
Recurrent Germline Variant in RAD21 Predisposes Children to Lymphoblastic Leukemia or Lymphoma

 , , ,
, , ,  , , , , , , , , and add
Show full author list
, , , , , , , , and add
Show full author list
Abstract
:1. Introduction
2. Results
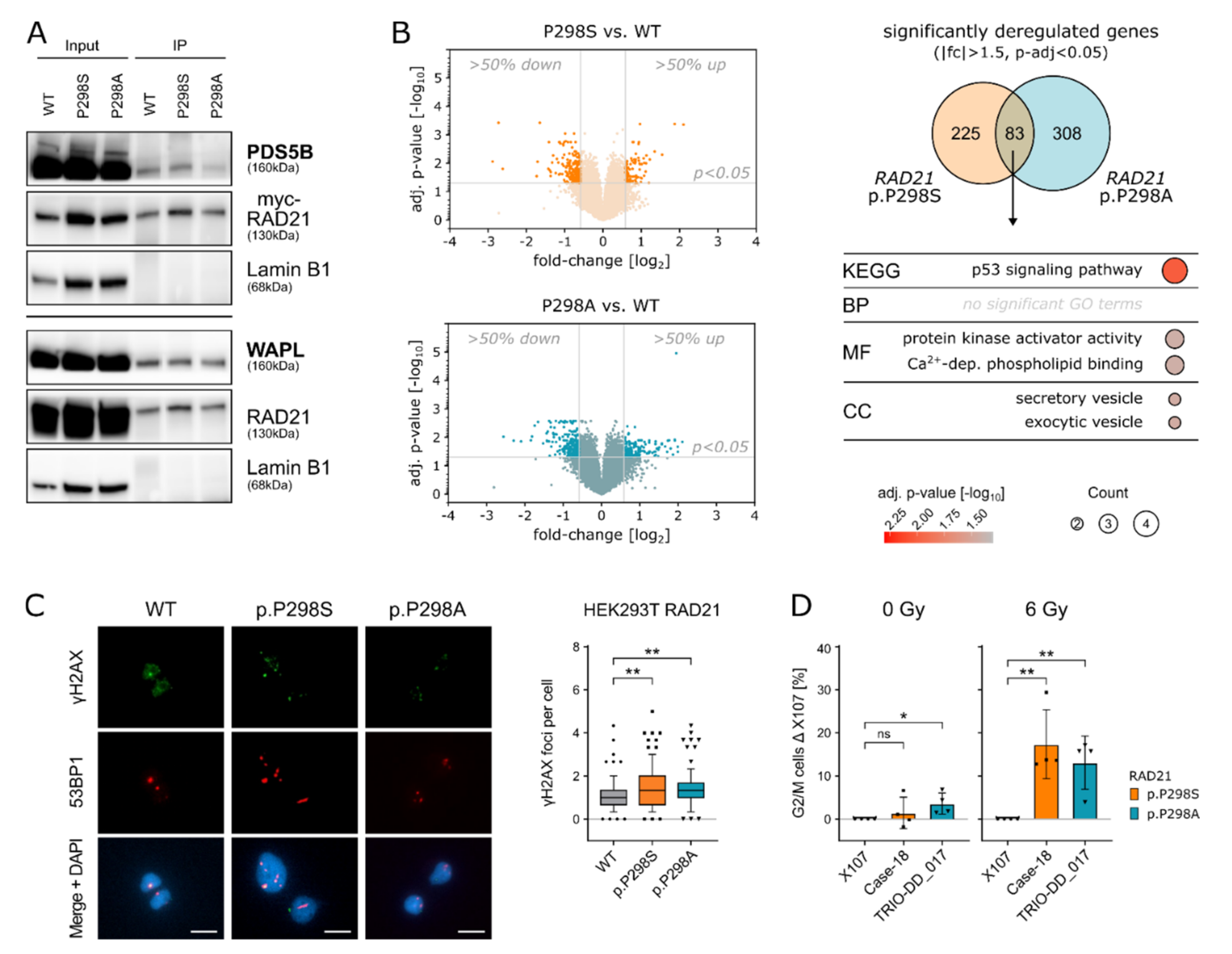
2.1. Identification of a Recurrent RAD21 Germline Alteration (p.P298S/A)
2.2. RAD21 p.P298S/A Alters Cell Cycle and DNA Damage Responses
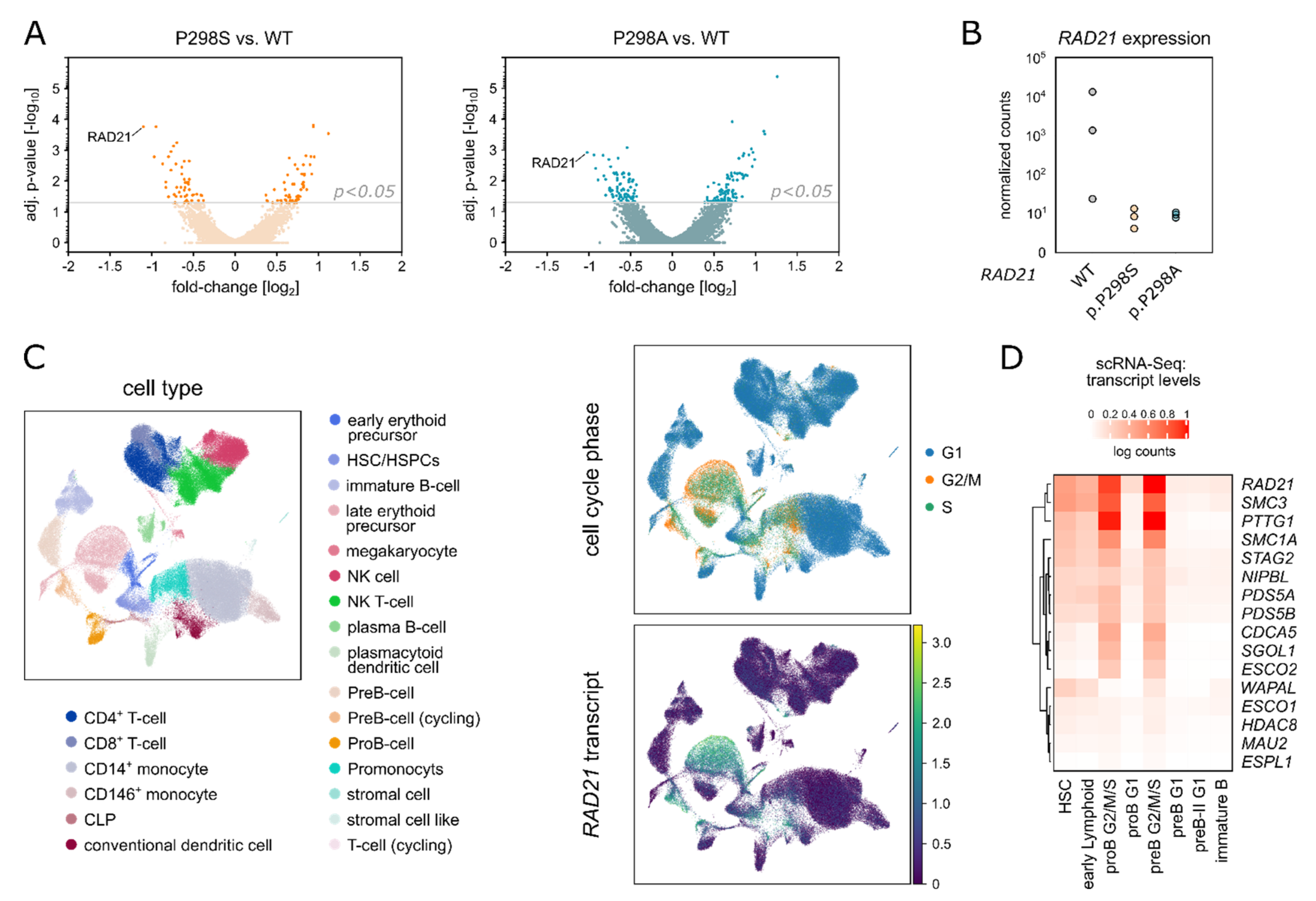
2.3. Amino Acid Replacements (S/A) at Position 298 of RAD21 Lead to Altered RAD21 Expression Levels
2.4. RAD21 p.P298S/A Is Recurrently Found in Pediatric Lymphoblastic Leukemia/Lymphoma
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Whole Exome Sequencing (WES)
4.3. Cell Culture
4.4. Cloning
4.5. HEK293T Cell Transfection
4.6. Microarray (R32-hRAD21)
4.7. Quantitative Real-Time (qRT)-PCR Analysis
4.8. GO-Term Analysis
4.9. Cell Sorting and RNA-Sequencing (pRTS-1-RAD21)
4.10. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Waldman, T. Emerging themes in cohesin cancer biology. Nat. Rev. Cancer 2020, 20, 504–515. [Google Scholar] [CrossRef]
- Haering, C.H.; Lowe, J.; Hochwagen, A.; Nasmyth, K. Molecular architecture of SMC proteins and the yeast cohesin complex. Mol. Cell 2002, 9, 773–788. [Google Scholar] [CrossRef]
- Losada, A.; Hirano, M.; Hirano, T. Identification of Xenopus SMC protein complexes required for sister chromatid cohesion. Genes Dev. 1998, 12, 1986–1997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michaelis, C.; Ciosk, R.; Nasmyth, K. Cohesins: Chromosomal proteins that prevent premature separation of sister chromatids. Cell 1997, 91, 35–45. [Google Scholar] [CrossRef] [Green Version]
- Busslinger, G.A.; Stocsits, R.R.; van der Lelij, P.; Axelsson, E.; Tedeschi, A.; Galjart, N.; Peters, J.M. Cohesin is positioned in mammalian genomes by transcription, CTCF and Wapl. Nature 2017, 544, 503–507. [Google Scholar] [CrossRef]
- Chan, K.L.; Roig, M.B.; Hu, B.; Beckouet, F.; Metson, J.; Nasmyth, K. Cohesin’s DNA exit gate is distinct from its entrance gate and is regulated by acetylation. Cell 2012, 150, 961–974. [Google Scholar] [CrossRef] [Green Version]
- Tedeschi, A.; Wutz, G.; Huet, S.; Jaritz, M.; Wuensche, A.; Schirghuber, E.; Davidson, I.F.; Tang, W.; Cisneros, D.A.; Bhaskara, V.; et al. Wapl is an essential regulator of chromatin structure and chromosome segregation. Nature 2013, 501, 564–568. [Google Scholar] [CrossRef]
- Losada, A.; Yokochi, T.; Hirano, T. Functional contribution of Pds5 to cohesin-mediated cohesion in human cells and Xenopus egg extracts. J. Cell Sci. 2005, 118, 2133–2141. [Google Scholar] [CrossRef] [Green Version]
- Guacci, V.; Koshland, D.; Strunnikov, A. A direct link between sister chromatid cohesion and chromosome condensation revealed through the analysis of MCD1 in S. cerevisiae. Cell 1997, 91, 47–57. [Google Scholar] [CrossRef] [Green Version]
- Nasmyth, K.; Peters, J.M.; Uhlmann, F. Splitting the chromosome: Cutting the ties that bind sister chromatids. Science 2000, 288, 1379–1385. [Google Scholar] [CrossRef]
- Tanaka, T.; Fuchs, J.; Loidl, J.; Nasmyth, K. Cohesin ensures bipolar attachment of microtubules to sister centromeres and resists their precocious separation. Nat. Cell Biol. 2000, 2, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Uhlmann, F.; Lottspeich, F.; Nasmyth, K. Sister-chromatid separation at anaphase onset is promoted by cleavage of the cohesin subunit Scc1. Nature 1999, 400, 37–42. [Google Scholar] [CrossRef]
- Heidinger-Pauli, J.M.; Mert, O.; Davenport, C.; Guacci, V.; Koshland, D. Systematic reduction of cohesin differentially affects chromosome segregation, condensation, and DNA repair. Curr. Biol. 2010, 20, 957–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sjogren, C.; Nasmyth, K. Sister chromatid cohesion is required for postreplicative double-strand break repair in Saccharomyces cerevisiae. Curr. Biol. 2001, 11, 991–995. [Google Scholar] [CrossRef] [Green Version]
- Parelho, V.; Hadjur, S.; Spivakov, M.; Leleu, M.; Sauer, S.; Gregson, H.C.; Jarmuz, A.; Canzonetta, C.; Webster, Z.; Nesterova, T.; et al. Cohesins functionally associate with CTCF on mammalian chromosome arms. Cell 2008, 132, 422–433. [Google Scholar] [CrossRef] [Green Version]
- Wendt, K.S.; Yoshida, K.; Itoh, T.; Bando, M.; Koch, B.; Schirghuber, E.; Tsutsumi, S.; Nagae, G.; Ishihara, K.; Mishiro, T.; et al. Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature 2008, 451, 796–801. [Google Scholar] [CrossRef]
- Canela, A.; Maman, Y.; Jung, S.; Wong, N.; Callen, E.; Day, A.; Kieffer-Kwon, K.R.; Pekowska, A.; Zhang, H.; Rao, S.S.P.; et al. Genome Organization Drives Chromosome Fragility. Cell 2017, 170, 507–521 e518. [Google Scholar] [CrossRef] [Green Version]
- Kon, A.; Shih, L.Y.; Minamino, M.; Sanada, M.; Shiraishi, Y.; Nagata, Y.; Yoshida, K.; Okuno, Y.; Bando, M.; Nakato, R.; et al. Recurrent mutations in multiple components of the cohesin complex in myeloid neoplasms. Nat. Genet. 2013, 45, 1232–1237. [Google Scholar] [CrossRef]
- Antic, Z.; Yu, J.; Bornhauser, B.C.; Lelieveld, S.H.; van der Ham, C.G.; van Reijmersdal, S.V.; Morgado, L.; Elitzur, S.; Bourquin, J.P.; Cazzaniga, G.; et al. Clonal dynamics in pediatric B-cell precursor acute lymphoblastic leukemia with very early relapse. Pediatr. Blood Cancer 2022, 69, e29361. [Google Scholar] [CrossRef]
- Moura-Castro, L.H.; Pena-Martinez, P.; Castor, A.; Galeev, R.; Larsson, J.; Jaras, M.; Yang, M.; Paulsson, K. Sister chromatid cohesion defects are associated with chromosomal copy number heterogeneity in high hyperdiploid childhood acute lymphoblastic leukemia. Genes Chromosomes Cancer 2021, 60, 410–417. [Google Scholar] [CrossRef]
- Horsfield, J.A.; Print, C.G.; Monnich, M. Diverse developmental disorders from the one ring: Distinct molecular pathways underlie the cohesinopathies. Front. Genet. 2012, 3, 171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fazio, G.; Massa, V.; Grioni, A.; Bystry, V.; Rigamonti, S.; Saitta, C.; Galbiati, M.; Rizzari, C.; Consarino, C.; Biondi, A.; et al. First evidence of a paediatric patient with Cornelia de Lange syndrome with acute lymphoblastic leukaemia. J. Clin. Pathol. 2019, 72, 558–561. [Google Scholar] [CrossRef]
- Wagener, R.; Taeubner, J.; Walter, C.; Yasin, L.; Alzoubi, D.; Bartenhagen, C.; Attarbaschi, A.; Classen, C.F.; Kontny, U.; Hauer, J.; et al. Comprehensive germline-genomic and clinical profiling in 160 unselected children and adolescents with cancer. Eur. J. Hum. Genet. 2021. [Google Scholar] [CrossRef]
- Krab, L.C.; Marcos-Alcalde, I.; Assaf, M.; Balasubramanian, M.; Andersen, J.B.; Bisgaard, A.M.; Fitzpatrick, D.R.; Gudmundsson, S.; Huisman, S.A.; Kalayci, T.; et al. Delineation of phenotypes and genotypes related to cohesin structural protein RAD21. Hum. Genet. 2020, 139, 575–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, J.; Enge, M.; Whitington, T.; Dave, K.; Liu, J.; Sur, I.; Schmierer, B.; Jolma, A.; Kivioja, T.; Taipale, M.; et al. Transcription factor binding in human cells occurs in dense clusters formed around cohesin anchor sites. Cell 2013, 154, 801–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merkenschlager, M.; Nora, E.P. CTCF and Cohesin in Genome Folding and Transcriptional Gene Regulation. Annu. Rev. Genom. Hum. Genet. 2016, 17, 17–43. [Google Scholar] [CrossRef] [PubMed]
- Zuin, J.; Dixon, J.R.; van der Reijden, M.I.; Ye, Z.; Kolovos, P.; Brouwer, R.W.; van de Corput, M.P.; van de Werken, H.J.; Knoch, T.A.; van IJcken, W.F.; et al. Cohesin and CTCF differentially affect chromatin architecture and gene expression in human cells. Proc. Natl. Acad. Sci. USA 2014, 111, 996–1001. [Google Scholar] [CrossRef] [Green Version]
- Rao, S.S.P.; Huang, S.C.; Glenn St Hilaire, B.; Engreitz, J.M.; Perez, E.M.; Kieffer-Kwon, K.R.; Sanborn, A.L.; Johnstone, S.E.; Bascom, G.D.; Bochkov, I.D.; et al. Cohesin Loss Eliminates All Loop Domains. Cell 2017, 171, 305-320.e324. [Google Scholar] [CrossRef] [Green Version]
- Uhlmann, F. SMC complexes: From DNA to chromosomes. Nat. Rev. Mol. Cell Biol. 2016, 17, 399–412. [Google Scholar] [CrossRef]
- Xu, H.; Balakrishnan, K.; Malaterre, J.; Beasley, M.; Yan, Y.; Essers, J.; Appeldoorn, E.; Tomaszewski, J.M.; Vazquez, M.; Verschoor, S.; et al. Rad21-cohesin haploinsufficiency impedes DNA repair and enhances gastrointestinal radiosensitivity in mice. PLoS ONE 2010, 5, e12112. [Google Scholar] [CrossRef]
- De Koninck, M.; Losada, A. Cohesin Mutations in Cancer. Cold Spring Harb. Perspect. Med. 2016, 6, a026476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin-Izquierdo, M.; Abaigar, M.; Hernandez-Sanchez, J.M.; Tamborero, D.; Lopez-Cadenas, F.; Ramos, F.; Lumbreras, E.; Madinaveitia-Ochoa, A.; Megido, M.; Labrador, J.; et al. Co-occurrence of cohesin complex and Ras signaling mutations during progression from myelodysplastic syndromes to secondary acute myeloid leukemia. Haematologica 2020, 106, 2215–2223. [Google Scholar] [CrossRef] [PubMed]
- Mullenders, J.; Aranda-Orgilles, B.; Lhoumaud, P.; Keller, M.; Pae, J.; Wang, K.; Kayembe, C.; Rocha, P.P.; Raviram, R.; Gong, Y.; et al. Cohesin loss alters adult hematopoietic stem cell homeostasis, leading to myeloproliferative neoplasms. J. Exp. Med. 2015, 212, 1833–1850. [Google Scholar] [CrossRef] [PubMed]
- Severin, D.M.; Leong, T.; Cassidy, B.; Elsaleh, H.; Peters, L.; Venter, D.; Southey, M.; McKay, M. Novel DNA sequence variants in the hHR21 DNA repair gene in radiosensitive cancer patients. Int. J. Radiat. Oncol. Biol. Phys. 2001, 50, 1323–1331. [Google Scholar] [CrossRef]
- Revenkova, E.; Focarelli, M.L.; Susani, L.; Paulis, M.; Bassi, M.T.; Mannini, L.; Frattini, A.; Delia, D.; Krantz, I.; Vezzoni, P.; et al. Cornelia de Lange syndrome mutations in SMC1A or SMC3 affect binding to DNA. Hum. Mol. Genet. 2009, 18, 418–427. [Google Scholar] [CrossRef] [Green Version]
- Vrouwe, M.G.; Elghalbzouri-Maghrani, E.; Meijers, M.; Schouten, P.; Godthelp, B.C.; Bhuiyan, Z.A.; Redeker, E.J.; Mannens, M.M.; Mullenders, L.H.; Pastink, A.; et al. Increased DNA damage sensitivity of Cornelia de Lange syndrome cells: Evidence for impaired recombinational repair. Hum. Mol. Genet. 2007, 16, 1478–1487. [Google Scholar] [CrossRef] [Green Version]
- Boamah, E.K.; White, D.E.; Talbott, K.E.; Arva, N.C.; Berman, D.; Tomasz, M.; Bargonetti, J. Mitomycin-DNA adducts induce p53-dependent and p53-independent cell death pathways. ACS Chem. Biol. 2007, 2, 399–407. [Google Scholar] [CrossRef]
- Sanders, J.T.; Freeman, T.F.; Xu, Y.; Golloshi, R.; Stallard, M.A.; Hill, A.M.; San Martin, R.; Balajee, A.S.; McCord, R.P. Radiation-induced DNA damage and repair effects on 3D genome organization. Nat. Commun. 2020, 11, 6178. [Google Scholar] [CrossRef]
- Cheng, H.; Zhang, N.; Pati, D. Cohesin subunit RAD21: From biology to disease. Gene 2020, 758, 144966. [Google Scholar] [CrossRef]
- Mazumdar, C.; Shen, Y.; Xavy, S.; Zhao, F.; Reinisch, A.; Li, R.; Corces, M.R.; Flynn, R.A.; Buenrostro, J.D.; Chan, S.M.; et al. Leukemia-Associated Cohesin Mutants Dominantly Enforce Stem Cell Programs and Impair Human Hematopoietic Progenitor Differentiation. Cell Stem Cell 2015, 17, 675–688. [Google Scholar] [CrossRef] [Green Version]
- Degner, S.C.; Verma-Gaur, J.; Wong, T.P.; Bossen, C.; Iverson, G.M.; Torkamani, A.; Vettermann, C.; Lin, Y.C.; Ju, Z.; Schulz, D.; et al. CCCTC-binding factor (CTCF) and cohesin influence the genomic architecture of the Igh locus and antisense transcription in pro-B cells. Proc. Natl. Acad. Sci. USA 2011, 108, 9566–9571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seitan, V.C.; Hao, B.; Tachibana-Konwalski, K.; Lavagnolli, T.; Mira-Bontenbal, H.; Brown, K.E.; Teng, G.; Carroll, T.; Terry, A.; Horan, K.; et al. A role for cohesin in T-cell-receptor rearrangement and thymocyte differentiation. Nature 2011, 476, 467–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panigrahi, A.K.; Pati, D. Road to the crossroads of life and death: Linking sister chromatid cohesion and separation to aneuploidy, apoptosis and cancer. Crit. Rev. Oncol. Hematol. 2009, 72, 181–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivas, M.A.; Meydan, C.; Chin, C.R.; Challman, M.F.; Kim, D.; Bhinder, B.; Kloetgen, A.; Viny, A.D.; Teater, M.R.; McNally, D.R.; et al. Smc3 dosage regulates B cell transit through germinal centers and restricts their malignant transformation. Nat. Immunol. 2021, 22, 240–253. [Google Scholar] [CrossRef] [PubMed]
- Padella, A.; Ghelli Luserna Di Rora, A.; Marconi, G.; Ghetti, M.; Martinelli, G.; Simonetti, G. Targeting PARP proteins in acute leukemia: DNA damage response inhibition and therapeutic strategies. J. Hematol. Oncol. 2022, 15, 10. [Google Scholar] [CrossRef] [PubMed]
- Worst, B.C.; van Tilburg, C.M.; Balasubramanian, G.P.; Fiesel, P.; Witt, R.; Freitag, A.; Boudalil, M.; Previti, C.; Wolf, S.; Schmidt, S.; et al. Next-generation personalised medicine for high-risk paediatric cancer patients—The INFORM pilot study. Eur. J. Cancer 2016, 65, 91–101. [Google Scholar] [CrossRef] [Green Version]
- Horak, P.; Klink, B.; Heining, C.; Groschel, S.; Hutter, B.; Frohlich, M.; Uhrig, S.; Hubschmann, D.; Schlesner, M.; Eils, R.; et al. Precision oncology based on omics data: The NCT Heidelberg experience. Int. J. Cancer 2017, 141, 877–886. [Google Scholar] [CrossRef] [Green Version]
- Linka, R.M.; Risse, S.L.; Bienemann, K.; Werner, M.; Linka, Y.; Krux, F.; Synaeve, C.; Deenen, R.; Ginzel, S.; Dvorsky, R.; et al. Loss-of-function mutations within the IL-2 inducible kinase ITK in patients with EBV-associated lymphoproliferative diseases. Leukemia 2012, 26, 963–971. [Google Scholar] [CrossRef]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef]
- Wu, T.D.; Nacu, S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics 2010, 26, 873–881. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ignatiadis, N.; Huber, W. Covariate powered cross-weighted multiple testing. J. R. Stat. Soc. Ser. B 2021, 83, 720–751. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| TRIO-DD | TRIO-D | R-ALL | INFORM | MASTER | ||
|---|---|---|---|---|---|---|
| Cohort | Number of patients | n = 60 | n = 158 | n = 150 | n = 114 | n = 2300 |
| pediatric | pediatric | pediatric | pediatric | adult | ||
| % Hematopoietic malignancies | 38.3% | 51.3% | 100% | 100% | 3.7% | |
| Inclusion criteria | Primary diagnosis | Primary diagnosis | IntReALL SR | Therapy refractory | Young adults < 51 y | |
| Patient | Sex | Male | Male | Male | - | Female |
| Age | 2 | 13 | 12 | - | 53 | |
| Tumor | pB-LBL | T-ALL | BCP-ALL | - | MPNST | |
| Risk group | SR | HR | SR | - | N/A | |
| RAD21 variant p.P298 | Protein exchange | ENSP00000297338.2 p.P298A | ENSP00000297338.2 p.P298S | ENSP00000297338.2 p.P298A | - | ENSP00000297338.2 p.P298A |
| Base exchange | ENST00000297338.2 c.892 C>G | ENST00000297338.2 c.892 C>T | ENST00000297338.2 c.892 C>G | - | ENST00000297338.2 c.892 C>G | |
| SNP ID | rs148308569 | rs148308569 | rs148308569 | - | rs148308569 | |
| MAF GnomAD | 10−5 | 10−6 | 10−5 | - | 10−5 | |
| MAF within the cohort | 1.7 × 10−2 | 6.5 × 10−3 | 6.7 × 10−3 | - | 4.3 × 10−4 | |
| Genetic history | Genetic counselling * | + | + | unknown | - | unknown |
| Family history | + | + | unknown | - | unknown | |
| 2nd Hit | Somatic Mutations | unknown | KRAS p.Q61R | KRAS p.G12C | - | PTCH2 p.A68V |
| Name | Sequence (5′ → 3′) |
|---|---|
| hRad21_MluI_F | GGCGCacgcgtgccaccATGTTCTACGCACATTTTGTTCTC |
| hRad21_SpeI_R | CCTCGactagtTATAATATGGAACCTTGGTCCAGGTGTTGC |
| hRad21_SwaI_F | GGCGCATTTAAATCATGTTCTACGCAC |
| hRad21_XhoI_R | CCTCGCTCGAGTCCATATAATATGGAACC |
| hRad21_P298S_F | GATCAAACAACACTTGTTtCAAATGAGGAAGAAGCATTTGC |
| hRad21_P298S_R | GCAAATGCTTCTTCCTCATTTGaAACAAGTGTTGTTTGATC |
| hRad21_P298A_F | GATCAAACAACACTTGTTgCAAATGAGGAAGAAGCATTTGC |
| hRad21_P298A_R | GCAAATGCTTCTTCCTCATTTGcAACAAGTGTTGTTTGATC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schedel, A.; Friedrich, U.A.; Morcos, M.N.F.; Wagener, R.; Mehtonen, J.; Watrin, T.; Saitta, C.; Brozou, T.; Michler, P.; Walter, C.; et al. Recurrent Germline Variant in RAD21 Predisposes Children to Lymphoblastic Leukemia or Lymphoma. Int. J. Mol. Sci. 2022, 23, 5174. https://doi.org/10.3390/ijms23095174
Schedel A, Friedrich UA, Morcos MNF, Wagener R, Mehtonen J, Watrin T, Saitta C, Brozou T, Michler P, Walter C, et al. Recurrent Germline Variant in RAD21 Predisposes Children to Lymphoblastic Leukemia or Lymphoma. International Journal of Molecular Sciences. 2022; 23(9):5174. https://doi.org/10.3390/ijms23095174
Chicago/Turabian StyleSchedel, Anne, Ulrike Anne Friedrich, Mina N. F. Morcos, Rabea Wagener, Juha Mehtonen, Titus Watrin, Claudia Saitta, Triantafyllia Brozou, Pia Michler, Carolin Walter, and et al. 2022. "Recurrent Germline Variant in RAD21 Predisposes Children to Lymphoblastic Leukemia or Lymphoma" International Journal of Molecular Sciences 23, no. 9: 5174. https://doi.org/10.3390/ijms23095174
APA StyleSchedel, A., Friedrich, U. A., Morcos, M. N. F., Wagener, R., Mehtonen, J., Watrin, T., Saitta, C., Brozou, T., Michler, P., Walter, C., Försti, A., Baksi, A., Menzel, M., Horak, P., Paramasivam, N., Fazio, G., Autry, R. J., Fröhling, S., Suttorp, M., ... Auer, F. (2022). Recurrent Germline Variant in RAD21 Predisposes Children to Lymphoblastic Leukemia or Lymphoma. International Journal of Molecular Sciences, 23(9), 5174. https://doi.org/10.3390/ijms23095174