco specific nitrosamines (TSNAs) in oral fluid as biomarkers of cancer risk: A population-based study. Environ. Res. 2016, 151, 635–641. [Google Scholar] [CrossRef]
- Hecht, S.S.; Carmella, S.G.; Murphy, S.E.; Akerkar, S.; Brunnemann, K.D.; Hoffmann, D. A tobacco-specific lung carcinogen in the urine of men exposed to cigarette smoke. N. Engl. J. Med. 1993, 329, 1543–1546. [Google Scholar] [CrossRef]
- Carmella, S.G.; Le Ka, K.A.; Upadhyaya, P.; Hecht, S.S. Analysis of N- and O-glucuronides of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol (NNAL) in human urine. Chem. Res. Toxicol. 2002, 15, 545–550. [Google Scholar] [CrossRef]
- Carmella, S.G.; Ming, X.; Olvera, N.; Brookmeyer, C.; Yoder, A.; Hecht, S.S. High throughput liquid and gas chromatography-tandem mass spectrometry assays for tobacco-specific nitrosamine and polycyclic aromatic hydrocarbon metabolites associated with lung cancer in smokers. Chem. Res. Toxicol. 2013, 26, 1209–1217. [Google Scholar] [CrossRef]
- Desai, D.; Kagan, S.S.; Amin, S.; Carmella, S.G.; Hecht, S.S. Identification of 4-(methylnitrosamino)-1-[3-(6-hydroxypyridyl)]-1-butanone as a urinary metabolite of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in rodents. Chem. Res. Toxicol. 1993, 6, 794–799. [Google Scholar] [CrossRef]
- Peterson, L.A.; Ng, D.K.; Stearns, R.A.; Hecht, S.S. Formation of NADP(H) analogs of tobacco-specific nitrosamines in rat liver and pancreatic microsomes. Chem. Res. Toxicol. 1994, 7, 599–608. [Google Scholar] [CrossRef]
- Castonguay, A.; Pepin, P.; Briere, N. Modulation of 4-(methylnitrosamino)-1-(3-pyridyl)-1 butanone demethylation and denitrosation by rat liver microsomes. Cancer Lett. 1991, 59, 67–74. [Google Scholar] [CrossRef]
- Lee, V.M.; Keefer, L.K.; Archer, M.C. An evaluation of the roles of metabolic denitrosation and alpha-hydroxylation in the hepatotoxicity of N-Nitrosodimethylamine. Chem. Res. Toxicol. 1996, 9, 1319–1324. [Google Scholar] [CrossRef]
- Carlson, E.S.; Upadhyaya, P.; Hecht, S.S. Evaluation of nitrosamide formation in the cytochrome P450-mediated metabolism of tobacco-specific nitrosamines. Chem. Res. Toxicol. 2016, 29, 2194–2205. [Google Scholar] [CrossRef]
- Hecht, S.S.; Young, R.; Chen, C.B. Metabolism in the F344 rat of 4-(N-methyl-N-nitrosamino)-1-(3-pyridyl)-1-butanone, a tobacco-specific carcinogen. Cancer Res. 1980, 40, 4144–4150. [Google Scholar]
- Hoffmann, D.; Castonguay, A.; Rivenson, A.; Hecht, S.S. Comparative carcinogenicity and metabolism of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and N′-nitrosonornicotine in Syrian Golden hamsters. Cancer Res. 1981, 41, 2386–2393. [Google Scholar]
- Stepanov, I.; Upadhyaya, P.; Carmella, S.G.; Feuer, R.; Jensen, J.; Hatsukami, D.K.; Hecht, S.S. Extensive metabolic activation of the tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in smokers. Cancer Epidemiol. Biomark. Prev. 2008, 17, 1764–1773. [Google Scholar] [CrossRef] [Green Version]
- Jing, M.; Wang, Y.; Upadhyaya, P.; Jain, V.; Yuan, J.M.; Hatsukami, D.K.; Hecht, S.S.; Stepanov, I. Liquid chromatography-electrospray ionization-tandem mass spectrometry quantitation of urinary [pyridine-D4]4-hydroxy-4-(3-pyridyl)butanoic acid, a biomarker of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone metabolic activation in smokers. Chem. Res. Toxicol. 2014, 27, 1547–1555. [Google Scholar] [CrossRef] [Green Version]
- Hecht, S.S.; Hatsukami, D.K.; Bonilla, L.E.; Hochalter, J.B. Quantitation of 4-oxo-4-(3-pyridyl)butanoic acid and enantiomers of 4-hydroxy-4-(3-pyridyl)butanoic acid in human urine: A substantial pathway of nicotine metabolism. Chem. Res. Toxicol. 1999, 12, 172–179. [Google Scholar] [CrossRef]
- Trushin, N.; Hecht, S.S. Stereoselective metabolism of nicotine and tobacco-specific N-nitrosamines to 4-hydroxy-4-(3-pyridyl)butanoic acid in rats. Chem. Res. Toxicol. 1999, 12, 164–171. [Google Scholar] [CrossRef]
- Upadhyaya, P.; Carmella, S.G.; Guengerich, F.P.; Hecht, S.S. Formation and metabolism of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol enantiomers in vitro in mouse, rat and human tissues. Carcinogenesis 2000, 21, 1233–1238. [Google Scholar] [CrossRef]
- Wu, Z.; Upadhyaya, P.; Carmella, S.G.; Hecht, S.S.; Zimmerman, C.L. Disposition of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol (NNAL) in bile duct-cannulated rats: Stereoselective metabolism and tissue distribution. Carcinogenesis 2002, 23, 171–179. [Google Scholar] [CrossRef] [Green Version]
- Hecht, S.S.; Carmella, S.G.; Ye, M.; Le, K.-a.; Jensen, J.A.; Zimmerman, C.L.; Hatsukami, D.K. Quantitation of metabolites of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone after cessation of smokeless tobacco use. Cancer Res. 2002, 62, 129–134. [Google Scholar] [PubMed]
- Carmella, S.G.; Ye, M.; Upadhyaya, P.; Hecht, S.S. Stereochemistry of metabolites of a tobacco-specific lung carcinogen in smokers’ urine. Cancer Res. 1999, 59, 3602–3605. [Google Scholar] [PubMed]
- Hecht, S.S.; Spratt, T.E.; Trushin, N. Corrigendum: Absolute configuration of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol formed metabolically from 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Carcinogenesis 2000, 21, 850. [Google Scholar] [CrossRef] [PubMed]
- Hecht, S.S.; Spratt, T.E.; Trushin, N. Absolute configuration of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol formed metabolically from 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Carcinogenesis 1997, 18, 1851–1854. [Google Scholar] [CrossRef] [Green Version]
- Morse, M.A.; Eklind, K.I.; Toussaint, M.; Amin, S.G.; Chung, F.L. Characterization of a glucuronide metabolite of 4-(methyl-nitrosamino)-1-(3-pyridyl)-1-butanone (NNK) and its dose-dependent excretion in the urine of mice and rats. Carcinogenesis 1990, 11, 1819–1823. [Google Scholar] [CrossRef]
- Dow, J.; Berg, C. Stereoselectivity of the carbonyl reduction of dolasetron in rats, dogs, and humans. Chirality 1995, 7, 342–348. [Google Scholar] [CrossRef]
- Hecht, S.S.; Hochalter, J.B.; Villalta, P.W.; Murphy, S.E. 2′-Hydroxylation of nicotine by cytochrome P450 2A6 and human liver microsomes: Formation of a lung carcinogen precursor. Proc. Natl. Acad. Sci. USA 2000, 97, 12493–12497. [Google Scholar] [CrossRef] [Green Version]
- Murphy, S.E. Biochemistry of nicotine metabolism and its relevance to lung cancer. J. Biol. Chem. 2021, 296, 100722. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. Fourth Report on Human Exposure to Environmental Chemicals, Updated Tables, (March 2021); U.S. Department of Health and Human Services: Atlanta, GA, USA, 2021.
- Kuiper, N.; Coats, E.M.; Crawford, T.N.; Gammon, D.G.; Loomis, B.; Watson, C.H.; Melstrom, P.C.; Lavinghouze, R.; Rogers, T.; King, B.A. Trends in manufacturer-reported nicotine yields in cigarettes sold in the United States, 2013–2016. Prev. Chronic Dis. 2020, 17, E148. [Google Scholar] [CrossRef]
- Lawler, T.S.; Stanfill, S.B.; de Castro, B.R.; Lisko, J.G.; Duncan, B.W.; Richter, P.; Watson, C.H. Surveillance of nicotine and pH in cigarette and cigar filler. Tob. Regul. Sci. 2017, 3 (Suppl. 1), 101–116. [Google Scholar] [CrossRef] [Green Version]
- Spratt, T.E.; Peterson, L.A.; Confer, W.L.; Hecht, S.S. Solvolysis of model compounds for alpha-hydroxylation of N′-nitrosonornicotine and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone: Evidence for a cyclic oxonium ion intermediate in the alkylation of nucleophiles. Chem. Res. Toxicol. 1990, 3, 350–356. [Google Scholar] [CrossRef]
- Hecht, S.S.; Spratt, T.E.; Trushin, N. Evidence for 4-(3-pyridyl)-4-oxobutylation of DNA in F344 rats treated with the tobacco-specific nitrosamines 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and N′-nitrosonornicotine. Carcinogenesis 1988, 9, 161–165. [Google Scholar] [CrossRef]
- Peterson, L.A.; Mathew, R.; SE, B.P.M.; Trushin, N.; Hecht, S.S. In vivo and in vitro persistence of pyridyloxobutyl DNA adducts from 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Carcinogenesis 1991, 12, 2069–2072. [Google Scholar] [CrossRef]
- Murphy, S.E.; Palomino, A.; Hecht, S.S.; Hoffmann, D. Dose-response study of DNA and hemoglobin adduct formation by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in F344 rats. Cancer Res. 1990, 50, 5446–5452. [Google Scholar]
- Hecht, S.S.; Han, S.; Kenney, P.M.; Wang, M.; Lindgren, B.; Wang, Y.; Lao, Y.; Hochalter, J.B.; Upadhyaya, P. Investigation of the reaction of myosmine with sodium nitrite in vitro and in rats. Chem. Res. Toxicol. 2007, 20, 543–549. [Google Scholar] [CrossRef] [Green Version]
- Peterson, L.A.; Hecht, S.S. A study of chemical carcinogenesis.141. O6-methylguanine is a critical determinant of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone tumorigenesis in A/J mouse lung. Cancer Res. 1991, 51, 5557–5564. [Google Scholar]
- Foiles, P.G.; Akerkar, S.A.; Carmella, S.G.; Kagan, M.; Stoner, G.D.; Resau, J.H.; Hecht, S.S. Mass spectrometric analysis of tobacco-specific nitrosamine-DNA adducts in smokers and nonsmokers. Chem. Res. Toxicol. 1991, 4, 364–368. [Google Scholar] [CrossRef]
- Spratt, T.E.; Trushin, N.; Lin, D.; Hecht, S.S. Analysis for N2-(pyridyloxobutyl)deoxyguanosine adducts in DNA of tissues exposed to tritium-labeled 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and N′-nitrosonornicotine. Chem. Res. Toxicol. 1989, 2, 169–173. [Google Scholar] [CrossRef]
- Stepanov, I.; Muzic, J.; Le, C.T.; Sebero, E.; Villalta, P.; Ma, B.; Jensen, J.; Hatsukami, D.; Hecht, S.S. Analysis of 4-hydroxy-1-(3-pyridyl)-1-butanone (HPB)-releasing DNA adducts in human exfoliated oral mucosa cells by liquid chromatography-electrospray ionization-tandem mass spectrometry. Chem. Res. Toxicol. 2013, 26, 37–45. [Google Scholar] [CrossRef] [Green Version]
- Ma, B.; Ruszczak, C.; Jain, V.; Khariwala, S.S.; Lindgren, B.; Hatsukami, D.K.; Stepanov, I. Optimized liquid chromatography nanoelectrospray-high-resolution tandem mass spectrometry method for the analysis of 4-hydroxy-1-(3-pyridyl)-1-butanone-releasing DNA adducts in human oral cells. Chem. Res. Toxicol. 2016, 29, 1849–1856. [Google Scholar] [CrossRef] [Green Version]
- Hölzle, D.; Schlöbe, D.; Tricker, A.R.; Richter, E. Mass spectrometric analysis of 4-hydroxy-1-(3-pyridyl)-1-butanone-releasing DNA adducts in human lung. Toxicology 2007, 232, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Schlöbe, D.; Hölzle, D.; Hatz, D.; von Meyer, L.; Tricker, A.R.; Richter, E. 4-Hydroxy-1-(3-pyridyl)-1-butanone-releasing DNA adducts in lung, lower esophagus and cardia of sudden death victims. Toxicology 2008, 245, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Heppel, C.W.; Heling, A.K.; Richter, E. Ultrasensitive method for the determination of 4-hydroxy-1-(3-pyridyl)-1-butanone-releasing DNA adducts by gas chromatography-high resolution mass spectrometry in mucosal biopsies of the lower esophagus. Anal. Bioanal. Chem. 2009, 393, 1525–1530. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ma, B.; Cao, Q.; Balbo, S.; Zhao, L.; Upadhyaya, P.; Hecht, S.S. Mass spectrometric quantitation of pyridyloxobutyl DNA phosphate adducts in rats chronically treated with N′-nitrosonornicotine. Chem. Res. Toxicol. 2019, 32, 773–783. [Google Scholar] [CrossRef]
- Liu, X.K.; Spratt, T.E.; Murphy, S.E.; Peterson, L.A. Pyridyloxobutylation of guanine residues by 4-[(acetoxymethyl)nitrosamino]-1-(3-pyridyl)-1-butanone generates substrates of O6-alkylguanine-DNA alkyltransferase. Chem. Res. Toxicol. 1996, 9, 949–953. [Google Scholar] [CrossRef]
- Wang, L.; Spratt, T.E.; Liu, X.K.; Hecht, S.S.; Pegg, A.E.; Peterson, L.A. Pyridyloxobutyl adduct O6-[4-oxo-4-(3-pyridyl)butyl]guanine is present in 4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanone-treated DNA and is a substrate for O6-alkylguanine-DNA alkyltransferase. Chem. Res. Toxicol. 1997, 10, 562–567. [Google Scholar] [CrossRef]
- Wang, M.; Cheng, G.; Sturla, S.J.; Shi, Y.; McIntee, E.J.; Villalta, P.W.; Upadhyaya, P.; Hecht, S.S. Identification of adducts formed by pyridyloxobutylation of deoxyguanosine and DNA by 4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanone, a chemically activated form of tobacco specific carcinogens. Chem. Res. Toxicol. 2003, 16, 616–626. [Google Scholar] [CrossRef]
- Hecht, S.S.; Villalta, P.W.; Sturla, S.J.; Cheng, G.; Yu, N.; Upadhyaya, P.; Wang, M. Identification of O2-substituted pyrimidine adducts formed in reactions of 4-(acetoxymethylnitrosamino)- 1-(3-pyridyl)-1-butanone and 4-(acetoxymethylnitros- amino)-1-(3-pyridyl)-1-butanol with DNA. Chem. Res. Toxicol. 2004, 17, 588–597. [Google Scholar] [CrossRef]
- Michel, A.K.; Zarth, A.T.; Upadhyaya, P.; Hecht, S.S. Identification of 4-(3-pyridyl)-4-oxobutyl-2′-deoxycytidine adducts formed in the reaction of DNA with 4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanone: A chemically activated form of tobacco-specific carcinogens. ACS Omega 2017, 2, 1180–1190. [Google Scholar] [CrossRef]
- Carlson, E.S.; Upadhyaya, P.; Villalta, P.W.; Ma, B.; Hecht, S.S. Analysis and identification of 2′-deoxyadenosine-derived adducts in lung and liver DNA of F-344 rats treated with the tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and enantiomers of its metabolite 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol. Chem. Res. Toxicol. 2018, 31, 358–370. [Google Scholar]
- Leng, J.; Wang, Y. Liquid chromatography-tandem mass spectrometry for the quantification of tobacco-specific nitrosamine-induced DNA adducts in mammalian cells. Anal. Chem. 2017, 89, 9124–9130. [Google Scholar] [CrossRef]
- Hecht, S.S.; Lin, D.; Chuang, J.; Castonguay, A. A study of chemical carcinogenesis.91. Reactions with deoxyguanosine of 4-(carbethoxynitrosamino)-1-(3-pyridyl)-1-butanone, a model-compound for α-hydroxylation of tobacco-specific nitrosamines. J. Am. Chem. Soc. 1986, 108, 1292–1295. [Google Scholar] [CrossRef]
- Lao, Y.; Villalta, P.W.; Sturla, S.J.; Wang, M.; Hecht, S.S. Quantitation of pyridyloxobutyl DNA adducts of tobacco-specific nitrosamines in rat tissue DNA by high-performance liquid chromatography-electrospray ionization-tandem mass spectrometry. Chem. Res. Toxicol. 2006, 19, 674–682. [Google Scholar] [CrossRef] [Green Version]
- Urban, A.M.; Upadhyaya, P.; Cao, Q.; Peterson, L.A. Formation and repair of pyridyloxobutyl DNA adducts and their relationship to tumor yield in A/J mice. Chem. Res. Toxicol. 2012, 25, 2167–2178. [Google Scholar] [CrossRef] [Green Version]
- Lao, Y.; Yu, N.; Kassie, F.; Villalta, P.W.; Hecht, S.S. Formation and accumulation of pyridyloxobutyl DNA adducts in F344 rats chronically treated with 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and enantiomers of its metabolite, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol. Chem. Res. Toxicol. 2007, 20, 235–245. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Wang, M.; Villalta, P.W.; Lindgren, B.R.; Upadhyaya, P.; Lao, Y.; Hecht, S.S. Analysis of pyridyloxobutyl and pyridylhydroxybutyl DNA adducts in extrahepatic tissues of F344 rats treated chronically with 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and enantiomers of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol. Chem. Res. Toxicol. 2009, 22, 926–936. [Google Scholar] [CrossRef] [Green Version]
- Jones, G.D.; Le Pla, R.C.; Farmer, P.B. Phosphotriester adducts (PTEs): DNA’s overlooked lesion. Mutagenesis 2010, 25, 3–16. [Google Scholar] [CrossRef] [Green Version]
- Haglund, J.; Henderson, A.P.; Golding, B.T.; Törnqvist, M. Evidence for phosphate adducts in DNA from mice treated with 4-(N-methyl-N-nitrosamino)-1-(3-pyridyl)-1-butanone (NNK). Chem. Res. Toxicol. 2002, 15, 773–779. [Google Scholar] [CrossRef]
- Ma, B.; Villalta, P.W.; Zarth, A.T.; Kotandeniya, D.; Upadhyaya, P.; Stepanov, I.; Hecht, S.S. Comprehensive high-resolution mass spectrometric analysis of DNA phosphate adducts formed by the tobacco-specific lung carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Chem. Res. Toxicol. 2015, 28, 2151–2159. [Google Scholar] [CrossRef]
- Ma, B.; Zarth, A.T.; Carlson, E.S.; Villalta, P.W.; Upadhyaya, P.; Stepanov, I.; Hecht, S.S. Identification of more than 100 structurally unique DNA-phosphate adducts formed during rat lung carcinogenesis by the tobacco-specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Carcinogenesis 2018, 39, 232–241. [Google Scholar] [CrossRef] [Green Version]
- Ma, B.; Zarth, A.T.; Carlson, E.S.; Villalta, P.W.; Stepanov, I.; Hecht, S.S. Pyridylhydroxybutyl and pyridyloxobutyl DNA phosphate adduct formation in rats treated chronically with enantiomers of the tobacco-specific nitrosamine metabolite 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol. Mutagenesis 2017, 32, 561–570. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Wang, Y. Replication of pyridyloxobutyl phosphotriester lesions in cells. Chem. Res. Toxicol. 2020, 33, 308–311. [Google Scholar] [CrossRef]
- Upadhyaya, P.; Sturla, S.J.; Tretyakova, N.; Ziegel, R.; Villalta, P.W.; Wang, M.; Hecht, S.S. Identification of adducts produced by the reaction of 4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanol with deoxyguanosine and DNA. Chem. Res. Toxicol. 2003, 16, 180–190. [Google Scholar] [CrossRef]
- Guo, S.; Leng, J.; Tan, Y.; Price, N.E.; Wang, Y. Quantification of DNA Lesions induced by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol in mammalian cells. Chem. Res. Toxicol. 2019, 32, 708–717. [Google Scholar] [CrossRef]
- Upadhyaya, P.; Kalscheuer, S.; Hochalter, J.B.; Villalta, P.W.; Hecht, S.S. Quantitation of pyridylhydroxybutyl-DNA adducts in liver and lung of F-344 rats treated with 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and enantiomers of its metabolite 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol. Chem. Res. Toxicol. 2008, 21, 1468–1476. [Google Scholar] [CrossRef] [Green Version]
- Stepanov, I.; Hecht, S.S. Mitochondrial DNA adducts in the lung and liver of F344 rats chronically treated with 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and (S)-4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol. Chem. Res. Toxicol. 2009, 22, 406–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foiles, P.G.; Trushin, N.; Castonguay, A. Measurement of O6-methyldeoxyguanosine in DNA methylated by the tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone using a biotin-avidin enzyme-linked immunosorbent assay. Carcinogenesis 1985, 6, 989–993. [Google Scholar] [CrossRef] [PubMed]
- Belinsky, S.A.; White, C.M.; Boucheron, J.A.; Richardson, F.C.; Swenberg, J.A.; Anderson, M. Accumulation and persistence of DNA adducts in respiratory tissue of rats following multiple administrations of the tobacco specific carcinogen 4-(N-methyl-N-nitrosamino)-1-(3-pyridyl)-1-butanone. Cancer Res. 1986, 46, 1280–1284. [Google Scholar] [PubMed]
- Upadhyaya, P.; Lindgren, B.R.; Hecht, S.S. Comparative levels of O6-methylguanine, pyridyloxobutyl-, and pyridylhydroxybutyl-DNA adducts in lung and liver of rats treated chronically with the tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Drug Metab. Dispos. 2009, 37, 1147–1151. [Google Scholar] [CrossRef] [Green Version]
- Ma, B.; Zarth, A.T.; Carlson, E.S.; Villalta, P.W.; Upadhyaya, P.; Stepanov, I.; Hecht, S.S. Methyl DNA phosphate adduct formation in rats treated chronically with 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and enantiomers of its metabolite 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol. Chem. Res. Toxicol. 2018, 31, 48–57. [Google Scholar] [CrossRef]
- Cheng, G.; Wang, M.; Upadhyaya, P.; Villalta, P.W.; Hecht, S.S. Formation of formaldehyde adducts in the reactions of DNA and deoxyribonucleosides with α-acetates of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK), 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol (NNAL), and N-nitrosodimethylamine (NDMA). Chem. Res. Toxicol. 2008, 21, 746–751. [Google Scholar] [CrossRef]
- Wang, M.; Cheng, G.; Villalta, P.W.; Hecht, S.S. Development of liquid chromatography electrospray ionization tandem mass spectrometry methods for analysis of DNA adducts of formaldehyde and their application to rats treated with N-nitrosodimethylamine or 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Chem. Res. Toxicol. 2007, 20, 1141–1148. [Google Scholar] [CrossRef]
- Peterson, L.A. Context matters: Contribution of specific DNA adducts to the genotoxic properties of the tobacco-specific nitrosamine NNK. Chem. Res. Toxicol. 2017, 30, 420–433. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Leng, J.; Wang, Y. DNA replication studies of N-nitroso compound-induced O6-alkyl-2′-deoxyguanosine lesions in Escherichia coli. J. Biol. Chem. 2019, 294, 3899–3908. [Google Scholar] [CrossRef]
- Pauly, G.T.; Peterson, L.A.; Moschel, R.C. Mutagenesis by O6-[4-oxo-4-(3-pyridyl)butyl]guanine in Escherichia coli and human cells. Chem. Res. Toxicol. 2002, 15, 165–169. [Google Scholar] [CrossRef]
- Du, H.; Leng, J.; Wang, P.; Li, L.; Wang, Y. Impact of tobacco-specific nitrosamine-derived DNA adducts on the efficiency and fidelity of DNA replication in human cells. J. Biol. Chem. 2018, 293, 11100–11108. [Google Scholar] [CrossRef] [Green Version]
- Jasti, V.P.; Spratt, T.E.; Basu, A.K. Tobacco-specific nitrosamine-derived O2-alkylthymidines are potent mutagenic lesions in SOS-induced Escherichia coli. Chem. Res. Toxicol. 2011, 24, 1833–1835. [Google Scholar] [CrossRef]
- Weerasooriya, S.; Jasti, V.P.; Bose, A.; Spratt, T.E.; Basu, A.K. Roles of translesion synthesis DNA polymerases in the potent mutagenicity of tobacco-specific nitrosamine-derived O2-alkylthymidines in human cells. DNA Repair 2015, 35, 63–70. [Google Scholar] [CrossRef] [Green Version]
- Wilson, K.A.; Garden, J.L.; Wetmore, N.T.; Wetmore, S.D. Computational insights into the mutagenicity of two tobacco-derived carcinogenic DNA lesions. Nucleic Acids Res. 2018, 46, 11858–11868. [Google Scholar] [CrossRef] [Green Version]
- Peterson, L.A.; Vu, C.; Hingerty, B.E.; Broyde, S.; Cosman, M. Solution structure of an O6-[4-oxo-4-(3-pyridyl)butyl]guanine adduct in an 11 mer DNA duplex: Evidence for formation of a base triplex. Biochemistry 2003, 42, 13134–13144. [Google Scholar] [CrossRef]
- Pegg, A.E. Repair of O6-alkylguanine by alkyltransferases. Mutat. Res. 2000, 462, 83–100. [Google Scholar] [CrossRef]
- Li, L.; Perdigao, J.; Pegg, A.E.; Lao, Y.; Hecht, S.S.; Lindgren, B.R.; Reardon, J.T.; Sancar, A.; Wattenberg, E.V.; Peterson, L.A. The influence of repair pathways on the cytotoxicity and mutagenicity induced by the pyridyloxobutylation pathway of tobacco-specific nitrosamines. Chem. Res. Toxicol. 2009, 22, 1464–1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Wang, P.; Wang, Y. Cytotoxic and mutagenic properties of alkyl phosphotriester lesions in Escherichia coli cells. Nucleic Acids Res. 2018, 46, 4013–4021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Yuan, J.; Price, N.E.; Wang, Y. Ada protein- and sequence context-dependent mutagenesis of alkyl phosphotriester lesions in Escherichia coli cells. J. Biol. Chem. 2020, 295, 8775–8783. [Google Scholar] [CrossRef]
- Hecht, S.S.; Jordan, K.G.; Choi, C.I.; Trushin, N. Effects of deuterium substitution on the tumorigenicity of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol in A/J mice. Carcinogenesis 1990, 11, 1017–1020. [Google Scholar] [CrossRef]
- Trushin, N.; Rivenson, A.; Hecht, S.S. Evidence supporting the role of DNA pyridyloxobutylation in rat nasal carcinogenesis by tobacco-specific nitrosamines. Cancer Res. 1994, 54, 1205–1211. [Google Scholar]
- Peterson, L.A.; Thomson, N.M.; Crankshaw, D.L.; Donaldson, E.E.; Kenney, P.J. Interactions between methylating and pyridyloxobutylating agents in A/J mouse lungs: Implications for 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone-induced lung tumorigenesis. Cancer Res. 2001, 61, 5757–5763. [Google Scholar]
- Mijal, R.S.; Loktionova, N.A.; Vu, C.C.; Pegg, A.E.; Peterson, L.A. O6-pyridyloxobutylguanine adducts contribute to the mutagenic properties of pyridyloxobutylating agents. Chem. Res. Toxicol. 2005, 18, 1619–1625. [Google Scholar] [CrossRef]
- Peterson, L.A.; Liu, X.K.; Hecht, S.S. Pyridyloxobutyl DNA adducts inhibit the repair of O6-methylguanine. Cancer Res. 1993, 53, 2780–2785. [Google Scholar]
- Castonguay, A.; Tjalve, H.; Hecht, S.S. Tissue distribution of the tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and its metabolites in F344 rats. Cancer Res. 1983, 43, 630–638. [Google Scholar]
- Hecht, S.S.; Lin, D.; Castonguay, A.; Rivenson, A. Effects of alpha-deuterium substitution on the tumorigenicity of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in F344 rats. Carcinogenesis 1987, 8, 291–294. [Google Scholar] [CrossRef]
- Ma, B.; Villalta, P.W.; Hochalter, J.B.; Stepanov, I.; Hecht, S.S. Methyl DNA phosphate adduct formation in lung tumor tissue and adjacent normal tissue of lung cancer patients. Carcinogenesis 2019, 40, 1387–1394. [Google Scholar] [CrossRef]
- Mirvish, S.S.; Sams, J.; Hecht, S.S. Kinetics of nornicotine and anabasine nitrosation in relation to N′-nitrosonornicotine occurrence in tobacco and to tobacco-induced cancer. J. Natl. Cancer Inst. 1977, 59, 1211–1213. [Google Scholar] [CrossRef]
- Knezevich, A.; Muzic, J.; Hatsukami, D.K.; Hecht, S.S.; Stepanov, I. Nornicotine nitrosation in saliva and its relation to endogenous synthesis of N′-nitrosonornicotine in humans. Nicotine Tob. Res. 2013, 15, 591–595. [Google Scholar] [CrossRef]
- Carmella, S.G.; Borukhova, A.; Desai, D.; Hecht, S.S. Evidence for endogenous formation of tobacco-specific nitrosamines in rats treated with tobacco alkaloids and sodium nitrite. Carcinogenesis 1997, 18, 587–592. [Google Scholar] [CrossRef] [Green Version]
- Stepanov, I.; Carmella, S.G.; Briggs, A.; Hertsgaard, L.; Lindgren, B.; Hatsukami, D.; Hecht, S.S. Presence of the carcinogen N′-nitrosonornicotine in the urine of some users of oral nicotine replacement therapy products. Cancer Res. 2009, 69, 8236–8240. [Google Scholar] [CrossRef] [Green Version]
- Stepanov, I.; Carmella, S.G.; Han, S.; Pinto, A.; Strasser, A.A.; Lerman, C.; Hecht, S.S. Evidence for endogenous formation of N′-nitrosonornicotine in some long-term nicotine patch users. Nicotine Tob. Res. 2009, 11, 99–105. [Google Scholar] [CrossRef] [Green Version]
- Bustamante, G.; Ma, B.; Yakovlev, G.; Yershova, K.; Le, C.; Jensen, J.; Hatsukami, D.K.; Stepanov, I. Presence of the carcinogen N′-nitrosonornicotine in saliva of e-cigarette users. Chem. Res. Toxicol. 2018, 31, 731–738. [Google Scholar] [CrossRef]
- Boyland, E.; Roe, F.J.C.; Gorrod, J.W. Induction of pulmonary tumours in mice by nitrosonornicotine, a possible constituent of tobacco smoke. Nature 1964, 202, 1126. [Google Scholar] [CrossRef]
- Hecht, S.S.; Young, R.; Maeura, Y. Comparative carcinogenicity in F344 rats and Syrian golden hamsters of N′-nitrosonornicotine and N′-nitrosonornicotine-1-N-oxide. Cancer Lett. 1983, 20, 333–340. [Google Scholar] [CrossRef]
- Hoffmann, D.; Raineri, R.; Hecht, S.S.; Maronpot, R.; Wynder, E.L. A study of tobacco carcinogenesis. XIV. Effects of N′-nitrosonornicotine and N′-nitrosonanabasine in rats. J. Natl. Cancer Inst. 1975, 55, 977–981. [Google Scholar] [CrossRef]
- Singer, G.M.; Taylor, H.W. Carcinogenicity of N′-nitrosonornicotine in Sprague-Dawley rats. J. Natl. Cancer Inst. 1976, 57, 1275–1276. [Google Scholar] [CrossRef]
- Castonguay, A.; Rivenson, A.; Trushin, N.; Reinhardt, J.; Spathopoulos, S.; Weiss, C.J.; Reiss, B.; Hecht, S.S. Effects of chronic ethanol consumption on the metabolism and carcinogenicity of N′-nitrosonornicotine in F344 rats. Cancer Res. 1984, 44, 2285–2290. [Google Scholar]
- Stoner, G.D.; Adams, C.; Kresty, L.A.; Amin, S.G.; Desai, D.; Hecht, S.S.; Murphy, S.E.; Morse, M.A. Inhibition of N′-nitrosonornicotine-induced esophageal tumorigenesis by 3-phenylpropyl isothiocyanate. Carcinogenesis 1998, 19, 2139–2143. [Google Scholar] [CrossRef] [Green Version]
- Balbo, S.; James-Yi, S.; Johnson, C.S.; O’Sullivan, M.G.; Stepanov, I.; Wang, M.; Bandyopadhyay, D.; Kassie, F.; Carmella, S.; Upadhyaya, P.; et al. (S)-N′-Nitrosonornicotine, a constituent of smokeless tobacco, is a powerful oral cavity carcinogen in rats. Carcinogenesis 2013, 34, 2178–2183. [Google Scholar] [CrossRef] [Green Version]
- Patten, C.J.; Smith, T.J.; Friesen, M.J.; Tynes, R.E.; Yang, C.S.; Murphy, S.E. Evidence for cytochrome P450 2A6 and 3A4 as major catalysts for N′-nitrosonornicotine alpha-hydroxylation by human liver microsomes. Carcinogenesis 1997, 18, 1623–1630. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.B.; Hecht, S.S.; Hoffmann, D. Metabolic α-hydroxylation of the tobacco-specific carcinogen, N′-nitrosonornicotine. Cancer Res. 1978, 38, 3639–3645. [Google Scholar]
- Upadhyaya, P.; Zimmerman, C.L.; Hecht, S.S. Metabolism and pharmacokinetics of N′-nitrosonornicotine in the patas monkey. Drug Metab. Dispos. 2002, 30, 1115–1122. [Google Scholar] [CrossRef] [Green Version]
- Wong, H.L.; Murphy, S.E.; Hecht, S.S. Cytochrome P450 2A-catalyzed metabolic activation of structurally similar carcinogenic nitrosamines: N′-nitrosonornicotine enantiomers, N-nitrosopiperidine, and N-nitrosopyrrolidine. Chem. Res. Toxicol. 2005, 18, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, H.; Inui, Y.; Yun, C.H.; Guengerich, F.P.; Shimada, T. Cytochrome P450 2E1 and 2A6 enzymes as major catalysts for metabolic activation of N-nitrosodialkylamines and tobacco-related nitrosamines in human liver microsomes. Carcinogenesis 1992, 13, 1789–1794. [Google Scholar] [CrossRef] [PubMed]
- Labuc, G.E.; Archer, M.C. Esophageal and hepatic microsomal metabolism of N-nitrosomethylbenzylamine and N-nitrosodimethylamine in the rat. Cancer Res. 1982, 42, 3181–3186. [Google Scholar] [PubMed]
- Von Hofe, E.; Schmerold, I.; Lijinsky, W.; Jeltsch, W.; Kleihues, P. DNA methylation in rat tissues by a series of homologous aliphatic nitrosamines ranging from N-nitrosodimethylamine to N-nitrosomethyldodecylamine. Carcinogenesis 1987, 8, 1337–1341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Upadhyaya, P.; Hecht, S.S. Quantitative analysis of 3’-hydroxynorcotinine in human urine. Nicotine Tob. Res. 2015, 17, 524–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, S.E.; Heiblum, R.; Trushin, N. Comparative metabolism of N′-nitrosonornicotine and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone by cultured F344 rat oral tissue and esophagus. Cancer Res. 1990, 50, 4685–4691. [Google Scholar]
- Huang, Q.; Stoner, G.; Resau, J.; Nickols, J.; Mirvish, S.S. Metabolism of N-Nitrosomethyl-n-amylamine by microsomes from human and rat esophagus. Cancer Res. 1992, 52, 3547–3551. [Google Scholar]
- Murphy, S.E.; Spina, D.A. Evidence for a high-affinity enzyme in rat esophageal microsomes which α-hydroxylates N′-nitrosonornicotine. Carcinogenesis 1994, 15, 2709–2713. [Google Scholar] [CrossRef]
- Hecht, S.S.; Lin, D.; Chen, C.B. Comprehensive analysis of urinary metabolites of N′-nitrosonornicotine. Carcinogenesis 1981, 2, 833–838. [Google Scholar] [CrossRef]
- Chen, C.H.; Fung, P.T.; Hecht, S.S. Assay for microsomal alpha-hydroxylation of N′-nitrosonornicotine and determination of the deuterium isotope effect for alpha-hydroxylation. Cancer Res. 1979, 39, 5057–5062. [Google Scholar]
- Hecht, S.S.; Reiss, B.; Lin, D.; Williams, G.M. Metabolism of N′-nitrosonornicotine by cultured rat esophagus. Carcinogenesis 1982, 3, 453–456. [Google Scholar] [CrossRef]
- Brittebo, E.B.; Castonguay, A.; Furuya, K.; Hecht, S.S. Metabolism of tobacco-specific nitrosamines by cultured rat nasal mucosa. Cancer Res. 1983, 43, 4343–4348. [Google Scholar]
- McCoy, G.; Katayama, S.; Young, R.; Wyatt, M.; Hecht, S. Influence of Chronic Ethanol Consumption on the Metabolism and Carcinogenicity of Tobacco-related Nitrosamines. In IARC Scientific Publication No. 41: N-Nitroso Compounds: Occurrence and Biological Effects; Bartsch, H., O’Neill, I., Castegnaro, M., Okada, M., Eds.; IARC: Lyon, France, 1982; pp. 635–642. [Google Scholar]
- Hecht, S.S.; Chen, C.H.; McCoy, G.D.; Hoffmann, D.; Domellof, L. α-hydroxylation of N-nitrosopyrrolidine and N′-nitrosonornicotine by human liver microsomes. Cancer Lett. 1979, 8, 35–41. [Google Scholar] [CrossRef]
- Harris, C.C.; Autrup, H.; Connor, R.; Barrett, L.A.; McDowell, E.M.; Trump, B.F. Interindividual variation in binding of benzo[a]pyrene to DNA in cultured human bronchi. Science 1976, 194, 1067–1069. [Google Scholar] [CrossRef]
- Hecht, S.S.; Chen, C.B.; Hoffmann, D. Metabolic ß-hydroxylation and N-oxidation of N′-nitrosonornicotine. J. Med. Chem. 1980, 23, 1175–1178. [Google Scholar] [CrossRef]
- Hecht, S.S.; Young, R. Regiospecificity in the metabolism of the homologous cyclic nitrosamines, N′-nitrosonornicotine and N′-nitrosoanabasine. Carcinogenesis 1982, 3, 1195–1199. [Google Scholar] [CrossRef]
- Yuan, J.M.; Knezevich, A.D.; Wang, R.; Gao, Y.T.; Hecht, S.S.; Stepanov, I. Urinary levels of the tobacco-specific carcinogen N′-nitrosonornicotine and its glucuronide are strongly associated with esophageal cancer risk in smokers. Carcinogenesis 2011, 32, 1366–1371. [Google Scholar] [CrossRef]
- Stepanov, I.; Yershova, K.; Carmella, S.; Upadhyaya, P.; Hecht, S.S. Levels of (S)-N′-nitrosonornicotine in U.S. tobacco products. Nicotine Tob. Res. 2013, 15, 1305–1310. [Google Scholar] [CrossRef] [Green Version]
- McIntee, E.J.; Hecht, S.S. Metabolism of N′-nitrosonornicotine enantiomers by cultured rat esophagus and in vivo in rats. Chem. Res. Toxicol. 2000, 13, 192–199. [Google Scholar] [CrossRef]
- Chen, G.; Dellinger, R.W.; Sun, D.; Spratt, T.E.; Lazarus, P. Glucuronidation of tobacco-specific nitrosamines by UGT2B10. Drug Metab. Dispos. 2008, 36, 824–830. [Google Scholar] [CrossRef] [Green Version]
- Stepanov, I.; Hecht, S.S. Tobacco-specific nitrosamines and their pyridine-N-glucuronides in the urine of smokers and smokeless tobacco users. Cancer Epidemiol. Biomark. Prev. 2005, 14, 885–891. [Google Scholar] [CrossRef] [Green Version]
- Hecht, S.S.; Chen, C.B.; Hoffmann, D. Evidence for metabolic α hydroxylation of N-nitrosopyrrolidine. Cancer Res. 1978, 38, 215–218. [Google Scholar]
- Carmella, S.G.; McIntee, E.J.; Chen, M.; Hecht, S.S. Enantiomeric composition of N′-nitrosonornicotine and N′-nitrosoanatabine in tobacco. Carcinogenesis 2000, 21, 839–843. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Carmella, S.G.; Hecht, S.S. Analysis of N′-nitrosonornicotine enantiomers in human urine by chiral stationary phase liquid chromatography-nanoelectrospray ionization-high resolution tandem mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2017, 1044–1045, 127–131. [Google Scholar] [CrossRef] [Green Version]
- Lao, Y.; Yu, N.; Kassie, F.; Villalta, P.W.; Hecht, S.S. Analysis of pyridyloxobutyl DNA adducts in F344 rats chronically treated with (R)- and (S)-N′-nitrosonornicotine. Chem. Res. Toxicol. 2007, 20, 246–256. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Wang, M.; Villalta, P.W.; Lindgren, B.R.; Lao, Y.; Hecht, S.S. Quantitation of pyridyloxobutyl DNA adducts in nasal and oral mucosa of rats treated chronically with enantiomers of N′-nitrosonornicotine. Chem. Res. Toxicol. 2009, 22, 949–956. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Balbo, S.; Wang, M.; Upadhyaya, P.; Khariwala, S.S.; Villalta, P.W.; Hecht, S.S. Quantitation of pyridyloxobutyl-DNA adducts in tissues of rats treated chronically with (R)- or (S)-N′-nitrosonornicotine (NNN) in a carcinogenicity study. Chem. Res. Toxicol. 2013, 26, 1526–1535. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Villalta, P.W.; Upadhyaya, P.; Hecht, S.S. Analysis of O6-[4-(3-pyridyl)-4-oxobut-1-yl]-2′-deoxyguanosine and other DNA adducts in rats treated with enantiomeric or racemic N′-ntrosonornicotine. Chem. Res. Toxicol. 2016, 29, 87–95. [Google Scholar] [CrossRef] [Green Version]
- Upadhyaya, P.; McIntee, E.J.; Villalta, P.W.; Hecht, S.S. Identification of adducts formed in the reaction of 5′-acetoxy-N′-nitrosonornicotine with deoxyguanosine and DNA. Chem. Res. Toxicol. 2006, 19, 426–435. [Google Scholar] [CrossRef] [Green Version]
- Zarth, A.T.; Upadhyaya, P.; Yang, J.; Hecht, S.S. DNA adduct formation from metabolic 5’-hydroxylation of the tobacco-specific carcinogen N′-nitrosonornicotine in human enzyme systems and in rats. Chem. Res. Toxicol. 2016, 29, 380–389. [Google Scholar] [CrossRef] [Green Version]
- Upadhyaya, P.; Hecht, S.S. Identification of adducts formed in the reactions of 5′-acetoxy-N′-nitrosonornicotine with deoxyadenosine, thymidine, and DNA. Chem. Res. Toxicol. 2008, 21, 2164–2171. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Carlson, E.S.; Zarth, A.T.; Upadhyaya, P.; Hecht, S.S. Investigation of 2’-deoxyadenosine-derived adducts specifically formed in rat liver and lung DNA by N′-nitrosonornicotine metabolism. Chem. Res. Toxicol. 2021, 34, 1004–1015. [Google Scholar] [CrossRef]
- Li, Y.; Hecht, S.S. Identification of an N′-nitrosonornicotine-specific deoxyadenosine adduct in rat liver and lung DNA. Chem. Res. Toxicol. 2021, 34, 992–1003. [Google Scholar] [CrossRef] [PubMed]
- Kotapati, S.; Wickramaratne, S.; Esades, A.; Boldry, E.J.; Quirk Dorr, D.; Pence, M.G.; Guengerich, F.P.; Tretyakova, N.Y. Polymerase bypass of N6-deoxyadenosine adducts derived from epoxide metabolites of 1,3-butadiene. Chem. Res. Toxicol. 2015, 28, 1496–1507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hecht, S.S. Approaches to cancer prevention based on an understanding of N-nitrosamine carcinogenesis. Proc. Soc. Exp. Biol. Med. 1997, 216, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hecht, S.S. Metabolic activation and DNA interactions of carcinogenic n-nitrosamines to which humans are commonly exposed. Int. J. Mol. Sci. 2022, 23, 4559. [Google Scholar] [CrossRef]
- Hecht, S.S.; Carmella, S.G.; Chen, M.; Koch, J.F.D.; Miller, A.T.; Murphy, S.E.; Jensen, J.A.; Zimmerman, C.L.; Hatsukami, D.K. Quantitation of urinary metabolites of a tobacco-specific lung carcinogen after smoking cessation. Cancer Res. 1999, 59, 590–596. [Google Scholar]
- Goniewicz, M.L.; Havel, C.M.; Peng, M.W.; Jacob, P., III; Dempsey, D.; Yu, L.; Zielinska-Danch, W.; Koszowski, B.; Czogala, J.; Sobczak, A.; et al. Elimination kinetics of the tobacco-specific biomarker and lung carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol. Cancer Epidemiol. Biomark. Prev. 2009, 18, 3421–3425. [Google Scholar] [CrossRef] [Green Version]
- Parsons, W.D.; Carmella, S.G.; Akerkar, S.; Bonilla, L.E.; Hecht, S.S. A metabolite of the tobacco-specific lung carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) in the urine of hospital workers exposed to environmental tobacco smoke. Cancer Epidemiol. Biomark. Prev. 1998, 7, 257–260. [Google Scholar]
- Anderson, K.E.; Carmella, S.G.; Ye, M.; Bliss, R.; Le, C.; Murphy, L.; Hecht, S.S. Metabolites of a tobacco-specific lung carcinogen in the urine of nonsmoking women exposed to environmental tobacco smoke in their homes. J. Natl. Cancer Inst. 2001, 93, 378–381. [Google Scholar] [CrossRef] [Green Version]
- Anderson, K.E.; Kliris, J.; Murphy, L.; Carmella, S.G.; Han, S.; Link, C.; Bliss, R.L.; Murphy, S.E.; Hecht, S.S. Metabolites of a tobacco-specific lung carcinogen in nonsmoking casino patrons. Cancer Epidemiol. Biomark. Prev. 2003, 12, 1544–1546. [Google Scholar]
- Xia, B.; Blount, B.C.; Guillot, T.; Brosius, C.; Li, Y.; Van Bemmel, D.M.; Kimmel, H.L.; Chang, C.M.; Borek, N.; Edwards, K.C.; et al. Tobacco-specific nitrosamines (NNAL, NNN, NAT, and NAB) exposures in the US Population Assessment of Tobacco and Health (PATH) Study Wave 1 (2013–2014). Nicotine Tob. Res. 2021, 23, 573–583. [Google Scholar] [CrossRef]
- Rostron, B.L.; Chang, C.M.; van Bemmel, D.M.; Xia, Y.; Blount, B.C. Nicotine and toxicant exposure among U.S. smokeless tobacco users: Results from 1999 to 2012 National Health and Nutrition Examination Survey data. Cancer Epidemiol. Biomark. Prev. 2015, 24, 1829–1837. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.M.; Nelson, H.H.; Carmella, S.G.; Wang, R.; Kuriger-Laber, J.; Jin, A.; Adams-Haduch, J.; Hecht, S.S.; Koh, W.P.; Murphy, S.E. CYP2A6 genetic polymorphisms and biomarkers of tobacco smoke constituents in relation to risk of lung cancer in the Singapore Chinese Health Study. Carcinogenesis 2017, 38, 411–418. [Google Scholar] [CrossRef]
- Yuan, J.M.; Nelson, H.H.; Butler, L.M.; Carmella, S.G.; Wang, R.; Kuriger-Laber, J.; Adams-Haduch, J.; Hecht, S.S.; Gao, Y.-T.; Murphy, S.E. Genetic determinants of cytochrome P450 2A6 and biomarkers of tobacco smoke exposure in relation to risk of lung cancer development in the Shanghai Cohort Study. Int. J. Cancer 2015, 138, 2161–2171. [Google Scholar] [CrossRef]













| DNA Adduct | Mutation | Cell | Ref. |
|---|---|---|---|
| POB DNA adduct | |||
| O6-POB-dGuo | G > A (~90%) and G > T (~2%) | E. coli | [117] |
| G > A (90%); G > T (~3%); other more complex types | HEK293 | [118] | |
| G > A (~75%) and G > T (~3%) | HEK293T | [119] | |
| O2-POB-Thd | T > G (37%) and T > A (12%) | E. coli | [120] |
| T > A (47%) | HEK293T | [121] | |
| T > A (~15%) | HEK293T | [119] | |
| O4-POB-Thd | T > C (~35%) | HEK293T | [119] |
| B1p(POB)B2 | Not mutagenic | E. coli | [105] |
| PHB DNA adduct | |||
| O6-PHB-dGuo | G > A (~95%) | E. coli | [117] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Hecht, S.S. Metabolism and DNA Adduct Formation of Tobacco-Specific N-Nitrosamines. Int. J. Mol. Sci. 2022, 23, 5109. https://doi.org/10.3390/ijms23095109
Li Y, Hecht SS. Metabolism and DNA Adduct Formation of Tobacco-Specific N-Nitrosamines. International Journal of Molecular Sciences. 2022; 23(9):5109. https://doi.org/10.3390/ijms23095109
Chicago/Turabian StyleLi, Yupeng, and Stephen S. Hecht. 2022. "Metabolism and DNA Adduct Formation of Tobacco-Specific N-Nitrosamines" International Journal of Molecular Sciences 23, no. 9: 5109. https://doi.org/10.3390/ijms23095109
APA StyleLi, Y., & Hecht, S. S. (2022). Metabolism and DNA Adduct Formation of Tobacco-Specific N-Nitrosamines. International Journal of Molecular Sciences, 23(9), 5109. https://doi.org/10.3390/ijms23095109